

Abstract
Following an infection or immunization, a primary CD8+ T cell response generally rises then falls rapidly before giving rise to a “memory” response. When we immunized mice with recombinant viral immunogens optimized to enhance the lytic capability of CD8+ T cells, we measured a profound depression in Ag-specific effector function after early restimulation. Indeed, a “mirror image” cytolytic capability was observed: the most powerful immunogens, as measured by cytolytic capacity 6 days after immunization, elicited the weakest secondary immune response when evaluated following an additional 6 days after restimulation. To understand the mechanism of this suppression, we examined the fate of splenocytes immunized with a vaccinia virus encoding Ag and IL-2 then restimulated ex vivo. We found that these splenocytes underwent an apoptotic cell death, upon early restimulation, that was not dependent on the engagement of the FasR (CD95). Unlike previously described mechanisms of “propriocidal cell death” and “clonal exhaustion,” the cell death we observed was not an inherent property of the CD8+ T cells but rather was due to a population of splenocytes that stained positive for both the Mac-1 and Gr-1 surface markers. Deletion of these cells in vitro or in vivo completely abrogated the observed suppression of cytolytic reactivity of Ag-specific CD8+ T cells. These observations could account for the apparent absence of Ag-specific immune responses after some current vaccination regimens employing powerful immunogens. Finally, our results may shed new light on a mechanism for the suppression of CD8+ T cell responses and its effect on vaccine efficacy and on immune memory.
Immunologists have long sought ways to enhance the speed and magnitude of the induction of Ag-specific cytotoxicity, but attention to the deactivation of the cytolytic response has been somewhat less intense. This deactivation must not occur before the work of the cellular immune response is complete (e.g., the eradication of a dangerous pathogen) and must not interfere with the establishment of the appropriate memory immune response. On the other hand, if deactivation never occurs, or is incomplete, the numbers of activated T cells will inevitably increase along with attendant risks of immune dysfunction, including autoimmunity.
The deactivation of immune responses is conducted, in part, by suppressive and regulatory circuits that result in apoptosis of lymphocytes (1). Mechanisms for the induction of apoptosis in T lymphocytes include “propriocidal cell death” and “clonal exhaustion.” The propriocidal form of death has been shown to occur upon TCR engagement in lymphocytes previously exposed to IL-2. This mechanism can control the extent of T cell activation by eliminating a portion of newly dividing, Ag-reactive lymphocytes (2, 3) and is mediated through the engagement of receptors for Fas and TNF (4-7).
Cytotoxic effector cells can be rapidly and specifically deleted in mice exposed to high doses of a particular strain of rapidly disseminating lymphocytic choriomeningitis virus (LCMV)4. A phenomenon termed “clonal exhaustion” is due to the apoptotic death of T cells after restimulation through their TCR, resulting in a chronic virus carrier state in which there is a complete absence of memory T cells (8-10). Thus, a functional state of tolerance may be the result of Ag persistence at a time when restimulation of lymphocytes programs them to die.
T cells may receive instructions to die from a population of cells called “natural suppressors” (NS). This poorly defined population of nonlymphoid cells may be derived from the monocyte-macrophage lineage. In experimental animal models, NS cells have been reported to suppress the generation of CTL independently of Ag and MHC restriction and may play a role in the inhibition of B cells and Th cells, as measured by allo- and Ag-specific proliferative responses (11-15). NS cells have been reported to be responsible for the suppression of immune responses associated with cancer as well as infectious diseases like Trypanosoma and Salmonella (11, 16, 17).
In this communication, we explore the immune deactivation that follows infection or immunization. We found that immunization strategies that have been optimized for their ability to elicit cytotoxicity elicit the weakest secondary immune responses upon early restimulation. We explore the immune sequelae after immunization with a vaccinia virus (VV)-encoding IL-2 and the model Ag, β-galactosidase (β-gal), an immunogen previously found to be highly effective in the generation of a potent primary immune response (18).
Materials and Methods
Cell lines
CT26.WT, the β-gal-expressing CT26.CL25, and EL4 thymoma and its β-gal-expressing (LacZ transfected) subclone, E22, have been described (18, 19). The first two cell lines are H-2d, while EL4 and E22 are H-2b. BSC-1 cells (CCL26, American Type Culture Collection, Manassas, VA) and HeLa S3 (CCL2.2, American Type Culture Collection) were used to prepare all the VV stocks. Cell lines were maintained in culture media consisting of RPMI 1640, 10% heat-inactivated FBS (Biofluids, Rockville, MD), 0.03% l-glutamine, 100 mg/ml streptomycin, 100 mg/ml penicillin, and 50 mg/ml gentamicin sulfate (National Institutes of Health Media Center, Bethesda, MD). CT26.CL25 and E22 were maintained in culture media containing 400 μg/ml G418 (Life Technologies, Grand Island, NY). BSC-1 and HeLa S3 were maintained in DMEM.
Recombinant vaccinia viruses
All rVV used in this study were generated by insertion of the foreign genes into the VV thymidine kinase gene by homologous recombination (20). Virus preparations were propagated from plaque-purified crude virus stocks, as described previously (21). Briefly, 175-cm2 flasks of HeLa S3 or BSC-1 cells were infected with 5 plaque-forming units (PFU)/cell and incubated at 37°C for about 72 h. Infected cells were harvested and centrifuged at 1000 × g for 10 min. Cells were resuspended in 10 mM Tris (pH 9.0) and lysed by 30 strokes of a dounce homogenizer. Nuclei and cell debris were partially removed by centrifugation for 5 min at 1000 × g, viral particles were collected after purification by centrifugation over a sucrose cushion, and stocks were aliquoted and stored at −80°C.
Viral concentrations were determined by plaque titration on BSC-1 cells. rVV used in a single experiment were titered concurrently to maximize accuracy. Preparation of rVV expressing the influenza A/PR/8/34 nucleoprotein (NP), NP-rVV, was previously described (22). Murine IL-2 cDNA was amplified by PCR from pBMGNeomIL2 and ligated into the SmaI-BamHI site of vaccinia expression vector, pMJ601, which contains the lacZ gene under the control of the natural p7.5 early promoter (18). IFN-γ was inserted into the VV genome using a similar procedure, as reported (23). In the VJS6 construct, the Escherichia coli lacZ gene was under the control of the natural p7.5 early/late promoter element from plasmid pSC65 (24); this construct was named β-gal-rVV for simplicity. Wild-type (WT) VV strain WR was kindly provided by J. Yewdell and J. Bennink (National Institute of Allergy and Infectious Diseases, Bethesda, MD).
Peptides
The following synthetic peptides were synthesized by Peptide Technologies (Washington, D.C.) to a purity of greater than 99% as determined by HPLC and amino acid analysis: TPHPARIGL (amino acids 876–884 of β-gal, H-2Ld-restricted (25)), DAPIYTNV (amino acids 96–103 of β-gal, H-2Kb-restricted, (26)).
