


Abstract
Proteins containing heme, iron(protoporphyrin IX) and its variants, continue to be one of the most-studied classes of biomolecules due to their diverse range of biological functions. The literature is abundant with reports of structural and functional characterization of individual heme proteins which demonstrate that heme protein reduction potential values, Em, span the range from –550 mV to +450 mV versus SHE. In order to unite these data for the purposes of global analysis, a new web-based resource of heme protein structure–function relationships is presented: the Heme Protein Database (HPD). This database is the first of its kind to combine heme protein structural classifications including protein fold, heme type and heme axial ligands, with heme protein reduction potential values in a web-searchable format. The HPD is located at http://heme.chem.columbia.edu/heme.php. The data illustrate that heme protein Em values are modulated over a 300 mV range by the type of global protein fold, a 600 mV range by the type of porphyrin and an 800 mV range by the axial ligands. Thus, the 1 V range observed in heme protein reduction potential values in biological systems arises from subtle combinations of these various factors.
INTRODUCTION
Since its inception in 1971, the Protein Data Bank (PDB) has been the sole public warehouse of biomolecular structures and has proven invaluable to the biochemical community (1). As the PDB has grown in size and complexity, several secondary databases have arisen from reduced subsets of protein structures (2). The secondary databases that classify proteins in terms of protein fold, the CATH Protein Structure Classification Database (CATH), and evolutionary and structural relationships, the Structural Classification of Proteins (SCOP), are two of the most widely accessed web-based protein structural analysis tools (3–5). There are also secondary databases based on a particular protein function, the Protein Kinase Resource, or protein type, the directory of P450-containing systems, which provide a more focused view of the structure–function relationships relevant to a specific biochemical function (6,7).
Proteins containing metal ions are well represented in the PDB and two secondary databases for metalloproteins have been previously assembled to facilitate research in bioinorganic chemistry. The Prosthetic Groups and Metal Ions in Protein Active Sites Database Version 2.0, the PROMISE Database, contains a survey of metalloproteins including the chemical structures of their metal cofactors (8). The list of cofactors in PROMISE includes chlorophylls, hemes, mono- and di-nuclear iron, iron–sulfur clusters, copper and molybdopterin. Each metalloprotein in the PROMISE database is categorized by function as well as by the number and type of metal–ion ligands. The PROMISE Database was an excellent resource, unfortunately it was last modified on 1 March 1999, has been unsupported since 2002, and lacks protein structural information beyond the primary coordination sphere of the bound metals. Another metal-centered secondary database is the Metalloprotein Database and Browser (MDB) that provides detailed structural information on the primary coordination sphere of bound metal ions (9). It is a searchable database of metal-site structural parameters that is an excellent resource for metal ion geometries including metal–ligand bond distances and angles. However, the MDB has not been updated since 2002 and is highly metal-site biased; there is no information on structure–function relationships between the protein and its metal cofactor.
In this contribution, we describe the construction and preliminary analysis of a secondary database devoted to heme proteins that make up nearly 5% of all the structurally characterized proteins in the PDB. The Heme Protein Database (HPD) is constructed to couple the structural information of a unique set of non-redundant heme proteins from the PDB with their electrochemical function collected from the primary literature. The HPD provides for a global analysis of the structural factors that influence the midpoint reduction potential of wild-type heme proteins near physiological pH and can be accessed at http://heme.chem.columbia.edu/heme.php. The data evince that heme reduction potentials in structurally characterized heme proteins span a 1 V range from −550 mV to +450 mV versus SHE. Furthermore, the data show that the distribution of redox activities in these proteins varies by protein secondary structure, heme coordination motif and heme porphyrin architecture.
