

Abstract
The past two decades have witnessed a dramatic accumulation of evidence indicating that the excitatory amino acid glutamate plays an important role in drug addiction and alcoholism. The purpose of this review is to summarize findings on glutamatergic substrates of addiction, surveying data from both human and animal studies. The effects of various drugs of abuse on glutamatergic neurotransmission are discussed, as are the effects of pharmacological or genetic manipulation of various components of glutamate transmission on drug reinforcement, conditioned reward, extinction, and relapse-like behavior. In addition, glutamatergic agents that are currently in use or are undergoing testing in clinical trials for the treatment of addiction are discussed, including acamprosate, N-acetylcysteine, modafinil, topiramate, lamotrigine, gabapentin and mematine. All drugs of abuse appear to modulate glutamatergic transmission, albeit by different mechanisms, and this modulation of glutamate transmission is believed to result in long-lasting neuroplastic changes in the brain that may contribute to the perseveration of drug-seeking behavior and drug-associated memories. In general, attenuation of glutamatergic transmission reduces drug reward, reinforcement, and relapse-like behavior. On the other hand, potentiation of glutamatergic transmission appears to facilitate the extinction of drug-seeking behavior. However, attempts at identifying genetic polymorphisms in components of glutamate transmission in humans have yielded only a limited number of candidate genes that may serve as risk factors for the development of addiction. Nonetheless, manipulation of glutamatergic neurotransmission appears to be a promising avenue of research in developing improved therapeutic agents for the treatment of drug addiction and alcoholism.
1. Introduction
Drug addiction is defined by several diagnostic criteria set forth by the American Psychiatric Association [1]. These criteria include a loss of control over drug intake, repeated unsuccessful attempts at quitting or reducing drug use, continued drug use despite negative consequences, a reduction in engagement in social, occupational and recreational activities in lieu of drug-seeking or self-administration behavior, and the emergence of symptoms of tolerance or withdrawal. Historically, research into the neurobiological substrates that underlie the rewarding and reinforcing effects of drugs of abuse has focused on the mesolimbic dopamine reward circuitry, comprised primarily of dopaminergic neurons in the ventral tegmental area (VTA) that project rostrally to forebrain and limbic regions such as the nucleus accumbens (NAcc), amygdala and frontal cortex [2]. However, as can be seen in Fig. 1, there has been a dramatic increase in attention that has been given to the role of the excitatory amino acid glutamate in drug addiction and alcoholism over the past two decades. The purpose of this review is to summarize the effects of drugs of abuse on glutamatergic neurotransmission as well as key findings on the role of glutamate transmission in drug reinforcement, the rewarding effects of drugs of abuse, extinction of drug-seeking behavior, and relapse. Various glutamatergic medications that are either approved for clinical use or are being examined in clinical trials for the treatment of addictive disorders will also be discussed. Finally, a brief summary of findings on potential genetic linkages between individual components of glutamate neurotransmission and addiction is presented.
Fig. 1.

Graph showing the increasing number of publications on the topic of glutamate and addiction over the past 20 years. Two separate PubMed searches were performed in April 2007, one using “drug addiction” and “glutamate” as key words (open circles) and the other using “alcoholism” and “glutamate” as key words (filled circles). The resulting number of publications (including review articles and commentaries) are plotted by year for 1987 to 2006.
2. Animal models of drug addiction and alcoholism
One of the most widely used methods to study drug addiction in animals is the intravenous self-administration (IVSA) paradigm [3]. In this model, experimental animals are implanted with indwelling intravenous catheters (most often the jugular vein in rodent studies) and are trained to perform an operant task (i.e., lever press or nose-poke) in order to receive an intravenous infusion of a cocaine, amphetamine, nicotine, etc. In the case of alcohol (ethanol), execution of the operant task results in presentation of a small amount of an alcohol-containing solution in a receptacle where the animal can consume the solution orally (some studies measure alcohol consumption in the home cage by presentation of two or more bottles containing ethanol solutions). By definition, if the delivery or presentation of the drug solution increases this behavior (i.e., appropriate lever pressing or nose-poking), the drug or alcohol solution is considered to be a positive reinforcer. Environmental cues, such as presentation of stimulus lights or auditory tones, are often paired with drug delivery or alcohol presentation to promote stimulus-drug associations, which enhance drug-taking behavior and can be utilized in subsequent reinstatement tests (see below). The effects of experimental manipulations (i.e., administration of test compounds either systemically or intracerebrally) on drug or alcohol self-administration behavior can then be assessed. However, the effects of any such manipulation must be interpreted with caution. For example, while it is tempting to interpret an observed decrease in self-administration behavior as signifying a reduction in the desire to self-administer the drug (and thus having possible therapeutic applications), there are equally plausible alternative explanations for the observed reduction in drug self-administration. For example, the experimental manipulation might have caused an overall reduction motor output, or an increase in sensitivity to the drug, resulting in less drug required to produce the same subjective effects. Therefore, in this review, to avoid the pitfalls of these alternative explanations, we will refer to alterations in operant drug IVSA or ethanol consumption as changes in reinforcement.
The operant self-administration paradigm is also amenable to studying the phenomenon of relapse. The most widely used animal model of relapse is the reinstatement paradigm [3]. While this model by no means perfectly mirrors the phenomenon of relapse in humans, it is considered to be the best model developed thus far [4]. In the reinstatement paradigm, following the achievement of stable levels of self-administration, animals undergo extinction training procedures, where the behavior that previously resulted in the delivery of the drug solution (i.e., lever press or nose-poke) no longer produces any consequences. As a result of this imposed drug unavailability (i.e., “forced abstinence”), animals will gradually extinguish (i.e., reduce) the behavior that previously resulted in drug delivery. Once predesignated extinction criteria have been obtained (for example, presses on the “active” drug-delivering lever are reduced to less than 20% of those observed when the drug was available), it is possible to “reinstate” operant responding by presenting one of the three main types of stimuli that are known to evoke relapse in humans: exposure to stressors, presentation of drug-associated environmental cues, or brief exposure to the drug itself. Upon presentation of one or more of these stimuli, animals will resume performing the operant task that previously led to drug delivery. This resumption, or “reinstatement”, of performing of the operant task is commonly interpreted as “drug-seeking behavior”. However, it should be noted that during reinstatement testing, the operant task does not result in actual drug delivery, so as to avoid the psychoactive effects produced by the drug which can confound interpretation of changes in drug-seeking behavior. This exemplifies one of the main divergences between the reinstatement model and relapse in humans, as the latter most often results in drug consumption.
Another animal model of drug addiction is the conditioned place preference (CPP) paradigm [3]. Although it is widely used, this paradigm does not measure active drug seeking or reinforcement; rather, it utilizes Pavlovian conditioning procedures to provide an index of the “rewarding” (or subjective pleasurable) effects of drugs of abuse based on the animal’s preference for a drug-paired environment over a non-drug paired environment. A typical CPP apparatus consists of two conditioning compartments with unique sensory characteristics (i.e., visually distinct wall patterns, flooring with unique tactile properties, or distinct olfactory cues). These two “conditioning” compartments are often connected by a neutral center “start” compartment. In a typical CPP experiment, an animal undergoes baseline preference testing and habituation, where it is placed in the center start compartment and allowed free access to both conditioning chambers for a specified amount of time. This allows for the animal to habituate to the testing environment as well as for the experimenter to determine if the animal exhibits any innate bias towards one of the two conditioning compartments (an ideal CPP apparatus would produce no innate preferences for either compartment). Then, the animal is subject to conditioning proceures where the conditioning drug (i.e., morphine, cocaine, amphetamine, etc.) is administered and the animal is confined to one of the two conditioning compartments for a specific amount of time. On alternate days, the animal is injected with saline or vehicle and then confined to the other conditioning compartment for the same amount of time. These conditioning sessions are repeated in an alternating fashion over a period of several days to allow the animal to form associations between the unique physical characteristics of the drug-paired compartment and the subjective effects of the conditioning drug. Finally, on the test day, the animal is placed back in the center compartment and allowed free access to both conditioning compartments. If the animal spends significantly more time in the drug-paired compartment compared to the saline-paired compartment, a CPP has been acquired. A conditioned place aversion (CPA) is observed if the animal spends significantly less time in the drug-paired compartment as compared with the saline-paired compartment. Drugs with aversive subjective properties, such as lithium chloride, or withdrawal from chronic drug administration reliably produce CPA.
The CPP paradigm has provided substantial information on the neural substrates of the rewarding effects of drugs of abuse. One advantage of this paradigm is that the procedures are relatively simple, inexpensive, and less time-consuming to conduct than intravenous drug self-administration. In addition, CPP paradigms can also be used to model various aspects of relapse. This is accomplished by extinguishing an established CPP by repeatedly pairing the previous drug-paired compartment with saline, or by allowing the CPP to dissipate over time with repeated testing of place preference. Then, pharmacological or other experimental manipulations can be introduced that result in a reinstatement of the original CPP. A disadvantage of the CPP technique, however, is that it does not directly measure drug-seeking behavior, but rather the motivation for secondary reinforcers (i.e., drug-associated environments) [3]. In addition, attempts to manipulate drug CPP via pharmacological or genetic methods do not always predict effects of those manipulations on drug self-administration behavior [3, 5, 6].
3. Glutamatergic neurotransmission
Glutamate is the most abundant excitatory neurotransmitter in the brain, mediating as much as 70% of synaptic transmission within the central nervous system and reaching extracellular concentrations in the low millimolar range. A diagram of a typical glutamatergic synapse is shown in Figure 2. Glutamate is packaged into synaptic vesicles in the presynaptic terminal by vesicular glutamate transporters (vGluTs) using a proton gradient generated by the hydrolysis of adenosine triphosphate (ATP). Thus far, three different vGluTs have been identified (vGluT1-3) [7]. Once released into the synaptic cleft, glutamate can bind to one of three different types of ionotropic glutamate receptors (iGluRs) located on the head of the postsynaptic spine: the N-methyl-D-aspartate (NMDA) receptor, the α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor, and the kainic acid (kainate, KA receptor). iGluRs are ligand-gated ion channels that mediate fast excitatory neurotransmission. Glutamate can also bind to metabotropic glutamate receptors (mGluRs) located in perisynaptic regions or on the presynaptic terminal (see Figure 2).
Fig. 2.

The glutamatergic synapse. Glutamate is packaged into synaptic vesicles in the presynaptic terminal by vGluT. When an action potential arrives at the terminal, glutamate is released by exocytosis into the synaptic cleft where it binds to and activates iGluRs (NMDA, AMPA and KA receptors) localized on the postsynaptic neuron, which results in cation influx and subsequent activation of VGCCs that propagate the action potential. The resulting cation influx can activate numerous second messenger systems, including PKA and CaMKII, which in turn interact with other signaling molecules or transcription factors can which modulate gene expression, local mRNA translation, or cytoskeletal remodeling. iGluR subunits can be phosphorylated by numerous kinases such as PKC, CaMKII, Fyn and others, altering the activity and functionality of these receptors. NMDA receptor subunits have recently been discovered to be expressed by glial cells and on presynaptic terminals (not shown). Glutamate can also be released into the extracellular space via nonexocytotic mechanisms such as cystine-glutamate-exchanger (xc) located on glial cells. Whether released from the presynaptic terminal or neighboring glial cells, extracellular glutamate binds and activates not only iGluRs but also postsynaptic mGluRs in the perisynaptic annulus, which are either positively coupled to PKC activity and mobilize intracellular Ca2+ from IP3-gated intracellular stores (as is the case for mGluR1 and 5) or negatively regulate AC (as is the case for mGluRs 2, 3, 4, 6, 7, and 8). Group I mGluRs positively regulate NMDA receptor function via PKC. Like iGluRs, mGluRs function can be altered by phosphorylation by various kinases. Glutamate release by the presynaptic terminal is negatively regulated by Group II or III mGluR autoreceptors, and is cleared from the extracellular space by EAATs located either on the presynaptic terminal, neighboring glial cells, or the postsynaptic neuron. In glia, glutamate is converted to glutamine, which is then transported back to the presynaptic terminal and converted back to glutamate. Although the numerous proteins that make up the postsynaptic density complex are not shown in this diagram, it should be noted that the Homer family of scaffolding of proteins links NMDA receptors to Group I mGluRs and IP3-gated intracellular Ca2+ stores, and several recent studies implicate Homer proteins in psychostimulant and alcohol addiction (see Sections 4 and 9).
NMDA receptors are heterotetrameric protein complexes that form ligand-gated ion channels composed of at least one NR1 subunit (for which there are at least 8 splice variants) and a combination of NR2A-D and NR3A or 3B subunits [8-10] (In mice, the NR1 subunit was previously been named ζ1 and the NR2A-D were previously named ε1-4). In addition to being stimulated by glutamate, amino acids such as D-serine and glycine act as co-agonists at the NMDA receptor. The NR2 subunits contain the glutamate binding domain, whereas the NR1 subunit contains the glycine-binding dopamine. Under resting conditions, the NMDA receptor channel pore is blocked by Mg2+ ions, but once sufficient membrane depolarization has been established (i.e., by opening of AMPA receptor channels), the Mg2+ block is removed, allowing the influx of cations (primarily Ca2+ ions, but the NMDA receptor is also permeable to K+ and Na+ ions). Activity of the NMDA receptor is modulated by polyamines and inhibited by Zn2+. The subunit composition of NMDA receptors are ontogenetically regulated and are neuroanatomically distinct. Once thought to be exclusively located on neurons, NMDA receptors have recently been shown to be expressed on glial cells including microglia, astrocytes and oligodendrocytes [10]. NMDA receptor subunits have also been found to exist on presynaptic terminals [11]. The NMDA receptor has been extensively implicated in mediating neural plasticity as well as learning and memory processes [12-14].
AMPA receptors are also heterotetrameric protein complexes that form ligand-gated ion channels composed of various subunits termed GluR1-4 (also termed GluRA-D) and GluRδ1 and 2 [8]. The mRNAs encoding AMPA subunits can be edited or alternatively spliced to form variants such as the flip and flop isoforms. Each GluR subunit contains a binding site for glutamate. Once activated, AMPA receptors are permeable to various cations including Ca2+, Na+ and K+, although the majority of AMPA receptors in the brain contain GluR2 subunits, which render the channel impermeable to Ca2+. Similar to NMDA receptors, AMPA receptor function can be modulated in the presence of polyamines. It is believed that both NMDA and AMPA receptors are necessary for the induction of many forms of synaptic plasticity such as long-term potentiation (LTP) and long-term depression (LTD) [15-21].
Like NMDA and AMPA receptors, kainic acid (kainate, KA) receptors are also tetrameric protein complexes that form ligand-gated ion channels composed of various subunits. These subunits are termed GluR5-7 and KA1 and 2 [8]. KA receptors can form homomeric tetramers composed entirely of GluR5, 6 or 7 subunits or heteromeric complexes containg GluR5 or KA subunits. KA receptors are permeable to Na+ and K+ ions and, like NMDA and AMPA receptors, contribute to excitatory postsynaptic currents. The role of KA receptors in synaptic plasticity is less well-defined, however, but KA receptors have been found to be localized presynaptically where they can modulate neurotransmitter release [22].
In addition to the iGluRs, glutamate can also bind to mGluRs, which are located either in the perisynaptic annulus or on presynaptic terminals. mGluRs are seven transmembrane domain G-protein coupled receptors (GPCRs) that mediate slower, modulatory glutamatergic transmission. mGluRs can be divided into three distinct groups, based on their pharmacological and signal transduction properties. Group I mGluR receptors (mGluR1 and mGluR5) activate the Gαq class of G-proteins, which stimulate one of several phospholipases (including phospholipase C), resulting in phosphoinositol hydrolysis and the formation of lipid signaling intermediates such as inositol triphosphate (IP3) and diacylglycerol (DAG), which in turn can activate various intracellular messengers including protein kinase C (PKC) [23-25]. Activation of Group I mGluR receptors also mobilizes calcium release from IP3 receptor-mediated intracellular stores, which can in turn activate other intracellular messengers such as calmodulin-dependent kinase II (CaMKII). Group I mGluRs, particularly mGluR5, are positively coupled to NMDA receptor function via PKC, and are structurally linked to these receptors as well as IP3-gated intracellular Ca2+ stores via the Homer family of proteins [26-30]. Group I mGluRs are rarely found presynaptically. Group II (mGluR2 and mGluR3) and Group III (mGluR4, mGluR6, mGluR7, and mGluR8) mGluRs, on the other hand, activate the Gαi class of G-proteins and are negatively coupled to adenylyl cyclase (AC) activity, and upon stimulation result in decreased intracellular levels of cyclic adenosine monophosphate (cAMP). Presynaptically localized Group II and Group III mGluRs, particularly mGluR2 and mGluR3, are thought to represent the classical inhibitory autoreceptor mechanism that suppresses excess glutamate release. mGluR3 and mGluR5 have been localized to glial cells such as astrocytes [31].
Activation of iGluRs alone is sufficient for the propagation of the action potential by the postsynaptic neuron, and can activate various intracellular signaling molecules including protein kinase A (PKA), mitogen-activated protein kinase (MAPK), and extracellular signal-related kinase (ERK) [32] (see Fig. 2). Activation of additional signaling molecules, such as PKC, is achieved by activation of mGluRs. Together, the simultanous activation of iGluRs and mGluRs activates a host of intracellular signaling pathways that result in protein phosphorylation of ion channels, other kinases, and transcription factors and eventually leads to the molecular events underlying neural plasticity. Such events include initiation and/or regulation of dendritic mRNA translation and de novo protein synthesis, changes in gene expression in the nucleus, and cytoskeletal remodeling (Fig. 2) [33]. Glutamate-mediated neural plasticity is also characterized by changes in iGluR subunit trafficking such as insertion of AMPA receptors into postsynaptic plasma membrane (see Section 11).
Glutamate is cleared from the extracellular environment by a family of sodium-dependent excitatory amino acid transporters (EAATs). To date, five separate EAATs have been idenfitied (EAAT1-5). EAAT1-3 are alternatively termed GLAST, GLT-1, and EAAC1, respectively. EAAT2 and EAAT5 are localized to the presynaptic terminal, EAAT3 and EAAT4 are localized to the postsynaptic neuron, and EAAT1 and EAAT2 are expressed in glial cells [7]. This family of EAATs provides numerous mechanisms to prevent an excessive accumulation of extracellular glutamate, which if high enough concentrations are reached, can result in excitotoxicity. Once inside glial cells, glutamate is converted to glutamate by glutamine synthetase, where it is secreted back outside the glia and taken up by the presynaptic terminal for conversion back to glutamate by glutaminase (Fig. 2). Conversely, glutamate can be transported from within glial cells to the extracellular environment by the cystine-glutamate exchanger (xc) [34-37].
As mentioned earlier, glutamatergic transmission accounts for up to 70% of synaptic transmission in the central nervous system. Thus, there are glutamatergic projections and/or neurons expressing glutamate receptors in numerous circuitries of the brain, including the mesolimbic dopamine reward circuitry. This “reward” circuitry is composed primarily of dopamine-synthesizing neurons in the VTA of the ventral midbrain that project rostrally to target regions such as the nucleus accumbens (NAcc), amygdaloid complex (Amyg) and frontal cortex (FC). Each of these regions receive substantial glutamatergic input [38, 39] (Fig. 3). For example, the VTA receives glutamatergic projections from the FC, Amyg, pendunculopontine tegmentum (PPT), and laterodorsal tegmentum (LDT) [40, 41]. A recent report indicated that a subpopulation of VTA neurons express vGluT2 mRNA [42], suggesting the existence of neurons intrinsic to this region that utilize glutamate as a neurotransmitter. The NAcc receives a convergence of glutamatergic input from the FC, Amyg, hippocampal formation (Hipp), and various nuclei of the thalamic (Thal). The FC cortex receives glutamatergic input from the Hipp, Amyg and Thal. Thus, there is a robust excitatory glutamatergic innervation of the mesolimbic dopamine reward circuitry which proves and anatomical basis for dopamine-glutamate interactions in regulating the addictive properties of drugs of abuse as well as synaptic plasticity [43-45] (see Section 11 for further discussion of this topic).
Fig. 3.
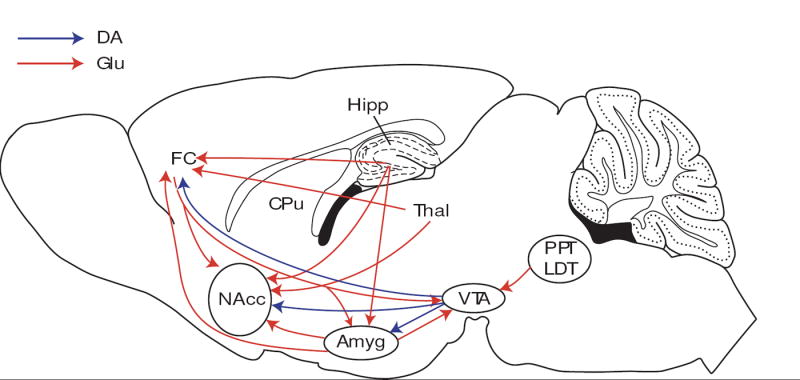
Sagittal section of the rodent brain showing neuroanatomical interactions between glutamate and mesolimbic dopamine systems. The “reward circuit” is hypothesized to consist of dopamine-synthesizing cell bodies in the VTA that project rostrally to innervate the NAcc, Amyg and FC, as well as other regions such as the portions of the CPu and ventral pallidum (not shown). This mesolimbic reward pathway is robustly innervated by glutamate-containing neurons. Dopamine-containing cell bodies in the VTA receive glutamatergic input from the PPT, LDT, Amyg and FC. The NAcc receives a host of glutamatergic innervation from the FC, Hipp, Thal, and Amyg. The FC receives glutamatergic input from the Hipp, Thal and Amyg. Drawing adapted from the atlas of Franklin and Paxinos [906]. Abbreviations: Amyg, amygdala; CPu, caudate-putamen (dorsal striatum); FC, frontal cortex; Hipp, hippocampus; LDT, laterodorsal tegmentum; NAcc, nucleus accumbens; PPT, pedunculopontine tegmentum; Thal, thalamus; VTA, ventral tegmental area.
In light of all the receptor proteins and transporters associated with glutamatergic neurotransmission, a substantial array of pharmacological ligands has become available to researchers attempting to examine the role of glutamatergic transmission in preclinical models of drug addiction. Table 1 details some of the more commonly used glutamatergic ligands that are employed in preclinical addiction research.
Table 1.
Commonly used glutamatergic ligands in preclinical addiction research
| Ligand | Chemical Name | Mode of Action |
|---|---|---|
| AP5 (APV) | 2-amino-5-phosphonovaleric acid | NMDA antagonist (competitive) |
| Dizocilpine (MK-801) | (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine | NMDA antagonist (non-competitive) |
| Ifenprodil | 2-(4-benzylpiperidino)-1-(4-hydroxyphenyl)-1-propanol | NMDA antagonist (polyamine site) (some preference for NR2B) |
| GYKI 52466 | 4-(8-methyl-9H-1,3-dioxolo[4,5-h][2,3]benzodiazepin-5-yl)-benzenamine | AMPA antagonist (non-competitive) |
| NBQX | 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide | AMPA/KA antagonist (competitive) |
| CNQX | 6-cyano-7-nitroquinoxaline-2,3-dione | AMPA/KA antagonist (competitive) (also NMDA glycine site antagonist) |
| DNQX | 6,7-dinitroquinoxaline-2,3-dione | AMPA/KA antagonist (competitive) |
| CPCCOEt | 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester | mGluR1 antagonist (allosteric) |
| LY367385 | (S)-(+)- α-amino-4-carboxy-2-methylbenzeneacetic acid | mGluR1 antagonist (allosteric) |
| EMQMCM | (3-ethyl-2-methyl-quinolin-6-yl)-(4-methoxy-cyclohexyl)-methanone methane sulfonate | mGluR1 antagonist (allosteric) |
| MPEP | 2-methyl-6-(phenylethynyl)pyridine | mGluR5 antagonist (allosteric) |
| MTEP | 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine | mGluR5 antagonist (allosteric) |
| LY379268 | (1R,4R,5S,6R)-4-amino-2-oxabicyclo[3.1.0]hexane-4,6-dicarboxylic acid | mGluR2/3 agonist (orthosteric) |
| DCPG | (S)-3,4-dicarboxyphenylglycine | mGluR8 agonist (orthosteric) |
| TBOA | DL-threo-β-benzyloxyaspartic acid | EAAT inhibitor |
| MS-153 | (R)-(-)-5-methyl-1-nicotinoyl-2-pyrazoline | EAAT activator |
4. Glutamate and cocaine
Cocaine is a potent inhibitor of presynaptic monoamine transporter function, and as a result produces substantial increases in extracellular levels of dopamine, serotonin and norepinephrine in the synaptic cleft, particularly in forebrain terminal fields that receive monoamineric projections. Cocaine also acts as a local anesthetic by blocking sodium channels and thereby inhibiting the propagation of action potentials. Some of the first observations that cocaine interacts with glutamatergic systems in the brain were reported in the late 1980’s and early 1990’s, when several groups of investigators showed that that sensitization to the locomotor stimulant effects of cocaine was blocked by pretreatment of the NMDA antagonist MK-801 [46], and that infusion of iGluR antagonists into the NAcc reduced the motor stimulant and reinforcing effects of cocaine [47-51]. Since these observations, a tremendous amount of evidence has accumulated indicating an important role for glutamate in the motor stimulant properties of cocaine and its role in locomotor sensitization, and the reviewer is directed to various reviews elsewhere for in depth details on this topic [52-60]. Here we will focus on the effects of cocaine on glutamatergic transmission as well as the effects of glutamatergic ligands on the rewarding and reinforcing effects of cocaine.
In addition to elevating extracellular levels of monoamines, in vitro release or in vivo microdialysis studies have shown that cocaine also increases extracellular levels of glutamate in various brain regions including the dorsal striatum [61], NAcc [62-71], septum [72], ventral pallidum [73, 74], VTA [75, 76] and cerebellum [77]. However, it should be noted that not all studies have observed cocaine-induced increases in extracellular glutamate, including studies in self-administering animals [78]. Others have found stimulatory effects of cocaine on extracellular glutamate only with high (i.e., 30 mg/kg i.p.) doses of cocaine [63, 65, 66], which may cause neurotoxicity. Thus, the ability of cocaine to induce increases in extracellular glutamate is not a universally observed phenomenon. Nonetheless, immunohistochemical studies have shown that cocaine decreases glutamate immunoreactivity in presynaptic terminals several brain regions including as the NAcc [79-81], suggesting that some of the observed elevations in extracellular glutamate in response to cocaine are derived from neuronal stores. Several reports suggest that environmental contexts and cues are important modulatory factors in the ability of cocaine to elevate extracellular levels of glutamate in the NAcc [67, 82] but not the frontal cortex [83].
Glutamate mediates numerous neuronal effects of acute exposure to cocaine. Low doses of acutely administered cocaine enhance glutamate-evoked neuronal firing in the cortex, striatum, or NAcc, whereas higher or self-administered doses tend to inhibit glutamate-evoked neuronal activity in these regions [84-86]. However, the ability of high concentrations of locally administered cocaine to suppress glutamate-induced neuronal activity should be interpreted with caution, since cocaine exerts local anesthetic effects via blockade of voltage-gated sodium channels. Drugs of abuse including cocaine can produce elevated resting neuronal membrane potentials (i.e., “up” states) in regions such as the dorsal striatum, NAcc, and FC, and this phenomenon requires coordinated interactions between dopaminergic and glutamatergic transmission [87, 88].
The ability of acute cocaine exposure to increase striatal neuropeptide expression and activate signaling molecules such as ERK is dependent on NMDA receptor activation [89-91]. Acute administration of cocaine has been shown to increase phosphorylation of the GluR1 subunit of the AMPA receptor in the striatum [92], induce a redistribution of AMPA and NMDA receptors in the VTA [93-95], and induce the formation of NR2B-D2 heteroreceptor complexes [96]. The expression of various iGluR subunits is reduced by acute cocaine. For example, in situ hybridization studies have shown that mRNA levels for GluR3, GluR4 and NR1 are decreased in the NAcc by acute cocaine exposure, as is NR1 mRNA expression in the striatum and VTA [97], perhaps as a compensatory response to increased glutamate overflow produced by cocaine. However, acute cocaine has been shown to increase NR1 mRNA expression in the hippocampus [98] and GluR2 mRNA in the striatum [99].
Repeated cocaine exposure can lead to a phenomenon called “behavioral sensitization” (sometimes termed “reverse tolerance”), which is a progressive increase in the behavioral (i.e., locomotor) response to cocaine in response to repeated exposure to the same dose. Behavioral sensitization to cocaine is paralleled by adaptive changes in mesolimbic dopamine system function as well as the responsiveness of this system to glutamate. For example, repeated exposure of rats to cocaine or amphetamine results in a transient enhanced responsiveness of VTA dopamine neurons to locally applied glutamate [100], and it was later determined that this phenomenon was a result of increased responsiveness of AMPA and not NMDA or mGluR receptors located on VTA dopamine neurons [101]. Such an effect is paralleled by an increase in expression of GluR1 receptor levels in the VTA, but not the substantia nigra, following repeated cocaine exposure [102-104]. Expression of NR1 in the VTA has also been reported to be increased following repeated cocaine, but the expression of NR2A/B, GluR2/3 or GluR6/7 is unaltered [102-105]. Some investigators have not observed changes in iGluR expression in the VTA [97, 106-108]. These discrepancies may be due to the type of detection methodology used (i.e., immunoblotting, radioimmunohistochemistry, or in situ hybridization), variations in cocaine dose, length of withdrawal period prior to analysis, or the inclusion of a final challenge administration of cocaine in order to demonstrate the maintenance of behavioral sensitization. Nonetheless, post-mortem analysis of human brain tissue samples from cocaine overdose victims did reveal an upregulation of numerous iGluR subunits in the VTA [109, 110], suggesting that perhaps cocaine, at least at high doses, alters iGluR subunit expression in the VTA.
In the primary target field of VTA DA neurons, the NAcc, it has been demonstrated that multiple cocaine exposures result in a sensitized increase in extracellular levels of glutamate [64, 65] but see [111], paralleled by an accumulation of presynaptic glutamate immunoreactivity in this region [112]. Changes in glutamate receptor expression in the NAcc following repeated cocaine exposure are, however, complex and inconsistent across studies. Some investigators have reported no changes in expression of NR1, NR2A/B, GluR1-2, or KA receptor subunits in the NAcc within 24 hr of discontinuation of cocaine treatment [102, 103], or have reported decreases in NR1 and GluR3/4 expression [97, 98]. However at later time points, others have observed increases in NR1 expression in this region [97, 103, 113], but only in rats that exhibited signs of behavioral sensitization. Reductions in NR2B expression in the NAcc were observed after 24 hr [114] but not 1 week [115] of cocaine withdrawal. In an elegant protein cross-linking study, it was demonstrated that repeated cocaine exposure increases the suface expression of GluR1-3 subunits in the NAcc at 3 weeks following discontinuation of treatment, but only in rats showing behavioral signs of sensitization [116]. Collectively, these data show that the effects of repeated cocaine exposure on iGluR subunit expression can vary considerably depending on many experimental factors, and that any lack of observed changes in overall protein expression may overlook more subtle adaptive changes such as increased surface expression of AMPA receptor subunits.
All of the aforementioned studies on the effects of repeated cocaine administration on iGluR expression utilized response non-contingent (i.e., passive, experimenter-administered) administration of the drug. Interestingly, a recent study by Hemby and colleagues examined the effect of active intravenous cocaine self-administration on changes in iGluR subunit expression in the NAcc [108]. It was found that during the early phases of cocaine withdrawal, NR1 and GluR5 expression was reduced in the VTA, no changes in any subunit examined were observed in the NAcc, and in the frontal cortex NR1 levels were increased and GluR2-6 and KA2 levels were decreased. Although it is difficult to compare these results with those reviewed above due to the many procedural differences involved, this study was one of the first to examine changes in iGluR subunit expression in addiction-related brain regions following active self-administration of the drug.