Antibodies
FITC- or phycoerythrin-labeled mAb recognizing mouse CD8, CD4, CD11b (Mac-1), Lyt-6G (Gr-1), and the isotype-matched controls were purchased from PharMingen (San Diego, CA). Concentrations used for cytofluorometry ranged between 1 and 12 μg/106 cells depending on the Ab. The mAb 24G.2 (CD16/CD23, PharMingen), which reacts with a common epitope of the extracellular domain of the mouse FcγRII/FcγRIII, was used to block the nonspecific binding of mAb during staining. For the in vitro and in vivo depletion studies, mAb were extensively dialyzed against PBS to remove the sodium azide.
Evaluation of CTL responses
Eight- to 12-wk-old female BALB/c, (Animal Production Colonies, Frederick Cancer Research Facility, National Institutes of Health, Frederick, MD), C57BL/6J, or MRL-lpr/lpr mice (The Jackson Laboratory, Bar Harbor, ME) were immunized with various doses (5 × 106 to 2 × 107 PFU/mouse) of different rVV. The spleens were collected on day 6 after immunization, separated into a single-cell suspension, and tested for their ability to lyse β-gal-positive targets in a 6-h 51Cr release assay as previously described (18). Briefly, 2 × 106 target cells were incubated with 200 μCi Na51CrO4 (51Cr) for 90 min (together with 1 μg/ml of peptide or 100 μl of crude VV-WT preparation, where designated). After labeling, the targets were washed and diluted to 105 viable cells/ml. Targets were then plated at 0.1 ml/well in 96-well plates (104 cells/well) and effectors were added at the indicated ratio. Plates were incubated for 6 h before harvesting. The amount of 51Cr released was determined by γ-counting and the percentage of specific lysis was calculated from triplicate samples using the formula: [(experimental cpm − spontaneous cpm)/(maximal cpm − spontaneous cpm)] × 100. In some experiments, splenocytes, homogenized to a single cell suspension, were cultured at 5 × 106 cells/ml in 75-cm2 flasks (Costar, Cambridge, MA) with 30 ml of RPMI 1640 containing 10% FCS (Biofluids), 0.1 mM nonessential amino acids, 1 mM sodium pyruvate (Biofluids), and 5 × 10−5 M 2-ME (Life Technologies, Rockville, MD). For the in vitro stimulation, the peptide (1 μg/ml) or irradiated tumor cells at a responder-to-stimulator ratio of 40:1 were added to the cultures. After 6 days, effectors were harvested and tested in a 6-h 51Cr release assay, as indicated above. In cell separation experiments, splenocytes were cultured at the same cell concentration in 24-well plates (Costar) containing a culture chamber insert with 0.4-μM pores (Millipore, Bedford, MA).
In vivo studies
In protection studies, BALB/c mice (5/group) were immunized with 5 × 106 PFU of rVV, boosted on day 6 with various Ags, and inoculated i.v. 21 days after the boosting with 5 × 105 tumor cells. Negative controls were always included and consisted of mice inoculated with only the vehicle used to resuspend the Ag. Mice were sacrificed on day 12 following tumor inoculation and lung metastases were enumerated in a coded, blind fashion.
Detection of apoptosis in lymphocyte subpopulations
A modification of the method described by Sherwood and Schimke (27) was employed. Briefly, 106 cells were stained with a FITC-conjugated anti-CD8 or anti-CD4 mAb (PharMingen) for 30 min at 4°C in FACS buffer (Ca2+- and Mg2+-free HBSS containing 0.5% BSA and 0.02% sodium azide). Cells were washed three times in cold FACS buffer and fixed with the addition of 70% ice-cold ethanol. After 1 h incubation at 4°C, cells were washed twice with PBS and resuspended in propidium iodide (PI) staining solution (PBS containing 100 μg/ml RNase A and 50 μg/ml PI, both from Sigma, St. Louis, MO). Flow cytometry was performed on a Becton Dickinson (San Jose, CA) FACScan using a 488 Argon Laser. Data analysis was performed on either the Cellfit (Becton Dickinson) or Modfit (Verity Software House, Topsham, ME) software packages. In some experiments, PI staining was compared with staining with the TUNEL method used by the in situ cell death detection kit (Boehringer Mannheim, Indianapolis, IN), obtaining comparable results (not shown).
Isolation of splenic populations
A panning technique employing flasks coated with mouse anti-rat Abs (T-25 AIS MicroCell, AIS, Santa Clara, CA) was used to deplete specific populations from spleens. Spleens were depleted of red cells with ACK lysis buffer (Biofluids) and resuspended in HBSS containing 1 mM EDTA and 10% mouse serum (HBSS-EDTA-MS). The remaining cells were placed on ice, incubated for 30 min with the primary Ab at a concentration of 10 μg/107 cells, and washed three times with cold HBSS-EDTA. Next, the cells were resuspended in ice-cold HBSS-EDTA-MS at a concentration of 4 × 107/ml, transferred to flasks coated with the secondary Ab (anti-rat), and incubated for 1 h at 4°C. Finally, the nonadherent cells were dislodged and collected as a negative fraction. Fluorescence labeling confirmed >95% depletion. The cells that remained attached to the flasks were provided with RPMI 1640 complete medium and incubated for further studies. Adherent cells were recovered by gentle scraping in Versene solution (1:5000, Biofluids). In some experiments, 2 h adherence to plastic or ingestion of carbonyl iron (Myloclear, Cedarlane Laboratories, Accurate Chemical and Scientific Corp., Westbury, NY) followed by magnetic sorting were used to deplete spleens of mature monocytes/macrophages. CD8+ lymphocytes were separated through affinity columns (R & D Systems, Minneapolis, MN) according to the manufacturer's instructions. The percentage of CD8+ cells after enrichment was usually 80–90%.
Statistical analysis
The Wilcoxon-Mann-Whitney U test was used to examine the null hypothesis of identity of ranks between two sets of data. All the p values were reported as two-sided.
Results
The “mirror image” result: the most powerful immunogens elicit the weakest secondary immune responses
In an effort to increase the immunogenicity of recombinant anti-cancer vaccines, we previously inserted a variety of cytokine genes into the genome of a β-gal-expressing rVV (18). Insertion of the gene-encoding IL-2 into a construct-encoding β-gal significantly augmented primary cytolytic T lymphocyte responses specific for VV. Conversely, insertion of the gene-encoding IFN-γ had the opposite effect of decreased CTL activity (Fig. 1A). However, when an aliquot of the same splenic preparation was evaluated for the generation of β-gal-specific cytotoxicity after a 6-day incubation in vitro with the β-gal-immunodominant peptide (TPHPARIGL), a nearly opposite result was obtained in which IL-2-rVV-treated mice showed greatly diminished CTL responses, while IFN-γ-rVV-treated mice exhibited potent cytotoxicity (Fig. 1C). Thus, while splenocyte cultures derived from mice that had been primed in vivo with rVV containing the genes for IL-2 and β-gal showed no cytotoxicity against β-gal-positive tumor cells upon restimulation with the β-gal peptide, cultures from mice infected with virus-encoding IFN-γ with β-gal displayed excellent killing capabilities. Mice immunized with β-gal-rVV, which did not encode any cytokine, showed only a weak cytolytic response.
FIGURE 1.