RESULTS
Assembly of the Heme Protein Database
To facilitate research efforts into heme protein structure and function, we have constructed the HPD from a list of all iron porphyrin-containing proteins contained in the PDB as of 1 May 2007. As shown in Figure 1, the HPD collects data on heme protein structure and function relationships into a single MySQL database. From the set of 1414 structurally characterized heme proteins in the PDB, a non-redundant set of entries was generated by eliminating duplicates due to the existence of mutant proteins, proteins with alternate substrates/exogenous ligands bound, and protein–protein complexes in which the individual partners were already included in the database. For a given heme protein structure, the highest resolution structure from each species of origin was chosen for inclusion in the non-redundant set as the representative example. Ultimately, a total of 738 hemes from 268 heme proteins comprise the non-redundant set used to construct the HPD. This non-redundant set was designed to analyze structural effects on wild-type heme protein reduction potentials near physiological pH using a PHP-based MySQL database search engine.
Figure 1.

The Heme Protein Databse web site.
From the structural biology perspective, each heme protein in the HPD is described by the protein name, the species of origin, the PDB ID code and its heme-binding domain CATH classification. The CATH system was selected to classify each heme protein domain structure in the HPD because it separates proteins into distinct domains with individual CATH codes (4). Thus, this system yields a more accurate assessment of the protein structure local to the heme cofactor. Of the 738 hemes in the HPD, 638 reside in 241 protein domains that are classified by CATH Version 3.0.0.5. The statistics of the HPD entries are given in Table 1.
Table 1.
Heme Protein Database statistics
| Proteins and domains | |
| Number of heme proteins in PDB | 1414 |
| Number of hemes in PDB | 3013 |
| Number of heme proteins in HPD | 268 |
| Number of hemes in HPD | 738 |
| Number of heme proteins in CATH | 241 |
| Number of hemes in CATH domains | 638 |
| Reduction potentials | |
| Number of reduction potentials | 153 |
| Most negative reduction potential | −550 mV |
| Most positive reduction potential | 450 mV |
| Dissociation constants | |
| Number of dissociation constants | 10 |
| Weakest dissociation constant | 2.5 μM |
| Tightest dissociation constant | 300 pM |
| Heme type abundance | |
| Heme a | 13.6% |
| Heme b | 53.5% |
| Heme c | 40.9% |
| Heme d1 | 1.9% |
From the bioinorganic chemistry perspective, the structure of each heme protein in the HPD is described by the heme type, the number and types of axial ligand(s) to the heme iron, and iron–ligand bond distances. Iron–axial ligand distance(s) were determined using the Ligand-Protein Contacts software (10). This structural information was augmented in the HPD with two key functional properties of heme proteins from the literature: the Fe(III)/Fe(II) reduction potential, Em value, and the heme protein dissociation constant, Kd value. The Em value is critical to the electrochemical function of heme proteins involved in electron transfer and redox catalysis, e.g. the cytochromes, peroxidases, catalases, monooxygenases and oxidases (11–17). An extensive search of the primary literature uncovered a total of 154 reduction potential values for structurally characterized heme proteins in buffered aqueous solution between pH 7.0 and 8.0. As important as the midpoint reduction potential value, the value of the heme protein dissociation constant, Kd value, is critical to heme protein structure, stability and function as heme loss typically leads to loss of biochemical function, although it can lead to catalytic activation as observed in soluble guanylate cyclase (18). An extensive search of the literature only uncovered 10 Kd values for the set of structurally characterized heme proteins in the HPD.
In toto, the HPD contains the following information for each entry: protein name, species of origin, PDB ID code, heme type, heme iron coordination number, identities of the ligands to the heme iron, metal–ligand bond distances, Em value, Kd value and CATH classification codes. In addition, links are provided to the PROMISE database when additional information is available. Finally, a MOLMOL representation of each heme protein is provided for download as a JPEG file (19). The HPD can be searched by any combination of these parameters using the search engines found on the web site. The Search By PDB ID function allows for the data on an individual heme protein to be retrieved using the PDB ID code. If the PDB ID code is not known, the detailed search page provides a search for information on individual heme proteins, e.g. the Em value of horse heart cytochrome c. The global view function on the HPD homepage provides for rapid searching for sets of heme proteins, e.g. the Em range of all α-helical heme proteins containing heme b.
