


Abstract
DOMINE is a database of known and predicted protein domain interactions compiled from a variety of sources. The database contains domain–domain interactions observed in PDB entries, and those that were predicted by eight different computational approaches. DOMINE contains a total of 20 513 unique domain–domain interactions among 4036 Pfam domains, out of which 4349 are inferred from PDB entries and 17 781 were predicted by at least one computational approach. This database will serve as a valuable resource to those working in the field of protein and domain interactions. DOMINE may not only serve as a reference to experimentalists who test for new protein and domain interactions, but also offers a consolidated dataset for analysis by bioinformaticians who seek to test ideas regarding the underlying factors that control the topological structure of interaction networks. DOMINE is freely available at http://domine.utdallas.edu.
INTRODUCTION
Identification of molecular interactions is an essential step towards a better understanding of various cellular processes. Recent advances in functional genomics have helped uncover thousands of protein–protein interactions (1–9). Studying interactions at the protein level, though extremely valuable towards a better understanding of the molecular machinery of a cell, do not provide insights on interaction specificity at the domain level. Most often, it is only a fraction of a protein that directly interacts with its biological partners. Since the majority of the proteins (two-thirds in prokaryotes and four-fifths in eukaryotes) are multi-domain proteins (10), an interaction between two proteins (either stably or transiently) often involves binding of two or more domains. Thus, understanding protein interactions at the domain level seems to be a logical step towards understanding precise atomic details of interactions.
Over the last few years, researchers have focused their attention on discovering and understanding protein domain (domain–domain) interactions. One way to infer domain–domain interactions is by studying three-dimensional (3D) structures. iPfam (11) and 3did (12) are two databases that contain information on known domain–domain interactions inferred from PDB entries (13). The number of known domain–domain interactions is still mostly limited by the availability of 3D structures. Although many thousands of protein interactions are known, the number of interactions with known protein structures is far fewer than the number of interactions. This limits us from uncovering all possible domain level interactions. Domain interactions inferred from structural data can only explain ∼5% of protein interactions in Saccharomyces cerevisiae and ∼19% of protein interactions in Homo sapiens (14). In recent years, several computational approaches have been proposed in an effort to unearth previously unrecognized domain–domain interactions on a genome scale. These include approaches based on correlated sequence signatures (15), maximum-likelihood estimation (16), phylogenetic profiling (17), statistical significance (18), domain pair exclusion analysis (19), random decision forest framework (20), sequence co-evolution (21), parsimony principle (22), domain fusion, GO (23) functional annotations and combination thereof (24,25).
While computational approaches have greatly contributed to the discovery and understanding of domain–domain interactions, the ever-increasing sets of predicted domain–domain interactions remains scattered under a variety of diverse formats and sources. This has created a need to develop a comprehensive resource that collates all known and predicted domain–domain interactions from various sources under one roof.
We present here DOMINE, a comprehensive database of protein domain interactions using Pfam-A (26) domain definitions, which collates known and predicted domain–domain interactions from 10 different sources. By making the existing datasets more accessible, this database will serve as a valuable resource to those working in the field of protein and domain interactions. DOMINE may not only serve as a reference to experimentalists who test for new protein and domain interactions, but also offers a consolidated dataset for analysis by bioinformaticians who seek to test ideas regarding the underlying factors that control the topological structure of interaction networks.
DATABASE CONTENTS
Data sources
DOMINE contains domain–domain interactions inferred from PDB entries (13), and those that were predicted by eight different computational approaches using Pfam-A (26) domain definitions. Interactions in the database were derived from the following sources.
iPfam—iPfam is a database of domain–domain interactions that are observed in PDB entries. The set of 4030 interactions (dated 17 February 2007) downloaded from ftp://ftp.sanger.ac.uk/pub/databases/Pfam/ was used.
3did—3did is a collection of domain–domain interactions in proteins for which high-resolution 3D structures are known (12). The set of 3034 interactions (August 2005) downloaded from http://gatealoy.pcb.ub.es/3did/ was used.