Immunization with powerful immunogens induces a profound loss of cytolytic capacity upon restimulation with peptide. IL-2-rVV immunization augments primary cytolytic responses. Two BALB/c mice were immunized i.v. with 5 × 106 PFU/mouse of different rVV. After 6 days, the spleens were removed, pooled, and tested in a 6-h 51Cr release assay against CT26.WT tumor cells infected with VV-WT (A) or pulsed with the β-gal peptide (B). Secondary responses are lost in mice immunized with IL-2-rVV. The splenocytes used for the previous experiment were further incubated in vitro with 1 μg/ml of the synthetic β-gal peptide for 6 days and then assayed in a 6-h 51Cr release assay against the CT26.CL25 β-gal-positive clone (C) or the CT26.WT cells pulsed with the β-gal peptide (D). Purification of CD8+ cells restores specific reactivity in mice immunized with IL-2-rVV. CD8+ cells were enriched through affinity columns from splenocytes used in Fig. 1C and D. This enriched population was admixed with naive, CD8-depleted splenocytes from syngeneic mice to constitute about 10% of total cells in culture. After a 6-day in vitro restimulation with β-gal peptide, the mixture was tested in a 6-h 51Cr release assay against CT26.WT cells pulsed with the β-gal peptide (E). Cytotoxicity toward the CT26.WT cells or the irrelevant E22 target cells was always <5% even at the highest E:T ratio (not shown). The E:T ratio was 33:1, then diluted threefold (11:1, 4:1, 1:1). The experiment was repeated four times with similar results.
Similar effects were observed using an Ld-restricted β-gal peptide pulsed onto target cells. While the addition of the gene-encoding IL-2 greatly enhanced peptide-specific cytolytic responses of fresh splenocytes (Fig. 1B), restimulation of these same splenocytes with peptide for 6 days in vitro resulted in the complete disappearance of cytotoxic activity (Fig. 1D). On the other hand, no primary β-gal-specific cytolysis was observed in mice primed with the IFN-γ-containing virus, but splenocytes from these mice were specifically cytolytic after identical stimulation with peptide (Fig. 1, B vs D). The phenomenon of apparent suppression of cytolytic activity was observed only upon “early” restimulation: when splenocytes were harvested 14 days after immunization with the panel of rVV-containing cytokine genes and stimulated with peptide in vitro, all groups mounted efficient CTL responses (data not shown).
A trivial explanation for the apparent lack of CD8+ T cell function was “fratricide,” i.e., the killing of one lymphocyte by another after the addition of soluble peptide. However, the cytolytic capacity of IL-2rVV-primed splenocytes was also eliminated by re-stimulation with tumor cells that had been transfected with the β-gal gene (CT26.CL25) rather than pulsed with peptide (not shown).
Lack of a cytolytic response is not associated with irreversible damage of CD8+ T cells
Induction of an unresponsive state by hyperstimulation of the immune response has been explained in other models by several mechanisms such as anergy, propriocidal apoptosis, and clonal exhaustion, which all focus on properties inherent to T lymphocytes. We thus set out to separate and characterize the CD8+ T lymphocytes using negative separation methods that eliminated other cells from the cultures, without ligation of differentiation Ags on the CD8+ cells. However, when purified populations of CD8+ lymphocytes derived from day 6 spleens of IL-2rVV-inoculated mice were cultured together with naïve splenocytes depleted of CD8+ cells and pulsed with β-gal peptide, excellent CTL responses were observed (Fig. 1E). Indeed, comparable responses were generated irrespective of the rVV inoculated 6 days before the cultures were established with the expected exception of the β-gal-negative virus, NP-rVV. Thus, the suppression observed was not a characteristic of the T cells, but was dependent on some other element(s) in the splenic population.
Suppression of CD8+ T lymphocyte activity occurs in vivo and is long-lived
To evaluate the effects of “early” boosting on the suppression of immune responses in vivo, we employed a tumor challenge model. We have previously established the importance of CD8+ T cells in the effective immune response to CT26.CL25 (19, 28). Mice were immunized with either carrier (HBSS) or with rVV-encoding β-gal in combination with IL-2 or IFN-γ, boosted 6 days later with β-gal protein, then challenged 3 weeks after the boost with a syngeneic murine carcinoma-expressing β-gal. The protection from tumor challenge was nearly complete in mice receiving an initial inoculum of IFN-γ-rVV (Fig. 2A). However, while the IL-2-rVV was also protective when boosted with an irrelevant protein, OVA, tumor protection was abrogated in mice that were initially primed with the IL-2-rVV and boosted with β-gal protein. Similar results were obtained in IL-2-rVV-immunized mice when a recombinant fowlpox virus (FPV)-expressing β-gal was used as a booster (Fig. 2B). FPV-β-gal and, to a lesser extent, the β-gal protein were immunogenic because they induced protection from tumor challenge when used alone (p = 0.001 and 0.005, respectively, Fig. 2, A and B). Thus, the suppression could be observed in vivo, was Ag-specific, and was relatively long-lived.
FIGURE 2.
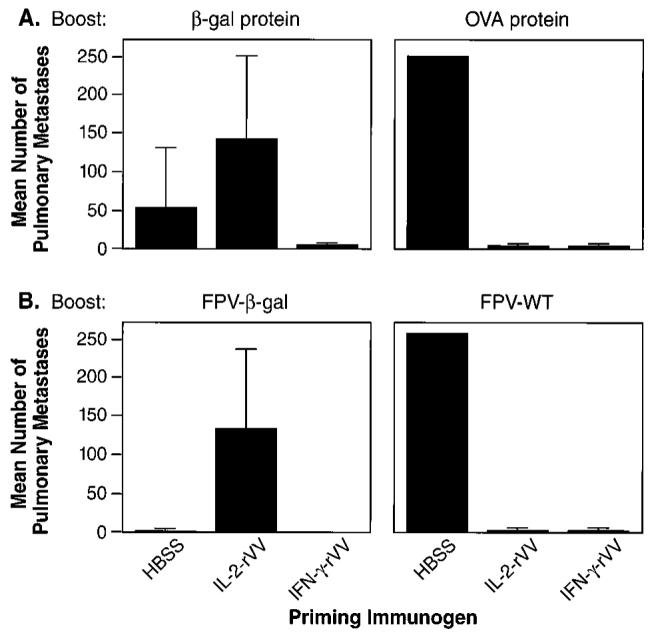
Impairment of immune response after boosting occurs in vivo and is long lasting. Five different BALB/c mice in each treatment group were injected i.v. with 5 × 106 PFU of the rVV indicated at the bottom of the boxes. HBSS alone was used as vehicle control. Six days later, mice were boosted with two i.v. injections of PBS alone (not shown) or PBS containing 400 μg of either OVA or β-gal protein (top). Alternatively, mice were boosted with a single i.v. inoculation of 107 PFU/mouse of wild-type (FPV-WT) or β-gal-expressing FPV (bottom). On day 21 following the in vivo restimulation, mice were challenged i.v. with 5 × 105 CT26.CL25 cells to establish pulmonary metastases. Twelve days later, mice were sacrificed, and pulmonary nodules were counted in a coded and blinded fashion. Two separate, independent experiments were performed under identical experimental conditions with overall similar results. An arbitrary value of 250 was assigned when metastases were too numerous to count. Data represent the mean ± SD of pulmonary metastases.