DISCUSSION
The HPD is a web-based research tool for heme proteins that couples functional biochemical properties with the available structural data on heme proteins. The combination of these two datasets provides for insight into the global structure–function relationships of heme proteins. The database was constructed to aid researchers in the fields of natural heme protein biochemistry as well as de novo metalloprotein design (20,21). The data evince that heme reduction potentials are modulated by a multitude of factors including, but not limited to, the type of porphyrin (shown in Figure 2), the axial ligand and the global protein fold as shown in Table 2. In toto, these structural factors act in concert to tune the reduction potentials of natural heme proteins over a 1 V range from –550 mV to +450 mV versus SHE.
Figure 2.

The chemical structures of the various hemes found in the Heme Protein Database. The basic structure at left is elaborated with the side chains shown in the box to yield a variety of hemes, in addition the structures of heme d1 and siroheme are shown.
Table 2.
Heme Protein Database midpoint potential statisticsa
| Entries | Range | Average | Median | |
|---|---|---|---|---|
| Heme domain CATH class | ||||
| All (1–4) | 153 | −550 to +450 | −14.6 | −6 |
| Mainly alpha (1) | 99 | −412 to +450 | 44.6 | 47 |
| Mainly beta (2) | 10 | −303 to +373 | 9.1 | 32.5 |
| Mixed alpha/beta (3) | 32 | −550 to +68 | −207.5 | −237.5 |
| Low 2° structure (4) | 2 | −172 to −25 | −98.5 | −98.5 |
| Heme type | ||||
| Heme a | 4 | +221 to +290 | 254.25 | 253 |
| Heme b | 42 | −550 to +250 | −46.1 | −4.5 |
| Heme c | 102 | −412 to +450 | −12.8 | −31 |
| Heme d1 | 1 | 287 | 287 | 287 |
| Heme o | 1 | 160 | 160 | 160 |
| siroheme | 1 | −340 | −340 | −340 |
| P460 | 1 | −260 | −260 | −260 |
| Fe(coproporphyrin) | 1 | 140 | 140 | 140 |
| Heme coordination motif | ||||
| All | 153 | −550 to +450 | −20.4 | −10 |
| His/His | 69 | −412 to +380 | −132.2 | −177 |
| His/Met | 36 | −60 to +450 | 242.1 | 263.5 |
| His/Tyr | 1 | −550 | −550 | −550 |
| His/NTerm | 3 | −260 to +373 | 156 | 355 |
| His/OH | 1 | 287 | 287 | 287 |
| His/Asn | 1 | −22 | −22 | −22 |
| Met/Met | 1 | 140 | 140 | 140 |
| Cys/– | 9 | −340 to −6 | −244.6 | −295 |
| His/– | 28 | −306 to +290 | 10 | 49 |
| Lys/– | 1 | −107 | −107 | −107 |
| Tyr/– | 1 | −303 | −303 | −303 |
| −/– | 1 | 100 | 100 | 100 |
aAll potentials reported in mV versus SHE.
As shown in Figure 3, the reduction potential of a heme bound to a protein, Em(bound), can be related to the reduction potential of the free heme, Em(free), by either the dissociation constants of the heme protein or the stability of the global fold towards unfolding in the two oxidation states. As shown mathematically below, the Em(bound) value is a function of the Em(free) value and the ratio of the ferric and ferrous heme dissociation constants, KdFe(III)/KdFe(II), or the difference in the stability of the global protein fold towards unfolding, ▵Gu, between the two oxidation states, ▵GuFe(II) – ▵GuFe(III) (22,23).
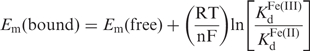 |
(1) |
| (2) |
As such, structural factors modulate the reduction potential of the bound heme by stabilizing or destabilizing the ferric and ferrous heme proteins. This Em modulation is reflected in changes in the ferric and ferrous heme protein Kd values as well as the corresponding global protein stabilities in the two oxidation states. A direct consequence of the observed 1 V range of reduction potentials in natural heme proteins is that the ratios of ferrous to ferric heme protein dissociation constants vary by 17 orders of magnitude and the difference between the natural values of (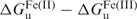 ) is 23 kcal/mol. Determining either the heme protein dissociation constants or the global protein stabilities for both oxidation states along with heme reduction potentials has proven invaluable in analyzing the factors that modulate Em values (23). In addition, delineation of the factors that influence the absolute values of the ferric and ferrous heme dissociation constants is critical to the rational design of heme proteins in both oxidation states (24,25).