During protracted cocaine withdrawal, extracellular levels of glutamate and presynaptic glutamate immunoreactivity are decreased in the NAcc [36, 37, 64, 67, 69, 79-81] but increased in the prefrontal cortex [117]. This reduction in extracellular levels of glutamate in the NAcc may be due to desensitization of presynaptic Group II mGluRs [118-121] as well as a down-regulation of xc function [36], which tightly regulate extracellular glutamate levels. As a result, increases in NR1, GluR1-3, and Group I mGluR expression have been observed in the NAcc [97, 103, 104, 122, 123], suggesting that adaptations to reduced extracellular glutamate levels may occur. The observed decreases in extracellular glutamate during cocaine withdrawal have been shown to be critically involved in relapse-like behavior, since pharmacological restoration of basal extracellular glutamate levels to those observed in non-withdrawn animals, as with agents such as N-acetylcysteine that promote the activity of xc (see Fig. 4 and Section 13.2), inhibit the ability of cocaine priming to further increase extracellular levels glutamate levels in the NAcc. The ability of N-acetylcysteine to inhibit cocaine-induced increases in NAcc glutamate overflow is paralleled by blockade of cocaine-induced reinstatement [37, 69], suggesting that an imbalance in glutamatergic transmission in the NAcc is may induce a predisposition towards relapse.
Fig. 4.

Hypothesized mechanism by which N-acetylcysteine (NAC) restores extracellular glutamate levels in the NAcc and prevents cocaine-induced reinstatement. (A) Cocaine self-administration induces glutamate release from prefrontal cortical neurons that synapse onto medium spiny neurons in the NAcc. (B) Repeated exposure to cocaine results in a desensitization of presynaptic Group II mGluRs and also down-regulates the efficacy of the cystine-glutamate exchanger (xc), resulting in decreased extracellular glutamate levels during cocaine withdrawal. (C) NAC, a cystine pro-drug, is converted to cystine and drives xc to transport glutamate from within glial cells into the extracellular environment. The resulting “normalization” of extracellular glutamate levels in the NAcc is believed to prevent further glutamate release produced by a priming injection of cocaine, thereby reducing subsequent reinstatement of cocaine-seeking behavior. See section 11.2 for more detail.
Other changes brought on by repeated cocaine exposure include:
an upregulation of NMDA receptor binding in the regions such as the cortex, striatum, amygdala and hippocampus has been reported following repeated cocaine exposure [124-126] but see [127]
a decrease in NR1 and/or NR2B/2C expression in regions such as globus pallidus, subiculum, striatum, and cerebellum [98, 105]
an up-regulation of NR1 and a down-regulation of GluR2-7 and KA2 expression in cerebral cortex [97, 108, 128]
increased phosphorylation of GluR1 in the prefrontal cortex [115]
an increase in mGluR5 expression in the hippocampus [129]
bidirectional alterations in the expression of NMDA and AMPA receptor subunit expression in the amygdala [130, 131]
A role for glutamatergic transmission in the rewarding and reinforcing effects of cocaine has been clearly demonstrated by pharmacological studies utilizing iGluR antagonists. Systemic administration of NMDA antagonists attenuate cocaine reinforcement [132-135] and the acquisition and/or expression of a cocaine CPP [136, 137]. Similar reductive effects on cocaine reinforcement have been reported following administration of inhibitors of glutamate carboxypeptidase II [138], which reduce free glutamate availability. The ability of iGluR antagonists to reduce cocaine reinforcement are likely mediated, at least in part, by NMDA and/or AMPA receptors in the NAcc and dorsal striatum, as evidenced by microinjection studies [49, 139, 140] but see [141].
Stimulation of AMPA receptors in the NAcc has been shown to reinstate previously extinguished cocaine-seeking behavior [141-143], and AMPA antagonists infused into the NAcc block reinstatement induced by priming injections of cocaine [142], cocaine-associated cues [144], or infusions of cocaine into the FC [145]. On the other hand, some studies have reported that NMDA receptor antagonists, whether administered systemically or into the NAcc shell, actually induce reinstatement of cocaine-seeking behavior [145-147]. The reason for the opposing effects of NMDA and AMPA antagonists on reinstatement are currently unclear, but may involve cocaine-induced increased AMPA receptor responsiveness (relative to NMDA) due to trafficking of AMPA subunits to the plasma membrane [116]. The FC has been identified as the primary source of glutamatergic afferents to the NAcc that mediate cocaine-primed reinstatement [68]. Together these data suggest a critical role for iGluRs in the NAcc in mediating the reinforcing effects of cocaine as well as the ability of exposure to cocaine or drug-associated cues to reinstate cocaine-seeking behavior (see also [148, 149]). However, further research into identifying the unique, and possibly opposing, contributions of AMPA and NMDA receptors in the NAcc to reinstatement of cocaine-seeking behavior is needed.
Glutamatergic transmission in the VTA also plays a role in cocaine reward, reinforcement and reinstatement. For example, electrical stimulation of glutamatergic fibers in the ventral subiculum reinstates cocaine-seeking behavior in a manner dependent on glutamate transmission in the VTA [150]. Blockade of NMDA or AMPA receptors in the VTA blocks the development of cocaine CPP [151] and cue-induced reinstatement [152]. Expression and phosphorylation of GluR1 in the VTA also mediates the reinforcing effects of cocaine [153].
The basolateral amygdaloid nucleus (BLA) also plays a role in cue-induced reinstatement of cocaine-seeking behavior (reviewed in [154]). However, conflicting results have emerged as the the role of glutamate transmission in this phenomenon. For example, infusion of NMDA into the BLA reinstates previously extinguished cocaine-seeking behavior [155], yet infusions of iGluR antagonists in this region apparently do not attenuate cue-induced reinstatement [156]. Thus, while glutamatergic transmission in this region may play a role in reinstatement per se, iGluRs apparently do no mediate the ability of drug-associated cues to induce reinstatement. In addition, AMPA receptors in the NAcc mediate cocaine-seeking behavior only when BLA dopamine receptors are blocked [157]
mGluRs, particularly Group I and Group II mGluRs, are also involved in cocaine reward and reinforcement. Following a pivotal study by Chiamulera and colleagues who showed that mice carrying a targeted deletion of the mGluR5 gene do not self-administer cocaine [158], numerous investigators have shown that mGluR5 antagonists reduce cocaine reinforcement and reinstatement [159-165]. mGluR5 receptor antagonists also reduce the development and/or expression of cocaine CPP [166, 167]. Dampening glutamate transmission via stimulation of presynaptic mGluR2/3 receptors or activating glutamate transporters attenuates cocaine reinforcement [168, 169], cue- and cocaine-induced reinstatement [168, 170], “incubation” of cocaine craving (i.e., a progressive increase in the magnitude of cue-induced reinstatement over time following cocaine self-administration) [171], and the development of cocaine CPP [172]. The NAcc and amygdala appear to be important mediators of some of these effects [144, 169, 171].
In addition to the aforementioned pharmacological evidence, studies utilizing genetically modified mice have also shed light on the role of glutamatergic transmission in cocaine addiction, as summarized below:
genetic deletion of GluR1 and/or GluR2 does not alter the acquisition of cocaine CPP [173, 174], but impairs extinction of cocaine-seeking behavior [174]; however, another group of investigators showed a failure of GluR1 null mutant mice to develop cocaine CPP [175]
cocaine CPP is attenuated in mice engineered to express an NR1 subunit with reduced cation flux properties explicitly in D1-containing neurons [176]
Mice carrying a targeted deletion of the mGluR5 gene do not self-administer cocaine and are indifferent to its locomotor stimulant effects [158]
genetic deletion of mGluR2 enhances cocaine CPP [71].
Altered behavioral and neurochemical responses to cocaine are observed in Homer1 or Homer2 knockout mice, as described below.
Homer proteins are a family of scaffolding proteins that link NMDA, Group I mGluRs and IP3-gated intracellular calcium stores to the postsynaptic density. A growing body of literature suggests that the expression of these proteins is regulated by cocaine and that they play an important role in the behavioral and neurochemical effects of this psychostimulant. Acute cocaine treatment induces a rapid but transient increase in Homer 1a but not Homer1b/c or Homer 2a/b expression in the dorsal striatum [177]. Chronic cocaine treatment, on the other hand, reduces Homer 1b/c levels in the NAcc [119]. In addition, antisense knockdown of Homer1 expression in this region sensitizes rats to the locomotor stimulant effects of cocaine and also downregulates GluR1 expression [178], suggesting that cocaine-induced down-regulation of Homer1 proteins may regulate its behavioral sensitizing effects. Subsequently, it was shown that mice carrying a targeted deletion of either the Homer1 or Homer2 gene demonstrate increased locomotor stimulant and rewarding effects (as measured by CPP) of cocaine as compared to wildtype controls [179-181], thus exhibiting a behavioral phenotype similar to that of cocaine-sensitized animals. In addition, Homer2 deficient mice exhibited a more rapid acquisition of cocaine IVSA and decreased basal extracellular glutamate levels in the NAcc (similar to cocaine withdrawn animals). Remarkably, many of these behavioral and neurochemical changes could be reversed by virally-mediated restoration of Homer2 in the NAcc [180]. These findings highlight the importance of Homer proteins and the functional consequences of alterations in their expression as a result of repeated cocaine exposure.
5. Glutamate and amphetamines
Amphetamines are psychomotor stimulant drugs that promote the release of monoamines by reversing the directionality of vesicular and plasmalemmal monoamine transporters located in presynaptic terminals, thereby promoting the release of dopamine, norepinephrine and serotonin into the synaptic cleft. Although there are many amphetamine-related compounds, the primary analogues that will be focused on in this review are d-amphetamine (herein referred to as “amphetamine”), methamphetamine, and methylenedioxymethamphetamine (MDMA, ‘Ecstasy’).
As with cocaine, the first observations that glutamate was involved in the psychomotor stimulant properties of amphetamine were reported in the late 1980’s, when it was demonstrated that sensitization to the locomotor stimulant effects of amphetamine was blocked by co-administration of the NMDA antagonist MK-801 [46], and that infusion of iGluR antagonists into the NAcc reduced the motor stimulant effects of amphetamine [47]. The reader is directed to various reviews elsewhere for in depth details on the role of glutamate in amphetamine-induced locomotor activity and behavioral sensitization [52-60]. In addition, the reader is also directed to several recent reviews on the role of glutamate in amphetamine-induced neurotoxicity [182-184].
In addition to being monoamine-releasing agents, amphetamines also elevate extracellular levels of glutamate, as demonstrated by in vitro release and in vivo microdialysis studies in the cerebral cortex [66, 185-190], dorsal striatum [187, 191] [192-202] but see [203], NAcc [66, 190, 204-207], hippocampus [187, 208, 209] and VTA [205, 210, 211]. However, it should be noted that some of these studies used doses of amphetamine that were in the neurotoxic range (i.e., 9-10 mg/kg i.p.) [193, 204]. On the other hand, high doses of methamphetamine are often intentionally utilized to explore the hypothesis that prolonged increases in extracellular glutamate levels underlie the neurotoxic effects of this drug (c.f., [192, 195, 196, 201, 202, 212]). Presynaptic glutamate immunoreactivity has been shown to be decreased in various brain regions following methamphetamine administration [213, 214], suggested that some of the elevations in extracellular glutamate induced by this drug are derived from neuronal stores. Elevation of extracellular glutamate by amphetamines in various brain regions is not likely a result of alterations in EAAT function [215, 216], although one study implicated EAATs in the ability of amphetamine to increase glutamate overflow in the VTA [211]. Some investigators have shown that local administration of high concentration of amphetamines actually reduce extracellular glutamate levels in certain brain regions [193, 217, 218] and suppress glutamate evoked neuronal activity in the NAcc and cerebral cortex [219, 220].
Acute administration of amphetamines has been consistently shown to increase the expression immediate early genes such as c-fos and Zif268, various neuropeptide precursors, and induce phosphorylation of various transcription factors in the dorsal striatum, all of which are dependent on iGluR- and/or mGluR-mediated mechanisms [89, 221-229]. Oddly, however, amphetamine-induced increases in the expression of immediate early genes in the NAcc appear to be NMDA-receptor independent [230]. Acute administration of methamphetamine has been shown to increase phosphorylation of the GluR1 subunit of the AMPA receptor in the striatum [92], which alters AMPA receptor channel conductance and promotes trafficking to the plasma membrane. With regards to VTA dopaminergic neurons, acute administration of amphetamines generally tends to inhibit the firing of these neurons by inducing somatodendritic release of dopamine, which in turn stimulates inhibitory D2-like autoreceptors. However, it has also been shown that acute amphetamine may actually excite VTA dopamine neurons by inhibition of mGluR-mediated inhibitory postsynaptic currents [231], Amphetamine can also induce cellular hallmarks of neural plasticity in these neurons, such as increasing the AMPA component of evoked excitatory postsynaptic currents [232-234]
Repeated exposure of amphetamines can have markedly different effects on glutamate-mediated neuronal activity and function as compared with acute exposure. For example, as mentioned above, acute administration of amphetamine can suppress glutamate evoked neuronal activity in the NAcc and cerebral cortex [219, 220]. However, repeated amphetamine administration results in enhanced neuronal responsiveness to locally applied glutamate in the VTA [100, 101] and frontal cortex [235] but not NAcc [100]. It is unclear if the mechanisms underlying the enhanced responsiveness to glutamate are the same across these regions.
Repeated amphetamine exposure also results in various changes in other components of glutamate transmission. For example, repeated methamphetamine administration produces a reduction in NMDA NR1, NR2A and NR2B protein levels in the striatum [236] but increases vGluT1 levels in this region, which may facilitate the incorporation of glutamate into synaptic vesicles to promote long-lasting methamphetamine-induced increases in extracellular glutamate [202]. Repeated exposure to amphetamine also produces changes in AMPA receptor expression which can be either short- or long-lasting. In a series of studies conducted by Wolf and colleagues, it was shown that 5 days of amphetamine treatment produced decreases in mRNA and protein levels of GluR1, GluR2 and NR1 in the NAcc and increased NR1 expression in the frontal cortex at 14 but not 3 days following that last amphetamine exposure [237-239]. However, changes in GluR1 in the frontal cortex were only transient, being increased at 3 but not 14 days following the last amphetamine administration [237, 238]. With regards to the VTA, it has been shown that repeated amphetamine treatment does not alter AMPA subunit expression [106, 240], but does sensitize the ability of intra-VTA applied AMPA to increase in extracellular levels of glutamate in this region [241], suggesting a sensitizing effect of repeated amphetamine on AMPA receptor function in the VTA without alteration in subunit expression. However, many of these studies examined receptor protein levels in tissue homogenates by immunoblotting, or immunoreactivity or mRNA expression at the level of the cell body. Thus, it is possible that more subtle changes in receptor levels, such as increased surface expression of AMPA receptor subunits as has been observed following cocaine treatment [116], may be overlooked by use of these techniques.
Repeated amphetamine also alters the expression of mGluRs, causing transient increases mGluR1 expression in the dorsal and ventral striatum, but more persistent reductions in mGluR5 expression in these regions [242]. Repeated amphetamine also increases hippocampal and cortical mGluR5 expression [243, 244]. Repeated amphetamine administration does not alter EAAT2 or EAAT3 expression in various regions including the midbrain, NAcc, dorsal striatum or FC [245, 246]; however, methamphetamine has been shown to increase the expression of EAAT2 in the striatum [247].
Glutamate appears to play an important role in the rewarding and reinforcing effects of amphetamines. Reductions in glutamate transmission by administration of the glutamate release inhibitor riluzole [248], the glutamate transporter activator MS-153 [172], infusion of an AMPA/KA antagonist into the NAcc [249], and virally-mediated overexpression of EAAT2 in the NAcc [250] all have been shown to attenuate the development of amphetamine CPP. However, it was also found that antagonism of mGluR2/3 receptors, which facilitates glutamate transmission, disrupts the ability of intra-NAcc amphetamine to establish a CPP [251], suggesting that excessive glutamatergic transmission may actually disrupt the neuronal communication that normally subserves the ability of amphetamine to establish a CPP. Our laboratory demonstrated that the development of amphetamine CPP was not attenuated by the mGluR5 antagonist MPEP [166]. However, others have shown that the expression of amphetamine CPP, but not MDMA CPP, is suppressed by MPEP [252].
With regards to amphetamine reinforcement and relapse-like behavior, surprisingly little attention has been given to the potential role of glutamate. Both dextromethorphan and the African tree shrub extract ibogaine, which have NMDA receptor antagonist properties, reduce methamphetamine reinforcement [253, 254] and the establishment of amphetamine CPP [255]. However, ibogaine has been reported to have numerous other neurochemical effects including nicotinic acetylcholine receptor (nAChR) antagonism [256], and current evidence from studies with ibogaine congeners that are devoid of NMDA antagonist activity indicate that the inhibitory effects of ibogaine on amphetamine reward and reinforcement are likely mediated by antagonism of nAChRs rather than NMDA receptor blockde [257]. However, one recent study did show that stimulation of mGluR2/3 receptors attenuates enhanced amphetamine reinforcement in amphetamine-sensitized rats [258], suggesting that glutamatergic transmission may indeed regulate the reinforcing effects of amphetamines. Clearly, more research in this area is needed.
6. Glutamate and opiates
There are dozens of opiate alkaloid compounds that are used clinically for pain management, including morphine, codeine, hydrocodone, oxycodone, meperidine, and fentanyl. Many of these compounds exhibit potential for abuse and addiction. For the purposes of this review, we will focus on interactions between glutamate and just two of these opiate drugs: morphine, which is considered the “gold standard” of narcotic analgesics, and its illegal and highly addictive diacetylated form, heroin.
Opiate alkaloids bind with high affinity to one or more of several opioid receptor proteins, including the μ, δ and κ subtypes [259]. Opioid receptors are GPCRs that are negatively coupled to AC activity and normally subserve neurotransmission mediated by endogenous opioid peptides such as enkephalins, endorphins and dynorphins. There is a substantial amount of literature suggesting that abused opiates such as morphine and heroin interact with glutamatergic transmission. While many of these opioid-glutamate interactions take place in the spinal cord and brainstem in the mediation of nociception, in this review we will focus primarily on interactions in supraspinal regions of the brain known to be involved in addiction.
Most in vitro and in vivo studies have shown that morphine suppresses basal and evoked increases in extracellular glutamate in regions such as the cerebral cortex [260-266], dorsal striatum [262, 267-269] but see [270, 271], NAcc [272, 273], globus pallidus and ventral pallidum [268, 274-277] and hippocampus [278], but not the VTA [279]. The ability of morphine to dampen extracellular glutamate levels is likely mediated by opioid receptors located presynaptically on glutamatergic terminals. Morphine can also act postsynaptically to suppress glutamate-evoked neuronal excitation [280-283]. The resulting decrease in neuronal activity is reflected by other studies showing that the stimulatory effects of acute morphine on the expression of immediate-early genes such as c-fos and c-jun in the dorsal and ventral striatum is blocked by NMDA and/or AMPA receptor antagonists [284-286].
Acute morphine administration induces neuronal plasticity in the VTA, as evidence by an increase in the AMPA/NMDA ratio of evoked excitatory postsynaptic currents [232]. In the NAcc core, a reduction in the expression of NR1, NR2B, NR2C, GluR1-4, and GluR6 was observed 3 days after acute morphine exposure; however, 21 days following acute morphine exposure, expression of all iGluR subunits was reported to be increased [287]. Transient changes in components of glutamatergic signaling have also been described in the hippocampus following acute morphine exposure, with the expression of mRNA encoding the NR1, NR2A and NR2B subunit proteins being reduced 4 hr after administration, returning to preinjection levels by 24 hr post-treatment [288].
More robust changes in glutamatergic signaling have been observed following repeated exposure to morphine. While acute exposure to morphine can suppress glutamate-evoked neuronal responses (see above), repeated exposure to morphine can result in tolerance to this effect, which can ultimately result in neuronal supersensitivity to glutamate [289-292]. Chronic morphine exposure reduces glutamate uptake in the FC, striatum and hippocampus [293], paralleled by decreases in striatal EAAT2 expression [294]. Reductions in [3H]MK-801 or [3H]glutamate binding in various brain regions have been reported following repeated morphine exposure [295-297], while [3H]MK-801 binding is increased in the hippocampus [298]. Along these lines, the induction of LTP at mossy fiber synapses in the hippocampus has been observed following repeated morphine exposure [299], which may be a result of altered AMPA receptor dynamics [300], an increase in glutamate receptor number or binding sites [298] or a molecular rearrangement of the postsynaptic density complex [301]. There are several reports that repeated morphine exposure does not alter NR1, NR2A or NR2B expression in the hippocampus [113, 130] or even decreases NR1 expression in this region [302]. These discrepancies are likely due to variations in dose, duration of morphine administration, and method of exposure (daily injections vs. continuous infusions via subcutaneous pellet or osmotic minipump implantation).
Rats self-administering morphine show an increase in plasma membrane levels of GluR1 in the basolateral amygdala [303], and during morphine withdrawal, expression of NR1 is increased in this region [130], which may contribute to the aversive motivational properties of opiate withdrawal [304]. Some investigators have reported that repeated morphine does not alter NR1, NR2A or NR2B expression in the FC [113] while others have reported that chronic morphine exposure decreases expression of these subunits in this region [302]. Similarly, some have reported that chronic morphine increases NR1 and NR2A expression in the NAcc [302, 305] and produces a shift towards NR2A-mediated NMDA function, such as decreased glycine binding and faster rates of receptor desensitization [306]. Yet others have reported that chronic morphine has no effect on NMDA subunit expression in the NAcc [113, 307]. As mentioned earlier, such discrepancies are more likely due to dose, duration, and method of morphine exposure. Chronic morphine upregulates mGluR5 expression in the limbic forebrain [308], which may be relevant to the ability of mGluR5 antagonists to attenuate the conditioned rewarding effects of morphine (see below).
One particular adaptation in glutamatergic signaling that may be important for the development of addiction to opiates is the reported upregulation of the expression of the AMPA subunit GluR1 in the VTA by repeated morphine exposure [102]. In an elegant pair of studies, Nestler and colleagues showed that behavioral correlates of repeated morphine exposure, such as locomotor sensitization and CPP, can be mimicked in drug-naïve animals by virally-mediated overexpression of GluR1 in the VTA [309], particularly in the rostral VTA [310]. Thus, the VTA may be a crucial site whereby repeated opiate exposure produces adaptive changes in glutamate neurotransmission that increase the propensity towards opiate addiction.
As opposed to the suppression of extracellular glutamate levels that is characteristic of acute morphine exposure, morphine withdrawal is characterized by increased overflow of glutamate in regions such as the locus coeruleus [311, 312], NAcc [272, 313], and hippocampus [278]. This withdrawal-induced increase in glutamate overflow is accompanied by reductions in Group II-mediated LTD in the NAcc [314], likely due to a desensitization of presynaptic mGluR2/3 receptors. However, in the dorsal striatum, extracellular levels of glutamate are reduced during morphine withdrawal [271], paralleled by an increased in EAAT2 expression [294]. Hippocampal synapses show increased surface expression of EAAT2 and parallel increases in glutamate uptake during morphine withdrawal [315]. Morphine withdrawal is also characterized by enhanced presynaptic inhibition of excitatory input into the VTA [316], which may result in reduced brain reward circuitry function that is characteristic of drug withdrawal [317].
Perhaps one of the most consistent findings in the literature on opiate-glutamate interactions is the ability of reduced glutamatergic signaling to attenuate the development of tolerance to the antinociceptive effects of morphine and to ameliorate the behavioral signs of morphine withdrawal. One of the first studies along these lines was published by Trujillo and Akil [318]. In this study, the NMDA receptor antagonist MK-801 not only blocked the development of tolerance to the antinociceptive effects of morphine, but also the development of physical dependence on morphine, as evidence by reduced naloxone-precipitated withdrawal symptoms. Since this study, numerous others have shown that morphine tolerance and/or withdrawal symptoms (including naloxone-precipitated CPA) are reduced by:
genetic deletion of NR2A [331]
genetic deletion of GluR1 [332]
administration of a glutamate release inhibitor [339]
activation of glutamate transporters [340]
inhibition of the processing of the putative glutamate precursor peptide N-acetyl-aspartylglutamate [341, 342]
On the contrary, facilitation of glutamatergic transmission, such as by administration of the glutamate transport inhibitor TBOA [343] or mGluR2/3 antagonists [344], actually exacerbates somatic signs of opiate withdrawal. There is a general consensus that the behavioral signs of opioid withdrawal are largely mediated by increased glutamatergic drive to the locus coeruleus [336, 344-350], an area that provides noradrenergic innervation to much of the brain. Reductions in glutamate transmission in this region reduce the expression of morphine withdrawal symptoms [348-350]. However, blockade of NMDA or AMPA receptors in the VTA also attenuates the behavioral signs of morphine withdrawal [351, 352], indicating that this region is also involved in generating symptoms of opiate withdrawal.
Glutamate is important for the rewarding properties of morphine, as measured by the CPP paradigm. Impairment of glutamatergic transmission by administration of NMDA and/or AMPA antagonists, either systemically [341, 353, 354] or into reward-related regions such as the NAcc [353, 355], ventral pallidum [356], central amygdala [357] or VTA [355, 358] attenuate the development and/or expression of morphine CPP. This is not due to drug substitution effects, since iGluR antagonists do not have morphine-like discriminative stimulus effects [359, 360]. Likewise, reductions in glutamatergic transmission by systemic administration of a glutamate transporter activator [172], a glutamate release inhibitor [248], genetic inactivation of the NR2A subunit of the NMDA receptor [331] or virally-mediated overexpression of EAAT2 in the NAcc [250] can attenuate the development of morphine CPP. Morphine-induced reinstatement of CPP can be attenuated by NMDA antagonists [361-363]. On the other hand, potentiation of glutamate transmission by the glutamate uptake inhibitor TBOA [343] or injection of NMDA into the CeA [357] facilitates the expression of morphine CPP. With regards to mGluRs, we found that doses of MPEP up to 20 mg/kg do not inhibit the development of morphine CPP in mice [166]. However, other investigators have found that higher doses of MPEP block the acquisition and/or expression of morphine CPP in rats [167, 308, 364]. Thus, there is ample evidence to support the notion that the functionality of glutamate transmission is positively correlated with the rewarding effects of morphine.
Heroin (diacetylmorphine) is abused primarily for its intense euphorigenic properties. There are, however, only a handful of studies examining interactions between this highly addictive opiate drug and glutamatergic signaling. One of the first studies demonstrating an interaction between heroin and glutamate was the observation that blockade of NMDA receptors in the NAcc with MK-801 attenuated the locomotor stimulant effects of heroin [48]. However, this same manipulation did not attenuate the reinforcing effects of heroin [49]. Infusions of NMDA antagonists into the VTA, in contrast, decrease heroin reinforcement, whereas administration of an AMPA/KA antagonist into this region actually increases heroin reinforcement [365], suggesting opposing roles of VTA NMDA and AMPA receptors in regulating the addictive properties of this opiate. Heroin administration is accompanied by delayed increases in extracellular levels of glutamate in the ventral pallidum [366], a region known to be involved in regulating heroin reinforcement [367]. Finally, cue-and/or context-induced reinstatement of heroin-seeking behavior is attenuated by stimulation of mGluR2/3 receptors, administered either systemically [368, 369] or directly into the VTA [368] or NAcc [370], but not the dorsal striatum [370]. mGluR2/3 agonists have no effect on baseline heroin reinforcement [369]. These studies indicate that Group II mGluRs in both the NAcc and VTA regulate the ability of contextual or other environmental cues to reinstate extinguished heroin-seeking behavior. Further research is clearly needed on the potential clinical utility of both iGluR and mGluR ligands in the treatment of heroin addiction.
7. Glutamate and nicotine
Although cigarette smoke contains several thousand different chemical compounds, nicotine is considered to be the primary component that promotes addiction to cigarettes. Nicotine binds with high affinity to nAChRs, which are pentameric ligand-gated cation channels comprised of various combinations of α and β type subunits. Nicotine is thought to exert is rewarding and reinforcing effects by activating VTA dopamine-containing neurons expressing nAChRs composed of the α4β2 subunit combination [371-374]. Alternatively, there is also evidence suggesting that nAChRs containing the α7 subunit are localized presynaptically on glutamatergic afferents to the VTA [375], and thus activation of these receptors by nicotine increases glutamate release in the VTA and activates iGluRs located postsynaptically on VTA dopamine neurons, with the end result of increasing the activity of the mesolimbic reward circuit [376-386].
Many studies have shown that nicotine elevates extracellular glutamate levels in a number of brain regions including the cerebral cortex [387-393], dorsal striatum[387, 388, 391, 394, 395], NAcc [376, 396-399], hippocampus [400], hypothalamus [387], locus coeruleus [401, 402] and cerebellum [403, 404]. As a result, excitatory postsynaptic currents and neural activity are increased by nicotine in many of these regions via iGluR-mediated mechanisms [382, 389, 405-423], including the induction of hippocampal LTP [424-426]. As a result, nicotine may induce long-lasting synaptic plasticity in numerous brain regions, which may promote addiction to this substance.
Repeated exposure to nicotine can produce adapative changes in the expression of various proteins related to glutamate neurotransmission. Rats and mice repeatedly administered nicotine, either passively or by active self-administration, show decreased levels of NR2A and NR2B levels in the striatum [427], and increased levels of these proteins in the FC [428]. Consistent with these latter findings, a microarray study revealed that in the post-mortem cerebral cortex of human cigarette smokers, a significant increase in the expression of GluR1 and NR2A is observed [429]. Chronic nicotine self-administration in rats also increases GluR2/3 expression in the VTA but does not induce changes in NR2A, NR2B, or GluR2/3 levels in the NAcc [428]. An upregulation of the glutamate transporter EAAT2 has been reported in the cerebellum in response to chronic nicotine exposure [430]. The expression of Group I mGluRs and Homer1 and Homer2 mRNA in the amygdala, NAcc and VTA are altered by nicotine [431], although many of these changes are only transient. Finally, a proton magnetic resonance spectroscopy analysis of humans smokers, former smokers and controls showed no differences in concentrations of glutamate in the hippocampus or anterior cingulate cortex [432]. Although the results of this study were negative, they represent a novel attempt at examining changes in glutamate transmission in the living human brain as a result of nicotine addiction.
Some of the adaptive changes in glutamate transmission produced by nicotine may be age-related, which may provide a neural basis for the enhanced vulnerability to nicotine addiction during adolescence in both humans and animals [433-435]. For example, repeated exposure of adolescent mice to nicotine produces a downregulation of the GluR2/3 levels in the striatum, whereas opposite effects are observed in adult exposed animals [427]. Thus, nicotine may produce age-dependent adaptations in glutamatergic transmission, and further research on this topic is needed to determine the glutamatergic substrates underlying enhanced vulnerability to nicotine addiction during the adolescent stage of development.
Pharmacological studies on nicotine reinforcement, relapse, and withdrawal have provided important developments in possible glutamate-based interventions for the treatment of nicotine addiction. NMDA antagonists such as MK-801 or memantine block the development and expression of locomotor sensitization to nicotine [436] as well the acquisition of nicotine IVSA [135]. Blockade of mGluR5 receptors has been shown to decrease the reinforcing effects of nicotine but not food [159, 160, 437, 438]. mGluR5 antagonists also decrease the break-point for nicotine reinforcement on a progressive ratio schedule [163]. Blockade of mGluR1 or mGluR5 receptors also attenuates cue- and nicotine-induced reinstatement of nicotine-seeking behavior [160, 439, 440]. However, stimulation of mGluR2/3 receptors or blockade of mGluR5 receptors does not block the ability of nicotine to potentiate ICSS thresholds [159, 441], and mGluR5 antagonism does not block the development of a nicotine CPP [166]. Thus, although glutamatergic ligands such as mGluR5 antagonists may be of some clinical benefit in reducing cigarette smoking or relapse during attempts to quit [442], they may not significantly attenuate the effects of nicotine on brain reward function.