CD8+ T cells from unresponsive cultures die an apoptotic death
We hypothesized that the unresponsive state observed above could be due to anergy or apoptosis. To explore the death rate under various conditions of restimulation, we employed a double-staining protocol designed to evaluate the percentage of hypodiploid (apoptotic) cells among lymphocytes that were positive for CD8 (Fig. 3). In splenocyte cultures from mice primed with V69 rVV, the mean percentage of apoptosis upon in vitro stimulation was 22.4%. This baseline value of apoptosis was not Ag specific, because V69 encoded the influenza NP gene in lieu of LacZ (NP-rVV) and did not prime CTL responses (Fig. 1). The number of apoptotic cells after stimulation with antigenic peptide rose to 46.0% in cultures derived from mice that had been immunized with VJS6, which encodes β-gal without the heterologous addition of cytokines. Levels of apoptosis comparable to negative controls were detected in cultures derived from IFN-γ-rVV immunized mice. Apoptosis of CD8+ T cells was significantly increased in mice immunized with the IL-2-rVV. Similar patterns of apoptotic death was observed in CD8+ cultures that were restimulated with tumor cells expressing the β-gal Ag; however, CD4+ cells present in the same experiment did not follow the same pattern of apoptosis (not shown). Thus, apoptotic death appeared to be limited to the CD8+ compartment and was not a generalized death of all lymphocytes present in the culture.
FIGURE 3.
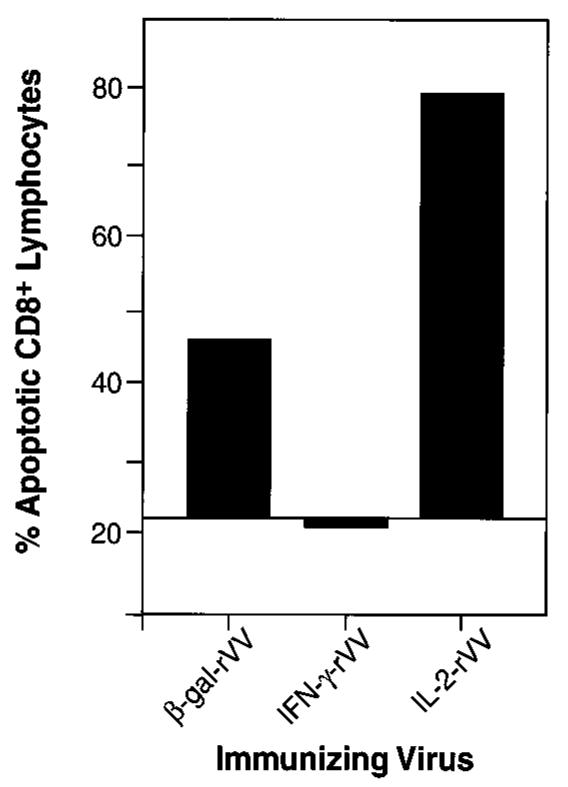
CD8+ T lymphocytes die an apoptotic death after immunization with IL-2-rVV in vivo and peptide restimulation in vitro. The cells used in this experiment are from the splenocytes used in the experiment shown in Fig. 1, C and D. Two spleens from BALB/c mice immunized 6 days earlier with different rVV were stimulated in vitro for 6 days with 1 μg/ml of β-gal peptide. One million cells were stained with FITC-conjugated mAb against the CD8 Ag. After staining, cells were fixed in ethanol and exposed to propidium iodide solution to allow detection of hypodiploid nuclei. Figures indicate the percentage of hypodiploid cells among CD8+ lymphocytes. The apoptosis percentage in cultures of spleens from mice immunized with NP-rVV, V69, was used as the background line. The experiment was repeated two additional times with similar results.
Suppression of CD8+ T cell function depends on membrane contact and is not Fas (CD95)-mediated
We next investigated the molecular mechanism causing apoptosis of CTL in acutely infected mice. The production of soluble factors secreted by the suppressive splenocytes was ruled out by mixing splenocytes from mice infected with different rVV in diffusion chambers. The experiments clearly showed that cell-cell contact was required for the immunosuppressive effect, because suppression of CTL generation only occurred when the suppressor-containing population was not separated by a membrane from the population responsive to peptide stimulation (Fig. 4).
FIGURE 4.
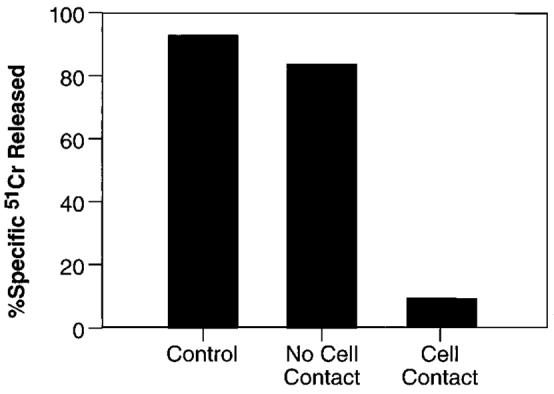
Suppression of CD8+ T cell function requires cell-cell contact. A suspension of three spleens from BALB/c mice infected 6 days earlier with 5 × 106 PFU/mouse of IFN-γ-rVV was incubated with a β-gal peptide either alone (control) or together with splenocytes of mice similarly infected with IL-2-rVV (1:1). The latter were either admixed in the same well (cell contact) or separated by a semipermeable membrane (no cell contact). After 6 days, CTL activity in the cultures was assayed against the same panel of target cells used in previous experiments. For simplicity, only the cytolytic activity against CT26.CL25 at an E:T ratio of 10:1 is shown. Control wells containing 1:1 mixture of splenocytes from IFN-γ-rVV-infected mice and normal mice did not show any difference from control culture whether they were admixed or separated by the insert membrane (not shown).
We tested whether Fas-Fas ligand (FasL) interactions were responsible for the observed cell membrane-associated suppression as this interaction is a primary inducer of apoptosis in activated lymphocytes. To elucidate this, we immunized MRL-lpr/lpr mice, which do not express a functional FasR (CD95) (Fig. 5, top), with IL-2-rVV or IFN-γ-rVV. As before, primary and secondary responses against the tumor cell line, EL-4, VV-WT infected EL-4, or the β-gal-expressing line, E22, were tested in 51Cr release assays (Fig. 5, bottom). An identical pattern of suppression was observed in the Fas-deficient mice as in normal C57BL/6J mice. This generation of the same IL-2-rVV-induced suppression in MRL-lpr/lpr mice indicated that the cell membrane-associated mechanism did not involve Fas-FasL interaction.
FIGURE 5.