ME—ME refers to Lee et al.'s (24) integrated approach to the prediction of domain–domain interactions. This method uses a Bayesian approach to integrate domain interactions predicted using a maximum-likelihood estimation (MLE) approach on yeast, worm, fruit-fly and human protein interaction networks with the gene ontology and domain fusion information. The set of 2391 high-confidence domain–domain interactions downloaded from http://www.biomedcentral.com/1471-2105/7/269 was used.
RCDP—Jothi et al.'s (21) Relative Co-evolution of Domain Pairs (RCDP) approach uses sequence co-evolution to predict the domain pair that is most likely to mediate a given protein–protein interaction. Given a protein–protein interaction, RCDP computes the degree of sequence co-evolution between all pairs of domains between the two proteins, and predicts the domain pair with the highest degree of co-evolution to be the mediating domain pair. The set of 960 unique domain–domain interactions (predicted from 1180 yeast protein–protein interactions) downloaded from http://www.rajajothi.com/RCDP/ was used.
P-value—Nye et al.'s (18) P-value method is a statistical approach that assigns P-values to pairs of domain superfamilies, measuring the strength of evidence within a set of protein interactions that domains from these superfamilies form contacts. A set of P-values is calculated for SCOP (27) superfamily pairs, based on a pooled dataset of interactions from yeast. These P-values were then used to predict which domains come into contact in an interacting protein pair. This scheme was applied on protein complexes in the Protein Quaternary Structure (PQS) database (28) to predict domain–domain contacts for 705 interacting protein pairs. Since interactions were predicted between SCOP domain families, for every yeast protein used, SGD (http://www.yeastgenome.org/) was used to map SCOP domains to Pfam-A (26) domains, and convert 705 interactions between SCOP domain families (http://www.mrc-bsu.cam.ac.uk/personal/thomas/) to 596 domain–domain interactions among Pfam domain families.
Fusion—2768 domain–domain interactions inferred using Ng et al.'s (25) domain fusion hypothesis was downloaded from http://interdom.i2r.a-star.edu.sg/download/version1.1/interdom_v1.2.zip (v1.2, 9 June 2004).
LP—Guimaraes et al.'s (22) Linear Programming (LP) approach is an optimization approach, which relies on the parsimony principle ‘domain–domain interaction partners are predicted by identifying the minimal weighted set of domain pairs that can justify a given protein–protein interaction network’. Given a protein–protein interaction network, the LP approach computes an LP-score, in the range (0,1), for every domain pair that could possibly justify interaction between two proteins. False positives in the protein–protein interaction network are handled using a probabilistic construction (P-scores). Domain pairs with an LP-score above a certain threshold are considered to be nteracting. A set of 3499 domain pairs with LP-score ≥0.5 and 0.0 ≤ P-score ≤ 0.1 downloaded from http://www.ncbi.nlm.nih.gov/CBBresearch/Przytycka/DDI/ was used. Since only interactions between Pfam-A domains are considered, 911 interactions in which at least one of the interacting partner is a Pfam-B domain were discarded, reducing the number of interactions to 2588.
DPEA—Riley et al.'s (19) Domain Pair Exclusion Analysis (DPEA) is a statistical approach to infer domain–domain interactions from the incomplete sets of protein–protein interactions from multiple organisms. It employs an expectation maximization algorithm to obtain a maximum-likelihood estimate or the probability of interaction of each potentially interacting domain pair. For each potential domain pair, a change in likelihood, expressed as a log odds score, is computed by excluding this domain pair from being considered as a potentially interacting domain pair. Domain pairs with log odds score above a certain threshold are considered to be interacting. A set of 3005 high-confidence interactions with log odds score ≥3.0 downloaded from http://genomebiology.com/2005/6/10/R89 was used. Since only interactions between Pfam-A domains are considered, 1193 interactions in which at least one of the interacting partner is a Pfam-B domain were discarded, reducing the number of interactions to 1812.