Glutamatergic ligands have also been shown to modulate the nicotine withdrawal syndrome. One early study showed that activation of mGluR2/3 receptors suppresses nicotine withdrawal symptoms [443]. However, other investigators have shown that mGluR2/3 agonists, administered either systemically or directly into the VTA, actually elicit withdrawal-like elevations in ICSS thresholds in nicotine-dependent rats [444]. Thus, there appears to be a disconnect between glutamatergic modulation of the behavioral signs of nicotine withdrawal and nicotine modulation of brain reward function. AMPA/KA antagonists such as NBQX can also precipitate withdrawal-like elevations in ICSS thresholds in nicotine-dependent rats [444]. In addition, mGluR5 antagonists such as MPEP can actually increase somatic signs of nicotine withdrawal [438]. This, dampening glutamate transmission may be beneficial in reducing the reinforcing effects of nicotine, but may actually exacerbate nicotine withdrawal symptoms.
8. Glutamate and cannabinoids
The primary psychoactive ingredient in marijuana is Δ9-tetrahydrocannabinol (THC). THC acts as an agonist at the type 1 cannabinoid (CB1) receptor, which is expressed primarily in the brain, and has lower affinity for CB2 receptors, which are expressed in more restricted areas of the brain but are abundant in the periphery. Both CB1 and CB2 receptors are GPCRs that are negatively coupled to AC activity and also modulate the function of various ion channels [445]. Endogenous CB receptor ligands include anandamide and 2-arachidonylglycerol. There is now substantial evidence from animal models that the endogenous cannabinoid system is involved in numerous aspects of drug addiction and alcoholism (see [446-448]) for recent reviews).
CB1 receptors are present at high densities on presynaptic terminals of glutamatergic synapses [449-451]. In vitro studies have shown that THC and other CB1 receptor agonists inhibit glutamate-mediated neurotransmission and/or decrease glutamate overflow in numerous brain regions including the cerebral cortex [452, 453], dorsal striatum [450, 454-457], NAcc [449, 458-461], globus pallidus [462], hippocampus [453, 463-473], amygdala [453, 474], hypothalamus [475, 476], substantia nigra [477], VTA [478], locus coeruleus [479] and cerebellum [480-482], primarily by inhibiting glutamate release from the presynaptic terminal. Cannabinoid-induced reductions in glutamatergic transmission are a candidate mechanism by which cannbinoids impair the induction of LTP [483, 484]. However, two microdialysis studies have shown that CB1 agonists actually increase extracellular levels of glutamate in the FC [485, 486]. These findings are in contrast to those cited above where cannabinoids suppress glutamatergic transmission in the cortex. The reasons for the discrepancy are currently unclear, but may be a result of the type of experimental technique used (slice preparation versus in vivo microdialysis) and route of drug administration (i.e., bath application versus systemic injection, the latter of which does not result in steady-state concentrations of the drug in the brain).
Little is known about the effects of chronic cannabinoid exposure on glutamatergic transmission. Few studies, if any, have examined the effect of chronic cannabinoid administration on the expression of elements of glutamate transmission, such as iGluR, mGluR and EAAT protein levels. However, tolerance to the ability of THC to alter synaptic plasticity in several brain regions has been demonstrated as a result of repeated THC administration [487, 488], suggesting the possibility of some degree of alteration of glutamatergic signaling by chronic cannabinoid exposure.
One of the first studies showing that THC may interact with mGluR function was published by Nah and colleagues, who showed that THC inhibited glutamate-stimulated increases in IP3 in cultured hippocampal neurons [489]. There is now substantial evidence that mGluR and CB1 receptor interact to modulate synaptic plasticity (reviewed in [490, 491]).
The effects of CB1 agonists such as THC on glutamatergic transmission in regions of the brain’s reward circuitry may mediate the addictive properties of THC. For example, in PFC slices, Auclair and colleagues found that CB1 receptor agonists inhibit glutamatergic synaptic transmission between layer V afferents and layer V, and favor LTD at the expense of LTP at the same synapses [452]. Given the prominent role of the PFC in addictive behaviors, the disruption of glutamatergic activity in this area may be associated with addiction to THC. Robbe and co-workers [449, 460] found that CB1 receptors are present on large fibers making synaptic- like contacts with GABAergic medium spiny neurons (MSNs) in the NAcc, and that the synthetic CB1 agonists WIN 55,212 and CP55940 inhibited glutamatergic transmission at the synapses between the prelimbic cortex and the NAcc. These authors suggested that cannabinoids can indirectly affect NAcc dopamine overflow via this mechanism. For instance, the glutamatergic afferents from the cortex to the NAcc control the firing of the GABAergic MSNs, which in turn inhibit the dopaminergic neurons of the VTA. Via the reduction of excitatory transmission in the NAcc, cannabinoids may disinhibit midbrain dopaminergic, thereby increase their firing rate and trigger an increase in extracellular levels of dopamine in the NAc [449, 460]. A similar reduction in glutamatergic transmission by THC has also been shown in the shell of the NAcc [459].
The effects of CB1 agonists have also been studied in the amygdala. Activation of CB1 via WIN 55,212 reduces basal synaptic transmission and pharmacologically isolated AMPA receptor-mediated postsynaptic currents in the lateral amygdala of mice [474]. This ability of cannabinoids to reduce glutamatergic transmission in the amygdala may underlie the ability of THC to alter emotional or drug-related memories.
To our knowledge, there are no studies published examining the effects of glutamatergic ligands on the reinforcing effects of THC or relapse-like behavior, likely because reliable self-administration of THC is difficult to obtain in laboratory animals [492]
9. Glutamate and alcohol
Ethanol was long thought to exert its actions on the brain solely via potentiation of GABAergic transmission and/or increases in plasma membrane fluidity. However, in the late 1980’s and early 1990’s, a series of reports were published indicating that ethanol also acts by inhibiting neuronal NMDA receptor function [493-499]. The NMDA receptor is now considered one of the primary molecular targets for the actions of ethanol in the brain. Studies with recombinant NMDA receptors have shown that NR2B-containing receptors are particularly sensitive to inhibition by ethanol, and NR2C- and NR3-containing receptors are slightly less sensitive to ethanol [500-506]. Ethanol appears to inhibit NMDA receptor function via a non-competitive mechanism [507] and induces the phosphorylation and internalization of NR2 subunits [508, 509]. NMDA receptors in many brain regions are sensitive to inhibition by ethanol, including the cerebral cortex [510, 511], NAcc [512, 513], septum [514, 515], amygdala [516, 517], hippocampus [511, 515, 518-521], locus coeruleus [522-524], VTA [525, 526] and cerebellum [511, 515, 519, 527]. As a result, ethanol inhibits the induction of several forms of neural plasticity such as LTP in the hippocampus [528-531], dorsal striatum [532] (but also see [509]) and bed nucleus of the stria terminalis [533] while enhancing LTD in the hippocampus [534]. The ability of ethanol to inhibit NMDA receptor function is dependent on various factors including the NR1 splice variant that is co-assembled with NR2 subunits [535], extracellular Mg2+ and glycine concentrations [536-540], intracellular Ca2+ concentrations [541], and phosphorylation by proteins kinases such as Fyn [542-544], PKA [545] but see [546], and PKC [547] as well as by the phosphorylation regulator DAARP-32 [548].
As a result of the ability of ethanol to inhibit NMDA receptor function, NMDA antagonists such as MK-801 can potentiate the some of effects of acute ethanol such as the duration of ethanol-induced loss-of-righting reflex [549-552] and stimulation of locomotor activity [553-555]. NMDA antagonists can also produce ethanol-like discriminative stimulus effects in both animals and humans [556-575].
In response to chronic inhibition of NMDA receptor function, repeated ethanol exposure induces an upregulation of various NMDA receptor subunits including the NR1, NR2A and NR2B subunits in the cerebral cortex and hippocampus [576-597]. However, the effects of chronic ethanol exposure on NR1 expression have been less consistent than those on NR2A and NR2B, likely a result of the numerous splice variants of the NR1 subunit that exist [590]. Changes in NR2B expression have been linked to methylation of the NR2B gene [598, 599]. Chronic ethanol upregulates NR1 expression in the VTA and amygdala [600, 601], regions that are critical for the reinforcing effects of ethanol. In addition to upregulating NMDA subunit expression, chronic ethanol also increases NMDA receptor functionality (i.e., conductance, cation influx, etc.) [587, 592, 595, 601-608] and synaptic clustering of the receptor [609].
As a result of ethanol-induced up-regulation of NMDA receptor expression, the central nervous system enters a state of hyperexcitability upon acute withdrawal from ethanol exposure [610, 611]. In animals and humans, this CNS hyperexcitability manifests itself as a propensity towards seizure-like activity, which can be suppressed by NMDA antagonists [550, 589, 602, 612-619]. Following prolonged ethanol withdrawal, NMDA receptor expression and functionality are reduced [620]
Ethanol also appears to inhibit the function of AMPA and KA receptors [507, 512, 523, 524, 608, 621-639], although these receptors appear to be less sensitive to inhibition by ethanol than NMDA receptors, requiring concentrations of 50 mM or greater. Like other drugs of abuse, ethanol increases the AMPA/NMDA ratio of excitatory postsynaptic currents in the VTA [232]; however, this effect was shown after an extremely low dose of ethanol (20 mg/kg i.p.), and it is not known if this effect is observed following administration of more pharmacologically relevant doses of ethanol. Nonetheless, chronic ethanol has been reported to upregulate the expression of the GluR1 subunit in the VTA [600] and the GluR2/3 subunit in the cerebral cortex [585] and hippocampus [640] without altering KA subunit expression. Chronic ethanol also upregulates Ca2+ influx mediated by AMPA receptors [641, 642]. Interestingly, AMPA/KA receptors in the central amygdala have recently been reported to be important for the conditioned rewarding effects of ethanol [517].
Although ethanol primarily acts on ion channels, there is also evidence that ethanol alters Group I mGluR function. Minami and co-workers showed a preferential inhibition of mGluR5-stimulated Ca2+-mediated Cl- currents by ethanol, albeit at high concentrations of 100-190 mM [643]. Chronic ethanol also inhibits mGluR-mediated phospholipase C activity [644] and down-regulates the expression of various mGluRs such as mGluR1, mGluR3, mGluR5 and mGluR7 in the hippocampus [645].
Numerous studies have shown that glutamate receptor ligands alter the reinforcing effects of ethanol. Infusion of NMDA receptor antagonists systemically [569, 617, 646-651], into the cerebral ventricles [652], or directly into regions such as the NAcc [653] or dorsal striatum [509] attenuates oral ethanol consumption in rats. Some, but not all, NMDA receptors antagonists also attenuate the alcohol deprivation effect [654], cue-induced reinstatement of alcohol-seeking behavior [655, 656], the acquisition of ethanol CPP [650, 657, 658] and sensitization to the locomotor stimulant effects of low doses of ethanol [659-663]. Surprisingly, NMDA antagonists such as MK-801, while producing ethanol-like discriminative stimulus effects, do not reinstate ethanol-seeking behavior [664]. AMPA/KA receptor antagonists also attenuate operant ethanol reinforcement [665] and cue-induced reinstatement [655, 666]. However, some of these studies have shown that NMDA or AMPA/KA ligands also attenuate sucrose or saccharin reinforcement [665], indicating that such compounds may not be selective for reducing ethanol intake, but may rather attenuate general appetitive responding.
Attenuation of glutamatergic transmission by mGluR ligands also appears to reduce the rewarding and reinforcing effects of ethanol as well as relapse-like behaviors. The mGluR1 antagonist CPCCOEt has been reported to reduce operant ethanol self-administration as well as acute ethanol-stimulated dopamine and glutamate release in the NAcc in mice [667]; however, another group of investigators found no effect of this ligand on ethanol self-administration in the same mouse strain [668]. The reason for this discrepancy are likely attributable to numerous procedural differences between the two studies, as discussed in [667]. The mGluR2/3 agonist LY379268 attenuates ethanol consumption and stress- and cue-induced reinstatement of ethanol-seeking behavior [669, 670]. However, locomotor suppressant effects of this ligand have been observed [669], and thus the reductive effects of this ligand on ethanol consumption and relapse-like behavior must be interpreted with caution. The mGluR5 antagonists MPEP and/or MTEP have been shown to reduce voluntary ethanol consumption, reinforcement and relapse in a variety of rat and mouse strains [667, 668, 671-676]. The ability of MPEP to reduce ethanol consumption is absent in mice lacking PKCε [674] indicating that this PKC isozyme is an important signaling target of mGluR5 and is involved in the regulation of ethanol consumption via this receptor. MPEP also attenuates the expression [667] but not the acquisition [166] of ethanol CPP, attenuates ethanol-stimulated dopamine and glutamate release in the NAcc [667], and can modulate the discriminative stimulus effects of self-administered ethanol [677]. Stimulation of mGluR8 receptors with DCPG also reduces ethanol consumption and cue-induced reinstatement of ethanol-seeking behavior [669]. However, as with LY379268, locomotor suppressant effects of this ligand have been observed [669], and thus the reductive effects of this ligand on ethanol consumption and relapse-like behavior must be interpreted with caution.
Mice carrying mutations in genes encoding various components of glutamatergic transmission have yielded novel insight into the mechanisms of action of ethanol and the role of various glutamatergic signaling components in ethanol-related behaviors [678], as summarized below:
mice carrying a gene encoding the NR1 subunit with reduced affinity for glycine demonstrate reduced anxiolytic and motor impairing effects of ethanol [679].
mice engineered to express NR2A subunits without the C-terminal tail (NR2AΔC/ΔC) were shown to display decreased ethanol-induced inhibition of NMDA receptor function in the hippocampus and evidence for increased behavioral sensitivity to ethanol [680].
mice lacking the NR2A subunit (previously known as ε1) do not develop tolerance to the hypnotic effects of high doses of ethanol and do not acquire ethanol CPP [681-683]
mice lacking the AMPA subunit GluR1 showed patterns of ethanol consumption, ethanol-induced depression of locomotor activity, and hypnotic effects of high doses of ethanol that are similar to that of wildtypes [684]
mice lacking the GluR3 subunit of the AMPA receptor show reduced cue-induced reinstatement of alcohol-seeking behavior as compared with wildtypes but normal basal levels of ethanol consumption [666]
mice lacking mGluR4 demonstrate normal ethanol consumption patterns, withdrawal severity, and hypnosis induced by high doses of ethanol, but do not demonstrate a locomotor stimulant effect of lower doses of ethanol [685]
mice lacking Fyn, a tyrosine kinase that phosphorylates (amongst other things) the NR2B subunit of the NMDA receptor, fail to show tolerance to the ability of ethanol to inhibit NMDA receptor function in the hippocampus, and are also hypersensitive to the hypnotic effects of ethanol [542, 686, 687] but do not seem to display altered ethanol consumption or CPP [687, 688], although one of these studies demonstrated reduced ethanol consumption in Fyn-deficient mice [686].
mice lacking the gene encoding the NMDA-Group I mGluR scaffolding protein Homer2 show a reduced preference for a 12% ethanol solution, an absence of CPP for higher doses of ethanol, an absence of sensitization to the locomotor stimulant effects of ethanol, a lack of ethanol-induced stimulation of extracellular dopamine and glutamate levels in the NAcc [689]. Many of these phenotypes were reversed by virally-induced restoration of Homer2 expression in the NAcc.
Microdialysis studies have shown that ethanol, particularly at low doses, elevates extracellular glutamate levels in the hippocampus [690], amygdala [516, 691] and NAcc [667, 690, 692, 693], whereas at higher doses ethanol can reduce glutamate overflow in these regions [690, 694-696] and in the cingulate cortex [697]. The precise mechanisms whereby ethanol alters extracellular glutamate levels remains uncertain, and may be perhaps due to ethanol-induced changes in glutamate uptake by glial cells [698-700]. However, not all investigators have found that acute ethanol increases extracellular glutamate in the NAcc [701-704]. Repeated exposure to ethanol may be necessary achieve this effect [516, 700], and it may be dependent on the strain of animal used [704, 705] or the effect may be delayed by several hours into the acute withdrawal phase [705]. More consistently, withdrawal from chronic ethanol exposure is characterized by elevated extracellular glutamate levels in regions such as the dorsal striatum [706, 707], NAcc [399, 708] and hippocampus [709, 710]. This phenomenon appears to increase over the course of repeated withdrawal periods [709], paralleling the “kindling” effect of multiple withdrawal episodes on seizure-like activity [711].
Thus, a paradoxical effect of ethanol on glutamatergic transmission exists, with acute exposure to low doses of ethanol as well as withdrawal from chronic exposure increasing extracellular levels of this neurotransmitter, while ethanol simultaneously acts to inhibit the function of one of its primary cognate receptors (i.e., the NMDA receptor).
10. Glutamate and abused inhalants
Abused inhalants comprise a wide variety of compounds that range from gasoline to industrial solvents and cleaning agents. Most members of this class of drugs are simple hydrocarbon or substituted hydrocarbon compounds such as toluene, hexane, benzene and trichloroethane [712, 713]. The use of inhalants for their intoxicating effects is widely recognized as a problem of drug abuse, and is particularly prevalent among teenagers and children because of the simplicity of obtaining these compounds. Recent research has shown that nearly 20 percent of children in middle school and high school have experimented with inhaled substances [714]. While there are numerous volatile compounds that are abused, toluene (methylbenzene) is considered the prototypical compound for this class of drugs. Thus, a considerable amount of research on the neurobiological substrates of inhalant abuse has focused on toluene.
Like other drugs of abuse, toluene increases the activity of the mesolimbic reward circuitry [715-717]. One of the first demonstrations of an interaction between toluene and glutamatergic transmission was reported by Woodward and colleagues [718], who revealed that toluene inhibited the function of recombinant NMDA receptors expressed in Xenopus ooctyes. Cells expressing the NR1/NR2B subunit combinations were the most sensitive to inhibition of NMDA-induced currents by toluene, with a reported EC50 value of 0.17 mM. Cells expressing NR1/NR2A or NR1/NR2C subunit combinations of the NMDA receptors showed less sensitivity to inhibition of NMDA-induced currents by toluene than those expressing the NR1/NR2B subunit combination. In contrast, cells expressing the AMPA/KA subunits GluR1, GluR1/2 or GluR6 showed no inhibition of kainate-induced currents by toluene at concentrations up to 9 mM. This was the first evidence that abused inhalants such as toluene act by inhibiting iGluRs function, particularly NMDA receptors. Acute toluene exposure has also been shown to reduce glutamate-stimulated increases in potassium channel activity via mGluR1 [719], indicating that toluene may also interact with mGluR function as well as iGluR function.
These effects were later replicated in cultured hippocampal neurons [720], and it was also demonstrated that prolonged (4-day) exposure of cultured hippocampal neurons to toluene produced an up-regulation of NMDA receptor function, with increased whole-cell current responses to NMDA, increased density of NR1 subunits, and an increase in expression of the NR2A and NR2B subunits. These results suggest that chronic exposure to the abused inhalant toluene induces compensatory responses in the functional expression of NMDA receptors, similar to that observed following chronic ethanol exposure.
Comparable effects of chronic toluene exposure have been demonstrated in vivo in rodents. Exposure of rats to 500 ppm toluene vapor for 16 hr per day for 3 months upregulated nonspecific glutamate binding in numerous brain regions [721]. In a study by Williams et al. [722], adult male rats that were exposed to toluene vapor (8000 ppm) for 30 min per day for 10 days demonstrated increased NR1, NR2B and GluR2/3 subunit levels in the medial prefrontal cortex, increased NR2B expression in the NAcc, and decreased NR1 subunit expression in the substantial nigra. Together, these findings suggest that, like ethanol, chronic toluene exposure upregulates NMDA (and possibly AMPA) receptor expression and function in many, but not all, regions of the brain.
To our knowledge, only one study to date has examined the effects of toluene exposure on extracellular glutamate levels in vivo. Using microdialysis techniques, Win-Shwe et al. [723] demonstrated that an acute administration of toluene (150 and 300 mg/kg i.p.) dose-dependently increased extracellular glutamate levels in the hippocampus of mice, with glutamate levels returning to baseline values within 1 hr of administration. Thus, as with ethanol, a paradoxical effect of toluene on glutamatergic transmission exists, with acute exposure of toluene increasing extracellular levels of this neurotransmitter while simultaneously acting to inhibit the function of one of its primary cognate receptors (i.e., NMDA receptors).
11. Drugs of abuse and synaptic plasticity
There is now overwhelming evidence that drugs of abuse induce various forms of synaptic plasticity, such as LTP and LTD, within the reward circuitry of the brain. Accordingly, there is a growing consensus that repeated exposure to drugs of abuse produces lasting neuroadaptive changes within this circuitry, which eventually result in the pathological drug seeking that is characteristic of addiction (see [43, 59, 724-730] for reviews). One of the first demonstrations that abused drugs induce electrophysiological indices of synaptic plasticity in reward-related brain regions was the finding that acute exposure to cocaine depressed excitatory transmission (as measured by the amplitude of glutamate-evoked excitatory postsynaptic currents, or EPSCs) in the NAcc [731, 732]. Similar effects were observed in the VTA following acute amphetamine exposure [733]. Jones and colleagues also showed that acute amphetamine also blocked the induction of LTD by glutamatergic synapses onto VTA dopamine neurons [734]. However, by analyzing the individual components of glutamate-evoked EPSCs (i.e., NMDA- and AMPA-mediated currents), Ungless and colleagues demonstrated that a single exposure to cocaine induced LTP in VTA dopamine neurons, as characterized by an increase in the AMPA-NMDA ratio of glutamate-evoked EPSCs [95]. This effect was transient, as it was evident at 5 but not 10 days following the exposure to cocaine. It was subsequently demonstrated that similar effects are observed following acute exposure to other drugs of abuse, including morphine, nicotine, amphetamine, ethanol, as well as acute exposure to stress [232, 233]. The increase in the AMPA component of the EPSCs is likely due to increased trafficking of AMPA receptor subunits to the plasma membrane, which is a well-known regulator of excitatory synaptic strength [20, 21, 735-739]. However, not all studies have observed increased membrane insertion of AMPA receptors following acute drug exposure [233]. Repeated exposure of animals to cocaine also facilitates the induction of LTP in VTA dopamine neurons, which is a possibly a result of cocaine-induced decreases in GABAergic inhibition of these neurons [740]. Similarly, it was recently reported that opiate drugs such a morphine inhibit the formation of LTP in local GABAergic inhibitory synapses onto VTA dopamine neurons [741].
Drug-induced synaptic plasticity has also been observed in the NAcc. Thomas and colleagues [742] demonstrated that repeated administration of cocaine to mice, which resulted in behavioral sensitization, resulted in LTD in the NAcc (measured one day after a final challenge with cocaine to demonstrate the continued expression of behavioral sensitization). This LTD was characterized by decreases in the AMPA-NMDA ratio of EPSCs evoked by stimulation of prefrontal glutamatergic afferents to the NAcc shell. These observed changes are important since AMPA receptors in the NAcc shell modulate brain reward function as assessed by ICSS [743]. Others have reported that repeated amphetamine attenuates the induction of PFC-Nacc LTP via dopaminergic mechanisms [744], but this effect is transient, as LTP in the NAcc could be established 8-10 days after discontinuation of amphetamine treatment.
While all of the aforementioned studies have examined changes in synaptic transmission after passive (i.e., experimenter-administered) exposure to drugs of abuse, a recent study examined the effects of active cocaine self-administration on synaptic plasticity in brain reward circuitry. Martin et al. [745] demonstrated that in rodents self-administering cocaine intravenously, LTD in the the NAcc core and shell was abolished following one day of abstinence, but this effect was still evident in the core 21 days into abstinence. These data are in contrast to those of Thomas et al. [742], who showed that repeated non response-contingent exposure to cocaine induced LTD in the NAcc shell. Thus, the effects of drugs of abuse on synaptic plasticity in reward-related regions of the brain are often transient, and may also be dependent on whether the drug is administered in a response-contigent or non-contingent manner.
While glutamatergic transmission is often considered a “prime mover” in terms of its role in synaptic plasticity, particularly drug-induced plasticity, it should not be construed that other neurotransmitter systems within the brain’s reward circuitry do not play an equal if not more important role. As the mesolimbic reward circuit is primarily dopaminergic and receives a significant amount of glutamatergic input (see Fig. 3), it should not be surprising that there is a great deal of convergence of dopamine and glutamate signaling within this system that mediates drug-induced synaptic plasticity [43, 87, 726, 728, 730, 746, 747]. For example, glutamatergic afferents (i.e., from the FC) make synaptic contacts into the “heads” of dendritic spines localized on MSNs in the NAcc, the primary type of neuron in this region. Dopaminergic afferents arising from the VTA form synapses onto these very same spines, although these synapses tend to be localized towards the “neck” of the spine. In accord with the close proximity of these converging dopaminergic and glutamatergic inputs, there is a great deal of biochemical cross-talk between these entities. For example, activation of D1 receptors on MSNs (i.e., due to drug-induced increases in extracellular dopamine in the NAcc) activates PKA, which phosphorylates GluR1 subunit and facilitates the insertion of AMPA receptors into the neuronal membrane [748-750]. Similar effects have been observed in the prefrontal cortex [751] and hippocampus [752]. This dopamine-induced increase in trafficking of AMPA receptors to the plasma membrane has a subsequent facilitatory effect on the induction of synaptic plasiticity in these neurons [20, 21, 735-739], which can lead to long-term adaptations in the brain reward circuitry.
12. Glutamate and extinction learning – relevance for addiction
It has become increasingly apparent that the brain circuits, neurotransmitters, and signal transduction mechanisms that underlie drug addiction have considerable overlap with those that underlie normal learning and memory processes. As a result, numerous theories have emerged that hypothesize drug addiction to be a disorder of learning and memory [43, 729, 753-760], where certain behaviors and drug-environment associations become “overlearned”. In other words, with repeated drug exposure, drug-taking behaviors become compulsive and automatic (i.e., instrumental overlearning) and the associations between drugs and specific environmental cues and contexts becomes overly salient (i.e., associative overlearning). These types of overlearning often lead to drug craving and relapse [148, 761, 762]. Attempts at extinguishing the salience of drug-associated cues by exposure therapy have been met with limited success [763, 764], likely because of the high degree of context specificity in extinction learning [765, 766]. In addition, most behavioral or pharmacological treatments for drug addiction are primarily aimed at reducing drug use, withdrawal symptoms, or relapse, with little attention being given to the process of extinction. Thus, there is a great need to develop therapeutic interventions whereby instrumental and associative “overlearning” that occurs in the process of addiction is subsequently extinguished.
Both learning and memory and drugs of abuse induce various forms of neuronal plasticity including LTP and LTD mediated by glutamatergic transmission [43, 724, 725, 728-730, 767-770]. Unfortunately, most of what is known about extinction learning has been derived from studies on aversive conditioning with footshock or appetitive conditioning with natural rewards. As a result, it would overly simplistic to assume that the neural substrates of extinction of drug-seeking behavior are synonymous with those that mediate extinction of other types of conditioned behaviors. There do, however, appear to be a number of commonalities. For example, extinction of conditioned fear has been reported to be facilitated by the NMDA receptor partial agonist D-cycloserine either administered systemically (reviewed in [771, 772]) or directly into the BLA [773-775]. Similarly, recent reports demonstrate that extinction of a cocaine CPP is also facilitated by D-cycloserine, administered either systemically [776, 777] or into the BLA [776]. Thus, it is possible that the neural substrates that subserve both extinction of drug-seeking behavior and extinction of other conditioned behaviors are somewhat overlapping.
Exinction has long been viewed as a process of “forgetting”, or a disintegration of the association between previously neutral stimuli and an emotional or subjective state. However, there is increasing evidence that extinction is not merely a process of forgetting, but actually a form of new and active learning. Although scientists investigating fear conditioning arrived at this realization as many as 10-15 years ago [778, 779], only recently has the notion that extinction of drug-seeking behavior is also a form of new and active learning come to light. In an eloquent series of studies, Self and colleagues have demonstrated that extinction training procedures following cocaine self-administration produce various hallmarks of neuronal plasticity in the NAcc. First, extinction training was shown to restore cocaine-induced deficits in tyrosine hydroylase immunoreactivity in the NAcc shell, whereas in animals that did not undergo extinction training, the levels of the enzyme remained reduced following cocaine self-administration [780]. Subsequently, these investigators showed that extinction training induces an upregulation in the expression of the GluR1 and GluR2/3 subunits of the AMPA receptor in the NAcc [781, 782] and that virally-mediated upregulation of these AMPA subunits in this region facilitates extinction learning [782]. Consistent with this, GluR1 deletion in mice results in resistance to extinction following cocaine or food self-administration [174]. Self and colleagues also demonstrated that extinction training also normalizes cocaine-induced deficits in levels of the NR1 subunit of the NMDA receptor in the NAcc core [781]. Another group of investigators led by Marshall showed that inhibition of extracellular signal-regulated kinase (ERK) in the NAcc core region results in a lasting attenuation of drug-induced reinstatement of cocaine CPP as well as cocaine-induced phosphorylation of several signaling molecules including ERK, CREB, Elk-1 and c-fos [783]. These authors suggest that these particular molecular substrates of learning and memory in the NAcc are necessary for reconsolidation of drug-associated memories. Finally, it was recently demonstrated that rats that have undergone extinction training show differences in Fos expression in various brain regions, particularly in AMPA receptor expressing cells, as compared with animals that have not undergone extinction training [784]. Collectively, these studies suggest that a considerable amount of neuroplasticity occurs in the brain during extinction following drug self-administration (or passive exposure, in the case of CPP), and that agents that potentiate glutamatergic transmission, such as D-cycloserine, might be of clinical benefit in facilitate extinction learning in drug addicts attempting to abstain from drug use.
13. Glutamatergic medications for the pharmacological management of drug addiction and alcoholism
13.1 Acamprosate
Acamprosate (calcium acetylhomotaurine) is a derivative of homotaurine (a nonspecific GABA agonist) that is N-acetylated to facilitate penetration across the blood-brain barrier, and is formulated as a calcium salt to increase absorption from the gastrointestinal tract (see Figure 5). Acamprosate was developed in Europe in the 1980’s as a pharmacological agent to reduce alcohol consumption, craving and relapse in alcoholic patients. Only one study in animals showing that acamprosate reduced voluntary ethanol consumption in rats [785] was published prior to the first demonstration of its clinical efficacy in reducing the incidence of relapse in alcoholics [786]. Over the years, acamprosate has demonstrated moderate efficacy in reducing overall alcohol consumption and subjective measures of alcohol craving, and promoting abstinence, as reviewed in recent meta-analyses [787-792]. It should be noted, however, that a large multicenter study of over 600 subjects found that acamprosate was no more effective than placebo in reducing the incidence of relapse in a medically managed care setting [793]. The reasons for these negative findings, particularly in light of the numerous previous clinical trials showing moderate efficacy of acamprosate, are still being debated.
Fig. 5.

Chemical structures of medications that act on glutamatergic transmission that have shown positive effects in the treatment of drug addiction and/or alcoholism in humans.