Inhibition of CD8+ T cell killing is not Fas-dependent. Lymphoblasts obtained from ConA-stimulated splenocytes from C57BL/6J and MRL-lpr/lpr mice were evaluated for CD95 (Fas/Apo1) expression by cytofluorometric analysis (A, left and right, respectively). Mice were immunized i.v. with 5 × 106 PFU/mouse of IL-2-rVV or IFN-γ-rVV (B). Six days later, the spleens from three different mice were removed, pooled, and tested in a 6-h 51Cr release assay against EL-4, EL-4 tumor cell line infected with VV-WT, or the β-gal-expressing line, E22 (Primary, top). The splenocytes used for the previous experiment where further incubated in vitro with 1 μg/ml of the H-2Kb-restricted, synthetic β-gal peptide for 6 days and then assayed in a 6-h 51Cr release assay against CT26.CL25, EL-4, or E22 (Secondary, bottom). The experiment was repeated with similar results.
An increase in Mac-1 and Gr-1 double-positive cells correlates with the induction of primary cytolytic T cells
The lack of response in the whole spleens taken from IL-2-rVV-infected mice could be explained with either the absence of competent APC or with the presence of regulatory suppressor elements distinct from CD8+ lymphocytes. The first possibility was addressed by mixing experiments in which we found that splenocytes from IL-2-rVV-infected mice could suppress the response of IFN-γ-rVV mice even when mixed at a 1:2 ratio before culture with β-gal peptide (not shown). Additionally, Fig. 4 shows a similar mixing experiment of splenocytes from IL-2-rVV-infected mice and IFN-γ-rVV-infected mice, which demonstrates the same pattern of suppression. These findings disfavored the hypothesis of insufficient APC function and supported the presence of suppressive elements.
To identify candidate cells with suppressive activity, we cytofluorometrically evaluated surface markers on the spleen cells of naive mice or mice immunized with IL-2-rVV or IFN-γ-rVV. As can be seen in the upper panels of Fig. 6, both Mac-1 and Gr-1 were present on the surfaces of a population of cells that was increased in the spleens of mice that had been immunized with IL-2-rVV. Gr-1 is a marker that is normally expressed by granulocytes, monocytes, and immature myeloid precursors in the bone marrow but is expressed at a very low level in the spleens of normal mice (29). This population of cells expressing both Ags was significantly increased (p < 0.01) in the spleens of IL-2-rVV-immunized mice but not in mice immunized with IFN-γ-rVV (means ± SD of three experiments were: 3.03 ± 0.87, 2.92 ± 0.22, and 7.93 ± 0.68 for naïve, IFN-γ-rVV- and IL-2-rVV-inoculated mice, respectively). Moreover, in vivo depletion with anti-Gr-1 mAb but not with the control mAb reduced the double-positive cells in the spleens of mice inoculated with IL-2-rVV to levels comparable to those detected in naïve or in IFN-γ-rVV-immunized mice. The depletion was even more effective in vitro (Fig. 6, bottom).
FIGURE 6.

Immunization with IL-2-rVV results in an increase in the percentage of Mac-1+ and Gr-1+-double-positive splenocytes. Splenocyte pools used in the experiment shown in Fig. 7 were stained with FITC-anti-Gr-1 and phycoerythrin-anti-Mac-1 mAbs in the presence of an Ab blocking the Fcγ-RII/Fcγ-RIII (2.4G2). Upper panels show unseparated splenocytes from unimmunized mice (naïve) or mice immunized 6 days earlier with either IFN-γ-rVV or IL-2-rVV. Lower panels show the IL-2-rVV-immunized splenocytes after depletion with the indicated mAbs. Not shown are the results of the in vitro depletion with IgG2b mAb used as negative control. As for the in vivo data, depletion with this mAb did not change the percentages of double-positive cells seen in untreated controls. A secondary anti-rat IgG Ab revealed the number of positive cells to be <1% after depletion, confirming that the negativity was not due to Ab competition. Data are representative of four separate, independently performed experiments.
Mac-1+/Gr-1+ cells mediate the elimination of CD8+ T cell function in vivo
To ascertain whether these cells were suppressing the generation of CTL in our cultures, different mAbs were used to deplete, in vitro or in vivo, specific populations before restimulation of the spleens of mice infected 6 days earlier with rVV immunogens. As shown in Fig. 7A, depletion of cells positive for Gr-1 or Mac-1 completely restored the capacity of cells to mount cytolytic responses. Cytotoxicity recovered in Ab-depleted cultures was similar to that obtained with a population of CD8+ lymphocytes enriched from the same spleens or with the other positive controls included in the assay (IFN-γ-rVV-immunized mice or mice immunized 14 days earlier with IL-2-rVV). An isotype-matched Ab (rat IgG2b) did not produce the same effect, ruling out an in vitro artifact related to the experimental protocol. Moreover, a second Ab directed against a mouse macrophage Ag, Mac-3, only restored 20% of the CTL activity seen in the cultures of CD8+ enriched populations (not shown). The data shown in Fig. 7A further supports the hypothesis that the suppression of CD8+ cell responses is not a characteristic of the T cells, but rather is a quality of some other splenocyte component. In vivo depletion of Gr-1+ cells by repeated i.p. inoculations of the Ab during the first days of infection with IL-2-rVV resulted in a complete recovery of the deficient CTL response, while no effect was observed with the control IgG2b (Fig. 7B). Thus, Mac-1+/Gr-1+ cells appear to mediate the suppression of CD8+ T cells in vitro and in vivo.
FIGURE 7.
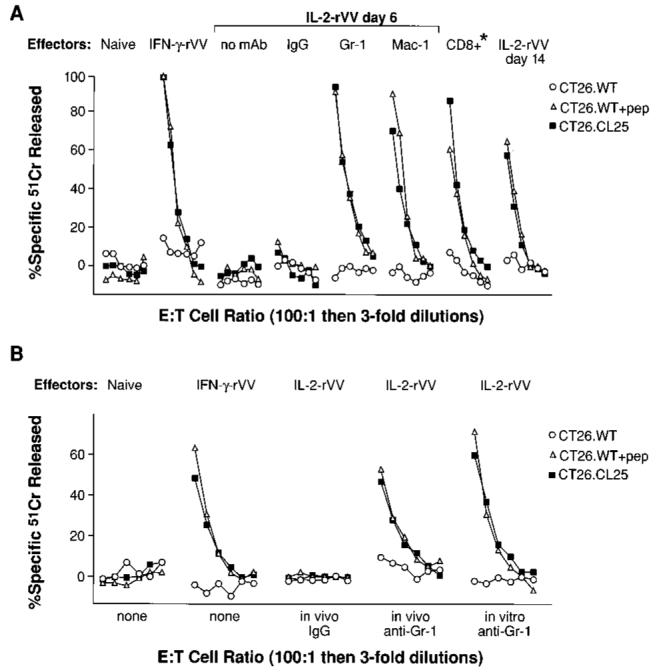
In vitro and in vivo depletion of either Mac-1+ or Gr-1+ cells restores CTL response in splenocyte cultures from mice immunized with IL-2-rVV. A, In vitro depletion of Mac-1+/Gr-1+ cells restores the immune response. Two to five BALB/c mice/group were inoculated with HBSS (naïve), 5 × 106 PFU of IL-2 rVV, or 5 × 106 PFU of IFN-γ-rVV. After 6 days, spleens were pooled together and depleted in vitro of Mac-1+ or Gr-1+ cells. IgG2b isotype-matched mAb was used as negative control of the depletion protocol. Splenocytes collected after depletion were cultured with β-gal peptide as described previously. The same panel of target cells was used to evaluate the cytolytic activity (A). As positive controls, the following experimental groups were also included: CD8+ splenocytes from mice inoculated with IL-2-rVV and cultured together with CD8-depleted naïve spleens (*); mice inoculated 6 days earlier with IFN-γ-rVV; and mice inoculated with IL-2-rVV 14 days before the in vitro restimulation. Data with E22 tumor cells were negative and are not shown for simplicity. The E:T ratio is 100:1 followed by threefold dilutions (33:1, 11:1, 4:1, 1:1, 0.3:1). The experiment was repeated four more times with similar results. B, In vivo depletion of Mac-1+/Gr-1+ cells restores the immune response. The experiment was performed as in A, with the exception that 200 μg Gr-1 mAb and the IgG2b controls were injected i.p. into mice on day 2 and 4 following immunization with the rVV (B).