) is 23 kcal/mol. Determining either the heme protein dissociation constants or the global protein stabilities for both oxidation states along with heme reduction potentials has proven invaluable in analyzing the factors that modulate Em values (23). In addition, delineation of the factors that influence the absolute values of the ferric and ferrous heme dissociation constants is critical to the rational design of heme proteins in both oxidation states (24,25).
Figure 3.

Thermodynamic cycles relating heme protein electrochemistry to (A) the heme protein dissociation constants and (B) the global protein stabilities in the ferric and ferrous oxidation states.
The literature has seen a vigorous debate on the role of heme solvent exposure in modulating heme reduction potential values. Early work from Kassner showed that moving a solvent-exposed heme ligand set into a protein core could shift the Em value by +300 mV (26). The reason for this shift was suggested to be that placing the heme in a low-polarity hydrophobic core destabilizes the formally charged ferric iron porphyrin core, [Fe(III)(porphyrin2−)]1+, relative to the formally neutral ferrous heme, [Fe(II)(porphyrin2−)]0 (26). Later work by Stellwagen supported this view by showing that heme solvent exposure, or conversely heme burial, could be correlated with heme reduction potentials, despite significant changes in the heme axial ligands (27). More recently, Tezcan et al. have shown a similar correlation for a series of c-type cytochromes after correction for the effects of different axial ligands (28).
While many heme protein papers discuss the role of heme solvent exposure on heme reduction potentials, one of the few papers in the literature that addresses the issue of protein fold and reduction potential is that of Gunner and co-workers (29). Using multiconformation continuum electrostatics calculations to determine the factors involved in the reduction potentials of cytochromes in four types of folds, they demonstrate that these protein folds raise the reduction potential of the heme–axial ligand complex by up to +260 mV compared to the fully solvated complex. However, they do not find a direct correlation between reduction potential and heme surface exposure. Overall, the data indicate that different folds influence the heme reduction potential in ways unique to that particular fold. In the case of the HPD, a cursory comparison of the solvent exposed heme b–(imidazole)2 complex Em value, –235 mV versus SHE, to those observed for natural bis-His b-type heme proteins, from –130 mV to +250 mV, appears to support the basic conclusion of Kassner (26). However, the range of values for bis-His c-type heme proteins in the HPD, from –412 mV to +380 mV, reveals that polarity/solvent exposure is not the only factor that sets heme reduction potential values. Thus, contributions from heme type and axial ligands also appear to influence Em values.
The porphyrin architecture that contains the iron is also a key factor in modulating the reduction potential. The data in the HPD indicate that hemes a, d1, o or Fe(coproporphyrin), generally have more positive reduction potentials than heme b. For hemes a and d1 the presence of electron-withdrawing substituents on the heme periphery result in the positive shifts in the Em values as observed in small-molecule complexes (30). This effect may be due to destabilization of the ferric state without altering the ferrous state as we have recently shown by comparing the absolute ferric and ferrous Kd values of heme a and Fe(diacetyldeuteroporphyrin IX) for a de novo designed bis-His heme protein (31). For hemes a and o, the burial of the porphyrin within a membrane protein may also be in part responsible for the positive shift in the reduction potential values. The data in the HPD also demonstrate that the reduction potentials between b-type and c-type hemes are quite similar because the electronic effects of vinyl and thioether groups are similar. This conclusion is also reflected in mutagenesis studies where b-type and c-type cytochromes are interconverted by incorporation or removal of the cysteine thioether bonds (32–35). Generally, these conversions result in a minimal change in heme reduction potential but significant changes in global protein stability. In other words, the ratio of the ferric and ferrous dissociation constants remains constant, but the individual dissociation constant values change significantly. At this point, more research is needed to clarify the structural and functional roles of the different types of iron porphyrins found in biochemical systems to address the fundamental question of why nature utilizes a range of heme structures when nearly all biological functions can be accomplished with b-type heme proteins (36).