Despite years of use as a treatment for alcoholism, the neuropharmacological mechanisms underlying the actions of acamprosate are still unclear. Originally, acamprosate was thought to exert its effects via a GABAergic mechanism, since the drug has a chemical structure similar to that of GABA (see Figure 5). However, subsequent studies have failed to find any direct evidence of acamprosate binding to recombinant GABAA receptors, or an ability of acamprosate to enhance GABAA receptor function [794-796]. Thus, acamprosate has not been classified as a GABAergic compound. Nonetheless, acamprosate may indirectly increase GABAergic neurotransmission by blocking inhibitory presynaptic GABAB autoreceptors [796] and/or by increasing extracellular levels of taurine [797, 798], an endogenous sulfonated amino acid that can potentiate GABAA receptor-mediated chloride flux.
The first studies suggesting that acamprosate exerts its actions through glutamatergic mechanisms were reported by Zeise and colleagues [799, 800]. These investigators demonstrated that locally applied acamprosate reduced the excitation of neuronal firing evoked by iontophoretic application of L-glutamate onto cortical neurons in vivo, and inhibited excitatory post-synaptic potentials evoked by glutamate and NMDA in rat forebrain slices. These data suggested that acamprosate may functionally antagonize excitatory amino acid neurotransmission and therefore reduce neuronal excitability. In support of this, subsequent studies have shown that acamprosate antagonizes NMDA-evoked postsynaptic currents in cultured hippocampal neurons and Xenopus oocytes expressing recombinant NMDA receptors [801], up-regulates NMDA receptor subunit expression in a pattern similar to that observed following treatment with the non-competitive NMDA antagonist MK-801 [801, 802], and reduces NMDA receptor-mediated Ca2+ influx in cultured rat midbrain neurons [803]. However, not all electrophysiological studies have provided evidence that acamprosate acts as an NMDA receptor antagonist, as some investigators have found no effect of acamprosate on NMDA-mediated synaptic transmission in the CA1 region of the hippocampus [804], while others have found that acamprosate actually potentiates NMDA receptor function in the CA1 region of the hippocampus [795] or in the NAcc [796]. Thus, the action of acamprosate on NMDA receptors appears to be inconsistent, and is perhaps dependent on factors such as brain region examined, NMDA receptor subunit composition, state of neuronal excitation, and the presence of various endogenous NMDA receptor neuromodulators such as polyamines [805]. Nonetheless, several binding studies have confirmed an interaction of acamprosate with the spermidine-, glutamate- and/or MK-801-sensitive binding site of the NMDA receptor [806-808]. Thus, there is a general consensus that acamprosate is an NMDA receptor modulator [805, 809].
In addition to an apparent interaction of acamprosate at ionotropic glutamate receptors, recent in vitro studies demonstrated that acamprosate inhibits the binding of and neurotoxic effects of trans-ACPD (a group I and II mGluR agonist), as well as the neurotoxic effects of ethanol withdrawal, in a manner similar to the mGluR5 antagonist SIB-1893 [808, 810]. These data suggest that acamprosate may act on metabotropic as well as ionotropic glutamate receptors. This is an important observation, given the important role of mGluR5 in regulating alcohol consumption and relapse (see Section 9). However, additional studies are needed to confirm a functional interaction between acamprosate and individual mGluR subtypes.
Despite its elusive mechanism of action, acamprosate is believed to restore the imbalance between excitatory and inhibitory neurontransmission caused by chronic alcohol exposure. Chronic exposure to alcohol produces an up-regulation of NMDA receptor function and a down-regulation of GABAA receptor function, resulting in an imbalance between excitatory and inhibitory amino acid transmission. During acute withdrawal from alcohol exposure, glutamate release is increased [709, 710] to further induce a state of CNS hyperexcitability. It is believed that through its modulatory effects on NMDA receptor function, suppression of glutamate release when the organism is in a hyperglutamatergic state (see also [811]), and possible enhancement of GABAergic transmission (though this remains controversial), acamprosate may restore the imbalance between excitatory and inhibitory amino acid transmission in the brain following chronic alcohol consumption [805, 812].
To date, very few animal or human studies have examined the potential efficacy of acamprosate in treating addiction to other drugs of abuse. Acamprosate was shown to be ineffective in preventing the development of morphine CPP in mice [813] and did not alter heroin reinforcement or stress- or heroin-induced reinstatement of heroin-seeking behavior in rats [814]. Thus, based on these animal studies, acamprosate is likely to have limited use in the treatment of opiate addiction. However, there have been several studies by our laboratory suggesting that acamprosate may be of potential benefit in the treatment of cocaine addiction. We found that acamprosate prevented the development [813] and cocaine-induced reinstatement [815] of a cocaine CPP in mice, and have recently demonstrated that acamprosate attenuates cocaine- and cue-induced reinstatement of cocaine-seeking behavior in rats without affecting basal levels of cocaine IVSA [816]. A pilot Phase II clinical trial was recently initiated to examine the effects of acamprosate on cocaine use, craving and acute withdrawal symptoms in humans (www.clinicaltrials.gov identifier: NCT00385268). However, rates of relapse following abstinence are not planned in this study, for which our animal studies indicate acamprosate might be most effective. The results of this clinical trial (and future trials on relapse) are eagerly awaited, since if they are positive, they may not only lead to approval of one of the first medications specifically for the treatment of cocaine addiction, but will also provide some predictive validity for the use of CPP, IVSA and reinstatement procedures in the screening of potential medications to treat drug addiction.
13.2 N-acetylcysteine
N-acetylcysteine (NAC) is an N-acetylated derivative of the naturally occurring amino acid cysteine (see Figure 5). NAC is used worldwide as a mucolytic agent and to treat acetaminophen overdose, and has numerous mechanisms of action including increasing glutathione synthesis. Because NAC is a cystine pro-drug, it can serve as a substrate for xc, which exchanges extracellular cystine molecules for intracellular glutamate molecules in glia, thereby elevating extracellular glutamate levels in many tissues including the brain [34, 817]. Numerous animals studies have shown that acute cocaine exposure elevates extracellular levels of glutamate in regions such as the NAcc, but during withdrawal from repeated cocaine exposure, extracellular levels of glutamate are decreased in this region relative to drug-naïve animals, likely due a desensitization of presynaptic mGluR2/3 autoreceptors and decreased activity of xc (see Section 4 and Figure 4). However, by providing a source of extracellular cysteine (which is converted to cystine) and increasing the activity of xc, administration of NAC “restores” extracellular levels of glutamate to those observed in drug-naïve animals and prevents the ability of a subsequent cocaine challenge to further increase glutamate levels in the NAcc (Figure 5). The resulting behavioral effect is a blockade of the ability of acute cocaine to reinstate cocaine-seeking behavior [37, 69].
Taking these findings from animal studies to the clinic, Kalivas and colleagues have conducted two preliminary investigations into the efficacy of NAC to reduce cocaine use, craving, withdrawal symptoms and relapse in human cocaine addicts. In a small safety and tolerability study, it was demonstrated that NAC was well-tolerated by cocaine addicts and produced slight trends in reductions in self-reports of cocaine craving and withdrawal symptoms [818]. A follow-up 4-week pilot open-label trial confirmed that NAC is well-tolerated by cocaine addicts and actually produces significant reductions in cocaine use [819]. Although these results provide encouraging data that NAC may be of potential use in the treatment of cocaine addiction, additional larger placebo-controlled clinical trials are needed to confirm these results.
13.3. Modafinil
Modafinil is a central nervous system stimulant that was originally designed as a wakefulness- and vigilance-enhancing drug for the treatment of narcolepsy and excessive daytime sleepiness caused by sleep apnea or shiftwork. Modafinil is sometimes prescribed as an off-label treatment for attention-deficit/hyperactivity disorder. Although its mechanism of action is not fully understood, it does not appear to act as a monoamine releaser as is the case for amphetamine-like stimulants. Modafinil may act by stimulating α-adrenoceptors, suppressing GABA release, weakly inhibiting the dopamine transporter, or stimulating hypothalamic orexin-containing neurons [820]. Modafinil also elevates extracellular levels of glutamate in numerous brain regions including the dorsal striatum, thalamus, hippocampus, and hypothalamus [821-823] without affecting glutamate synthesis [824]. Modafinil is not considered to have abuse potential, although reports of non-medical use are increasing [820], and as a result modafinil is currently classified as a Schedule IV controlled substance.
Two clinical reports have shown some potential efficacy of modafinil in the treatment of cocaine addiction. In a small placebo-controlled drug-interaction study by Dackis and colleagues, it was reported that modafinil actually blunted the euphorigenic effects of intravenous cocaine in cocaine addicts [825], a findings that was later independently replicated [826]. A double-blind placebo-controlled study of treatment-seeking cocaine-dependent outpatients showed that modafinil significantly reduced daily cocaine use and prolonged abstinence [827]. Although these data provide potentially promising evidence that modafinil might be of use in the treatment of cocaine addiction, it is possible that some of the effects observed might be due to decreases in peak plasma concentrations of cocaine in the presence of modafinil [828]. Studies on the efficacy of modafinil in preventing relapse to cocaine use are needed. Nonetheless, these results show promise for modafinil as a potential treatment for cocaine addiction.
It is somewhat counterintuitive that a drug like modafinil, which increases extracellular glutamate levels similar to other psychostimulants (see Sections 4 and 5), results in reductions in cocaine intake, whereas numerous animal studies have shown that blockade of glutamatergic neurotransmission (i.e., by administration of iGluRs or mGluR5 receptor antagonists) reduces cocaine reinforcement. It is possible that the increases in extracellular glutamate levels produced by modafinil “normalize” the reduced extracellular glutamate observed during cocaine withdrawal and therefore attenuate the ability of cocaine to further increase glutamate release, akin to the effect observed with NAC (see section 13.2 and Figure 4). Further studies are needed to test this hypothesis.
13.4. Topiramate
Topiramate has been used as an anticonvulsant for over a decade, and has more recently been approved for the treatment of migraine. Topiramate is also sometimes prescribed for psychiatric conditions such as bipolar disorder and post-traumatic stress disorder. Like many anticonvulsant drugs, however, topiramate has multiple mechanisms of action, including inhibition of voltage-gated Na+ and Ca2+ channels and activation of GABAA receptors [829-831]. In addition, it has recently been observed that topiramate also blocks GluR5-containing AMPA receptors [832, 833].
Within the past few years it has become apparent that topiramate may be a novel therapeutic agent for the treatment of drug addiction. Topiramate attenuates the somatic symptoms of withdrawal from various drugs of abuse [834, 835]. Several recent clinical trials have demonstrated efficacy of topiramate in attenuating alcohol craving or consumption [836-839], and has been shown to promote abstinence from cocaine [840], nicotine [841, 842] and MDMA [843]. Since topiramate has numerous mechanisms of action, it is unknown whether its reductive effects on drug and alcohol use are a result of its interactions with AMPA receptors. However, it should be noted that since AMPA receptors play a critical role in mediating cocaine-seeking behavior and relapse (see Section 4), this mechanism of action may provide a possible neurochemical basis for the ability of topiramate to reduce relapse to cocaine use.
13.5. Lamotrigine
Similar to topiramate, lagotrimine is an anticonvulsant that inhibits voltage-gated Na+ and Ca2+ channels [829-831]. Recently lamotrigine has shown some promise as a mood stabilizer. Through inhibition of presynaptic Na+ and Ca2+ channels, lamotrigine prevents the release of various neurotransmitters, including glutamate [844-853]. Lamotrigine inhibits the somatic signs of withdrawal from various drugs of abuse [834, 835]. Recent clinical studies show that lamotrigine also appears to exhibit efficacy in reducing craving for and use of cocaine [854-857], although it does not seem to alter the subjective effects of cocaine [858]. Similar reductive effects of lamotrigine on craving for and use of alcohol [859] and abused inhalants [860] have also been reported. These findings suggest that lamotrigine may be of clinical benefit in the treatment of addiction to cocaine, alcohol or abused inhalants. As with the multimodal actions of other anticonvulsants, it is difficult to determine whether the positive clinical effects of lamotrigine are due to its glutamate release-inhibiting properties, but this mechanism of action would fit well with the preclinical literature reviewed presently.
13.6. Gabapentin
Similar to its fellow anticonvulsants topiramate and lagotrimine, gabapentin inhibits presynaptic voltage-gated Na+ and Ca2+ channels [829-831], and as such prevents the release of various neurotransmitters, including glutamate [632, 861-866]. Gabapentin has been used successfully in the alleviation of somatic symptoms of drug withdrawal, particularly alcohol withdrawal [867-873]. Similar protective effects of gabapentin on animals or in vitro models of CNS hyperexcitability during alcohol withdrawal have been observed [874, 875].
The results of clinical studies on possible therapeutic uses of gabapentin for cocaine addiction have been mixed. Some studies demonstrated that gabapentin does not reduce the use of cocaine [856, 876, 877], while other studies have shown some positive results [878-880]. Gabapentin has been reported to attenuate the discriminative stimulus effects of cocaine [881]. However, gabapentin does not reduce methamphetamine use [882], has limited effects on promoting abstinence from smoking [883], and does not appear to reduce craving for or the subjective effects of alcohol [884, 885]. Thus, despite its glutamate release-inhibiting properties, gabapentin may not be of much clinical use in the treatment of drug addiction or alcoholism other than alleviating the symptoms of alcohol withdrawal.
13.7. Memantine
Memantine is a noncompetitive NMDA receptor antagonist that is used for the treatment of cognitive decline in Alzheimer’s disease. In addition to its actions at NMDA receptors, memantine also blocks 5-HT3 receptors as well as nAChRs. Memantine is one of the few NMDA receptor antagonists that is generally well-tolerated by humans and does not appear to have abuse potential [886]. Clinical studies have shown that memantine is efficacious in reducing withdrawal symptoms in detoxified alcoholics [835] and opiate addicts [887], consistent the NMDA hyperactivity hypothesis of alcohol withdrawal (see Section 9). Several clinical trials have reported that memantine was superior to placebo in attenuating on-going drinking and/or craving for alcohol in alcoholics [888-890]. This amelioration of craving for alcohol may be a result of the ethanol-like subjective effects that are produced by memantine [888, 889]. However, a larger placebo-controlled study indicated that memantine does not appear to reduce on-going drinking behavior in alcohol-dependent patients [891]. Also, memantine has been reported to increase the subjective and cardiovascular effects of cocaine without altering the choice to self-administer cocaine [892, 893]. These data suggest that memantine may be of use in the treatment of alcohol or opiate withdrawal, but the disparate results that have been reported on its ability to reduce on-going alcohol consumption and/or alcohol craving need to be further evaluated. In addition, based on its inability to reduce the choice to self-administer cocaine and its potentiation of the subjective effects of this psychostimulant, memantine is likely to be ineffective in the treatment of cocaine-dependence, though large-scale clinical trials are needed to verify this.
14. Genetic linkages between glutamatergic neurotransmission and addiction in humans
Despite overwhelming evidence that glutamatergic transmission is involved in drug addiction and alcoholism, and the widely accepted notion that addiction has a strong genetic component, only a handful of genetic alterations (such as single nucleotide polymorphisms, SNPs), have been successfully linked to or associated with addictive behaviors. One of the first findings in this area was reported by Sander and colleagues, who found an increased allelic frequency of a silent SNP in exon 5 of the EAAT2 gene (G603A) in a population of German alcoholics with co-morbid antisocial personality disorder, but not in alcoholics without the co-morbid psychiatric diagnosis [894]. Thus, in this population, the EAAT2 SNP may not have been associated with alcoholism per se, but in tendencies towards risk-taking behaviors that are occasionally found in alcoholic individuals. An additional study found an association between the G603A allele and alcoholic cirrhosis [895]
As reviewed in Section 9, one of the molecular targets of alcohol is the NMDA receptor, the function of which is inhibited by alcohol. Indeed, non-alcoholics with a family history of alcoholism have altered subjective responses to NMDA antagonists such as ketamine as compared with non-alcoholics without a family history of alcoholism [896]. Accordingly, several groups of investigators have attempted to identify allelic variations in the genes encoding one or more of the NMDA receptor subunit proteins that may confer susceptibility to alcoholism. However, the results of these studies have been mixed. Two groups of investigators have shown that alcoholics with a history of alcohol withdrawal seizures and delirium tremens were more likely to carry a G2108A SNP in exon 7 of the NR1 subunit gene than controls [897, 898]. An association of delirium tremens was also demonstrated to be associated with a Ser310Ala polymorphism in the GluR7 KA receptor gene [899], although this same polymorphism was not associated with alcoholism per se [900]. With regards to other iGluR subunits, findings have been less consistent. For example, a decreased allelic frequency of a C2664T SNP in exon 13 of the NR2B subunit gene in early-onset alcoholics has been demonstrated [897], while other groups of investigators have shown no association between alcoholism and a C2873T SNP in the NR2B gene, even in early onset alcoholics [901], or a C366G SNP in this gene [895]. Given the present set of data, it appears that genetic variations in iGluR subunit genes may be related to the presence of delirium tremens or alcohol-withdrawal seizures in alcoholic patients, but further research is needed to clarify whether such polymorphisms are associated with risk for alcoholism itself.
Group II and Group III mGluRs are often localized to presynaptic glutamatergic terminals where they regulate glutamate release via classic inhibitory autoreceptor mechanisms (see Figure 2). Therefore, genetic mutations in these mGluRs may result in a lack of inhibitory feedback tone on the presynaptic glutamatergic terminal, resulting in excessive glutamate release and the possibility of seizures. Preuss and colleagues hypothesized that since mice carrying a targeted deletion of the mGluR7 gene show increased seizure susceptibility [902], polymorphisms in one or more presynaptic mGluRs might confer susceptibility to delirium tremens during alcohol withdrawal. However, these investigators found no association of a Tyr433Phe polymorphism in the mGluR7 gene or a C2756T polymorphism in mGluR8 gene and seizures or delirium tremens in a population of alcoholic patients [903].
In addition to alcoholism, there is evidence for a genetic component of cocaine addiction. As discussed in Section 4, the postsynaptic scaffolding Homer family of proteins plays a role in the behavioral and neurochemical responses to cocaine [30, 904]. A recent genetic linkage study attempted to determine the presence of polymorphisms in Homer genes in African-American cocaine addicts [905]. Of the 7 polymorphisms analyzed (4 in the Homer1 gene, 3 in the Homer2 gene), only one SNP (ID #rs6871510, localized to the Homer1 gene) was detected to occur more frequently in cocaine addicts. While this study represents a potentially exciting and important link between cocaine addiction and glutamatergic signaling, replication studies in other cocaine addict populations are needed, as are studies to determine the precisely how this polymorphism alters signal transduction by the NMDA-Group I mGluR-Homer complex.
15. Summary, conclusions and future directions
All drugs of abuse alter glutamate transmission via one mechanism or another. Psychostimulants and nicotine enhance extracellular levels of glutamate, opiates and cannabinoids reduce synaptic overflow of glutamate, and alcohol has mixed effects on extracellular levels of this amino acid. Pharmacological agents that attenuate glutamatergic signaling, either by receptor antagonism, release inhibition, or enhancement of cellular uptake, tend to reduce the reinforcing and rewarding effects of most drugs of abuse, and can also attenuate the reinstatement of drug-seeking behavior, an established animal model of relapse. On the contrary, enhancement of glutamatergic transmission appears to promote the extinction of drug-seeking behavior, likely by facilitating new learning about drug availability, expectancies, or drug-cue associations. Thus, pharmacological compounds that antagonize glutamate transmission, including those already being used or tested in humans, are likely candidates that may be of use in the pharmacological management of excessive drug intake and relapse prevention. On the contrary, drugs that potentiate glutamatergic transmission and therefore promote synaptic plasticity and “new” learning may be of benefit in the facilitation of extinction learning.
Given that glutamate transmission is one of the primary neurochemical substrates of synaptic plasticity, and the overwhelming evidence reviewed here that all drugs of abuse interact with glutamate transmission, it is not surprising that drugs of abuse can cause long-lasting neuroadaptions of glutamate systems in the brain. These adaptations somehow lead in compulsive drug use, loss of volitional control over drug intake, and hypersalience of drug-associated environmental cues or contexts, all of which are characteristic of addiction. The question is – where to we go from here? From a scientific perspective, we are of the opinion that much more needs to be done to fully characterize the changes in glutamate systems that are caused by volitional intake of drugs of abuse in experimental animals, particularly in the context of drug intake patterns that mimic those taken by drug addicted humans. From treatment perspective, we feel that future research should focus on which changes in glutamate transmission can be most safely and effectively targeted by pharmacological or genetic interventions that either counteract the adaptive changes in the brain produced by prolonged intake of drugs of abuse, or promote new synaptic plasticity that can help the individual regain volitional control over drug intake and extinguish drug-associated memories and cravings.
Abbreviations
- 5-HT
5-hydroxytryptamine (serotonin)
- AC
adenylyl cyclase
- AMPA
α-amino-3-hydroxy-5-methylisoxazole-4- propionic acid
- Amyg
amygdaloid complex
- ATP
adenosine triphosphate
- BLA
basolateral amygdala
- cAMP
cyclic adenosine monophosphate
- CB
cannabinoid
- CPP
conditioned place preference
- CPu
caudate-putamen
- DARPP-32
dopamine and cAMP-regulated phosphoprotein-32 kD
- EAAT
excitatory amino acid transporter
- EPSC
excitatory postsynaptic current
- ERK
extracellular signal-related kinase
- FC
frontal cortex
- GABA
gamma-aminobutyric acid
- GPCR
G-protein coupled receptor
- Hipp
hippocampus
- ICSS
intracranial self-stimulation
- iGluR
ionotropic glutamate receptor
- IP3
inositol triphosphate
- IVSA
intravenous self-administration
- KA
kainic acid
- MAPK
mitogen-activated protein kinase
- LTD
long-term depression
- LTP
long-term potentiation
- MDMA
methylenedioxymethamphetamine
- mGluR
metabotropic glutamate receptor
- MSN
medium spiny neuron
- NAcc
nucleus accumbens
- nAChR
nicotinic acetylcholine receptor
- NMDA
N-methyl-D-aspartate
- PKA
protein kinase A
- PKC
protein kinase C
- PPT
pedunculopontine tegmentum
- SNP
single nucleotide polymorphism
- Thal
thalamus
- THC
Δ9-tetrahydrocannabinol
- VGCC
voltage-gated calcium channel
- vGluT
vesicular glutamate transporter
- VTA
ventral tegmental area
- xc
cystine-glutamate exchanger
Footnotes
This work was supported by PHS grant AA013582 (MFO) and AA007474 (JTG) from the National Institute on Alcohol Abuse and Alcoholism, and a grant from the Alcoholic Beverage Medical Research Foundation (MFO).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Association AP. Diagnostic and Statistical Manual of Mental Disorders. Washington DC: American Psychiatric Press; 1994. [Google Scholar]
- 2.Bjorklund A, Dunnett SB. Dopamine neuron systems in the brain: an update. Trends Neurosci. 2007;30:194–202. doi: 10.1016/j.tins.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Sanchis-Segura C, Spanagel R. Behavioural assessment of drug reinforcement and addictive features in rodents: an overview. Addict Biol. 2006;11:2–38. doi: 10.1111/j.1369-1600.2006.00012.x. [DOI] [PubMed] [Google Scholar]
- 4.Epstein DH, Preston KL, Stewart J, Shaham Y. Toward a model of drug relapse: an assessment of the validity of the reinstatement procedure. Psychopharmacology. 2006;189:1–16. doi: 10.1007/s00213-006-0529-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tzschentke TM. Measuring reward with the conditioned place preference paradigm: a comprehensive review of drug effects, recent progress and new issues. Prog Neurobiol. 1998;56:613–672. doi: 10.1016/s0301-0082(98)00060-4. [DOI] [PubMed] [Google Scholar]
- 6.Bardo MT, Bevins RA. Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology. 2000;153:31–43. doi: 10.1007/s002130000569. [DOI] [PubMed] [Google Scholar]
- 7.Shigeri Y, Seal RP, Shimamoto K. Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res Rev. 2004;45:250–65. doi: 10.1016/j.brainresrev.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 8.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–62. [PubMed] [Google Scholar]
- 9.Stephenson FA. Structure and trafficking of NMDA and GABAA receptors. Biochem Soc Trans. 2006;34:877–81. doi: 10.1042/BST0340877. [DOI] [PubMed] [Google Scholar]
- 10.Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Junco-Clemente P, Linares-Clemente P, Fernandez-Chacon R. Active zones for presynaptic plasticity in the brain. Mol Psychiatry. 2005;10:185–200. doi: 10.1038/sj.mp.4001628. image 131. [DOI] [PubMed] [Google Scholar]
- 12.Castellano C, Cestari V, Ciamei A. NMDA receptors and learning and memory processes. Curr Drug Targets. 2001;2:273–83. doi: 10.2174/1389450013348515. [DOI] [PubMed] [Google Scholar]
- 13.Riedel G, Platta B, Micheaub J. Glutamate receptor function in learning and memory. Behavioural Brain Res. 2003;140:1–47. doi: 10.1016/s0166-4328(02)00272-3. [DOI] [PubMed] [Google Scholar]
- 14.Perez-Otano I, Ehlers MD. Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci. 2005;28:229–38. doi: 10.1016/j.tins.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 16.Malenka RC, Nicoll RA. Long-term potentiation--a decade of progress? Science. 1999;285:1870–4. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 17.Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25:578–88. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- 18.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 19.Cooke SF, Bliss TV. Plasticity in the human central nervous system. Brain. 2006;129:1659–73. doi: 10.1093/brain/awl082. [DOI] [PubMed] [Google Scholar]