Discussion
In this report, we describe the induction of apoptosis of CD8+ T cells by rVV inoculation followed by “early” boosting. An apparently similar state has been described with LCMV (8). We found that viral-induced suppression correlated with the intensity of the immune response. A partial suppression was seen with low doses of β-gal-rVV (Fig. 1C) and was almost complete at higher doses of 2 × 107 PFU/mouse (data not shown). The apoptotic death of CD8+ T cells and coincident loss of cytolytic capacity was not invariably associated with IL-2-rVV infection as it was absent when the dose was lowered to 5 × 104 PFU/mouse (not shown). Moreover, a Mac-1+/Gr-1+ cell-dependent suppression of the generation of alloreactive CTL was observed in the spleens of mice acutely infected with VV-WT (data not shown). We have shown that Ag has been cleared by day 6 after infection (N.P.R. and V.B., unpublished observation). The artificial reintroduction of the same Ag at a time when Ag has been cleared by the ongoing immune response may mimic the long-term persistence of Ag seen when a particular strain of LCMV is used (8). In our experiments, the reintroduction of Ag was accomplished either by in vivo reinfection with a recombinant virus or purified protein or by in vitro restimulation with an immunodominant peptide. “Exhaustion” of the immune response may not be due to a failure of the T cell compartment but instead may be a consequence of the feedback circuit that regulates the intensity of immune stimulation in the face of persistent Ag.
It is well documented that, at the apex of their increase, lymphocytes are unable to respond to mitogenic activation, as seen in the paradigmatic example of LCMV infection (30). In fact, further stimulation is associated with the induction of apoptosis (9). Based on our data and on the work of others, the following scenario seems likely: during the first encounter with an infectious agent, professional APC present the Ag to naïve lymphocytes. Activated lymphocytes migrate from lymphoid organs to the peripheral tissues via the circulation and start eliminating the antigenic challenge. Activation of silent, autoreactive lymphocytes recognizing cross-reactive epitopes can occur (31-33). Local tissue destruction, and the systemic toxicities of cytokines may be harmful and could potentially trigger a circuit leading to autoimmune destruction of tissues. Proportional to the intensity of the immune response, a population of granulocyte-macrophage precursors is mobilized from the bone marrow that is capable of differentiating into a suppressive population. Through a contact-dependent mechanism, the macrophage-like regulatory cells induce apoptosis of previously activated lymphocytes, down-modulating the immune response.
The kinetics of the induction of Mac-1+/Gr-1+ cells following inoculation with rVV were coincident with the functional suppression of the generation of cytolytic responses upon antigenic re-stimulation. The earliest detection of an increase of Mac-1+/Gr-1+ cells was on day 4, and baseline levels of splenic Mac-1+/Gr-1+ cells were measured by day 14, although some minor variability in these kinetics was observed (data not shown). The delay observed between the peak of CTL activity and the induction of the Mac-1+/Gr-1+ suppressor cells could allow for the destruction of viable pathogen and elimination of the antigenic stimulation for the future “memory” pathogen-specific T cells.
CD8+ lymphocytes activated in the presence of Mac-1+/Gr-1+ cells are induced to die and show clear signs of apoptosis, such as the appearance of hypodiploid nuclei (Fig. 3) or the incorporation of labeled-nucleotides by the enzyme terminal deoxynucleotidyl-transferase (TdT), employed in the TUNEL method (not shown). The percentage of CD8+ lymphocytes undergoing apoptosis after in vitro exposure to β-gal peptide was unexpectedly high (nearly 60% after the background is subtracted in the β-gal-expressing IL-2-rVV group). The most likely explanation for this high apoptotic rate could be the extremely high numbers of precursors that have been revealed by recent studies employing ELISPOT assays and class I tetramers (34, 35). In addition, there may also be nonspecific apoptotic death of “bystander” cells interacting with the Mac-1+/Gr-1+ cells.
The molecular mechanism causing apoptosis of CD8+ T cells is not known at present. In experiments with diffusion chambers, we found that cell-cell contact was required for loss of cytolytic capability and apoptosis to occur (Fig. 4). Fas Ag (CD95) seemed a likely candidate for the observed cell contact-dependent immunosuppression. The FasR is a cell surface protein that is a member of the TNFR family, and it is expressed on many cells in the immune system and other tissues. Its main function is to trigger apoptosis when complexed with FasL (CD95L) (36, 37). The activation of mature human T cells sensitizes them to Fas-mediated apoptosis (38, 39). This is important in the regulation of the immune response and the maintenance of self-tolerance as illustrated by patients with a deficiency in FasR known as autoimmune lymphoproliferative syndrome (ALPS). These patients exhibit massive T cell expansions resulting in lymphadenopathy and splenomegaly as well as varied autoimmune responses (40, 41). However, the cell membrane-associated mechanism responsible for apoptosis of CD8+ T cells in our system did not involve Fas-FasL as indicated by the observation of the same IL-2-rVV-induced suppression in MRL-lpr/lpr mice, the murine counterpart to ALPS (Fig. 5). This finding is consistent with the observations of Lohman et al. (42) that in vivo there were significant numbers of apoptotic cells in the spleens of lpr/lpr mice after LCMV infection. This indicates that Fas is not required for the immune down-regulation of the CD8+ T lymphocyte response after acute LCMV infection. Further, TNF/TNFR, and CTLA-4/B7-1/B7-2 interactions were not responsible for the effect as a wide range of concentrations of blocking Abs against TNF-α or CTLA-4 were completely ineffective or detrimental to the generation of a CTL response (V.B. and N.P.R., unpublished observations).
The Mac-1+/Gr-1+ cells we describe here are currently being further characterized and are comprised mostly of monocytes and a population of myeloid precursors. These latter cells closely resemble the cells previously designated “natural suppressor” (NS) cells. Comparison of the functional qualities of these cells with those of NS cells is interesting. Phenotypically, NS cells lack the usual markers of mature B cells, T cells, or macrophages and do not kill classical NK targets. They have been found in several environments involving intense hematopoiesis such as neonatal/ newborn spleens, adult bone marrow, adult spleens after total lymphoid irradiation, during growth of some tumors, after treatment with cyclophosphamide, or during graft-vs-host-disease (43-46). We have recently characterized an identical population of suppressor elements in mice bearing large tumor burdens (V.B. and N.P.R., manuscript in preparation).