RDFF—Chen and Liu's Random Decision Forest Framework (RDFF) approach explores all possible domain–domain interactions and predicts protein–protein interactions based on protein domains (20). The decision tree-based model is used to infer domain–domain interactions for each correctly predicted protein–protein interaction pair. The set of 2475 domain–domain interactions between Pfam-A domains downloaded from http://www.ittc.ku.edu/~xwchen/PPI/random_forest_PPI was used.
DIMA—Domain Interaction MAp (DIMA) approach uses phylogenetic profiling to predict functional and physical associations between domains. The set of 8012 interactions reported in Pagel et al. (17) was used.
Data characteristics
The set of known domain–domain interactions, collected from iPfam and 3did, is considered as gold-standard positives against which computational predictions can be evaluated against. This set contains a total of 4349 unique interactions. While this set of gold-standard positives derived from protein structures clearly represents the most reliable data source, it is not entirely free of false positives (non-biological contacts in some structures). The plot in Figure 1 shows, for each computational approach, the percentage of predictions known to be true against the percentage of predictions in which both the domains (hetero-domain) are known to be part of the same biological process (based on GO classification). Since the predictions are validated against the set of gold-standard positives, which may not be complete by any chance, one need to be extremely careful in interpreting this plot, and should refrain from reaching to conclusions on the predictive powers of individual approaches. The two main reasons for the superior performance of ME are (i) its integration of multiple sources of information (knowledge gained from protein interaction networks of four different organisms, domain fusion and gene ontology information), and (ii) the performance-influenced choice of likelihood ratio cutoff value, which was used to select the set of 2391 high-confidence predictions. Other methods did not let performance numbers dictate the choice of predictor variables, which could be one of the reasons for their poor showing in the plot. In other words, by choosing a stringent (predictor variable) cutoff, a method may boost its performance at the expense of reduced number of predictions.
Figure 1.

Validation of computational predictions against the set of known interactions. Two interacting (hetero) domains are considered to be part of the same biological process if they are classified as such by GO (23).
Table 1 contains the percentage of overlap of predictions between any two different computational approaches. Note that 1748 out of 1812 (97%) of DPEA's predictions are confirmed by LP, and 1748 out of 2588 (68%) of LP's predictions are confirmed by DPEA. However, only ∼11% of LP's predictions and 12% of DPEA's predictions are known to be in the set of gold-standard positives. Since both LP and DPEA are based on optimization frameworks (parsimony principle and maximum-likelihood estimation, respectively), it was decided that the predictions by LP and DPEA be merged into a single pool, referred to as LP + DPEA, containing 2652 unique interactions. Since ME uses domain fusion as a source of information in its integrated approach, as expected, a good fraction of predictions by ME and Fusion are confirmed by each other. To our surprise, only a very small fraction of DIMA's predictions is confirmed by any other method, and vice-versa.
Table 1.
Extent of overlap of predictions between any two different computational approaches
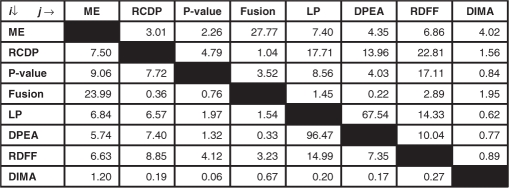 |
Entry (i, j) in the table represents the percentage of predictions by the method in row i confirmed by the method in column j (see Supplementary Table 1 for actual numbers).
Data integration
The interaction data collected from abovementioned 10 sources were collated to obtain a total of 20 513 unique domain–domain interactions among 4036 Pfam-A domains, out of which 4349 are inferred from PDB entries (the union of the sets of interactions from iPfam and 3did), and 17 781 were predicted by at least one computational approach.