- 20.Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30:126–34. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 21.Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–13. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- 22.Huettner JE. Kainate receptors and synaptic transmission. Prog Neurobiol. 2003;70:387–407. doi: 10.1016/s0301-0082(03)00122-9. [DOI] [PubMed] [Google Scholar]
- 23.Conn PJ, Pin J-P. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 24.Hermans E, Challiss RA. Structural, signalling and regulatory properties of the group I metabotropic glutamate receptors: prototypic family C G-protein-coupled receptors. Biochem J. 2001;359:465–84. doi: 10.1042/0264-6021:3590465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coutinho V, Knopfel T. Metabotropic glutamate receptors: electrical and chemical signaling properties. Neuroscientist. 2002;8:551–561. doi: 10.1177/1073858402238514. [DOI] [PubMed] [Google Scholar]
- 26.Brakeman PR, Lanahan AA, O’Brien R, Roche K, Barnes CA, Huganir RL, et al. Homer: a protein that selectively binds metabotropic glutamate receptors. Nature. 1997;386:284–8. doi: 10.1038/386284a0. [DOI] [PubMed] [Google Scholar]
- 27.Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583–92. doi: 10.1016/s0896-6273(00)80810-7. [DOI] [PubMed] [Google Scholar]
- 28.Xiao B, Tu JC, Worley PF. Homer: a link between neural activity and glutamate receptor function. Curr Opin Neurobiol. 2000;10:370–4. doi: 10.1016/s0959-4388(00)00087-8. [DOI] [PubMed] [Google Scholar]
- 29.de Bartolomeis A, Iasevoli F. The Homer family and the signal transduction system at glutamatergic postsynaptic density: potential role in behavior and pharmacotherapy. Psychopharmacol Bull. 2003;37:51–83. [PubMed] [Google Scholar]
- 30.Szumlinski KK, Kalivas PW, Worley PF. Homer proteins: implications for neuropsychiatric disorders. Curr Opin Neurobiol. 2006;16:251–7. doi: 10.1016/j.conb.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Mudo G, Trovato-Salinaro A, Caniglia G, Cheng Q, Condorelli DF. Cellular localization of mGluR3 and mGluR5 mRNAs in normal and injured rat brain. Brain Res. 2007 doi: 10.1016/j.brainres.2007.02.041. [DOI] [PubMed] [Google Scholar]
- 32.Ron D, Jurd R. The “ups and downs” of signaling cascades in addiction. Sci STKE. 2005;2005:re14. doi: 10.1126/stke.3092005re14. [DOI] [PubMed] [Google Scholar]
- 33.Rao VR, Finkbeiner S. NMDA and AMPA receptors: old channels, new tricks. Trends Neurosci. 2007;30:284–91. doi: 10.1016/j.tins.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 34.Melendez RI, Vuthiganon J, Kalivas PW. Regulation of extracellular glutamate in the prefrontal cortex: focus on the cystine glutamate exchanger and group I metabotropic glutamate receptors. J Pharmacol Exp Ther. 2005;314:139–47. doi: 10.1124/jpet.104.081521. [DOI] [PubMed] [Google Scholar]
- 35.McBean GJ. Cerebral cystine uptake: a tale of two transporters. Trends Pharmacol Sci. 2002;23:299–302. doi: 10.1016/s0165-6147(02)02060-6. [DOI] [PubMed] [Google Scholar]
- 36.Baker DA, Shen H, Kalivas PW. Cystine/glutamate exchange serves as the source for extracellular glutamate: modifications by repeated cocaine administration. Amino Acids. 2002;23:161–2. doi: 10.1007/s00726-001-0122-6. [DOI] [PubMed] [Google Scholar]
- 37.Moran MM, McFarland K, Melendez RI, Kalivas PW, Seamans JK. Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci. 2005;25:6389–6393. doi: 10.1523/JNEUROSCI.1007-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heimer L, Alheid GF, de Olmos JS, Groenewegen HJ, Haber SN, Harlan RE, et al. The accumbens: beyond the core-shell dichotomy. J Neuropsychiatry Clin Neurosci. 1997;9:354–381. doi: 10.1176/jnp.9.3.354. [DOI] [PubMed] [Google Scholar]
- 39.Groenewegen HJ, Wright CI, Beijer AV, Voorn P. Convergence and segregation of ventral striatal inputs and outputs. Ann N Y Acad Sci. 1999;877:49–63. doi: 10.1111/j.1749-6632.1999.tb09260.x. [DOI] [PubMed] [Google Scholar]
- 40.Omelchenko N, Sesack SR. Glutamate synaptic inputs to ventral tegmental area neurons in the rat derive primarily from subcortical sources. Neuroscience. 2007;146:1259–1274. doi: 10.1016/j.neuroscience.2007.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geisler S, Derst C, Veh RW, Zahm DS. Glutamatergic afferents of the ventral tegmental area in the rat. J Neurosci. 2007;27:5730–5743. doi: 10.1523/JNEUROSCI.0012-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamaguchi T, Sheen W, Morales M. Glutamatergic neurons are present in the rat ventral tegmental area. Eur J Neurosci. 2007;25:106–18. doi: 10.1111/j.1460-9568.2006.05263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pulvirenti L, Diana M. Drug dependence as a disorder of neural plasticity: focus on dopamine and glutamate. Rev Neurosci. 2001;12:141–58. doi: 10.1515/revneuro.2001.12.2.141. [DOI] [PubMed] [Google Scholar]
- 44.Tzschentke TM, Schmidt WJ. Glutamatergic mechanisms in addiction. Mol Psychiatry. 2003;8:373–82. doi: 10.1038/sj.mp.4001269. [DOI] [PubMed] [Google Scholar]
- 45.Lapish CC, Seamans JK, Judson Chandler L. Glutamate-dopamine cotransmission and reward processing in addiction. Alcohol Clin Exp Res. 2006;30:1451–65. doi: 10.1111/j.1530-0277.2006.00176.x. [DOI] [PubMed] [Google Scholar]
- 46.Karler R, Calder LD, Chaudhry IA, Turkanis SA. Blockade of “reverse tolerance” to cocaine and amphetamine by MK-801. Life Sci. 1989;45:599–606. doi: 10.1016/0024-3205(89)90045-3. [DOI] [PubMed] [Google Scholar]
- 47.Pulvirenti L, Swerdlow NR, Koob GF. Microinjection of a gluatamate antagonist into the nucleus accumbens reduces psychostimulant locomotion in rats. Neurosci Lett. 1989;103:213–218. doi: 10.1016/0304-3940(89)90578-8. [DOI] [PubMed] [Google Scholar]
- 48.Pulvirenti L, Swerdlow NR, Koob GF. Nucleus accumbens NMDA antagonist decreases locomotor activity produced by cocaine, heroin or accumbens dopamine, but not caffeine. Pharmacol Biochem Behav. 1991;40:841–5. doi: 10.1016/0091-3057(91)90095-j. [DOI] [PubMed] [Google Scholar]
- 49.Pulvirenti L, Maldonado-Lopez R, Koob GF. NMDA receptors in the nucleus accumbens modulate intravenous cocaine but not heroin self-administration in the rat. Brain Res. 1992;594:327–30. doi: 10.1016/0006-8993(92)91145-5. [DOI] [PubMed] [Google Scholar]
- 50.Witkin JM. Blockade of the locomotor stimulant effects of cocaine and methamphetamine by glutamate antagonists. Life Sci. 1993;53:L405–10. doi: 10.1016/0024-3205(93)90496-p. [DOI] [PubMed] [Google Scholar]
- 51.Pulvirenti L, Berrier R, Kreifeldt M, Koob GF. Modulation of locomotor activity by NMDA receptors in the nucleus accumbens core and shell regions of the rat. Brain Res. 1994;664:231–236. doi: 10.1016/0006-8993(94)91977-1. [DOI] [PubMed] [Google Scholar]
- 52.Pierce RC, Kalivas PW. A circuitry model of the expression of behavioral sensitization to amphetamine-like psychostimulants. Brain Res Rev. 1997;25:192–216. doi: 10.1016/s0165-0173(97)00021-0. [DOI] [PubMed] [Google Scholar]
- 53.Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:679–720. doi: 10.1016/s0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]
- 54.Rockhold RW. Glutamatergic involvement in psychomotor stimulant action. Prog Drug Res. 1998;50:155–92. doi: 10.1007/978-3-0348-8833-2_4. [DOI] [PubMed] [Google Scholar]
- 55.White FJ, Kalivas PW. Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend. 1998;51:141–154. doi: 10.1016/s0376-8716(98)00072-6. [DOI] [PubMed] [Google Scholar]
- 56.Clark D, Overton PG. Alterations in excitatory amino acid-mediated regulation of midbrain dopaminergic neurones induced by chronic psychostimulant administration and stress: relevance to behavioural sensitization and drug addiction. Addict Biol. 1998;3:109–135. doi: 10.1080/13556219872191. [DOI] [PubMed] [Google Scholar]
- 57.Vanderschuren LJ, Kalivas PW. Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology. 2000;151:99–120. doi: 10.1007/s002130000493. [DOI] [PubMed] [Google Scholar]
- 58.Steketee JD. Neurotransmitter systems of the medial prefrontal cortex: potential role in sensitization to psychostimulants. Brain Res Rev. 2003;41:203–28. doi: 10.1016/s0165-0173(02)00233-3. [DOI] [PubMed] [Google Scholar]
- 59.Wolf ME, Sun X, Mangiavacchi S, Chao SZ. Psychomotor stimulants and neuronal plasticity. Neuropharmacology. 2004;47(Suppl 1):61–79. doi: 10.1016/j.neuropharm.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 60.Steketee JD. Cortical mechanisms of cocaine sensitization. Crit Rev Neurobiol. 2005;17:69–86. doi: 10.1615/critrevneurobiol.v17.i2.20. [DOI] [PubMed] [Google Scholar]
- 61.McKee BL, Meshul CK. Time-dependent changes in extracellular glutamate in the rat dorsolateral striatum following a single cocaine injection. Neuroscience. 2005;133:605–13. doi: 10.1016/j.neuroscience.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 62.Robinson SE, Maher JR, McDowell KP, Kunko PM. Effects of cocaine and the cocaine analog CFT on glutamatergic neurons. Pharmacol Biochem Behav. 1995;50:627–33. doi: 10.1016/0091-3057(94)00355-6. [DOI] [PubMed] [Google Scholar]
- 63.Smith JA, Mo Q, Guo H, Kunko PM, Robinson SE. Cocaine increases extraneuronal levels of aspartate and glutamate in the nucleus accumbens. Brain Res. 1995;683:264–9. doi: 10.1016/0006-8993(95)00383-2. [DOI] [PubMed] [Google Scholar]
- 64.Pierce RC, Bell K, Duffy P, Kalivas PW. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci. 1996;16:1550–1560. doi: 10.1523/JNEUROSCI.16-04-01550.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reid MS, Berger SP. Evidence for sensitization of cocaine-induced nucleus accumbens glutamate release. Neuroreport. 1996;7:1325–9. doi: 10.1097/00001756-199605170-00022. [DOI] [PubMed] [Google Scholar]
- 66.Reid MS, Hsu KJ, Berger SP. Cocaine and amphetamine preferentially stimulate glutamate release in the limbic system: studies on the involvement of dopamine. Synapse. 1997;27:95–105. doi: 10.1002/(SICI)1098-2396(199710)27:2<95::AID-SYN1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 67.Bell K, Duffy P, Kalivas PW. Context-specific enhancement of glutamate transmission by cocaine. Neuropsychopharmacology. 2000;23:335–344. doi: 10.1016/S0893-133X(00)00100-7. [DOI] [PubMed] [Google Scholar]
- 68.McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23:3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, et al. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci. 2003;6:743–749. doi: 10.1038/nn1069. [DOI] [PubMed] [Google Scholar]
- 70.Bowers MS, McFarland K, Lake RW, Peterson YK, Lapish CC, Gregory ML, et al. Activator of G protein signaling 3: a gatekeeper of cocaine sensitization and drug seeking. Neuron. 2004;42:269–81. doi: 10.1016/s0896-6273(04)00159-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morishima Y, Miyakawa T, Furuyashiki T, Tanaka Y, Mizuma H, Nakanishi S. Enhanced cocaine responsiveness and impaired motor coordination in metabotropic glutamate receptor subtype 2 knockout mice. Proc Natl Acad Sci U S A. 2005;102:4170–5. doi: 10.1073/pnas.0500914102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shoji S, Simms D, McDaniel WC, Gallagher JP. Chronic cocaine enhances gamma-aminobutyric acid and glutamate release by altering presynaptic and not postsynaptic gamma-aminobutyric acidB receptors within the rat dorsolateral septal nucleus. J Pharmacol Exp Ther. 1997;280:129–37. [PubMed] [Google Scholar]
- 73.Sizemore GM, Co C, Smith JE. Ventral pallidal extracellular fluid levels of dopamine, serotonin, gamma amino butyric acid, and glutamate during cocaine self-administration in rats. Psychopharmacology. 2000;150:391–8. doi: 10.1007/s002130000456. [DOI] [PubMed] [Google Scholar]
- 74.Smith JE, Koves TR, Co C. Brain neurotransmitter turnover rates during rat intravenous cocaine self-administration. Neuroscience. 2003;117:461–75. doi: 10.1016/s0306-4522(02)00819-9. [DOI] [PubMed] [Google Scholar]
- 75.Kalivas PW, Duffy P. D1 receptors modulate glutamate transmission in the ventral tegmental area. J Neurosci. 1995;15:5379–88. doi: 10.1523/JNEUROSCI.15-07-05379.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kalivas PW, Duffy P. Repeated cocaine administration alters extracellular glutamate in the ventral tegmental area. J Neurochem. 1998;70:1497–502. doi: 10.1046/j.1471-4159.1998.70041497.x. [DOI] [PubMed] [Google Scholar]
- 77.Dworkin SI, Co C, Smith JE. Rat brain neurotransmitter turnover rates altered during withdrawal from chronic cocaine administration. Brain Res. 1995;682:116–26. doi: 10.1016/0006-8993(95)00327-m. [DOI] [PubMed] [Google Scholar]
- 78.Howes SR, Dalley JW, Morrison CH, Robbins TW, Everitt BJ. Leftward shift in the acquisition of cocaine self-administration in isolation-reared rats: relationship to extracellular levels of dopamine, serotonin and glutamate in the nucleus accumbens and amygdala-striatal FOS expression. Psychopharmacology. 2000;151:55–63. doi: 10.1007/s002130000451. [DOI] [PubMed] [Google Scholar]
- 79.Meshul CK, Noguchi N, Emre N, Ellison G. Cocaine-induced changes in glutamate and GABA immunolabeling within rat habenula and nucleus accumbens. Synapse. 1998;30:211–220. doi: 10.1002/(SICI)1098-2396(199810)30:2<211::AID-SYN11>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 80.Keys AS, Mark GP, Emre N, Meshul CK. Reduced glutamate immunolabeling in the nucleus accumbens following extended withdrawal from self-administered cocaine. Synapse. 1998;30:393–401. doi: 10.1002/(SICI)1098-2396(199812)30:4<393::AID-SYN6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 81.Kozell B, Meshul K. Alterations in nerve terminal glutamate immunoreactivity in the nucleus accumbens and ventral tegmental area following single and repeated doses of cocaine. Psychopharmacology. 2003;165:337–45. doi: 10.1007/s00213-002-1296-7. [DOI] [PubMed] [Google Scholar]
- 82.Hotsenpiller G, Giorgetti M, Wolf ME. Alterations in behaviour and glutamate transmission following presentation of stimuli previously associated with cocaine exposure. Eur J Neurosci. 2001;14:1843–1855. doi: 10.1046/j.0953-816x.2001.01804.x. [DOI] [PubMed] [Google Scholar]
- 83.Hotsenpiller G, Wolf ME. Extracellular glutamate levels in prefrontal cortex during the expression of associative responses to cocaine related stimuli. Neuropharmacology. 2002;43:1218–29. doi: 10.1016/s0028-3908(02)00308-8. [DOI] [PubMed] [Google Scholar]
- 84.Jimenez-Rivera CA, Waterhouse BD. Effects of systemically and locally applied cocaine on cerebrocortical neuron responsiveness to afferent synaptic inputs and glutamate. Brain Res. 1991;546:287–96. doi: 10.1016/0006-8993(91)91493-k. [DOI] [PubMed] [Google Scholar]
- 85.Kiyatkin EA, Rebec GV. Dopamine-independent action of cocaine on striatal and accumbal neurons. Eur J Neurosci. 2000;12:1789–1800. doi: 10.1046/j.1460-9568.2000.00066.x. [DOI] [PubMed] [Google Scholar]
- 86.Schramm-Sapyta NL, Olsen CM, Winder DG. Cocaine self-administration reduces excitatory responses in the mouse nucleus accumbens shell. Neuropsychopharmacology. 2006;31:1444–51. doi: 10.1038/sj.npp.1300918. [DOI] [PubMed] [Google Scholar]
- 87.Nicola SM, Surmeier DJ, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- 88.O’Donnell P. Dopamine gating of forebrain neural ensembles. Eur J Neurosci. 2003;17:429–35. doi: 10.1046/j.1460-9568.2003.02463.x. [DOI] [PubMed] [Google Scholar]
- 89.Hanson GR, Singh N, Merchant K, Johnson M, Gibb JW. The role of NMDA receptor systems in neuropeptide responses to stimulants of abuse. Drug Alcohol Depend. 1995;37:107–10. doi: 10.1016/0376-8716(94)01065-s. [DOI] [PubMed] [Google Scholar]
- 90.Valjent E, Corvol JC, Pages C, Besson MJ, Maldonado R, Caboche J. Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J Neurosci. 2000;20:8701–8709. doi: 10.1523/JNEUROSCI.20-23-08701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jenab S, Festa ED, Nazarian A, Wu HB, Sun WL, Hazim R, et al. Cocaine induction of ERK proteins in dorsal striatum of Fischer rats. Mol Brain Res. 2005;142:134–8. doi: 10.1016/j.molbrainres.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 92.Snyder GL, Allen PB, Fienberg AA, Valle CG, Huganir RL, Nairn AC, et al. Regulation of phosphorylation of the GluR1 AMPA receptor in the neostriatum by dopamine and psychostimulants in vivo. J Neurosci. 2000;20:4480–8. doi: 10.1523/JNEUROSCI.20-12-04480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bellone C, Luscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci. 2006;9:636–41. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- 94.Schilstrom B, Yaka R, Argilli E, Suvarna N, Schumann J, Chen BT, et al. Cocaine enhances NMDA receptor-mediated currents in ventral tegmental area cells via dopamine D5 receptor-dependent redistribution of NMDA receptors. J Neurosci. 2006;26:8549–58. doi: 10.1523/JNEUROSCI.5179-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–7. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- 96.Liu XY, Chu XP, Mao LM, Wang M, Lan HX, Li MH, et al. Modulation of D2R-NR2B interactions in response to cocaine. Neuron. 2006;52:897–909. doi: 10.1016/j.neuron.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 97.Ghasemzadeh MB, Nelson LC, Lu XY, Kalivas PW. Neuroadaptations in ionotropic and metabotropic glutamate receptor mRNA produced by cocaine treatment. J Neurochem. 1999;72:157–65. doi: 10.1046/j.1471-4159.1999.0720157.x. [DOI] [PubMed] [Google Scholar]
- 98.Yamaguchi M, Suzuki T, Abe S, Hori T, Kurita H, Asada T, et al. Repeated cocaine administration differentially affects NMDA receptor subunit (NR1, NR2A-C) mRNAs in rat brain. Synapse. 2002;46:157–169. doi: 10.1002/syn.10132. [DOI] [PubMed] [Google Scholar]
- 99.Yuferov V, Kroslak T, Laforge KS, Zhou Y, Ho A, Kreek MJ. Differential gene expression in the rat caudate putamen after “binge” cocaine administration: advantage of triplicate microarray analysis. Synapse. 2003;48:157–69. doi: 10.1002/syn.10198. [DOI] [PubMed] [Google Scholar]
- 100.White FJ, Hu XT, Zhang XF, Wolf ME. Repeated administration of cocaine or amphetamine alters neuronal responses to glutamate in the mesoaccumbens dopamine system. J Pharmacol Exp Ther. 1995;273:445–54. [PubMed] [Google Scholar]
- 101.Zhang XF, Hu XT, White FJ, Wolf ME. Increased responsiveness of ventral tegmental area dopamine neurons to glutamate after repeated administration of cocaine or amphetamine is transient and selectively involves AMPA receptors. J Pharmacol Exp Ther. 1997;281:699–706. [PubMed] [Google Scholar]
- 102.Fitzgerald L, Ortiz J, Hamedani A, Nestler E. Drugs of abuse and stress increase the expression of GluR1 and NMDAR1 glutamate receptor subunits in the rat ventral tegmental area: common adaptations among cross-sensitizing agents. J Neurosci. 1996;16:274–282. doi: 10.1523/JNEUROSCI.16-01-00274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Churchill L, Swanson CJ, Urbina M, Kalivas PW. Repeated cocaine alters glutamate receptor subunit levels in the nucleus accumbens and ventral tegmental area of rats that develop behavioral sensitization. J Neurochem. 1999;72:2397–403. doi: 10.1046/j.1471-4159.1999.0722397.x. [DOI] [PubMed] [Google Scholar]
- 104.Lu L, Grimm JW, Shaham Y, Hope BT. Molecular neuroadaptations in the accumbens and ventral tegmental area during the first 90 days of forced abstinence from cocaine self-administration in rats. J Neurochem. 2003;85:1604–13. doi: 10.1046/j.1471-4159.2003.01824.x. [DOI] [PubMed] [Google Scholar]
- 105.Loftis JM, Janowsky A. Cocaine treatment- and withdrawal-induced alterations in the expression and serine phosphorylation of the NR1 NMDA receptor subunit. Psychopharmacology. 2002;164:349–59. doi: 10.1007/s00213-002-1209-9. [DOI] [PubMed] [Google Scholar]
- 106.Lu W, Monteggia LM, Wolf ME. Repeated administration of amphetamine or cocaine does not alter AMPA receptor subunit expression in the rat midbrain. Neuropsychopharmacology. 2002;26:1–13. doi: 10.1016/S0893-133X(01)00272-X. [DOI] [PubMed] [Google Scholar]
- 107.Backes E, Hemby SE. Discrete cell gene profiling of ventral tegmental dopamine neurons after acute and chronic cocaine self-administration. J Pharmacol Exp Ther. 2003;307:450–9. doi: 10.1124/jpet.103.054965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hemby SE, Horman B, Tang W. Differential regulation of ionotropic glutamate receptor subunits following cocaine self-administration. Brain Res. 2005;1064:75–82. doi: 10.1016/j.brainres.2005.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tang W-X, Fasulo WH, Mash DC, Hemby SE. Molecular profiling of midbrain dopamine regions in cocaine overdose victims. J Neurochem. 2003;85:911–924. doi: 10.1046/j.1471-4159.2003.01740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hemby SE, Tang W, Muly EC, Kuhar MJ, Howell L, Mash DC. Cocaine-induced alterations in nucleus accumbens ionotropic glutamate receptor subunits in human and non-human primates. J Neurochem. 2005 doi: 10.1111/j.1471-4159.2005.03517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Robinson SE, Kunko PM, Smith JA, Wallace MJ, Mo Q, Maher JR. Extracellular aspartate concentration increases in nucleus accumbens after cocaine sensitization. Eur J Pharmacol. 1997;319:31–6. doi: 10.1016/s0014-2999(96)00923-5. [DOI] [PubMed] [Google Scholar]
- 112.Kozell LB, Meshul CK. Nerve terminal glutamate immunoreactivity in the rat nucleus accumbens and ventral tegmental area after a short withdrawal from cocaine. Synapse. 2004;51:224–32. doi: 10.1002/syn.10304. [DOI] [PubMed] [Google Scholar]
- 113.Scheggi S, Mangiavacchi S, Masi F, Gambarana C, Tagliamonte A, De Montis MG. Dizocilpine infusion has a different effect in the development of morphine and cocaine sensitization: behavioral and neurochemical aspects. Neuroscience. 2002;109:267–74. doi: 10.1016/s0306-4522(01)00483-3. [DOI] [PubMed] [Google Scholar]
- 114.Loftis JM, Janowsky A. Regulation of NMDA receptor subunits and nitric oxide synthase expression during cocaine withdrawal. J Neurochem. 2000;75:2040–50. doi: 10.1046/j.1471-4159.2000.0752040.x. [DOI] [PubMed] [Google Scholar]
- 115.Zhang X, Lee TH, Davidson C, Lazarus C, Wetsel WC, Ellinwood EH. Reversal of cocaine-induced behavioral sensitization and associated phosphorylation of the NR2B and GluR1 subunits of the NMDA and AMPA receptors. Neuropsychopharmacology. 2007;32:377–87. doi: 10.1038/sj.npp.1301101. [DOI] [PubMed] [Google Scholar]
- 116.Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25:9144–51. doi: 10.1523/JNEUROSCI.2252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Williams JM, Steketee JD. Cocaine increases medial prefrontal cortical glutamate overflow in cocaine-sensitized rats: a time course study. Eur J Neurosci. 2004;20:1639–46. doi: 10.1111/j.1460-9568.2004.03618.x. [DOI] [PubMed] [Google Scholar]
- 118.Neugebauer V, Zinebi F, Russell R, Gallagher JP, Shinnick-Gallagher P. Cocaine and kindling alter the sensitivity of group II and III metabotropic glutamate receptors in the central amygdala. J Neurophysiol. 2000;84:759–70. doi: 10.1152/jn.2000.84.2.759. [DOI] [PubMed] [Google Scholar]
- 119.Swanson CJ, Baker DA, Carson D, Worley PF, Kalivas PW. Repeated cocaine administration attenuates group I metabotropic glutamate receptor-mediated glutamate release and behavioral activation: a potential role for Homer. J Neurosci. 2001;21:9043–52. doi: 10.1523/JNEUROSCI.21-22-09043.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xi ZX, Ramamoorthy S, Baker DA, Shen H, Devadoss J, Kalivas PW. Modulation of group II metabotropic glutamate receptor signaling by chronic cocaine. J Pharmacol Exp Ther. 2002;308:608–615. doi: 10.1124/jpet.102.039735. [DOI] [PubMed] [Google Scholar]
- 121.Huang CC, Yang PC, Lin HJ, Hsu KS. Repeated cocaine administration impairs group II metabotropic glutamate receptor-mediated long-term depression in rat medial prefrontal cortex. J Neurosci. 2007;27:2958–2968. doi: 10.1523/JNEUROSCI.4247-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tang W, Wesley M, Freeman WM, Liang B, Hemby SE. Alterations in ionotropic glutamate receptor subunits during binge cocaine self-administration and withdrawal in rats. J Neurochem. 2004;89:1021–33. doi: 10.1111/j.1471-4159.2004.02392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Grignaschi G, Burbassi S, Zennaro E, Bendotti C, Cervo L. A single high dose of cocaine induces behavioural sensitization and modifies mRNA encoding GluR1 and GAP-43 in rats. Eur J Neurosci. 2004;20:2833–7. doi: 10.1111/j.1460-9568.2004.03712.x. [DOI] [PubMed] [Google Scholar]
- 124.Itzhak Y, Stein I. Sensitization to the toxic effects of cocaine in mice is associated with the regulation of N-methyl-D-aspartate receptors in the cortex. J Pharmacol Exp Ther. 1992;262:464–70. [PubMed] [Google Scholar]
- 125.Itzhak Y. Modulation of the PCP/NMDA receptor complex and sigma binding sites by psychostimulants. Neurotoxicol Teratol. 1994;16:363–8. doi: 10.1016/0892-0362(94)90024-8. [DOI] [PubMed] [Google Scholar]
- 126.Itzhak Y, Martin JL. Cocaine-induced kindling is associated with elevated NMDA receptor binding in discrete mouse brain regions. Neuropharmacology. 2000;39:32–9. doi: 10.1016/s0028-3908(99)00073-8. [DOI] [PubMed] [Google Scholar]
- 127.Szumlinski KK, Herrick-Davis K, Teitler M, Maisonneuve IM, Glick SD. Behavioural sensitization to cocaine is dissociated from changes in striatal NMDA receptor levels. Neuroreport. 2000;11:2785–8. doi: 10.1097/00001756-200008210-00035. [DOI] [PubMed] [Google Scholar]
- 128.Toda S, McGinty JF, Kalivas PW. Repeated cocaine administration alters the expression of genes in corticolimbic circuitry after a 3-week withdrawal: a DNA macroarray study. J Neurochem. 2002;82:1290–1299. doi: 10.1046/j.1471-4159.2002.01083.x. [DOI] [PubMed] [Google Scholar]
- 129.Freeman WF, Brebner K, Lynch WJ, Robertson DJ, Roberts DCS, Vrana KE. Cocaine-responsive gene expression changes in rat hippocampus. Neuroscience. 2001;108:371–380. doi: 10.1016/s0306-4522(01)00432-8. [DOI] [PubMed] [Google Scholar]
- 130.Turchan J, Maj M, Przewlocka B. The effect of drugs of abuse on NMDAR1 receptor expression in the rat limbic system. Drug Alcohol Depend. 2003;72:193–6. doi: 10.1016/s0376-8716(03)00193-5. [DOI] [PubMed] [Google Scholar]
- 131.Lu L, Dempsey J, Shaham Y, Hope BT. Differential long-term neuroadaptations of glutamate receptors in the basolateral and central amygdala after withdrawal from cocaine self-administration in rats. J Neurochem. 2005;94:161–8. doi: 10.1111/j.1471-4159.2005.03178.x. [DOI] [PubMed] [Google Scholar]
- 132.Pulvirenti L, Balducci C, Koob GF. Dextromethorphan reduces intravenous cocaine self-administration in the rat. Eur J Pharmacol. 1997;321:279–83. doi: 10.1016/s0014-2999(96)00970-3. [DOI] [PubMed] [Google Scholar]
- 133.Hyytia P, Backstrom P, Liljequist S. Site-specific NMDA receptor antagonists produce differential effects on cocaine self-administration in rats. Eur J Pharmacol. 1999;378:9–16. doi: 10.1016/s0014-2999(99)00446-x. [DOI] [PubMed] [Google Scholar]
- 134.Papp M, Gruca P, Willner P. Selective blockade of drug-induced place preference conditioning by ACPC, a functional NDMA-receptor antagonist. Neuropsychopharmacology. 2002;27:727–43. doi: 10.1016/S0893-133X(02)00349-4. [DOI] [PubMed] [Google Scholar]
- 135.Blokhina EA, Kashkin VA, Zvartau EE, Danysz W, Bespalov AY. Effects of nicotinic and NMDA receptor channel blockers on intravenous cocaine and nicotine self-administration in mice. Eur Neuropsychopharmacol. 2005;15:219–25. doi: 10.1016/j.euroneuro.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 136.Kotlinska J, Biala G. Memantine and ACPC affect conditioned place preference induced by cocaine in rats. Pol J Pharmacol. 2000;52:179–85. [PubMed] [Google Scholar]
- 137.Maldonado C, Rodriguez-Arias M, Castillo A, Aguilar MA, Minarro J. Effect of memantine and CNQX in the acquisition, expression and reinstatement of cocaine-induced conditioned place preference. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:932–939. doi: 10.1016/j.pnpbp.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 138.Slusher BS, Thomas A, Paul M, Schad CA, Ashby CR., Jr Expression and acquisition of the conditioned place preference response to cocaine in rats is blocked by selective inhibitors of the enzyme N-acetylated-alpha-linked-acidic dipeptidase (NAALADASE) Synapse. 2001;41:22–8. doi: 10.1002/syn.1056. [DOI] [PubMed] [Google Scholar]
- 139.Di Ciano P, Everitt BJ. Dissociable effects of antagonism of NMDA and AMPA/KA receptors in the nucleus accumbens core and shell on cocaine-seeking behavior. Neuropsychopharmacology. 2001;25:341–360. doi: 10.1016/S0893-133X(01)00235-4. [DOI] [PubMed] [Google Scholar]
- 140.Vanderschuren LJ, Di Ciano P, Everitt BJ. Involvement of the dorsal striatum in cue-controlled cocaine seeking. J Neurosci. 2005;25:8665–70. doi: 10.1523/JNEUROSCI.0925-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cornish JL, Duffy P, Kalivas PW. A role for nucleus accumbens glutamate transmission in the relapse to cocaine-seeking behavior. Neuroscience. 1999;92:1359–1367. doi: 10.1016/s0306-4522(99)00214-6. [DOI] [PubMed] [Google Scholar]
- 142.Cornish JL, Kalivas PW. Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J Neurosci. 2000;20:RC89. doi: 10.1523/JNEUROSCI.20-15-j0006.2000. 1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Suto N, Tanabe LM, Austin JD, Creekmore E, Pham CT, Vezina P. Previous exposure to psychostimulants enhances the reinstatement of cocaine seeking by nucleus accumbens AMPA. Neuropsychopharmacology. 2004;29:2149–59. doi: 10.1038/sj.npp.1300533. [DOI] [PubMed] [Google Scholar]