Classifying the heme reduction potentials by coordination motif reveals a general trend in the heme protein reduction potential values with His/Met > His/OH > Met/Met > His/N-terminus > –/– (no axial ligands) > His/– > His/Asn > Lys/– > His/His > Cys/– > Tyr/– > His/Tyr. Thus, the heme with the most positive midpoint reduction potential value, +450 versus SHE, is the His/Met heme c from diheme cytochrome c peroxidase with the most negative Em observed in the His/Tyr coordinated heme b of the hemophore HasA (37). This trend follows the expectations of Hard-Soft Acid-Base theory coupled with Equation (1) which predicts that hard bases, e.g. tyrosinate, cysteinate and histidine, preferentially stabilize ferric heme that leads to negative shifts in reduction potentials. Conversely, soft ligands such as methionine stabilize ferrous heme relative to ferric heme which results in positive shifts in Em values. In other words, ligand motifs that favor binding to Fe(III) heme generally result in more negative reduction potential values than ligand motifs that favor binding to Fe(II) heme. The 375 mV difference in the average Em values of bis-His and His-Met ligated hemes is significantly larger than the +200 mV reduction potential shift commonly observed in mutagenesis studies of natural heme proteins where a bis-His heme protein is converted into His-Met protein. This is not surprising since the HPD analysis also includes contributions from differences in the type of heme and global protein fold. This is also the reason that examples can be found of individual proteins that do not appear to obey the predictions of Hard-Soft Acid-Base theory, e.g. the +380 mV value for a bis-His c-type heme more positive than the –60 mV value for a His/Met c-type heme in the same protein, the photosynthetic reaction center protein from Blastochloris viridis (38). In addition, an analysis of the amino acids ligands bound to heme shows several unique coordination motifs, mono-Lys (39), and His/Asn (40), as well as the notable absence of aspartate coordination to heme iron.
CONCLUSION
In conclusion, a web-based resource for wild-type heme protein structure–function analysis has been constructed that couples available structural data with electrochemical data on a non-redundant set of heme proteins. Designed as a resource for researchers investigating natural and designed heme proteins, the HPD provides a link between heme protein structure and function that provides greater insight into this long-studied cofactor. The HPD illustrates the influence of various structural factors on the reduction potential of heme bound to protein scaffolds on a global scale and highlights areas where more data is needed to fully understand structure–function relationships in heme proteins.
ACKNOWLEDGEMENTS
The authors wish to thank Dr Jinyou Zhuang, Ms Jennifer Amoroso and Mr Koon-Cheung Ching for technical assistance in assembling the HPD. B.R.G. is a Camille Dreyfus Teacher-Scholar. This work is supported by a grant from the American Heart Association (0755879T). The Open Access publication charges were waived by Oxford University Press.
Conflict of interest statement. None declared.