Interactions predicted by computational approaches are classified into three categories using a simple classification scheme. Putative interactions predicted by an approach using multiple sources of evidence or those predicted by more than two sufficiently different approaches are considered as high-confidence predictions (HCP). Putative interactions with support from just one approach, but whose constituent domains (hetero) are known to be part of the same biological process (based on GO classification), are considered as medium-confidence predictions (MCP). The rest of the predictions is considered as low-confidence predictions. A schematic overview of the classification is shown in Figure 2. Of the 17 781 predictions, 3143 interactions are HCP (predicted by ME or at least two sufficiently different approaches), 730 interactions are medium-confidence predictions (hetero-domain interactions in which both domains are a part of the same biological process as per GO classification), and the remaining 13 908 are low-confidence predictions. The sets of high-, medium- and low-confidence predictions are enriched with 42.3, 5.8 and 1.8% of known interactions, respectively. The list of 55 predicted domain–domain interactions, confirmed by at least four sufficiently different computational approaches, is given in the Supplementary Table 2.
Figure 2.

A schematic overview of the DOMINE database.
DATABASE INTERFACE AND ACCESS
Availability
DOMINE is freely available at http://domine.utdallas.edu. A user-friendly web-interface was developed on Linux and Windows, and was tested during development using Internet Explorer and Firefox web browsers. MySQL is used to store the database content.
Searching DOMINE
Domain interaction information contained in the database can be accessed by either the ‘Browse’ or the ‘Search’ option on the menu. Using the former, users can glance through interactions of any domain for which the interaction data is available. An option to browse the list of Pfam domain IDs based on their gene ontology (GO) classification is also available. Using the search option, users can query the database by searching for a keyword (e.g. ATPase), Pfam ID (e.g. AAA) or accession (e.g. PF00004 or 00004 or 4). Users may also query the database using Interpro ID (e.g. IPR004825 or 004825 or 4,825) or GO term (e.g. phosphorylation or GO:0006468 or 0006468 or 6468).
Clicking on a domain name (Pfam ID) from anywhere on the Web site displays interaction information, if available, for that domain (see Figure 3). For each interacting domain, the list of domains that it is known/predicted to interact with are displayed along with external links to the Pfam, Interpro and GO databases. For each predicted interaction, information on whether DOMINE considers it to be a high-, medium- or low-confidence prediction is provided in addition to the source(s) of evidence.
Figure 3.

Screen shot of query result for 14-3-3 domain.
Data download
End users with adequate computational capabilities can download the entire content of the database in the form of a zip-compressed file, which includes a README file. To enable easy parsing, the data are presented in simple tab-delimited text files.
CONCLUSIONS AND FUTURE DEVELOPMENTS
The DOMINE database has been developed as a repository for protein domain interactions compiled from a variety of sources. The database contains domain–domain interactions inferred from experimental data (PDB entries) as well as those predicted by eight different computational approaches. DOMINE can serve as a directory to domain-specific information contained in Pfam (26), Interpro (29) and GO (23) databases. Links to these external databases are provided for each domain so that users can learn more about their domain of interest with just one click.
The currently employed classification scheme for assigning confidence levels to predicted interactions is simple. In the near future, we plan to manually examine the predicted interaction data to assign confidence levels, or at least identify obvious false positives and flag them as such. Currently, we did not take into account those inferences from small-scale studies reported in literature. In the future, we plan to add interactions inferred from small-scale studies through a controlled literature mining.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Daniel Barrell, Evelyn Camon and Nicky Mulder from EBI for providing us with the Pfam-GO-Interpro mappings. We thank Anantharaman V, Balaji S and Aravind L from NCBI for letting us use their signaling helix image in DOMINE's web site banner. T.M.P. and R.J. were supported by the Intramural Research Program of the National Library of Medicine, NIH. Funding to pay the Open Access publication charges for this article was provided by NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1.Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 2.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl Acad. Sci. USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rain JC, Selig L, De Reuse H, Battaglia V, Reverdy C, Simon S, Lenzen G, Petel F, Wojcik J, et al. The protein-protein interaction map of Helicobacter pylori. Nature. 2001;409:211–215. doi: 10.1038/35051615. [DOI] [PubMed] [Google Scholar]