- 144.Backstrom P, Hyytia P. Involvement of AMPA/kainate, NMDA, and mGlu5 receptors in the nucleus accumbens core in cue-induced reinstatement of cocaine seeking in rats. Psychopharmacology. 2007;192:571–580. doi: 10.1007/s00213-007-0753-8. [DOI] [PubMed] [Google Scholar]
- 145.Park W-K, Bari AA, Jey AR, Anderson SM, Spealman RD, Rowlett JK, et al. Cocaine administered into the medial prefrontal cortex reinstates cocaine-seeking behavior by increasing AMPA receptor-mediated glutamate transmission in the nucleus accumbens. J Neurosci. 2002;22:2916–2925. doi: 10.1523/JNEUROSCI.22-07-02916.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.De Vries TJ, Schoffelmeer AN, Binnekade R, Mulder AH, Vanderschuren LJ. MK-801 reinstates drug-seeking behaviour in cocaine-trained rats. Neuroreport. 1998;9:637–40. doi: 10.1097/00001756-199803090-00014. [DOI] [PubMed] [Google Scholar]
- 147.Famous KR, Schmidt HD, Pierce RC. When administered into the nucleus accumbens core or shell, the NMDA receptor antagonist AP-5 reinstates cocaine-seeking behavior in the rat. Neurosci Lett. 2007;420:169–173. doi: 10.1016/j.neulet.2007.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162:1403–13. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- 149.Kalivas PW, Volkow N, Seamans J. Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron. 2005;45:647–50. doi: 10.1016/j.neuron.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 150.Vorel SR, Liu X, Hayes RJ, Spector JA, Gardner EL. Relapse to cocaine-seeking after hippocampal theta burst stimulation. Science. 2001;292:1175–8. doi: 10.1126/science.1058043. [DOI] [PubMed] [Google Scholar]
- 151.Harris GC, Aston-Jones G. Critical role for ventral tegmental glutamate in preference for a cocaine-conditioned environment. Neuropsychopharmacology. 2003;28:73–76. doi: 10.1038/sj.npp.1300011. [DOI] [PubMed] [Google Scholar]
- 152.Sun W, Akins CK, Mattingly AE, Rebec GV. Ionotropic glutamate receptors in the ventral tegmental area regulate cocaine-seeking behavior in rats. Neuropsychopharmacology. 2005;30:2073–2081. doi: 10.1038/sj.npp.1300744. [DOI] [PubMed] [Google Scholar]
- 153.Choi KH, Rahman Z, Edwards S, Hall S, Neve RL, Self DW. Opposite effects of GluR1 and PKA-resistant GluR1 overexpression in the ventral tegmental area on cocaine reinforcement. Ann N Y Acad Sci. 2003;1003:372–4. doi: 10.1196/annals.1300.029. [DOI] [PubMed] [Google Scholar]
- 154.See RE. Neural substrates of cocaine-cue associations that trigger relapse. Eur J Pharmacol. 2005;526:140–6. doi: 10.1016/j.ejphar.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 155.Hayes RJ, Vorel SR, Spector J, Liu X, Gardner EL. Electrical and chemical stimulation of the basolateral complex of the amygdala reinstates cocaine-seeking behavior in the rat. Psychopharmacology. 2003;168:75–83. doi: 10.1007/s00213-002-1328-3. [DOI] [PubMed] [Google Scholar]
- 156.See RE, Kruzich PJ, Grimm JW. Dopamine, but not glutamate, receptor blockade in the basolateral amygdala attenuates conditioned reward in a rat model of relapse to cocaine-seeking behavior. Psychopharmacology. 2001;154:301–10. doi: 10.1007/s002130000636. [DOI] [PubMed] [Google Scholar]
- 157.Di Ciano P, Everitt BJ. Direct interactions between the basolateral amygdala and nucleus accumbens core underlie cocaine-seeking behavior by rats. J Neurosci. 2004;24:7167–7173. doi: 10.1523/JNEUROSCI.1581-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chiamulera C, Epping-Jordan MP, Zocchi A, Marcon C, Cottiny C, Tacconi S, et al. Reinforcing and locomotor stimulant effects of cocaine are absent in mGluR5 null mutant mice. Nat Neurosci. 2001;4:873–874. doi: 10.1038/nn0901-873. [DOI] [PubMed] [Google Scholar]
- 159.Kenny PJ, Paterson NE, Boutrel B, Semenova S, Harrison AA, Gasparini F, et al. Metabotropic glutamate 5 receptor antagonist MPEP decreased nicotine and cocaine self-administration but not nicotine and cocaine-induced facilitation of brain reward function in rats. Ann N Y Acad Sci. 2003;1003:415–8. doi: 10.1196/annals.1300.040. [DOI] [PubMed] [Google Scholar]
- 160.Tessari M, Pilla M, Andreoli M, Hutcheson DM, Heidbreder CA. Antagonism at metabotropic glutamate 5 receptors inhibits nicotine- and cocaine-taking behaviours and prevents nicotine-triggered relapse to nicotine-seeking. Eur J Pharmacol. 2004;499:121–33. doi: 10.1016/j.ejphar.2004.07.056. [DOI] [PubMed] [Google Scholar]
- 161.Kenny PJ, Boutrel B, Gasparini F, Koob GF, Markou A. Metabotropic glutamate 5 receptor blockade may attenuate cocaine self-administration by decreasing brain reward function in rats. Psychopharmacology. 2005;179:247–54. doi: 10.1007/s00213-004-2069-2. [DOI] [PubMed] [Google Scholar]
- 162.Lee B, Platt DM, Rowlett JK, Adewale AS, Spealman RD. Attenuation of behavioral effects of cocaine by the metabotropic glutamate receptor 5 antagonist 2-methyl-6-(phenylethynyl)-pyridine in squirrel monkeys: comparison with dizocilpine. J Pharmacol Exp Ther. 2005;312:1232–1240. doi: 10.1124/jpet.104.078733. [DOI] [PubMed] [Google Scholar]
- 163.Paterson NE, Markou A. The metabotropic glutamate receptor 5 antagonist MPEP decreased break points for nicotine, cocaine and food in rats. Psychopharmacology. 2005;179:255–61. doi: 10.1007/s00213-004-2070-9. [DOI] [PubMed] [Google Scholar]
- 164.Iso Y, Grajkowska E, Wroblewski JT, Davis J, Goeders NE, Johnson KM, et al. Synthesis and structure-activity relationships of 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine analogues as potent, noncompetitive metabotropic glutamate receptor subtype 5 antagonists; search for cocaine medications. J Med Chem. 2006;49:1080–1100. doi: 10.1021/jm050570f. [DOI] [PubMed] [Google Scholar]
- 165.Backstrom P, Hyytia P. Ionotropic and metabotropic glutamate receptor antagonism attenuates cue-induced cocaine seeking. Neuropsychopharmacology. 2006;31:778–786. doi: 10.1038/sj.npp.1300845. [DOI] [PubMed] [Google Scholar]
- 166.Mcgeehan AJ, Olive MF. The mGluR5 antagonist MPEP reduces the conditioned rewarding effects of cocaine but not other drugs of abuse. Synapse. 2003;47:240–242. doi: 10.1002/syn.10166. [DOI] [PubMed] [Google Scholar]
- 167.Herzig V, Schmidt WJ. Effects of MPEP on locomotion, sensitization and conditioned reward induced by cocaine or morphine. Neuropharmacology. 2004;47:973–984. doi: 10.1016/j.neuropharm.2004.07.037. [DOI] [PubMed] [Google Scholar]
- 168.Adewale AS, Platt DM, Spealman RD. Pharmacological stimulation of group II metabotropic glutamate receptors reduces cocaine self-administration and cocaine-induced reinstatement of drug seeking in squirrel monkeys. J Pharmacol Exp Ther. 2006;318:922–931. doi: 10.1124/jpet.106.105387. [DOI] [PubMed] [Google Scholar]
- 169.Peters J, Kalivas PW. The group II metabotropic glutamate receptor agonist, LY379268, inhibits both cocaine- and food-seeking behavior in rats. Psychopharmacology. 2006;186:143–9. doi: 10.1007/s00213-006-0372-9. [DOI] [PubMed] [Google Scholar]
- 170.Baptista MAS, Martin-Fardon R, Weiss F. Preferential effects of the metabotropic glutamate 2/3 receptor agonist LY379268 on conditioned reinstatement versus primary reinforcement: comparison between cocaine and a potent conventional reinforcer. J Neurosci. 2004;24:4723–4727. doi: 10.1523/JNEUROSCI.0176-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lu L, Uejima JL, Gray SM, Bossert JM, Shaham Y. Systemic and central amygdala injections of the mGluR2/3 agonist LY379268 attenuate the expression of incubation of cocaine craving. Biol Psychiatry. 2007;61:591–598. doi: 10.1016/j.biopsych.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 172.Nakagawa T, Fujio M, Ozawa T, Minami M, Satoh M. Effect of MS-153, a glutamate transporter activator, on the conditioned rewarding effects of morphine, methamphetamine and cocaine in mice. Behav Brain Res. 2005;156:233–9. doi: 10.1016/j.bbr.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 173.Mead AN, Brown G, Le Merrer J, Stephens DN. Effects of deletion of gria1 or gria2 genes encoding glutamatergic AMPA-receptor subunits on place preference conditioning in mice. Psychopharmacology. 2005;179:164–71. doi: 10.1007/s00213-004-2071-8. [DOI] [PubMed] [Google Scholar]
- 174.Mead AN, Zamanillo D, Becker N, Stephens DN. AMPA-receptor GluR1 subunits are involved in the control over behavior by cocaine-paired cues. Neuropsychopharmacology. 2007;32:343–53. doi: 10.1038/sj.npp.1301045. [DOI] [PubMed] [Google Scholar]
- 175.Dong Y, Saal D, Thomas M, Faust R, Bonci A, Robinson T, et al. Cocaine-induced potentiation of synaptic strength in dopamine neurons: behavioral correlates in GluRA(-/-) mice. Proc Natl Acad Sci U S A. 2004;101:14282–7. doi: 10.1073/pnas.0401553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Heusner CL, Palmiter RD. Expression of mutant NMDA receptors in dopamine D1 receptor-containing cells prevents cocaine sensitization and decreases cocaine preference. J Neurosci. 2005;25:6651–7. doi: 10.1523/JNEUROSCI.1474-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhang G, Mao L, Liu X, Parelkar N, Arora A, Yang L, et al. In vivo regulation of Homer1a expression in the striatum by cocaine. Mol Pharmacol. 2007;71:1148–1158. doi: 10.1124/mol.106.028399. [DOI] [PubMed] [Google Scholar]
- 178.Ghasemzadeh MB, Permenter LK, Lake RW, Kalivas PW. Nucleus accumbens Homer proteins regulate behavioral sensitization to cocaine. Ann N Y Acad Sci. 2003;1003:395–7. doi: 10.1196/annals.1300.034. [DOI] [PubMed] [Google Scholar]
- 179.Szumlinski KK, Toda S, Middaugh LD, Worley PF, Kalivas PW. Evidence for a relationship between Group 1 mGluR hypofunction and increased cocaine and ethanol sensitivity in Homer2 null mutant mice. Ann N Y Acad Sci. 2003;1003:468–71. doi: 10.1196/annals.1300.055. [DOI] [PubMed] [Google Scholar]
- 180.Szumlinski KK, Dehoff MH, Kang SH, Frys KA, Lominac KD, Klugmann M, et al. Homer proteins regulate sensitivity to cocaine. Neuron. 2004;43:401–413. doi: 10.1016/j.neuron.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 181.Lominac KD, Oleson EB, Pava M, Klugmann M, Schwarz MK, Seeburg PH, et al. Distinct roles for different Homer1 isoforms in behaviors and associated prefrontal cortex function. J Neurosci. 2005;25:11586–94. doi: 10.1523/JNEUROSCI.3764-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yamamoto BK, Bankson MG. Amphetamine neurotoxicity: cause and consequence of oxidative stress. Crit Rev Neurobiol. 2005;17:87–117. doi: 10.1615/critrevneurobiol.v17.i2.30. [DOI] [PubMed] [Google Scholar]
- 183.Riddle EL, Fleckenstein AE, Hanson GR. Mechanisms of methamphetamine-induced dopaminergic neurotoxicity. AAPS J. 2006;8:E413–8. doi: 10.1007/BF02854914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Cadet JL, Krasnova IN, Jayanthi S, Lyles J. Neurotoxicity of substituted amphetamines: molecular and cellular mechanisms. Neurotox Res. 2007;11:183–202. doi: 10.1007/BF03033567. [DOI] [PubMed] [Google Scholar]
- 185.Blagoeva P, Masi I, De Carolis AS, Longo VG. Amino acid release from the cerebral cortex of the rabbit, its relationship with the electrocorticogram. Physiol Behav. 1972;9:307–13. doi: 10.1016/0031-9384(72)90150-3. [DOI] [PubMed] [Google Scholar]
- 186.Moroni F, Corradetti R, Casamenti F, Moneti G, Pepeu G. The release of endogenous GABA and glutamate from the cerebral cortex in the rat. Naunyn Schmiedebergs Arch Pharmacol. 1981;316:235–9. doi: 10.1007/BF00505655. [DOI] [PubMed] [Google Scholar]
- 187.Kim JS, Kornhuber HH, Brand U, Menge HG. Effects of chronic amphetamine treatment on the glutamate concentration in cerebrospinal fluid and brain: implications for a theory of schizophrenia. Neurosci Lett. 1981;24:93–6. doi: 10.1016/0304-3940(81)90365-7. [DOI] [PubMed] [Google Scholar]
- 188.Stephans SE, Yamamoto BY. Effect of repeated methamphetamine administrations on dopamine and glutamate efflux in rat prefrontal cortex. Brain Res. 1995;700:99–106. doi: 10.1016/0006-8993(95)00938-m. [DOI] [PubMed] [Google Scholar]
- 189.Del Arco A, Martínez R, Mora F. Amphetamine increases extracellular concentrations of glutamate in the prefrontal cortex of the awake rat: a microdialysis study. Neurochem Res. 1998;23:1153–1158. doi: 10.1023/a:1020769816332. [DOI] [PubMed] [Google Scholar]
- 190.Shoblock JR, Sullivan EB, Maisonneuve IM, Glick SD. Neurochemical and behavioral differences between d-methamphetamine and d-amphetamine in rats. Psychopharmacology. 2003;165:359–369. doi: 10.1007/s00213-002-1288-7. [DOI] [PubMed] [Google Scholar]
- 191.Earle ML, Davies JA. The effect of methamphetamine on the release of glutamate from striatal slices. J Neural Transm Gen Sect. 1991;86:217–22. doi: 10.1007/BF01250707. [DOI] [PubMed] [Google Scholar]
- 192.Nash JF, Yamamoto BK. Methamphetamine neurotoxicity and striatal glutamate release: comparison to 3,4-methylenedioxymethamphetamine. Brain Res. 1992;581:237–43. doi: 10.1016/0006-8993(92)90713-j. [DOI] [PubMed] [Google Scholar]
- 193.Nash JF, Yamamoto BK. Effect of D-amphetamine on the extracellular concentrations of glutamate and dopamine in iprindole-treated rats. Brain Res. 1993;627:1–8. doi: 10.1016/0006-8993(93)90741-5. [DOI] [PubMed] [Google Scholar]
- 194.Mora F, Porras A. Effects of amphetamine on the release of excitatory amino acid neurotransmitters in the basal ganglia of the conscious rat. Can J Physiol Pharmacol. 1993;71:348–51. doi: 10.1139/y93-054. [DOI] [PubMed] [Google Scholar]
- 195.Abekawa T, Ohmori T, Koyama T. Effects of repeated administration of a high dose of methamphetamine on dopamine and glutamate release in rat striatum and nucleus accumbens. Brain Res. 1994;643:276–81. doi: 10.1016/0006-8993(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 196.Stephans SE, Yamamoto BK. Methamphetamine-induced neurotoxicity: roles for glutamate and dopamine efflux. Synapse. 1994;17:203–9. doi: 10.1002/syn.890170310. [DOI] [PubMed] [Google Scholar]
- 197.Del Arco A, González-Mora JL, Armas VR, Mora F. Amphetamine increases the extracellular concentration of glutamate in striatum of the awake rat: involvement of high affinity transporter mechanisms. Neuropharmacology. 1999;38:943–954. doi: 10.1016/s0028-3908(99)00043-x. [DOI] [PubMed] [Google Scholar]
- 198.Badiani A, Oates MM, Fraioli S, Browman KE, Ostrander MM, Xue CJ, et al. Environmental modulation of the response to amphetamine: dissociation between changes in behavior and changes in dopamine and glutamate overflow in the rat striatal complex. Psychopharmacology. 2000;151:166–74. doi: 10.1007/s002139900359. [DOI] [PubMed] [Google Scholar]
- 199.Anderzhanova E, Rayevsky KS, Saransaari P, Riitamaa E, Oja SS. Effects of sydnocarb and D-amphetamine on the extracellular levels of amino acids in the rat caudate-putamen. Eur J Pharmacol. 2001;428:87–95. doi: 10.1016/s0014-2999(01)01285-7. [DOI] [PubMed] [Google Scholar]
- 200.Bustamante D, You ZB, Castel MN, Johansson S, Goiny M, Terenius L, et al. Effect of single and repeated methamphetamine treatment on neurotransmitter release in substantia nigra and neostriatum of the rat. J Neurochem. 2002;83:645–54. doi: 10.1046/j.1471-4159.2002.01171.x. [DOI] [PubMed] [Google Scholar]
- 201.Mark KA, Soghomonian JJ, Yamamoto BK. High-dose methamphetamine acutely activates the striatonigral pathway to increase striatal glutamate and mediate long-term dopamine toxicity. J Neurosci. 2004;24:11449–56. doi: 10.1523/JNEUROSCI.3597-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Mark KA, Quinton MS, Russek SJ, Yamamoto BK. Dynamic changes in vesicular glutamate transporter 1 function and expression related to methamphetamine-induced glutamate release. J Neurosci. 2007;27:6823–6831. doi: 10.1523/JNEUROSCI.0013-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Miele M, Mura MA, Enrico P, Esposito G, Serra PA, Migheli R, et al. On the mechanism of d-amphetamine-induced changes in glutamate, ascorbic acid and uric acid release in the striatum of freely moving rats. Br J Pharmacol. 2000;129:582–588. doi: 10.1038/sj.bjp.0703066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Labarca R, Gajardo MI, Seguel M, Silva H, Jerez S, Ruiz A, et al. Effects of D-amphetamine administration on the release of endogenous excitatory amino acids in the rat nucleus accumbens. Prog Neuropsychopharmacol Biol Psychiatry. 1995;19:467–73. doi: 10.1016/0278-5846(94)00027-f. [DOI] [PubMed] [Google Scholar]
- 205.Xue CJ, Ng JP, Li Y, Wolf ME. Acute and repeated systemic amphetamine administration: effects on extracellular glutamate, aspartate, and serine levels in rat ventral tegmental area and nucleus accumbens. J Neurochem. 1996;67:352–63. doi: 10.1046/j.1471-4159.1996.67010352.x. [DOI] [PubMed] [Google Scholar]
- 206.Dalia A, Uretsky NJ, Wallace LJ. Dopaminergic agonists administered into the nucleus accumbens: effects on extracellular glutamate and on locomotor activity. Brain Res. 1998;788:111–7. doi: 10.1016/s0006-8993(97)01518-7. [DOI] [PubMed] [Google Scholar]
- 207.Ito K, Abekawa T, Koyama T. Relationship between development of cross-sensitization to MK-801 and delayed increases in glutamate levels in the nucleus accumbens induced by a high dose of methamphetamine. Psychopharmacology. 2006;187:293–302. doi: 10.1007/s00213-006-0423-2. [DOI] [PubMed] [Google Scholar]
- 208.Rocher C, Gardier AM. Effects of repeated systemic administration of d-Fenfluramine on serotonin and glutamate release in rat ventral hippocampus: comparison with methamphetamine using in vivo microdialysis. Naunyn Schmiedebergs Arch Pharmacol. 2001;363:422–8. doi: 10.1007/s002100000381. [DOI] [PubMed] [Google Scholar]
- 209.Raudensky J, Yamamoto BK. Effects of chronic unpredictable stress and methamphetamine on hippocampal glutamate function. Brain Res. 2007;1135:129–35. doi: 10.1016/j.brainres.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Wolf ME, Xue CJ. Amphetamine-induced glutamate efflux in the rat ventral tegmental area is prevented by MK-801, SCH 23390, and ibotenic acid lesions of the prefrontal cortex. J Neurochem. 1999;73:1529–38. doi: 10.1046/j.1471-4159.1999.0731529.x. [DOI] [PubMed] [Google Scholar]
- 211.Wolf ME, Xue CJ, Li Y, Wavak D. Amphetamine increases glutamate efflux in the rat ventral tegmental area by a mechanism involving glutamate transporters and reactive oxygen species. J Neurochem. 2000;75:1634–1644. doi: 10.1046/j.1471-4159.2000.0751634.x. [DOI] [PubMed] [Google Scholar]
- 212.Sonsalla PK, Nicklas WJ, Heikkila RE. Role for excitatory amino acids in methamphetamine-induced nigrostriatal dopaminergic toxicity. Science. 1989;243:398–400. doi: 10.1126/science.2563176. [DOI] [PubMed] [Google Scholar]
- 213.Burrows KB, Meshul CK. Methamphetamine alters presynaptic glutamate immunoreactivity in the caudate nucleus and motor cortex. Synapse. 1997;27:133–44. doi: 10.1002/(SICI)1098-2396(199710)27:2<133::AID-SYN4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 214.Burrows KB, Meshul CK. High-dose methamphetamine treatment alters presynaptic GABA and glutamate immunoreactivity. Neuroscience. 1999;90:833–50. doi: 10.1016/s0306-4522(98)00506-5. [DOI] [PubMed] [Google Scholar]
- 215.Kokoshka JM, Metzger RR, Wilkins DG, Gibb JW, Hanson GR, Fleckenstein AE. Methamphetamine treatment rapidly inhibits serotonin, but not glutamate, transporters in rat brain. Brain Res. 1998;799:78–83. doi: 10.1016/s0006-8993(98)00472-7. [DOI] [PubMed] [Google Scholar]
- 216.Bogen IL, Haug KH, Myhre O, Fonnum F. Short- and long-term effects of MDMA (“ecstasy”) on synaptosomal and vesicular uptake of neurotransmitters in vitro and ex vivo. Neurochem Int. 2003;43:393–400. doi: 10.1016/s0197-0186(03)00027-5. [DOI] [PubMed] [Google Scholar]
- 217.Kalivas PW, Duffy P. Dopamine regulation of extracellular glutamate in the nucleus accumbens. Brain Res. 1997;761:173–7. doi: 10.1016/s0006-8993(97)00464-2. [DOI] [PubMed] [Google Scholar]
- 218.Wolf ME, Xue CJ. Amphetamine and D1 dopamine receptor agonists produce biphasic effects on glutamate efflux in rat ventral tegmental area: modification by repeated amphetamine administration. J Neurochem. 1998;70:198–209. doi: 10.1046/j.1471-4159.1998.70010198.x. [DOI] [PubMed] [Google Scholar]
- 219.White SR, Duffy P, Kalivas PW. Methylenedioxymethamphetamine depresses glutamate-evoked neuronal firing and increases extracellular levels of dopamine and serotonin in the nucleus accumbens in vivo. Neuroscience. 1994;62:41–50. doi: 10.1016/0306-4522(94)90313-1. [DOI] [PubMed] [Google Scholar]
- 220.Mair RD, Kauer JA. Amphetamine depresses excitatory synaptic transmission at prefrontal cortical layer V synapses. Neuropharmacology. 2007;52:193–9. doi: 10.1016/j.neuropharm.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 221.Wang JQ, Daunais JB, McGinty JF. NMDA receptors mediate amphetamine-induced upregulation of zif/268 and preprodynorphin mRNA expression in rat striatum. Synapse. 1994;18:343–53. doi: 10.1002/syn.890180410. [DOI] [PubMed] [Google Scholar]
- 222.Wang JQ, Daunais JB, McGinty JF. Role of kainate/AMPA receptors in induction of striatal zif/268 and preprodynorphin mRNA by a single injection of amphetamine. Mol Brain Res. 1994;27:118–26. doi: 10.1016/0169-328x(94)90192-9. [DOI] [PubMed] [Google Scholar]
- 223.Konradi C, Leveque JC, Hyman SE. Amphetamine and dopamine-induced immediate early gene expression in striatal neurons depends on postsynaptic NMDA receptors and calcium. J Neurosci. 1996;16:4231–9. doi: 10.1523/JNEUROSCI.16-13-04231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wang JQ, McGinty JF. Intrastriatal injection of the metabotropic glutamate receptor antagonist MCPG attenuates acute amphetamine-stimulated neuropeptide mRNA expression in rat striatum. Neurosci Lett. 1996;218:13–6. doi: 10.1016/0304-3940(96)13107-4. [DOI] [PubMed] [Google Scholar]
- 225.Mao L, Wang JQ. Activation of metabotropic glutamate receptor mediates upregulation of transcription factor mRNA expression in rat striatum induced by acute administration of amphetamine. Brain Res. 2002;924:167–75. doi: 10.1016/s0006-8993(01)03230-9. [DOI] [PubMed] [Google Scholar]
- 226.Choe ES, Chung KT, Mao L, Wang JQ. Amphetamine increases phosphorylation of extracellular signal-regulated kinase and transcription factors in the rat striatum via group I metabotropic glutamate receptors. Neuropsychopharmacology. 2002;27:565–75. doi: 10.1016/S0893-133X(02)00341-X. [DOI] [PubMed] [Google Scholar]
- 227.Ferguson SM, Norton CS, Watson SJ, Akil H, Robinson TE. Amphetamine-evoked c-fos mRNA expression in the caudate-putamen: the effects of DA and NMDA receptor antagonists vary as a function of neuronal phenotype and environmental context. J Neurochem. 2003;86:33–44. doi: 10.1046/j.1471-4159.2003.01815.x. [DOI] [PubMed] [Google Scholar]
- 228.Mao L, Wang JQ. Contribution of ionotropic glutamate receptors to acute amphetamine-stimulated preproenkephalin mRNA expression in the rat striatum in vivo. Neurosci Lett. 2003;346:17–20. doi: 10.1016/s0304-3940(03)00542-1. [DOI] [PubMed] [Google Scholar]
- 229.Parelkar NK, Wang JQ. mGluR5-dependent increases in immediate early gene expression in the rat striatum following acute administration of amphetamine. Mol Brain Res. 2004;122:151–7. doi: 10.1016/j.molbrainres.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 230.Dalia A, Wallace LJ. Amphetamine induction of c-fos in the nucleus accumbens is not inhibited by glutamate antagonists. Brain Res. 1995;694:299–307. doi: 10.1016/0006-8993(95)00794-q. [DOI] [PubMed] [Google Scholar]
- 231.Paladini CA, Fiorillo CD, Morikawa H, Williams JT. Amphetamine selectively blocks inhibitory glutamate transmission in dopamine neurons. Nat Neurosci. 2001;4:275–281. doi: 10.1038/85124. [DOI] [PubMed] [Google Scholar]
- 232.Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 233.Faleiro LJ, Jones S, Kauer JA. Rapid synaptic plasticity of glutamatergic synapses on dopamine neurons in the ventral tegmental area in response to acute amphetamine injection. Neuropsychopharmacology. 2004;29:2115–25. doi: 10.1038/sj.npp.1300495. [DOI] [PubMed] [Google Scholar]
- 234.Prieto-Gomez B, Vazquez-Alvarez AM, Martinez-Pena JL, Reyes-Vazquez C, Yang PB, Dafny N. Methylphenidate and amphetamine modulate differently the NMDA and AMPA glutamatergic transmission of dopaminergic neurons in the ventral tegmental area. Life Sci. 2005;77:635–49. doi: 10.1016/j.lfs.2004.10.076. [DOI] [PubMed] [Google Scholar]
- 235.Peterson JD, Wolf ME, White FJ. Altered responsiveness of medial prefrontal cortex neurons to glutamate and dopamine after withdrawal from repeated amphetamine treatment. Synapse. 2000;36:342–4. doi: 10.1002/(SICI)1098-2396(20000615)36:4<342::AID-SYN11>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 236.Yamamoto H, Kitamura N, Lin XH, Ikeuchi Y, Hashimoto T, Shirakawa O, et al. Differential changes in glutamatergic transmission via N-methyl-D-aspartate receptors in the hippocampus and striatum of rats behaviourally sensitized to methamphetamine. Int J Neuropsychopharmcol. 1999;2:155–163. doi: 10.1017/S1461145799001480. [DOI] [PubMed] [Google Scholar]
- 237.Lu W, Chen H, Xue CJ, Wolf ME. Repeated amphetamine administration alters the expression of mRNA for AMPA receptor subunits in rat nucleus accumbens and prefrontal cortex. Synapse. 1997;26:269–80. doi: 10.1002/(SICI)1098-2396(199707)26:3<269::AID-SYN8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 238.Lu W, Wolf ME. Repeated amphetamine administration alters AMPA receptor subunit expression in rat nucleus accumbens and medial prefrontal cortex. Synapse. 1999;32:119–31. doi: 10.1002/(SICI)1098-2396(199905)32:2<119::AID-SYN5>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 239.Lu W, Monteggia LM, Wolf ME. Withdrawal from repeated amphetamine administration reduces NMDAR1 expression in the rat substantia nigra, nucleus accumbens and medial prefrontal cortex. Eur J Neurosci. 1999;11:3167–77. doi: 10.1046/j.1460-9568.1999.00736.x. [DOI] [PubMed] [Google Scholar]
- 240.Bardo MT, Robinet PM, Mattingly BA, Margulies JE. Effect of 6-hydroxydopamine or repeated amphetamine treatment on mesencephalic mRNA levels for AMPA glutamate receptor subunits in the rat. Neurosci Lett. 2001;302:133–6. doi: 10.1016/s0304-3940(01)01681-0. [DOI] [PubMed] [Google Scholar]
- 241.Giorgetti M, Hotsenpiller G, Ward P, Teppen T, Wolf ME. Amphetamine-induced plasticity of AMPA receptors in the ventral tegmental area: effects on extracellular levels of dopamine and glutamate in freely moving rats. J Neurosci. 2001;21:6362–9. doi: 10.1523/JNEUROSCI.21-16-06362.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Mao L, Wang JQ. Differentially altered mGluR1 and mGluR5 mRNA expression in rat caudate nucleus and nucleus accumbens in the development and expression of behavioral sensitization to repeated amphetamine administration. Synapse. 2001;41:230–40. doi: 10.1002/syn.1080. [DOI] [PubMed] [Google Scholar]
- 243.Yu MF, Lin TY, Ho WH, Yin HS. Amphetamine induces differential changes in the gene expression of metabotropic glutamate receptor 5 in cultured cortical and hippocampal neurons. J Mol Neurosci. 2001;17:13–24. doi: 10.1385/JMN:17:1:13. [DOI] [PubMed] [Google Scholar]
- 244.Yu MF, Lin WW, Li LT, Yin HS. Activation of metabotropic glutamate receptor 5 is associated with effect of amphetamine on brain neurons. Synapse. 2003;50:334–344. doi: 10.1002/syn.10275. [DOI] [PubMed] [Google Scholar]
- 245.Sidiropoulou K, Chao S, Lu W, Wolf ME. Amphetamine administration does not alter protein levels of the GLT-1 and EAAC1 glutamate transporter subtypes in rat midbrain, nucleus accumbens, striatum, or prefrontal cortex. Mol Brain Res. 2001;90:187–92. doi: 10.1016/s0169-328x(01)00110-3. [DOI] [PubMed] [Google Scholar]
- 246.Armstrong V, Reichel CM, Doti JF, Crawford CA, McDougall SA. Repeated amphetamine treatment causes a persistent elevation of glial fibrillary acidic protein in the caudate-putamen. Eur J Pharmacol. 2004;488:111–5. doi: 10.1016/j.ejphar.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 247.Shirai Y, Shirakawa O, Nishino N, Saito N, Nakai H. Increased striatal glutamate transporter by repeated intermittent administration of methamphetamine. Psychiatry Clin Neurosci. 1996;50:161–4. doi: 10.1111/j.1440-1819.1996.tb01682.x. [DOI] [PubMed] [Google Scholar]
- 248.Tzschentke TM, Schmidt WJ. Blockade of morphine- and amphetamine-induced conditioned place preference in the rat by riluzole. Neurosci Lett. 1998;242:114–6. doi: 10.1016/s0304-3940(98)00023-8. [DOI] [PubMed] [Google Scholar]
- 249.Layer RT, Uretsky NJ, Wallace LJ. Effects of the AMPA/kainate receptor antagonist DNQX in the nucleus accumbens on drug-induced conditioned place preference. Brain Res. 1993;617:267–73. doi: 10.1016/0006-8993(93)91094-9. [DOI] [PubMed] [Google Scholar]
- 250.Fujio M, Nakagawa T, Sekiya Y, Ozawa T, Suzuki Y, Minami M, et al. Gene transfer of GLT-1, a glutamate transporter, into the nucleus accumbens shell attenuates methamphetamine- and morphine-induced conditioned place preference in rats. Eur J Neurosci. 2005;22:2744–54. doi: 10.1111/j.1460-9568.2005.04467.x. [DOI] [PubMed] [Google Scholar]