REFERENCES
- 1.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weissig H, Bourne PE. Protein structure resources. Acta Crystallogr. D Biol. Crystallogr. 2002;58:908–915. doi: 10.1107/s0907444902003906. [DOI] [PubMed] [Google Scholar]
- 3.Orengo CA, Thornton JM. Protein families and their evolution – a structural perspective. Annu. Rev. Biochem. 2005;74:867–900. doi: 10.1146/annurev.biochem.74.082803.133029. [DOI] [PubMed] [Google Scholar]
- 4.Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH – a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 5.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP – a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 6.Smith CM, Shindyalov IN, Veretnik S, Gribskov M, Taylor SS, TenEyck LF, Bourne PE. The protein kinase resource. TIBS. 1997;22:444–446. doi: 10.1016/s0968-0004(97)01131-6. [DOI] [PubMed] [Google Scholar]
- 7.Degtyarenko KN. Structural domains of P450-containing monooxygenase systems. Protein Eng. 1995;8:737–747. doi: 10.1093/protein/8.8.737. [DOI] [PubMed] [Google Scholar]
- 8.Degtyarenko KN, North ACT, Findlay JBC. PROMISE: a database of bioinorganic motifs. Nucleic Acids Res. 1999;27:233–236. doi: 10.1093/nar/27.1.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castagnetto JM, Hennessy SW, Roberts VA, Getzoff ED, Tainer JA, Pique ME. MDB: the metalloprotein database and browser at the Scripps Research Institute. Nucleic Acids Res. 2002;30:379–382. doi: 10.1093/nar/30.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. Automated analysis of interatomic contacts in proteins. Bioinformatics. 1999;15:327–332. doi: 10.1093/bioinformatics/15.4.327. [DOI] [PubMed] [Google Scholar]
- 11.Keilin D. On cytochrome, a respiratory pigment, common in animals, yeast and higher plants. Proc. R. Soc. Lond. B. 1925;98:312–339. [Google Scholar]
- 12.Scott RA, Mauk AG. (1996) Cytochrome c - A Multidisciplinary Approach. University Science Books, Sausalito, p. 738. [Google Scholar]
- 13.Sono M, Roach MP, Coulter ED, Dawson JH. Heme-containing oxygenases. Chem. Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 14.Yoshikawa S. Cytochrome. c oxidase. Adv. Protein Chem. 2002;60:341–395. doi: 10.1016/s0065-3233(02)60057-9. [DOI] [PubMed] [Google Scholar]
- 15.Dunford HB. (1999) Heme Peroxidases. John Wiley, New York, p. 507. [Google Scholar]
- 16.Maehly AC. Plant peroxidase. Methods Enzymol. 1955;2:801–813. [Google Scholar]
- 17.Rodgers KR. Heme-based sensors in biological systems. Curr. Opin. Chem. Biol. 1999;3:158–167. doi: 10.1016/S1367-5931(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 18.Denninger JW, Marletta MA. Guanylate cyclase and the •NO/cGMP signaling pathway. Biochim. Biophys. Acta. 1999;1411:334–350. doi: 10.1016/s0005-2728(99)00024-9. [DOI] [PubMed] [Google Scholar]
- 19.Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 20.Chapman SK, Daff S, Munro AW. Heme: the most versatile redox centre in biology? Struct. Bond. 1997;88:39–70. [Google Scholar]
- 21.Reedy CJ, Gibney BR. Heme protein assemblies. Chem. Rev. 2004;104:617–650. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- 22.Nesset MJM, Shokhirev NV, Enemark PD, Jacobson SE, Walker FA. Models of the cytochromes. Redox properties and thermodynamic stabilities of complexes of “hindered” iron(III) and iron(II) tetraphenylporphyrinates with substituted pyridines and imidazoles. Inorg. Chem. 1996;35:5188–5200. [Google Scholar]
- 23.Telford JR, Wittung-Stafshede P, Gray HB, Winkler JR. Protein folding triggered by electron transfer. Acc. Chem. Res. 1998;31:755–763. [Google Scholar]
- 24.Reedy CJ, Kennedy ML, Gibney BR. Thermodynamic characterization of ferric and ferrous haem binding to a designed four-α-helix protein. Chem. Commun. 2003:570–571. doi: 10.1039/b211488g. [DOI] [PubMed] [Google Scholar]