- 4.Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 5.Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 6.Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- 7.Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, Vidalain PO, Han JD, Chesneau A, et al. A map of the interactome network of the metazoan C. elegans. Science. 2004;303:540–543. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butland G, Peregrin-Alvarez JM, Li J, Yang W, Yang X, Canadien V, Starostine A, Richards D, Beattie B, et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433:531–537. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- 9.Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 10.Apic G, Gough J, Teichmann SA. Domain combinations in archaeal, eubacterial and eukaryotic proteomes. J. Mol. Biol. 2001;310:311–325. doi: 10.1006/jmbi.2001.4776. [DOI] [PubMed] [Google Scholar]
- 11.Finn RD, Marshall M, Bateman A. iPfam: visualization of protein-protein interactions in PDB at domain and amino acid resolutions. Bioinformatics. 2005;21:410–412. doi: 10.1093/bioinformatics/bti011. [DOI] [PubMed] [Google Scholar]
- 12.Stein A, Russell RB, Aloy P. 3did: interacting protein domains of known three-dimensional structure. Nucleic Acids Res. 2005;33:D413–D417. doi: 10.1093/nar/gki037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schuster-Bockler B, Bateman A. Reuse of structural domain-domain interactions in protein networks. BMC Bioinformatics. 2007;8:259. doi: 10.1186/1471-2105-8-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sprinzak E, Margalit H. Correlated sequence-signatures as markers of protein-protein interaction. J. Mol. Biol. 2001;311:681–692. doi: 10.1006/jmbi.2001.4920. [DOI] [PubMed] [Google Scholar]
- 16.Deng M, Mehta S, Sun F, Chen T. Inferring domain-domain interactions from protein-protein interactions. Genome Res. 2002;12:1540–1548. doi: 10.1101/gr.153002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pagel P, Wong P, Frishman D. A domain interaction map based on phylogenetic profiling. J. Mol. Biol. 2004;344:1331–1346. doi: 10.1016/j.jmb.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 18.Nye TM, Berzuini C, Gilks WR, Babu MM, Teichmann SA. Statistical analysis of domains in interacting protein pairs. Bioinformatics. 2005;21:993–1001. doi: 10.1093/bioinformatics/bti086. [DOI] [PubMed] [Google Scholar]
- 19.Riley R, Lee C, Sabatti C, Eisenberg D. Inferring protein domain interactions from databases of interacting proteins. Genome Biol. 2005;6:R89. doi: 10.1186/gb-2005-6-10-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen XW, Liu M. Prediction of protein-protein interactions using random decision forest framework. Bioinformatics. 2005;21:4394–4400. doi: 10.1093/bioinformatics/bti721. [DOI] [PubMed] [Google Scholar]
- 21.Jothi R, Cherukuri PF, Tasneem A, Przytycka TM. Co-evolutionary analysis of domains in interacting proteins reveals insights into domain-domain interactions mediating protein-protein interactions. J. Mol. Biol. 2006;362:861–875. doi: 10.1016/j.jmb.2006.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guimaraes KS, Jothi R, Zotenko E, Przytycka TM. Predicting domain-domain interactions using a parsimony approach. Genome Biol. 2006;7:R104. doi: 10.1186/gb-2006-7-11-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee H, Deng M, Sun F, Chen T. An integrated approach to the prediction of domain-domain interactions. BMC Bioinformatics. 2006;7:269. doi: 10.1186/1471-2105-7-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng SK, Zhang Z, Tan SH, Lin K. InterDom: a database of putative interacting protein domains for validating predicted protein interactions and complexes. Nucleic Acids Res. 2003;31:251–254. doi: 10.1093/nar/gkg079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finn RD, Mistry J, Schuster-Bockler B, Griffiths-Jones S, Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A, et al. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34:D247–D251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 28.Henrick K, Thornton JM. PQS: a protein quaternary structure file server. Trends Biochem. Sci. 1998;23:358–361. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]
- 29.Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Buillard V, Cerutti L, et al. New developments in the InterPro database. Nucleic Acids Res. 2007;35:D224–D228. doi: 10.1093/nar/gkl841. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.