- 251.Gerdjikov TV, Beninger RJ. Place preference induced by nucleus accumbens amphetamine is impaired by local blockade of Group II metabotropic glutamate receptors in rats. BMC Neurosci. 2006;7:43. doi: 10.1186/1471-2202-7-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Herzig V, Capuani EM, Kovar KA, Schmidt WJ. Effects of MPEP on expression of food-, MDMA- or amphetamine-conditioned place preference in rats. Addict Biol. 2005;10:243–9. doi: 10.1080/13556210500223272. [DOI] [PubMed] [Google Scholar]
- 253.Glick SD, Maisonneuve IM. Development of novel medications for drug addiction. The legacy of an African shrub. Ann N Y Acad Sci. 2000;909:88–103. doi: 10.1111/j.1749-6632.2000.tb06677.x. [DOI] [PubMed] [Google Scholar]
- 254.Glick SD, Maisonneuve IM, Dickinson HA, Kitchen BA. Comparative effects of dextromethorphan and dextrorphan on morphine, methamphetamine, and nicotine self-administration in rats. Eur J Pharmacol. 2001;422:87–90. doi: 10.1016/s0014-2999(01)01066-4. [DOI] [PubMed] [Google Scholar]
- 255.Moroz I, Parker LA, Siegel S. Ibogaine interferes with the establishment of amphetamine place preference learning. Exp Clin Psychopharmacol. 1997;5:119–22. doi: 10.1037//1064-1297.5.2.119. [DOI] [PubMed] [Google Scholar]
- 256.Glick SD, Maisonneuve IM, Szumlinski KK. 18-Methoxycoronaridine (18-MC) and ibogaine: comparison of antiaddictive efficacy, toxicity, and mechanisms of action. Ann N Y Acad Sci. 2000;914:369–86. doi: 10.1111/j.1749-6632.2000.tb05211.x. [DOI] [PubMed] [Google Scholar]
- 257.Pace CJ, Glick SD, Maisonneuve IM, He LW, Jokiel PA, Kuehne ME, et al. Novel iboga alkaloid congeners block nicotinic receptors and reduce drug self-administration. Eur J Pharmacol. 2004;492:159–67. doi: 10.1016/j.ejphar.2004.03.062. [DOI] [PubMed] [Google Scholar]
- 258.Kim JH, Austin JD, Tanabe L, Creekmore E, Vezina P. Activation of group II mGlu receptors blocks the enhanced drug taking induced by previous exposure to amphetamine. Eur J Neurosci. 2005;21:295–300. doi: 10.1111/j.1460-9568.2004.03822.x. [DOI] [PubMed] [Google Scholar]
- 259.Raynor K, Kong H, Chen Y, Tasuda K, Yu L, Bell GI, et al. Pharmacological characterization of the cloned κ-, δ-, and μ-opioid receptors. Mol Pharmacol. 1994;45:330–334. [PubMed] [Google Scholar]
- 260.Coutinho-Netto J, Abdul-Ghani AS, Bradford HF. Suppression of evoked and spontaneous release of neurotransmitters in vivo by morphine. Biochem Pharmacol. 1980;29:2777–80. doi: 10.1016/0006-2952(80)90011-8. [DOI] [PubMed] [Google Scholar]
- 261.Coutinho-Netto J, Abdul-Ghani AS, Bradford HF. Morphine suppression of neurotransmitter release evoked by sensory stimulation in vivo. Biochem Pharmacol. 1982;31:1019–23. doi: 10.1016/0006-2952(82)90337-9. [DOI] [PubMed] [Google Scholar]
- 262.Desole MS, Esposito G, Fresu L, Migheli R, Enrico P, Mura MA, et al. Effects of morphine treatment and withdrawal on striatal and limbic monoaminergic activity and ascorbic acid oxidation in the rat. Brain Res. 1996;723:154–61. doi: 10.1016/0006-8993(96)00235-1. [DOI] [PubMed] [Google Scholar]
- 263.Nicol B, Rowbotham DJ, Lambert DG. μ- and κ-opioids inhibit K+ evoked glutamate release from rat cerebrocortical slices. Neurosci Lett. 1996;218:79–82. doi: 10.1016/s0304-3940(96)13104-9. [DOI] [PubMed] [Google Scholar]
- 264.Vlaskovska M, Schramm M, Nylander I, Kasakov L, You ZB, Herrera-Marschitz M, et al. Opioid effects on 45Ca2+ uptake and glutamate release in rat cerebral cortex in primary culture. J Neurochem. 1997;68:517–24. doi: 10.1046/j.1471-4159.1997.68020517.x. [DOI] [PubMed] [Google Scholar]
- 265.Yang TT, Hung CF, Lee YJ, Su MJ, Wang SJ. Morphine inhibits glutamate exocytosis from rat cerebral cortex nerve terminals (synaptosomes) by reducing Ca2+ influx. Synapse. 2004;51:83–90. doi: 10.1002/syn.10290. [DOI] [PubMed] [Google Scholar]
- 266.Hao Y, Yang JY, Guo M, Wu CF, Wu MF. Morphine decreases extracellular levels of glutamate in the anterior cingulate cortex: an in vivo microdialysis study in freely moving rats. Brain Res. 2005;1040:191–6. doi: 10.1016/j.brainres.2005.01.072. [DOI] [PubMed] [Google Scholar]
- 267.Cummins JT, Morin AM. The release of glutamic acid from isolated brain tissues. Psychopharmacol Commun. 1975;1:383–91. [PubMed] [Google Scholar]
- 268.Smith JE, Co C, Freeman ME, Lane JD. Brain neurotransmitter turnover correlated with morphine-seeking behavior of rats. Pharmacol Biochem Behav. 1982;16:509–19. doi: 10.1016/0091-3057(82)90460-9. [DOI] [PubMed] [Google Scholar]
- 269.Enrico P, Mura MA, Esposito G, Serra P, Migheli R, De Natale G, et al. Effect of naloxone on morphine-induced changes in striatal dopamine metabolism and glutamate, ascorbic acid and uric acid release in freely moving rats. Brain Res. 1998;797:94–102. doi: 10.1016/s0006-8993(98)00371-0. [DOI] [PubMed] [Google Scholar]
- 270.Tokuyama S, Wakabayashi H, Ho IK. Direct evidence for a role of glutamate in the expression of the opioid withdrawal syndrome. Eur J Pharmacol. 1996;295:123–9. doi: 10.1016/0014-2999(95)00645-1. [DOI] [PubMed] [Google Scholar]
- 271.Huang NK, Tseng CJ, Wong CS, Tung CS. Effects of acute and chronic morphine on DOPAC and glutamate at subcortical DA terminals in awake rats. Pharmacol Biochem Behav. 1997;56:363–71. doi: 10.1016/s0091-3057(96)00236-5. [DOI] [PubMed] [Google Scholar]
- 272.Sepulveda MJ, Hernandez L, Rada P, Tucci S, Contreras E. Effect of precipitated withdrawal on extracellular glutamate and aspartate in the nucleus accumbens of chronically morphine-treated rats: an in vivo microdialysis study. Pharmacol Biochem Behav. 1998;60:255–62. doi: 10.1016/s0091-3057(97)00550-9. [DOI] [PubMed] [Google Scholar]
- 273.Martin G, Przewlocki R, Siggins GR. Chronic morphine treatment selectively augments metabotropic glutamate receptor-induced inhibition of N-methyl-D-aspartate receptor-mediated neurotransmission in nucleus accumbens. J Pharmacol Exp Ther. 1999;288:30–5. [PubMed] [Google Scholar]
- 274.Huffman RD, Felpel LP. A microiontophoretic study of morphine on single neurons in the rat globus pallidus. Neurosci Lett. 1981;22:195–9. doi: 10.1016/0304-3940(81)90087-2. [DOI] [PubMed] [Google Scholar]
- 275.Frey JM, Huffman RD. Effects of enkephalin and morphine on rat globus pallidus neurons. Brain Res Bull. 1985;14:251–9. doi: 10.1016/0361-9230(85)90090-5. [DOI] [PubMed] [Google Scholar]
- 276.Huffman RD, Frey JM. Response of rat globus pallidus neurons to microintophoretically applied mu and kappa opioid receptor agonists. Eur J Pharmacol. 1989;170:179–91. doi: 10.1016/0014-2999(89)90538-4. [DOI] [PubMed] [Google Scholar]
- 277.Johnson PI, Napier TC. Morphine modulation of GABA- and glutamate-induced changes of ventral pallidal neuronal activity. Neuroscience. 1997;77:187–97. doi: 10.1016/s0306-4522(96)00482-4. [DOI] [PubMed] [Google Scholar]
- 278.Guo M, Xu NJ, Li YT, Yang JY, Wu CF, Pei G. Morphine modulates glutamate release in the hippocampal CA1 area in mice. Neurosci Lett. 2005;381:12–5. doi: 10.1016/j.neulet.2005.01.071. [DOI] [PubMed] [Google Scholar]
- 279.Ojanen SP, Palmen M, Hyytia P, Kiianmaa K. Extracellular glutamate and GABA in the ventral tegmental area of alcohol-preferring AA and alcohol-avoiding ANA rats treated repeatedly with morphine. Eur J Pharmacol. 2007;559:38–45. doi: 10.1016/j.ejphar.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 280.Satoh M, Zieglgansberger W, Herz A. Interaction between morphine and putative excitatory neurotransmitters in cortical neurons in naive and tolerant rats. Life Sci. 1975;17:75–80. doi: 10.1016/0024-3205(75)90239-8. [DOI] [PubMed] [Google Scholar]
- 281.Bioulac B, Lund JP, Puil E. Morphine excitation in the cerebral cortex. Can J Physiol Pharmacol. 1975;53:683–7. doi: 10.1139/y75-096. [DOI] [PubMed] [Google Scholar]
- 282.Satoh M, Zieglgansberger W, Herz A. Actions of opiates upon single unit activity in the cortex of naive and tolerant rats. Brain Res. 1976;115:99–110. doi: 10.1016/0006-8993(76)90825-8. [DOI] [PubMed] [Google Scholar]
- 283.Giacchino JL, Henriksen SJ. Opioid effects on activation of neurons in the medial prefrontal cortex. Prog Neuropsychopharmacol Biol Psychiatry. 1998;22:1157–78. doi: 10.1016/s0278-5846(98)00053-0. [DOI] [PubMed] [Google Scholar]
- 284.Liu J, Nickolenko J, Sharp FR. Morphine induces c-fos and junB in striatum and nucleus accumbens via D1 and N-methyl-D-aspartate receptors. Proc Natl Acad Sci U S A. 1994;91:8537–41. doi: 10.1073/pnas.91.18.8537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Bontempi B, Sharp FR. Systemic morphine-induced Fos protein in the rat striatum and nucleus accumbens is regulated by mu opioid receptors in the substantia nigra and ventral tegmental area. J Neurosci. 1997;17:8596–612. doi: 10.1523/JNEUROSCI.17-21-08596.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Garcia MM, Anderson AT, Edwards R, Harlan RE. Morphine induction of c-fos expression in the rat forebrain through glutamatergic mechanisms: role of non-n-methyl-D-aspartate receptors. Neuroscience. 2003;119:787–94. doi: 10.1016/s0306-4522(02)00975-2. [DOI] [PubMed] [Google Scholar]
- 287.Jacobs EH, Wardeh G, Smit AB, Schoffelmeer AN. Morphine causes a delayed increase in glutamate receptor functioning in the nucleus accumbens core. Eur J Pharmacol. 2005;511:27–30. doi: 10.1016/j.ejphar.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 288.Le Greves P, Huang W, Zhou Q, Thornwall M, Nyberg F. Acute effects of morphine on the expression of mRNAs for NMDA receptor subunits in the rat hippocampus, hypothalamus and spinal cord. Eur J Pharmacol. 1998;341:161–4. doi: 10.1016/s0014-2999(97)01400-3. [DOI] [PubMed] [Google Scholar]
- 289.Satoh M, Zieglgansberger W, Herz A. Supersensitivity of cortical neurones of the rat to acetylcholine and L-glutamate following chronic morphine treatment. Naunyn Schmiedebergs Arch Pharmacol. 1976;293:101–3. doi: 10.1007/BF00498877. [DOI] [PubMed] [Google Scholar]
- 290.Fry JP, Herz A, Zieglgansberger W. A demonstration of naloxone-precipitated opiate withdrawal on single neurones in the morphine-tolerant/dependent rat brain. Br J Pharmacol. 1980;68:585–92. doi: 10.1111/j.1476-5381.1980.tb14574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Haberny KA, Young GA. Interactive effects of MK-801 and morphine on EEG, EEG power spectra and behavior in rats: I. Morphine tolerance development. Eur J Pharmacol. 1994;261:1–9. doi: 10.1016/0014-2999(94)90293-3. [DOI] [PubMed] [Google Scholar]
- 292.McDaid J, Dallimore JE, Mackie AR, Napier TC. Changes in accumbal and pallidal pCREB and ΔFosB in morphine-sensitized rats: correlations with receptor-evoked electrophysiological measures in the ventral pallidum. Neuropsychopharmacology. 2006;31:1212–26. doi: 10.1038/sj.npp.1300854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Ronnback L, Hansson E, Cupello A, Rapallino MV, Zeuchner J, Rosengren L. Neurotransmitter uptake in various brain regions of chronically morphinized rats. Neurochem Res. 1986;11:317–26. doi: 10.1007/BF00967978. [DOI] [PubMed] [Google Scholar]
- 294.Ozawa T, Nakagawa T, Shige K, Minami M, Satoh M. Changes in the expression of glial glutamate transporters in the rat brain accompanied with morphine dependence and naloxone-precipitated withdrawal. Brain Res. 2001;905:254–8. doi: 10.1016/s0006-8993(01)02536-7. [DOI] [PubMed] [Google Scholar]
- 295.Gudehithlu KP, Reddy PL, Bhargava HN. Effect of morphine tolerance and abstinence on the binding of [3H]MK-801 to brain regions and spinal cord of the rat. Brain Res. 1994;639:269–74. doi: 10.1016/0006-8993(94)91740-x. [DOI] [PubMed] [Google Scholar]
- 296.Bhargava HN, Reddy PL, Gudehithlu KP. Down-regulation of N-methyl-D-aspartate (NMDA) receptors of brain regions and spinal cord of rats treated chronically with morphine. Gen Pharmacol. 1995;26:131–6. doi: 10.1016/0306-3623(94)00147-f. [DOI] [PubMed] [Google Scholar]
- 297.Koyuncuoglu H, Nurten A, Yamanturk P, Nurten R. The importance of the number of NMDA receptors in the development of supersensitivity or tolerance to and dependence on morphine. Pharmacol Res. 1999;39:311–9. doi: 10.1006/phrs.1998.0443. [DOI] [PubMed] [Google Scholar]
- 298.Gudehithlu KP, Bhargava HN. Differential binding of [3H]MK-801 to brain regions and spinal cord of mice treated chronically with morphine. Gen Pharmacol. 1996;27:91–4. doi: 10.1016/0306-3623(95)00110-7. [DOI] [PubMed] [Google Scholar]
- 299.Harrison JM, Allen RG, Pellegrino MJ, Williams JT, Manzoni OJ. Chronic morphine treatment alters endogenous opioid control of hippocampal mossy fiber synaptic transmission. J Neurophysiol. 2002;87:2464–70. doi: 10.1152/jn.2002.87.5.2464. [DOI] [PubMed] [Google Scholar]
- 300.Zhong W, Dong Z, Tian M, Cao J, Xu T, Xu L, et al. Opiate withdrawal induces dynamic expressions of AMPA receptors and its regulatory molecule CaMKII alpha in hippocampal synapses. Life Sci. 2006;79:861–9. doi: 10.1016/j.lfs.2006.02.040. [DOI] [PubMed] [Google Scholar]
- 301.Moron JA, Abul-Husn NS, Rozenfeld R, Dolios G, Wang R, Devi LA. Morphine administration alters the profile of hippocampal postsynaptic density-associated proteins: a proteomics study focusing on endocytic proteins. Mol Cell Proteomics. 2007;6:29–42. doi: 10.1074/mcp.M600184-MCP200. [DOI] [PubMed] [Google Scholar]
- 302.Murray F, Harrison NJ, Grimwood S, Bristow LJ, Hutson PH. Nucleus accumbens NMDA receptor subunit expression and function is enhanced in morphine-dependent rats. Eur J Pharmacol. 2007;562:191–7. doi: 10.1016/j.ejphar.2007.01.027. [DOI] [PubMed] [Google Scholar]
- 303.Glass MJ, Kruzich PJ, Colago EE, Kreek MJ, Pickel VM. Increased AMPA GluR1 receptor subunit labeling on the plasma membrane of dendrites in the basolateral amygdala of rats self-administering morphine. Synapse. 2005;58:1–12. doi: 10.1002/syn.20176. [DOI] [PubMed] [Google Scholar]
- 304.Watanabe T, Nakagawa T, Yamamoto R, Maeda A, Minami M, Satoh M. Involvement of glutamate receptors within the central nucleus of the amygdala in naloxone-precipitated morphine withdrawal-induced conditioned place aversion in rats. Jpn J Pharmacol. 2002;88:399–406. doi: 10.1254/jjp.88.399. [DOI] [PubMed] [Google Scholar]
- 305.Inoue M, Mishina M, Ueda H. Locus-specific rescue of GluRε1 NMDA receptors in mutant mice identifies the brain regions important for morphine tolerance and dependence. J Neurosci. 2003;23:6529–36. doi: 10.1523/JNEUROSCI.23-16-06529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Martin G, Guadano-Ferraz A, Morte B, Ahmed S, Koob GF, De Lecea L, et al. Chronic morphine treatment alters N-methyl-D-aspartate receptors in freshly isolated neurons from nucleus accumbens. J Pharmacol Exp Ther. 2004;311:265–73. doi: 10.1124/jpet.104.067504. [DOI] [PubMed] [Google Scholar]
- 307.Zhu H, Jang CG, Ma T, Oh S, Rockhold RW, Ho IK. Region specific expression of NMDA receptor NR1 subunit mRNA in hypothalamus and pons following chronic morphine treatment. Eur J Pharmacol. 1999;365:47–54. doi: 10.1016/s0014-2999(98)00861-9. [DOI] [PubMed] [Google Scholar]
- 308.Aoki T, Narita M, Shibasaki M, Suzuki T. Metabotropic glutamate receptor 5 localized in the limbic forebrain is critical for the development of morphine-induced rewarding effect in mice. Eur J Neurosci. 2004;20:1633–1638. doi: 10.1111/j.1460-9568.2004.03609.x. [DOI] [PubMed] [Google Scholar]
- 309.Carlezon WA, Jr, Boundy VA, Haile CN, Lane SB, Kalb RG, Neve RL, et al. Sensitization to morphine induced by viral-mediated gene transfer. Science. 1997;277:812–4. doi: 10.1126/science.277.5327.812. [DOI] [PubMed] [Google Scholar]
- 310.Carlezon WAJ, Haile CN, Coppersmith R, Hayashi Y, Malinow R, Neve RL, et al. Distinct sites of opiate reward and aversion within the midbrain identified using a herpes simplex virus vector expressing GluR1. J Neurosci. 2000;20:RC62. doi: 10.1523/JNEUROSCI.20-05-j0002.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Aghajanian GK, Kogan JH, Moghaddam B. Opiate withdrawal increases glutamate and aspartate efflux in the locus coeruleus: an in vivo microdialysis study. Brain Res. 1994;636:126–30. doi: 10.1016/0006-8993(94)90186-4. [DOI] [PubMed] [Google Scholar]
- 312.Hoshi K, Yamamoto A, Ishizuki S, Fujihira E, Ichihara K. Excitatory amino acid release in the locus coeruleus during naloxone-precipitated morphine withdrawal in adjuvant arthritic rats. Inflamm Res. 2000;49:36–41. doi: 10.1007/PL00000201. [DOI] [PubMed] [Google Scholar]
- 313.Sepulveda J, Oliva P, Contreras E. Neurochemical changes of the extracellular concentrations of glutamate and aspartate in the nucleus accumbens of rats after chronic administration of morphine. Eur J Pharmacol. 2004;483:249–58. doi: 10.1016/j.ejphar.2003.10.037. [DOI] [PubMed] [Google Scholar]
- 314.Robbe D, Bockaert J, Manzoni OJ. Metabotropic glutamate receptor 2/3-dependent long-term depression in the nucleus accumbens is blocked in morphine withdrawn mice. Eur J Neurosci. 2002;16:2231–5. doi: 10.1046/j.1460-9568.2002.02273.x. [DOI] [PubMed] [Google Scholar]
- 315.Xu NJ, Bao L, Fan HP, Bao GB, Pu L, Lu YJ, et al. Morphine withdrawal increases glutamate uptake and surface expression of glutamate transporter GLT1 at hippocampal synapses. J Neurosci. 2003;23:4775–84. doi: 10.1523/JNEUROSCI.23-11-04775.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Manzoni OJ, Williams JT. Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. J Neurosci. 1999;19:6629–36. doi: 10.1523/JNEUROSCI.19-15-06629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Kenny PJ. Brain reward systems and compulsive drug use. Trends Pharmacol Sci. 2007;28:135–41. doi: 10.1016/j.tips.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 318.Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–7. doi: 10.1126/science.1824728. [DOI] [PubMed] [Google Scholar]
- 319.Tanganelli S, Antonelli T, Morari M, Bianchi C, Beani L. Glutamate antagonists prevent morphine withdrawal in mice and guinea pigs. Neurosci Lett. 1991;122:270–2. doi: 10.1016/0304-3940(91)90875-t. [DOI] [PubMed] [Google Scholar]
- 320.Marek P, Ben-Eliyahu S, Gold M, Liebeskind JC. Excitatory amino acid antagonists (kynurenic acid and MK-801) attenuate the development of morphine tolerance in the rat. Brain Res. 1991;547:77–81. doi: 10.1016/0006-8993(91)90576-h. [DOI] [PubMed] [Google Scholar]
- 321.Tiseo PJ, Inturrisi CE. Attenuation and reversal of morphine tolerance by the competitive N-methyl-D-aspartate receptor antagonist, LY274614. J Pharmacol Exp Ther. 1993;264:1090–6. [PubMed] [Google Scholar]
- 322.Cappendijk SL, de Vries R, Dzoljic MR. Excitatory amino acid receptor antagonists and naloxone-precipitated withdrawal syndrome in morphine-dependent mice. Eur Neuropsychopharmacol. 1993;3:111–6. doi: 10.1016/0924-977x(93)90262-k. [DOI] [PubMed] [Google Scholar]
- 323.Fundytus ME, Coderre TJ. Effect of activity at metabotropic, as well as ionotropic (NMDA), glutamate receptors on morphine dependence. Br J Pharmacol. 1994;113:1215–20. doi: 10.1111/j.1476-5381.1994.tb17127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Popik P, Skolnick P. The NMDA antagonist memantine blocks the expression and maintenance of morphine dependence. Pharmacol Biochem Behav. 1996;53:791–7. doi: 10.1016/0091-3057(95)02163-9. [DOI] [PubMed] [Google Scholar]
- 325.Gonzalez P, Cabello P, Germany A, Norris B, Contreras E. Decrease of tolerance to, and physical dependence on morphine by, glutamate receptor antagonists. Eur J Pharmacol. 1997;332:257–62. doi: 10.1016/s0014-2999(97)01099-6. [DOI] [PubMed] [Google Scholar]
- 326.Kozela E, Pilc A, Popik P. Inhibitory effects of MPEP, and mGluR5 antagonist, and memantine, and N-methyl-D-aspartate receptor antagonist, on morphine antinociceptive tolerance in mice. Psychopharmacology. 2003;165:245–251. doi: 10.1007/s00213-002-1287-8. [DOI] [PubMed] [Google Scholar]
- 327.Leal MB, Michelin K, Souza DO, Elisabetsky E. Ibogaine attenuation of morphine withdrawal in mice: role of glutamate N-methyl-D-aspartate receptors. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:781–5. doi: 10.1016/S0278-5846(03)00109-X. [DOI] [PubMed] [Google Scholar]
- 328.Danysz W, Kozela E, Parsons CG, Sladek M, Bauer T, Popik P. Peripherally acting NMDA receptor/glycineB site receptor antagonists inhibit morphine tolerance. Neuropharmacology. 2005;48:360–71. doi: 10.1016/j.neuropharm.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 329.Kawasaki Y, Jin C, Suemaru K, Kawasaki H, Shibata K, Choshi T, et al. Effect of glutamate receptor antagonists on place aversion induced by naloxone in single-dose morphine-treated rats. Br J Pharmacol. 2005;145:751–7. doi: 10.1038/sj.bjp.0706228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Rasmussen K, Kendrick WT, Kogan JH, Aghajanian GK. A selective AMPA antagonist, LY293558, suppresses morphine withdrawal-induced activation of locus coeruleus neurons and behavioral signs of morphine withdrawal. Neuropsychopharmacology. 1996;15:497–505. doi: 10.1016/S0893-133X(96)00094-2. [DOI] [PubMed] [Google Scholar]
- 331.Miyamoto Y, Yamada K, Nagai T, Mori H, Mishina M, Furukawa H, et al. Behavioural adaptations to addictive drugs in mice lacking the NMDA receptor ε1 subunit. Eur J Neurosci. 2004;19:151–8. doi: 10.1111/j.1460-9568.2004.03086.x. [DOI] [PubMed] [Google Scholar]
- 332.Vekovischeva OY, Zamanillo D, Echenko O, Seppala T, Uusi-Oukari M, Honkanen A, et al. Morphine-induced dependence and sensitization are altered in mice deficient in AMPA-type glutamate receptor-A subunits. J Neurosci. 2001;21:4451–9. doi: 10.1523/JNEUROSCI.21-12-04451.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Palucha A, Branski P, Pilc A. Selective mGlu5 receptor antagonist MTEP attenuates naloxone-induced morphine withdrawal symptoms. Pol J Pharmacol. 2004;56:863–6. [PubMed] [Google Scholar]
- 334.Rasmussen K, Martin H, Berger JE, Seager MA. The mGlu5 receptor antagonists MPEP and MTEP attenuate behavioral signs of morphine withdrawal and morphine-withdrawal-induced activation of locus coeruleus neurons in rats. Neuropharmacology. 2005;48:173–80. doi: 10.1016/j.neuropharm.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 335.Fundytus ME, Coderre TJ. Attenuation of precipitated morphine withdrawal symptoms by acute i.c.v administration of a group II mGluR agonist. Br J Pharmacol. 1997;121:511–4. doi: 10.1038/sj.bjp.0701174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Vandergriff J, Rasmussen K. The selective mGlu2/3 receptor agonist LY354740 attenuates morphine-withdrawal-induced activation of locus coeruleus neurons and behavioral signs of morphine withdrawal. Neuropharmacology. 1999;38:217–22. doi: 10.1016/s0028-3908(98)00196-8. [DOI] [PubMed] [Google Scholar]
- 337.Klodzinska A, Chojnacka-Wojcik E, Palucha A, Branski P, Popik P, Pilc A. Potential anti-anxiety, anti-addictive effects of LY 354740, a selective group II glutamate metabotropic receptors agonist in animal models. Neuropharmacology. 1999;38:1831–9. doi: 10.1016/s0028-3908(99)00066-0. [DOI] [PubMed] [Google Scholar]
- 338.Popik P, Kozela E, Pilc A. Selective agonist of group II glutamate metabotropic receptors, LY354740, inhibits tolerance to analgesic effects of morphine in mice. Br J Pharmacol. 2000;130:1425–31. doi: 10.1038/sj.bjp.0703438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Jin C, Araki H, Kawasaki Y, Nagata M, Suemaru K, Shibata K, et al. The glutamate release inhibitor riluzole attenuates the formation of conditioned place aversion induced by naloxone in rats undergoing a single morphine exposure. Brain Res. 2006;1069:120–6. doi: 10.1016/j.brainres.2005.11.058. [DOI] [PubMed] [Google Scholar]
- 340.Nakagawa T, Ozawa T, Shige K, Yamamoto R, Minami M, Satoh M. Inhibition of morphine tolerance and dependence by MS-153, a glutamate transporter activator. Eur J Pharmacol. 2001;419:39–45. doi: 10.1016/s0014-2999(01)00965-7. [DOI] [PubMed] [Google Scholar]
- 341.Popik P, Kozela E, Wrobel M, Wozniak KM, Slusher BS. Morphine tolerance and reward but not expression of morphine dependence are inhibited by the selective glutamate carboxypeptidase II (GCP II, NAALADase) inhibitor, 2-PMPA. Neuropsychopharmacology. 2003;28:457–67. doi: 10.1038/sj.npp.1300048. [DOI] [PubMed] [Google Scholar]
- 342.Kozela E, Wrobel M, Kos T, Wojcikowski J, Daniel WA, Wozniak KM, et al. 2-MPPA, a selective glutamate carboxypeptidase II inhibitor, attenuates morphine tolerance but not dependence in C57/Bl mice. Psychopharmacology. 2005;183:275–84. doi: 10.1007/s00213-005-0182-5. [DOI] [PubMed] [Google Scholar]
- 343.Sekiya Y, Nakagawa T, Ozawa T, Minami M, Satoh M. Facilitation of morphine withdrawal symptoms and morphine-induced conditioned place preference by a glutamate transporter inhibitor DL-threo-beta-benzyloxyaspartate in rats. Eur J Pharmacol. 2004;485:201–10. doi: 10.1016/j.ejphar.2003.11.062. [DOI] [PubMed] [Google Scholar]
- 344.Rasmussen K, Hsu MA, Vandergriff J. The selective mGlu2/3 receptor antagonist LY341495 exacerbates behavioral signs of morphine withdrawal and morphine-withdrawal-induced activation of locus coeruleus neurons. Neuropharmacology. 2004;46:620–8. doi: 10.1016/j.neuropharm.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 345.Akaoka H, Aston-Jones G. Opiate withdrawal-induced hyperactivity of locus coeruleus neurons is substantially mediated by augmented excitatory amino acid input. J Neurosci. 1991;11:3830–9. doi: 10.1523/JNEUROSCI.11-12-03830.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Hong M, Milne B, Jhamandas K. Evidence for the involvement of excitatory amino acid pathways in the development of precipitated withdrawal from acute and chronic morphine: an in vivo voltammetric study in the rat locus coeruleus. Brain Res. 1993;623:131–41. doi: 10.1016/0006-8993(93)90020-n. [DOI] [PubMed] [Google Scholar]
- 347.Tokuyama S, Zhu H, Wakabayashi H, Feng YZ, Ho IK. The role of glutamate in the locus coeruleus during opioid withdrawal and effects of H-7, a protein kinase inhibitor, on the action of glutamate in rats. J Biomed Sci. 1998;5:45–53. doi: 10.1007/BF02253355. [DOI] [PubMed] [Google Scholar]
- 348.Taylor JR, Punch LJ, Elsworth JD. A comparison of the effects of clonidine and CNQX infusion into the locus coeruleus and the amygdala on naloxone-precipitated opiate withdrawal in the rat. Psychopharmacology. 1998;138:133–42. doi: 10.1007/s002130050655. [DOI] [PubMed] [Google Scholar]
- 349.Tokuyama S, Zhu H, Oh S, Ho IK, Yamamoto T. Further evidence for a role of NMDA receptors in the locus coeruleus in the expression of withdrawal syndrome from opioids. Neurochem Int. 2001;39:103–9. doi: 10.1016/s0197-0186(01)00019-5. [DOI] [PubMed] [Google Scholar]
- 350.Ozawa T, Nakagawa T, Sekiya Y, Minami M, Satoh M. Effect of gene transfer of GLT-1, a glutamate transporter, into the locus coeruleus by recombinant adenoviruses on morphine physical dependence in rats. Eur J Neurosci. 2004;19:221–6. doi: 10.1111/j.1460-9568.2004.03101.x. [DOI] [PubMed] [Google Scholar]
- 351.Wang HL, Zhao Y, Xiang XH, Wang HS, Wu WR. Blockade of ionotropic glutamatergic transmission in the ventral tegmental area attenuates the physical signs of morphine withdrawal in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:1079–87. doi: 10.1016/j.pnpbp.2004.05.043. [DOI] [PubMed] [Google Scholar]
- 352.Wang HL, Xiang XH, Guo Y, Wu WR, Cao DY, Wang HS, et al. Ionotropic glutamatergic neurotransmission in the ventral tegmental area modulates ΔFosB expression in the nucleus accumbens and abstinence syndrome in morphine withdrawal rats. Eur J Pharmacol. 2005;527:94–104. doi: 10.1016/j.ejphar.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 353.Popik P, Kolasiewicz W. Mesolimbic NMDA receptors are implicated in the expression of conditioned morphine reward. Naunyn Schmied Arch Pharmacol. 1999;359:288–294. doi: 10.1007/pl00005354. [DOI] [PubMed] [Google Scholar]
- 354.Yonghui L, Xigeng Z, Yunjing B, Xiaoyan Y, Nan S. Opposite effects of MK-801 on the expression of food and morphine-induced conditioned place preference in rats. J Psychopharmacol. 2006;20:40–6. doi: 10.1177/0269881105057250. [DOI] [PubMed] [Google Scholar]
- 355.Lue WM, Huang EY, Yang SN, Wong CS, Tao PL. Post-treatment of dextromethorphan reverses morphine effect on conditioned place preference in rats. Synapse. 2007;61:420–8. doi: 10.1002/syn.20391. [DOI] [PubMed] [Google Scholar]
- 356.Dallimore JE, Mickiewicz AL, Napier TC. Intra-ventral pallidal glutamate antagonists block expression of morphine-induced place preference. Behav Neurosci. 2006;120:1103–14. doi: 10.1037/0735-7044.120.5.1103. [DOI] [PubMed] [Google Scholar]
- 357.Rezayof A, Golhasani-Keshtan F, Haeri-Rohani A, Zarrindast MR. Morphine-induced place preference: involvement of the central amygdala NMDA receptors. Brain Res. 2007;1133:34–41. doi: 10.1016/j.brainres.2006.11.049. [DOI] [PubMed] [Google Scholar]
- 358.Harris GC, Wimmer M, Byrne R, Aston-Jones G. Glutamate-associated plasticity in the ventral tegmental area is necessary for conditioning environmental stimuli with morphine. Neuroscience. 2004;129:841–7. doi: 10.1016/j.neuroscience.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 359.Jackson A, Brown G, Stephens DN. N-methyl-D-aspartate (NMDA) and alpha-amino-3-hydroxy-5-methyl-4- isoxazoleproprionate (AMPA) glutamate-receptor antagonists have different interactions with the discriminative stimuli of abused drugs. Psychopharmacology. 1996;128:320–7. doi: 10.1007/s002130050140. [DOI] [PubMed] [Google Scholar]