We and others have recently demonstrated that IL-2 can profoundly enhance the activity of CD8+ T cells after immunization (18). These findings have been extended to the clinic in cancer immunotherapy trials where the addition of adjuvant IL-2 to synthetic peptide immunogens results in enhanced tumor destruction. However, at the same time we observed an apparent disappearance of Ag-specific T cells from the circulation (47). Based on the data presented, one might speculate that the disappearance of these anti-tumor T cells may be due to a suppressive population of Mac-1+/Gr-1+ cells.
In conclusion, we have characterized the function of a population of cells involved in a novel feedback pathway that directs the death of T lymphocytes. Immunizations that are separated by intervals that are too short to allow for the activities of regulatory cells to subside may be detrimental for the immune responses that we seek to elicit. The induction or isolation of these cells could represent a powerful tool useful in the control of autoimmunity and transplant rejection.
Acknowledgments
We thank P. Spiess and D. Jones for help with the animal experiments, M. Blalock for assistance with graphics, E. P. Shulman for tissue culture preparations, R. Kirken for the purification of anti-Gr-1 mAb, and A. Mixon for the flow cytometry experiments.
Footnotes
This work was supported, in part, by the Italian Association for Cancer Research, AIRC (V.B.) and the Strong Children's Research Center, University of Rochester School of Medicine, Rochester, NY (M.W.).
Abbreviations used in this paper: LCMV, lymphocytic choriomeningitis virus; VV, vaccinia virus; FPV, fowlpox virus; NS, natural suppressor; NP, nucleoprotein; β-gal, β-galactosidase; ALPS, autoimmune lymphoproliferative syndrome; FasL, Fas ligand; PFU, plaque-forming unit; WT, wild-type.
References
- 1.Scott DW, Grdina T, Shi Y. T cells commit suicide, but B cells are murdered! J. Immunol. 1996;156:2352. [PubMed] [Google Scholar]
- 2.Lenardo MJ. Interleukin-2 programs mouse αβ T lymphocytes for apoptosis. Nature. 1991;353:858. doi: 10.1038/353858a0. [DOI] [PubMed] [Google Scholar]
- 3.Critchfield JM, Racke MK, Zuniga-Pflucker JC, Cannella B, Raine CS, Goverman J, Lenardo MJ. T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science. 1994;263:1139. doi: 10.1126/science.7509084. [DOI] [PubMed] [Google Scholar]
- 4.Welsh RM, Selin LK, Razvi ES. Role of apoptosis in the regulation of virus-induced T cell responses, immune suppression, and memory. J. Cell. Biochem. 1995;59:135. doi: 10.1002/jcb.240590202. [DOI] [PubMed] [Google Scholar]
- 5.Abbas AK. Die and let live: eliminating dangerous lymphocytes. Cell. 1996;84:655. doi: 10.1016/s0092-8674(00)81042-9. [DOI] [PubMed] [Google Scholar]
- 6.Van Parijs L, Abbas AK. Role of Fas-mediated cell death in the regulation of immune responses. Curr. Opin. Immunol. 1996;8:321. doi: 10.1016/s0952-7915(96)80125-7. [DOI] [PubMed] [Google Scholar]
- 7.Wong B, Choi Y. Pathways leading to cell death in T cells. Curr. Opin. Immunol. 1997;9:358. doi: 10.1016/s0952-7915(97)80082-9. [DOI] [PubMed] [Google Scholar]
- 8.Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 1993;362:758. doi: 10.1038/362758a0. [DOI] [PubMed] [Google Scholar]
- 9.Razvi ES, Welsh RM. Programmed cell death of T lymphocytes during acute viral infection: a mechanism for virus-induced immune deficiency. J. Virol. 1993;67:5754. doi: 10.1128/jvi.67.10.5754-5765.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Razvi ES, Jiang Z, Woda BA, Welsh RM. Lymphocyte apoptosis during the silencing of the immune response to acute viral infections in normal, lpr, and Bcl-2-transgenic mice. Am. J. Pathol. 1995;147:79. [PMC free article] [PubMed] [Google Scholar]
- 11.Young MR, Wright MA, Lozano Y, Matthews JP, Benefield J, Prechel MM. Mechanisms of immune suppression in patients with head and neck cancer: influence on the immune infiltrate of the cancer. Int. J. Cancer. 1996;67:333. doi: 10.1002/(SICI)1097-0215(19960729)67:3<333::AID-IJC5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 12.Sulitzeanu D. Immunosuppressive factors in human cancers. Adv. Cancer Res. 1993;60:247. doi: 10.1016/s0065-230x(08)60827-1. [DOI] [PubMed] [Google Scholar]
- 13.Tsuchiya Y, Igarashi M, Suzuki R, Kumagai K. Production of colony-stimulating factor by tumor cells and the factor-mediated induction of suppressor cells. J. Immunol. 1988;141:699. [PubMed] [Google Scholar]
- 14.Haskill S, Koren H, Becker S, Fowler W, Walton L. Mononuclear-cell infiltration in ovarian cancer. III. Suppressor-cell and ADCC activity of macrophages from ascitic and solid ovarian tumours. Br. J. Cancer. 1982;45:747. doi: 10.1038/bjc.1982.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alleva DG, Walker TM, Elgert KD. Induction of macrophage suppressor activity by fibrosarcoma-derived transforming growth factor-β 1: contrasting effects on resting and activated macrophages. J. Leukocyte Biol. 1995;57:919. doi: 10.1002/jlb.57.6.919. [DOI] [PubMed] [Google Scholar]
- 16.Schleifer KW, Mansfield JM. Suppressor macrophages in African trypanosomiasis inhibit T cell proliferative responses by nitric oxide and prostaglandins. J. Immunol. 1993;151:5492. [PubMed] [Google Scholar]
- 17.al-Ramadi BK, Brodkin MA, Mosser DM, Eisenstein TK. Immunosuppression induced by attenuated Salmonella: evidence for mediation by macrophage precursors. J. Immunol. 1991;146:2737. [PubMed] [Google Scholar]
- 18.Bronte V, Tsung K, Rao JB, Chen PW, Wang M, Rosenberg SA, Restifo NP. IL-2 enhances the function of recombinant poxvirus-based vaccines in the treatment of established pulmonary metastases. J. Immunol. 1995;154:5282. [PMC free article] [PubMed] [Google Scholar]
- 19.Wang M, Bronte V, Chen PW, Gritz L, Panicali D, Rosenberg SA, Restifo NP. Active immunotherapy of cancer with a nonreplicating recombinant fowlpox virus encoding a model tumor-associated antigen. J. Immunol. 1995;154:4685. [PMC free article] [PubMed] [Google Scholar]
- 20.Chakrabarti S, Brechling K, Moss B. Vaccinia virus expression vector: coexpression of β-galactosidase provides visual screening of recombinant virus plaques. Mol. Cell. Biol. 1985;5:3403. doi: 10.1128/mcb.5.12.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Earl PL, Moss B. Generation of vaccinia viruses and their products. In: Ausubel FM, Kinston R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. Greene Publishing Associates and Wiley Interscience; New York: 1991. p. 16.18.1. [Google Scholar]