- 25.Reddi AR, Reedy CJ, Mui S, Gibney BR. Thermodynamic investigation into the mechanisms of proton-coupled electron transfer in heme protein maquettes. Biochemistry. 2007;46:291–305. doi: 10.1021/bi061607g. [DOI] [PubMed] [Google Scholar]
- 26.Kassner RJ. A theoretical model for the effects of local nonpolar heme environments on the redox potentials in cytochromes. J. Am. Chem. Soc. 1973;95:2675–2677. doi: 10.1021/ja00789a044. [DOI] [PubMed] [Google Scholar]
- 27.Stellwagen E. Haem exposure as the determinate of oxidation-reduction potential of haem proteins. Nature. 1978;275:73–74. doi: 10.1038/275073a0. [DOI] [PubMed] [Google Scholar]
- 28.Tezcan FA, Winkler JR, Gray HB. Effects of ligation and folding on reduction potentials of heme proteins. J. Am. Chem. Soc. 1998;120:13383–13388. [Google Scholar]
- 29.Mao JJ, Hauser K, Gunner MR. How cytochromes with different folds control heme redox potentials. Biochemistry. 2003;42:9829–9840. doi: 10.1021/bi027288k. [DOI] [PubMed] [Google Scholar]
- 30.Vanderkooi G, Stotz E. Oxidation-reduction potentials of heme a hemochromes. J. Biol. Chem. 1996;241:3316–3323. [PubMed] [Google Scholar]
- 31.Zhuang J, Amoroso JH, Kinloch R, Dawson JH, Baldwin MJ, Gibney BR. Evaluation of electron-withdrawing group effects on heme binding in designed proteins: implications for heme a in cytochrome c oxidase. Inorg. Chem. 2006;45:4685–4694. doi: 10.1021/ic060072c. [DOI] [PubMed] [Google Scholar]
- 32.Barker PD, Ferrer JC, Mylrajan M, Loehr TM, Feng R, Konishi Y, Funk WD, MacGillivray RT, Mauk AG. Transmutation of a heme protein. Proc. Natl Acad. Sci. USA. 1993;90:6542–6546. doi: 10.1073/pnas.90.14.6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomlinson EJ, Ferguson SJ. Conversion of a c type cytochrome to b type that spontaneously forms in vitro from apo protein and heme: implications for c type cytochrome biogenesis and folding. Proc. Natl Acad. Sci. USA. 2000;97:5156–5160. doi: 10.1073/pnas.090089397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Faraone-Mennella J, Tezcan FA, Gray HB, Winkler JR. Stability and folding kinetics of structurally characterized cytochrome c-b562. Biochemistry. 2006;45:10504–10511. doi: 10.1021/bi060242x. [DOI] [PubMed] [Google Scholar]
- 35.Cowley AB, Lukat-Rodgers GS, Rodgers KR, Benson DR. A possible role for the covalent heme-protein linkage in cytochrome c revealed via comparison of N-Acetylmicroperoxidase-8 and a synthetic, monohistidine-coordinated heme peptide. Biochemistry. 2004;43:1656–1666. doi: 10.1021/bi035531p. [DOI] [PubMed] [Google Scholar]
- 36.Allen JWA, Barker PD, Daltrop O, Stevens JM, Tomlinson EJ, Sinha N, Sambongi Y, Ferguson SJ. Why isn’t ‘standard’ heme good enough for c-type and d1-type cytochromes? Dalton Trans. 2005;21:3410–3418. doi: 10.1039/b508139b. [DOI] [PubMed] [Google Scholar]
- 37.Arciero DM, Hooper AB. A di-heme cytochrome-c peroxidase from Nitrosomonas europaea catalytically active in both the oxidized and half-reduced states. J. Biol. Chem. 1994;268:11878–11886. [PubMed] [Google Scholar]
- 38.Dracheva SM, Drachev LA, Konstantinov AA, Semenov AY, Skulachev VP, Arutjunjan AM, Shuvalov VA, Zaberezhnaya SM. Electrogenic steps in the redox reactions catalyzed by photosynthetic reaction-centre complex from Rhodopseudomonas viridis. Eur. J. Biochem. 1998;171:253–264. doi: 10.1111/j.1432-1033.1988.tb13784.x. [DOI] [PubMed] [Google Scholar]
- 39.Bamford VA, Angove HC, Seward HE, Thomson AJ, Cole JA, Butt JN, Hemmings AM, Richardson DJ. Structure and spectroscopy of the periplasmic cytochrome c nitrite reductase from Escherichia coli. Biochemistry. 2002;41:2921–2931. doi: 10.1021/bi015765d. [DOI] [PubMed] [Google Scholar]
- 40.Leys D, Backers K, Meyer TE, Hagen WR, Cusanovich MA, Van Beeumen JJ. Crystal structures of an oxygen-binding cytochrome c from Rhodobacter sphaeroides. J. Biol. Chem. 2000;275:16050–16056. doi: 10.1074/jbc.275.21.16050. [DOI] [PubMed] [Google Scholar]