- 22.Smith GL, Levin JZ, Palese P, Moss B. Synthesis and cellular location of the ten influenza polypeptides individually expressed by recombinant vaccinia viruses. Virology. 1987;160:336. doi: 10.1016/0042-6822(87)90004-3. [DOI] [PubMed] [Google Scholar]
- 23.Whitman ED, Tsung K, Paxson J, Norton JA. In vitro and in vivo kinetics of recombinant vaccinia virus cancer-gene therapy. Surgery. 1994;116:183. [PubMed] [Google Scholar]
- 24.Bronte V, Carroll MW, Goletz TJ, Wang M, Overwijk WW, Marincola F, Rosenberg SA, Moss B, Restifo NP. Antigen expression by dendritic cells correlates with the therapeutic effectiveness of a model recombinant poxvirus tumor vaccine. Proc. Natl. Acad. Sci. USA. 1997;94:3183. doi: 10.1073/pnas.94.7.3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gavin MA, Gilbert MJ, Riddell SR, Greenberg PD, Bevan MJ. Alkali hydrolysis of recombinant proteins allows for the rapid identification of class I MHC-restricted CTL epitopes. J. Immunol. 1993;151:3971. [PubMed] [Google Scholar]
- 26.Overwijk WW, Surman DR, Tsung K, Restifo NP. Identification of a Kb-restricted CTL epitope of β-galactosidase: potential use in development of immunization protocols for “self” antigens. Methods. 1997;12:117. doi: 10.1006/meth.1997.0461. [DOI] [PubMed] [Google Scholar]
- 27.Sherwood SW, Schimke RT. Cell cycle analysis of apoptosis using flow cytometry. Methods Cell Biol. 1995;46:77. doi: 10.1016/s0091-679x(08)61925-1. [DOI] [PubMed] [Google Scholar]
- 28.Wang M, Chen PW, Bronte V, Zhai Y, Rosenberg SA, Restifo NP. Anti-tumor activity of cytotoxic T lymphocytes elicited with recombinant and synthetic forms of a model tumor antigen. J. Immunother. 1995;18:139. doi: 10.1097/00002371-199510000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hestdal K, Ruscetti FW, Ihle JN, Jacobsen SE, Dubois CM, Kopp WC, Longo DL, Keller JR. Characterization and regulation of RB6-8C5 antigen expression on murine bone marrow cells. J. Immunol. 1991;147:22. [PubMed] [Google Scholar]
- 30.Saron MF, Shidani B, Nahori MA, Guillon JC, Truffa-Bachi P. Lymphocytic choriomeningitis virus-induced immunodepression: inherent defect of B and T lymphocytes. J. Virol. 1990;64:4076. doi: 10.1128/jvi.64.9.4076-4083.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Selin LK, Nahill SR, Welsh RM. Cross-reactivities in memory cytotoxic T lymphocyte recognition of heterologous viruses. J. Exp. Med. 1994;179:1933. doi: 10.1084/jem.179.6.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zarozinski CC, Welsh RM. Minimal bystander activation of CD8 T cells during the virus-induced polyclonal T cell response. J. Exp. Med. 1997;185:1629. doi: 10.1084/jem.185.9.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ehl S, Hombach J, Aichele P, Hengartner H, Zinkernagel RM. Bystander activation of cytotoxic T cells: studies on the mechanism and evaluation of in vivo significance in a transgenic mouse model. J. Exp. Med. 1997;185:1241. doi: 10.1084/jem.185.7.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butz EA, Bevan MJ. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity. 1998;8:167. doi: 10.1016/s1074-7613(00)80469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McMichael A, O'Callaghan CA. A new look at T cells. J. Exp. Med. 1998;187:1367. doi: 10.1084/jem.187.9.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lynch DH, Ramsdell F, Alderson MR. Fas and FasL in the homeostatic regulation of immune responses. Immunol. Today. 1995;16:569. doi: 10.1016/0167-5699(95)80079-4. [DOI] [PubMed] [Google Scholar]
- 37.Matiba B, Mariani SM, Krammer PH. The CD95 system and the death of a lymphocyte. Semin. Immunol. 1997;9:59. doi: 10.1006/smim.1996.0054. [DOI] [PubMed] [Google Scholar]
- 38.Owen-Schaub LB, Yonehara S, Crump WL, Grimm EA. DNA fragmentation and cell death is selectively triggered in activated human lymphocytes by Fas antigen engagement. Cell Immunol. 1992;140:197. doi: 10.1016/0008-8749(92)90187-t. [DOI] [PubMed] [Google Scholar]
- 39.Ju ST, Panka DJ, Cui H, Ettinger R, el-Khatib M, Sherr DH, Stanger BZ, Marshak-Rothstein A. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature. 1995;373:444. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- 40.Sneller MC, Wang J, Dale JK, Strober W, Middelton LA, Choi Y, Fleisher TA, Lim MS, Jaffe ES, Puck JM, Lenardo MJ, Straus SE. Clincial, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood. 1997;89:1341. [PubMed] [Google Scholar]
- 41.Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middleton LA, Lin AY, Strober W, Lenardo MJ, Puck JM. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 42.Lohman BL, Razvi ES, Welsh RM. T-lymphocyte downregulation after acute viral infection is not dependent on CD95 (Fas) receptor-ligand interactions. J. Virol. 1996;70:8199. doi: 10.1128/jvi.70.11.8199-8203.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strober S. Natural suppressor (NS) cells, neonatal tolerance, and total lymphoid irradiation: exploring obscure relationships. Annu. Rev. Immunol. 1984;2:219. doi: 10.1146/annurev.iy.02.040184.001251. [DOI] [PubMed] [Google Scholar]
- 44.Oseroff A, Okada S, Strober S. Natural suppressor (NS) cells found in the spleen of neonatal mice and adult mice given total lymphoid irradiation (TLI) express the null surface phenotype. J. Immunol. 1984;132:101. [PubMed] [Google Scholar]
- 45.Maier T, Holda JH, Claman HN. Graft-vs-host reactions (GVHR) across minor murine histocompatibility barriers. II. Development of natural suppressor cell activity. J. Immunol. 1985;135:1644. [PubMed] [Google Scholar]
- 46.Jaffe ML, Arai H, Nabel GJ. Mechanisms of tumor-induced immunosuppression: evidence for contact-dependent T cell suppression by monocytes. Mol. Med. 1996;2:692. [PMC free article] [PubMed] [Google Scholar]
- 47.Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, Restifo NP, Dudley ME, Schwarz SL, Spiess PJ, Wunderlich JR, Parkhurst MR, Kawakami Y, Seipp CA, Einhorn JH, White DE. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat. Med. 1998;4:321. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]