- 360.Bespalov AY, Beardsley PM, Balster RL. Interactions between N-methyl-D-aspartate receptor antagonists and the discriminative stimulus effects of morphine in rats. Pharmacol Biochem Behav. 1998;60:507–17. doi: 10.1016/s0091-3057(98)00006-9. [DOI] [PubMed] [Google Scholar]
- 361.Ribeiro Do, Couto B, Aguilar MA, Manzanedo C, Rodriguez-Arias M, Minarro J. NMDA glutamate but not dopamine antagonists blocks drug-induced reinstatement of morphine place preference. Brain Res Bull. 2005;64:493–503. doi: 10.1016/j.brainresbull.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 362.Popik P, Wrobel M, Bisaga A. Reinstatement of morphine-conditioned reward is blocked by memantine. Neuropsychopharmacology. 2006;31:160–70. doi: 10.1038/sj.npp.1300760. [DOI] [PubMed] [Google Scholar]
- 363.Ma YY, Chu NN, Guo CY, Han JS, Cui CL. NR2B-containing NMDA receptor is required for morphine-but not stress-induced reinstatement. Exp Neurol. 2007;203:309–19. doi: 10.1016/j.expneurol.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 364.Popik P, Wrobel M. Morphine conditioned reward is inhibited by MPEP, the mGluR5 antagonist. Neuropharmacology. 2002;43:1210–1217. doi: 10.1016/s0028-3908(02)00309-x. [DOI] [PubMed] [Google Scholar]
- 365.Xi ZX, Stein EA. Blockade of ionotropic glutamatergic transmission in the ventral tegmental area reduces heroin reinforcement in rat. Psychopharmacology. 2002;164:144–150. doi: 10.1007/s00213-002-1190-3. [DOI] [PubMed] [Google Scholar]
- 366.Caille S, Parsons LH. Intravenous heroin self-administration decreases GABA efflux in the ventral pallidum: an in vivo microdialysis study in rats. Eur J Neurosci. 2004;20:593–596. doi: 10.1111/j.1460-9568.2004.03497.x. [DOI] [PubMed] [Google Scholar]
- 367.Hubner CB, Koob GF. The ventral pallidum plays a role in mediating cocaine and heroin self-administration in the rat. Brain Res. 1990;508:20–29. doi: 10.1016/0006-8993(90)91112-t. [DOI] [PubMed] [Google Scholar]
- 368.Bossert JM, Liu SY, Lu L, Shaham Y. A role of ventral tegmental area glutamate in contextual cue-induced relapse to heroin seeking. J Neurosci. 2004;24:10726–10730. doi: 10.1523/JNEUROSCI.3207-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Bossert JM, Busch RF, Gray SM. The novel mGluR2/3 agonist LY379268 attenuates cue-induced reinstatement of heroin seeking. Neuroreport. 2005;16:1013–6. doi: 10.1097/00001756-200506210-00026. [DOI] [PubMed] [Google Scholar]
- 370.Bossert JM, Gray SM, Lu L, Shaham Y. Activation of group II metabotropic glutamate receptors in the nucleus accumbens shell attenuates context-induced relapse to heroin seeking. Neuropsychopharmacology. 2006;31:2197–209. doi: 10.1038/sj.npp.1300977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Laviolette SR, van der Kooy D. The neurobiology of nicotine addiction: bridging the gap from molecules to behavior. Nat Rev Neurosci. 2004;5:55–65. doi: 10.1038/nrn1298. [DOI] [PubMed] [Google Scholar]
- 372.Wonnacott S, Sidhpura N, Balfour DJ. Nicotine: from molecular mechanisms to behaviour. Curr Opin Pharmacol. 2005;5:53–9. doi: 10.1016/j.coph.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 373.Picciotto MR, Corrigall WA. Neuronal systems underlying behaviors related to nicotine addiction: neural circuits and molecular genetics. J Neurosci. 2002;22:3338–3341. doi: 10.1523/JNEUROSCI.22-09-03338.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Watkins SS, Koob GF, Markou A. Neural mechanisms underlying nicotine addiction: acute positive reinforcement and withdrawal. Nicotine Tob Res. 2000;2:19–37. doi: 10.1080/14622200050011277. [DOI] [PubMed] [Google Scholar]
- 375.Jones IW, Wonnacott S. Precise localization of α7 nicotinic acetylcholine receptors on glutamatergic axon terminals in the rat ventral tegmental area. J Neurosci. 2004;24:11244–52. doi: 10.1523/JNEUROSCI.3009-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Fu Y, Matta SG, Gao W, Brower VG, Sharp BM. Systemic nicotine stimulates dopamine release in nucleus accumbens: re-evaluation of the role of N-methyl-D-aspartate receptors in the ventral tegmental area. J Pharmacol Exp Ther. 2000;294:458–465. [PubMed] [Google Scholar]
- 377.Schilstrom B, Fagerquist MV, Zhang X, Hertel P, Panagis G, Nomikos GG, et al. Putative role of presynaptic α7 nicotinic receptors in nicotine stimulated increases of extracellular levels of glutamate and aspartate in the ventral tegmental area. Synapse. 2000;38:375–83. doi: 10.1002/1098-2396(20001215)38:4<375::AID-SYN2>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 378.Schilstrom B, Nomikos GG, Nisell M, Hertel P, Svensson TH. N-methyl-D-aspartate receptor antagonism in the ventral tegmental area diminishes the systemic nicotine-induced dopamine release in the nucleus accumbens. Neuroscience. 1998;82:781–789. doi: 10.1016/s0306-4522(97)00243-1. [DOI] [PubMed] [Google Scholar]
- 379.Schilstrom B, Svensson HM, Svensson TH, Nomikos GG. Nicotine and food induced dopamine release in the nucleus accumbens of the rat: putative role of α7 nicotinic receptors in the ventral tegmental area. Neuroscience. 1998;85:1005–9. doi: 10.1016/s0306-4522(98)00114-6. [DOI] [PubMed] [Google Scholar]
- 380.Svensson TH, Mathe JM, Nomikos GG, Schilstrom B. Role of excitatory amino acids in the ventral tegmental area for central actions of non-competitive NMDA-receptor antagonists and nicotine. Amino Acids. 1998;14:51–6. doi: 10.1007/BF01345242. [DOI] [PubMed] [Google Scholar]
- 381.Grillner P, Svensson TH. Nicotine-induced excitation of midbrain dopamine neurons in vitro involves ionotropic glutamate receptor activation. Synapse. 2000;38:1–9. doi: 10.1002/1098-2396(200010)38:1<1::AID-SYN1>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 382.Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–57. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- 383.Sziraki I, Sershen H, Hashim A, Lajtha A. Receptors in the ventral tegmental area mediating nicotine-induced dopamine release in the nucleus accumbens. Neurochem Res. 2002;27:253–61. doi: 10.1023/a:1014844823534. [DOI] [PubMed] [Google Scholar]
- 384.Schilstrom B, Rawal N, Mameli-Engvall M, Nomikos GG, Svensson TH. Dual effects of nicotine on dopamine neurons mediated by different nicotinic receptor subtypes. Int J Neuropsychopharmacol. 2003;6:1–11. doi: 10.1017/S1461145702003188. [DOI] [PubMed] [Google Scholar]
- 385.Kosowski AR, Cebers G, Cebere A, Swanhagen AC, Liljequist S. Nicotine-induced dopamine release in the nucleus accumbens is inhibited by the novel AMPA antagonist ZK200775 and the NMDA antagonist CGP39551. Psychopharmacology. 2004;175:114–23. doi: 10.1007/s00213-004-1797-7. [DOI] [PubMed] [Google Scholar]
- 386.Kosowski AR, Liljequist S. The NR2B-selective N-methyl-D-aspartate receptor antagonist Ro 25-6981 [(+/-)-(R,S)-alpha-(4-hydroxyphenyl)-beta-methyl-4-(phenylmethyl)-1-pipe ridine propanol] potentiates the effect of nicotine on locomotor activity and dopamine release in the nucleus accumbens. J Pharmacol Exp Ther. 2004;311:560–7. doi: 10.1124/jpet.104.070235. [DOI] [PubMed] [Google Scholar]
- 387.Perez de la Mora M, Mendez-Franco J, Salceda R, Aguirre JA, Fuxe K. Neurochemical effects of nicotine on glutamate and GABA mechanisms in the rat brain. Acta Physiol Scand. 1991;141:241–50. doi: 10.1111/j.1748-1716.1991.tb09074.x. [DOI] [PubMed] [Google Scholar]
- 388.Toth E, Vizi ES, Lajtha A. Effect of nicotine on levels of extracellular amino acids in regions of the rat brain in vivo. Neuropharmacology. 1993;32:827–32. doi: 10.1016/0028-3908(93)90192-6. [DOI] [PubMed] [Google Scholar]
- 389.Gioanni Y, Rougeot C, Clarke PB, Lepouse C, Thierry AM, Vidal C. Nicotinic receptors in the rat prefrontal cortex: increase in glutamate release and facilitation of mediodorsal thalamo-cortical transmission. Eur J Neurosci. 1999;11:18–30. doi: 10.1046/j.1460-9568.1999.00403.x. [DOI] [PubMed] [Google Scholar]
- 390.Lopez E, Arce C, Vicente S, Oset-Gasque MJ, Gonzalez MP. Nicotinic receptors mediate the release of amino acid neurotransmitters in cultured cortical neurons. Cereb Cortex. 2001;11:158–63. doi: 10.1093/cercor/11.2.158. [DOI] [PubMed] [Google Scholar]
- 391.Marchi M, Risso F, Viola C, Cavazzani P, Raiteri M. Direct evidence that release-stimulating α7 nicotinic cholinergic receptors are localized on human and rat brain glutamatergic axon terminals. J Neurochem. 2002;80:1071–8. doi: 10.1046/j.0022-3042.2002.00805.x. [DOI] [PubMed] [Google Scholar]
- 392.Lambe EK, Picciotto MR, Aghajanian GK. Nicotine induces glutamate release from thalamocortical terminals in prefrontal cortex. Neuropsychopharmacology. 2003;28:216–25. doi: 10.1038/sj.npp.1300032. [DOI] [PubMed] [Google Scholar]
- 393.Wang BW, Liao WN, Chang CT, Wang SJ. Facilitation of glutamate release by nicotine involves the activation of a Ca2+/calmodulin signaling pathway in rat prefrontal cortex nerve terminals. Synapse. 2006;59:491–501. doi: 10.1002/syn.20267. [DOI] [PubMed] [Google Scholar]
- 394.Toth E, Sershen H, Hashim A, Vizi ES, Lajtha A. Effect of nicotine on extracellular levels of neurotransmitters assessed by microdialysis in various brain regions: role of glutamic acid. Neurochem Res. 1992;17:265–71. doi: 10.1007/BF00966669. [DOI] [PubMed] [Google Scholar]
- 395.Meshul CK, Kamel D, Moore C, Kay TS, Krentz L. Nicotine alters striatal glutamate function and decreases the apomorphine-induced contralateral rotations in 6-OHDA-lesioned rats. Exp Neurol. 2002;175:257–74. doi: 10.1006/exnr.2002.7900. [DOI] [PubMed] [Google Scholar]
- 396.Reid MS, Fox L, Ho LB, Berger SP. Nicotine stimulation of extracellular glutamate levels in the nucleus accumbens: neuropharmacological characterization. Synapse. 2000;35:129–136. doi: 10.1002/(SICI)1098-2396(200002)35:2<129::AID-SYN5>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 397.Lallemand F, Ward RJ, Dravolina O, De Witte P. Nicotine-induced changes of glutamate and arginine in naive and chronically alcoholized rats: an in vivo microdialysis study. Brain Res. 2006;1111:48–60. doi: 10.1016/j.brainres.2006.06.083. [DOI] [PubMed] [Google Scholar]
- 398.Kashkin VA, De Witte P. Nicotine increases microdialysate brain amino acid concentrations and induces conditioned place preference. Eur Neuropsychopharmacol. 2005;15:625–32. doi: 10.1016/j.euroneuro.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 399.Saellstroem Baum S, Huebner A, Krimphove M, Morgenstern R, Badawy AA, Spies CD. Nicotine stimulation on extracellular glutamate levels in the nucleus accumbens of ethanol-withdrawn rats in vivo. Alcohol Clin Exp Res. 2006;30:1414–21. doi: 10.1111/j.1530-0277.2006.00169.x. [DOI] [PubMed] [Google Scholar]
- 400.Fedele E, Varnier G, Ansaldo MA, Raiteri M. Nicotine administration stimulates the in vivo N-methyl-D-aspartate receptor/nitric oxide/cyclic GMP pathway in rat hippocampus through glutamate release. Br J Pharmacol. 1998;125:1042–8. doi: 10.1038/sj.bjp.0702130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Engberg G. Nicotine induced excitation of locus coeruleus neurons is mediated via release of excitatory amino acids. Life Sci. 1989;44:1535–40. doi: 10.1016/0024-3205(89)90446-3. [DOI] [PubMed] [Google Scholar]
- 402.Erhardt S, Hajos M, Lindberg A, Engberg G. Nicotine-induced excitation of locus coeruleus neurons is blocked by elevated levels of endogenous kynurenic acid. Synapse. 2000;37:104–8. doi: 10.1002/1098-2396(200008)37:2<104::AID-SYN4>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 403.Lim DK, Park SH, Choi WJ. Subacute nicotine exposure in cultured cerebellar cells increased the release and uptake of glutamate. Arch Pharm Res. 2000;23:488–94. doi: 10.1007/BF02976578. [DOI] [PubMed] [Google Scholar]
- 404.Reno LA, Zago W, Markus RP. Release of [3H]-L-glutamate by stimulation of nicotinic acetylcholine receptors in rat cerebellar slices. Neuroscience. 2004;124:647–53. doi: 10.1016/j.neuroscience.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 405.Vidal C, Changeux JP. Nicotinic and muscarinic modulations of excitatory synaptic transmission in the rat prefrontal cortex in vitro. Neuroscience. 1993;56:23–32. doi: 10.1016/0306-4522(93)90558-w. [DOI] [PubMed] [Google Scholar]
- 406.Kiba H, Jayaraman A. Nicotine induced c-fos expression in the striatum is mediated mostly by dopamine D1 receptor and is dependent on NMDA stimulation. Mol Brain Res. 1994;23:1–13. doi: 10.1016/0169-328x(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 407.McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–6. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- 408.Garcia-Munoz M, Patino P, Young SJ, Groves PM. Effects of nicotine on dopaminergic nigrostriatal axons requires stimulation of presynaptic glutamatergic receptors. J Pharmacol Exp Ther. 1996;277:1685–93. [PubMed] [Google Scholar]
- 409.Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–6. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- 410.Schilstrom B, De Villiers S, Malmerfelt A, Svensson TH, Nomikos GG. Nicotine-induced Fos expression in the nucleus accumbens and the medial prefrontal cortex of the rat: role of nicotinic and NMDA receptors in the ventral tegmental area. Synapse. 2000;36:314–21. doi: 10.1002/(SICI)1098-2396(20000615)36:4<314::AID-SYN8>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 411.Radcliffe KA, Dani JA. Nicotinic stimulation produces multiple forms of increased glutamatergic synaptic transmission. J Neurosci. 1998;18:7075–83. doi: 10.1523/JNEUROSCI.18-18-07075.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Aramakis VB, Metherate R. Nicotine selectively enhances NMDA receptor-mediated synaptic transmission during postnatal development in sensory neocortex. J Neurosci. 1998;18:8485–95. doi: 10.1523/JNEUROSCI.18-20-08485.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Radcliffe KA, Fisher JL, Gray R, Dani JA. Nicotinic modulation of glutamate and GABA synaptic transmission of hippocampal neurons. Ann N Y Acad Sci. 1999;868:591–610. doi: 10.1111/j.1749-6632.1999.tb11332.x. [DOI] [PubMed] [Google Scholar]
- 414.Fisher JL, Dani JA. Nicotinic receptors on hippocampal cultures can increase synaptic glutamate currents while decreasing the NMDA-receptor component. Neuropharmacology. 2000;39:2756–69. doi: 10.1016/s0028-3908(00)00102-7. [DOI] [PubMed] [Google Scholar]
- 415.Barazangi N, Role LW. Nicotine-induced enhancement of glutamatergic and GABAergic synaptic transmission in the mouse amygdala. J Neurophysiol. 2001;86:463–74. doi: 10.1152/jn.2001.86.1.463. [DOI] [PubMed] [Google Scholar]
- 416.Matsubayashi H, Amano T, Amano H, Sasa M. Excitation of rat striatal large neurons by dopamine and/or glutamate released from nerve terminals via presynaptic nicotinic receptor (α4β2 type) stimulation. Jpn J Pharmacol. 2001;86:429–36. doi: 10.1254/jjp.86.429. [DOI] [PubMed] [Google Scholar]
- 417.Kawa K. Acute synaptic modulation by nicotinic agonists in developing cerebellar Purkinje cells of the rat. J Physiol. 2002;538:87–102. doi: 10.1113/jphysiol.2001.012885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Erhardt S, Schwieler L, Engberg G. Excitatory and inhibitory responses of dopamine neurons in the ventral tegmental area to nicotine. Synapse. 2002;43:227–37. doi: 10.1002/syn.10044. [DOI] [PubMed] [Google Scholar]
- 419.Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–919. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 420.Wu M, Hajszan T, Leranth C, Alreja M. Nicotine recruits a local glutamatergic circuit to excite septohippocampal GABAergic neurons. Eur J Neurosci. 2003;18:1155–68. doi: 10.1046/j.1460-9568.2003.02847.x. [DOI] [PubMed] [Google Scholar]
- 421.Fagen ZM, Mansvelder HD, Keath JR, McGehee DS. Short- and long-term modulation of synaptic inputs to brain reward areas by nicotine. Ann N Y Acad Sci. 2003;1003:185–95. doi: 10.1196/annals.1300.011. [DOI] [PubMed] [Google Scholar]
- 422.Teo MY, van Wyk M, Lin J, Lipski J. Differential effects of nicotine on the activity of substantia nigra and ventral tegmental area dopaminergic neurons in vitro. Acta Neurobiol Exp (Wars) 2004;64:119–30. doi: 10.55782/ane-2004-1498. [DOI] [PubMed] [Google Scholar]
- 423.Giocomo LM, Hasselmo ME. Nicotinic modulation of glutamatergic synaptic transmission in region CA3 of the hippocampus. Eur J Neurosci. 2005;22:1349–56. doi: 10.1111/j.1460-9568.2005.04316.x. [DOI] [PubMed] [Google Scholar]
- 424.Fujii S, Ji Z, Morita N, Sumikawa K. Acute and chronic nicotine exposure differentially facilitate the induction of LTP. Brain Res. 1999;846:137–43. doi: 10.1016/s0006-8993(99)01982-4. [DOI] [PubMed] [Google Scholar]
- 425.Yamazaki Y, Jia Y, Hamaue N, Sumikawa K. Nicotine-induced switch in the nicotinic cholinergic mechanisms of facilitation of long-term potentiation induction. Eur J Neurosci. 2005;22:845–60. doi: 10.1111/j.1460-9568.2005.04259.x. [DOI] [PubMed] [Google Scholar]
- 426.Yamazaki Y, Jia Y, Niu R, Sumikawa K. Nicotine exposure in vivo induces long-lasting enhancement of NMDA receptor-mediated currents in the hippocampus. Eur J Neurosci. 2006;23:1819–28. doi: 10.1111/j.1460-9568.2006.04714.x. [DOI] [PubMed] [Google Scholar]
- 427.Adriani W, Granstrem O, Macri S, Izykenova G, Dambinova S, Laviola G. Behavioral and neurochemical vulnerability during adolescence in mice: studies with nicotine. Neuropsychopharmacology. 2004;29:869–78. doi: 10.1038/sj.npp.1300366. [DOI] [PubMed] [Google Scholar]
- 428.Wang F, Chen H, Steketee JD, Sharp BM. Upregulation of ionotropic glutamate receptor subunits within specific mesocorticolimbic regions during chronic nicotine self-administration. Neuropsychopharmacology. 2007;32:103–9. doi: 10.1038/sj.npp.1301033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Mexal S, Frank M, Berger R, Adams CE, Ross RG, Freedman R, et al. Differential modulation of gene expression in the NMDA postsynaptic density of schizophrenic and control smokers. Mol Brain Res. 2005;139:317–32. doi: 10.1016/j.molbrainres.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 430.Lim DK, Kim HS. Chronic exposure of nicotine modulates the expressions of the cerebellar glial glutamate transporters in rats. Arch Pharm Res. 2003;26:321–9. doi: 10.1007/BF02976963. [DOI] [PubMed] [Google Scholar]
- 431.Kane JK, Hwang Y, Konu O, Loughlin SE, Leslie FM, Li MD. Regulation of Homer and group I metabotropic glutamate receptors by nicotine. Eur J Neurosci. 2005;21:1145–54. doi: 10.1111/j.1460-9568.2005.03945.x. [DOI] [PubMed] [Google Scholar]
- 432.Gallinat J, Schubert F. Regional cerebral glutamate concentrations and chronic tobacco consumption. Pharmacopsychiatry. 2007;40:64–7. doi: 10.1055/s-2007-970144. [DOI] [PubMed] [Google Scholar]
- 433.Barron S, White A, Swartzwelder HS, Bell RL, Rodd ZA, Slawecki CJ, et al. Adolescent vulnerabilities to chronic alcohol or nicotine exposure: findings from rodent models. Alcohol Clin Exp Res. 2005;29:1720–5. doi: 10.1097/01.alc.0000179220.79356.e5. [DOI] [PubMed] [Google Scholar]
- 434.Fournier ME, Levy S. Recent trends in adolescent substance use, primary care screening, and updates in treatment options. Curr Opin Pediatr. 2006;18:352–8. doi: 10.1097/01.mop.0000236381.33907.9d. [DOI] [PubMed] [Google Scholar]
- 435.Deas D, Brown ES. Adolescent substance abuse and psychiatric comorbidities. J Clin Psychiatry. 2006;67:e02. doi: 10.4088/jcp.0706e02. [DOI] [PubMed] [Google Scholar]
- 436.Kelsey JE, Beer T, Lee E, Wagner A. Low doses of dizocilpine block the development and subsequent expression of locomotor sensitization to nicotine in rats. Psychopharmacology. 2002;161:370–8. doi: 10.1007/s00213-002-1015-4. [DOI] [PubMed] [Google Scholar]
- 437.Paterson NE, Semenova S, Gasparini F, Markou A. The mGluR5 antagonist MPEP decreased nicotine self-administration in rats and mice. Psychopharmacology. 2003;167:257–264. doi: 10.1007/s00213-003-1432-z. [DOI] [PubMed] [Google Scholar]
- 438.Liechti ME, Markou A. Interactive effects of the mGlu5 receptor antagonist MPEP and the mGlu2/3 receptor antagonist LY341495 on nicotine self-administration and reward deficits associated with nicotine withdrawal in rats. Eur J Pharmacol. 2006;554:164–174. doi: 10.1016/j.ejphar.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Bespalov AY, Dravolina OA, Sukhanov I, Zakharova E, Blokhina E, Zvartau E, et al. Metabotropic glutamate receptor (mGluR5) antagonist MPEP attenuated cue- and schedule-induced reinstatement of nicotine self-administration behavior in rats. Neuropharmacology. 2005;49(Suppl 1):167–78. doi: 10.1016/j.neuropharm.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 440.Dravolina OA, Zakharova ES, Shekunova EV, Zvartau EE, Danysz W, Bespalov AY. mGlu1 receptor blockade attenuates cue- and nicotine-induced reinstatement of extinguished nicotine self-administration behavior in rats. Neuropharmacology. 2007;52:263–9. doi: 10.1016/j.neuropharm.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 441.Harrison AA, Gasparini F, Markou A. Nicotine potentiation of brain stimulation reward reversed by DHβE and SCH 23390, but not by eticlopride, LY 314582 or MPEP in rats. Psychopharmacology. 2002;160:56–66. doi: 10.1007/s00213-001-0953-6. [DOI] [PubMed] [Google Scholar]
- 442.Markou A. Metabotropic glutamate receptor antagonists: novel therapeutics for nicotine dependence and depression? Biol Psychiatry. 2006;61:17–22. doi: 10.1016/j.biopsych.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 443.Helton DR, Tizzano JP, Monn JA, Schoepp DD, Kallman MJ. LY354740: a metabotropic glutamate receptor agonist which ameliorates symptoms of nicotine withdrawal in rats. Neuropharmacology. 1997;36:1511–6. doi: 10.1016/s0028-3908(97)00170-6. [DOI] [PubMed] [Google Scholar]
- 444.Kenny PJ, Gasparini F, Markou A. Group II metabotropic and alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA)/kainate glutamate receptors regulate the deficit in brain reward function associated with nicotine withdrawal in rats. J Pharmacol Exp Ther. 2003;306:1068–76. doi: 10.1124/jpet.103.052027. [DOI] [PubMed] [Google Scholar]
- 445.Howlett AC, Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Porrino LJ. Cannabinoid physiology and pharmacology: 30 years of progress. Neuropharmacology. 2004;47(Suppl 1):345–358. doi: 10.1016/j.neuropharm.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 446.Parolaro D, Vigano D, Rubino T. Endocannabinoids and drug dependence. Curr Drug Targets CNS Neurol Disord. 2005;4:643–55. doi: 10.2174/156800705774933014. [DOI] [PubMed] [Google Scholar]
- 447.Maldonado R, Valverde O, Berrendero F. Involvement of the endocannabinoid system in drug addiction. Trends Neurosci. 2006;29:225–232. doi: 10.1016/j.tins.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 448.Colombo G, Serra S, Vacca G, C MAM, Gessa GL. Endocannabinoid system and alcohol addiction: pharmacological studies. Pharmacol Biochem Behav. 2005;81:369–380. doi: 10.1016/j.pbb.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 449.Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ. Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci. 2001;21:109–16. doi: 10.1523/JNEUROSCI.21-01-00109.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Kofalvi A, Rodrigues RJ, Ledent C, Mackie K, Vizi ES, Cunha RA, et al. Involvement of cannabinoid receptors in the regulation of neurotransmitter release in the rodent striatum: a combined immunochemical and pharmacological analysis. J Neurosci. 2005;25:2874–84. doi: 10.1523/JNEUROSCI.4232-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Katona I, Urban GM, Wallace M, Ledent C, Jung KM, Piomelli D, et al. Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci. 2006;26:5628–37. doi: 10.1523/JNEUROSCI.0309-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Auclair N, Otani S, Soubrie P, Crepel F. Cannabinoids modulate synaptic strength and plasticity at glutamatergic synapses of rat prefrontal cortex pyramidal neurons. J Neurophysiol. 2000;83:3287–93. doi: 10.1152/jn.2000.83.6.3287. [DOI] [PubMed] [Google Scholar]
- 453.Domenici MR, Azad SC, Marsicano G, Schierloh A, Wotjak CT, Dodt HU, et al. Cannabinoid receptor type 1 located on presynaptic terminals of principal neurons in the forebrain controls glutamatergic synaptic transmission. J Neurosci. 2006;26:5794–9. doi: 10.1523/JNEUROSCI.0372-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Gerdeman G, Lovinger DM. CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol. 2001;85:468–71. doi: 10.1152/jn.2001.85.1.468. [DOI] [PubMed] [Google Scholar]
- 455.Huang CC, Lo SW, Hsu KS. Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J Physiol. 2001;532:731–48. doi: 10.1111/j.1469-7793.2001.0731e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Brown TM, Brotchie JM, Fitzjohn SM. Cannabinoids decrease corticostriatal synaptic transmission via an effect on glutamate uptake. J Neurosci. 2003;23:11073–7. doi: 10.1523/JNEUROSCI.23-35-11073.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Narushima M, Hashimoto K, Kano M. Endocannabinoid-mediated short-term suppression of excitatory synaptic transmission to medium spiny neurons in the striatum. Neurosci Res. 2006;54:159–64. doi: 10.1016/j.neures.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 458.Hoffman AF, Lupica CR. Direct actions of cannabinoids on synaptic transmission in the nucleus accumbens: a comparison with opioids. J Neurophysiol. 2001;85:72–83. doi: 10.1152/jn.2001.85.1.72. [DOI] [PubMed] [Google Scholar]
- 459.Pistis M, Muntoni AL, Pillolla G, Gessa GL. Cannabinoids inhibit excitatory inputs to neurons in the shell of the nucleus accumbens: an in vivo electrophysiological study. Eur J Neurosci. 2002;15:1795–802. doi: 10.1046/j.1460-9568.2002.02019.x. [DOI] [PubMed] [Google Scholar]
- 460.Robbe D, Alonso G, Manzoni OJ. Exogenous and endogenous cannabinoids control synaptic transmission in mice nucleus accumbens. Ann N Y Acad Sci. 2003;1003:212–25. doi: 10.1196/annals.1300.013. [DOI] [PubMed] [Google Scholar]
- 461.Schoffelmeer AN, Hogenboom F, Wardeh G, De Vries TJ. Interactions between CB1 cannabinoid and mu opioid receptors mediating inhibition of neurotransmitter release in rat nucleus accumbens core. Neuropharmacology. 2006;51:773–81. doi: 10.1016/j.neuropharm.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 462.Freiman I, Szabo B. Cannabinoids depress excitatory neurotransmission between the subthalamic nucleus and the globus pallidus. Neuroscience. 2005;133:305–13. doi: 10.1016/j.neuroscience.2005.01.058. [DOI] [PubMed] [Google Scholar]
- 463.Shen M, Piser TM, Seybold VS, Thayer SA. Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J Neurosci. 1996;16:4322–34. doi: 10.1523/JNEUROSCI.16-14-04322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Shen M, Thayer SA. Delta9-tetrahydrocannabinol acts as a partial agonist to modulate glutamatergic synaptic transmission between rat hippocampal neurons in culture. Mol Pharmacol. 1999;55:8–13. doi: 10.1124/mol.55.1.8. [DOI] [PubMed] [Google Scholar]
- 465.Sullivan JM. Mechanisms of cannabinoid-receptor-mediated inhibition of synaptic transmission in cultured hippocampal pyramidal neurons. J Neurophysiol. 1999;82:1286–94. doi: 10.1152/jn.1999.82.3.1286. [DOI] [PubMed] [Google Scholar]
- 466.Misner DL, Sullivan JM. Mechanism of cannabinoid effects on long-term potentiation and depression in hippocampal CA1 neurons. J Neurosci. 1999;19:6795–805. doi: 10.1523/JNEUROSCI.19-16-06795.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Hajos N, Ledent C, Freund TF. Novel cannabinoid-sensitive receptor mediates inhibition of glutamatergic synaptic transmission in the hippocampus. Neuroscience. 2001;106:1–4. doi: 10.1016/s0306-4522(01)00287-1. [DOI] [PubMed] [Google Scholar]
- 468.Ohno-Shosaku T, Tsubokawa H, Mizushima I, Yoneda N, Zimmer A, Kano M. Presynaptic cannabinoid sensitivity is a major determinant of depolarization-induced retrograde suppression at hippocampal synapses. J Neurosci. 2002;22:3864–72. doi: 10.1523/JNEUROSCI.22-10-03864.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Wang SJ. Cannabinoid CB1 receptor-mediated inhibition of glutamate release from rat hippocampal synaptosomes. Eur J Pharmacol. 2003;469:47–55. doi: 10.1016/s0014-2999(03)01734-5. [DOI] [PubMed] [Google Scholar]
- 470.Hoffman AF, Riegel AC, Lupica CR. Functional localization of cannabinoid receptors and endogenous cannabinoid production in distinct neuron populations of the hippocampus. Eur J Neurosci. 2003;18:524–34. doi: 10.1046/j.1460-9568.2003.02773.x. [DOI] [PubMed] [Google Scholar]
- 471.Kofalvi A, Vizi ES, Ledent C, Sperlagh B. Cannabinoids inhibit the release of [3H]glutamate from rodent hippocampal synaptosomes via a novel CB1 receptor-independent action. Eur J Neurosci. 2003;18:1973–8. doi: 10.1046/j.1460-9568.2003.02897.x. [DOI] [PubMed] [Google Scholar]
- 472.Cannizzaro C, D’Amico M, Preziosi P, Martire M. Presynaptic effects of anandamide and WIN55,212-2 on glutamatergic nerve endings isolated from rat hippocampus. Neurochem Int. 2006;48:159–65. doi: 10.1016/j.neuint.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 473.Takahashi KA, Castillo PE. The CB1 cannabinoid receptor mediates glutamatergic synaptic suppression in the hippocampus. Neuroscience. 2006;139:795–802. doi: 10.1016/j.neuroscience.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 474.Azad SC, Eder M, Marsicano G, Lutz B, Zieglgansberger W, Rammes G. Activation of the cannabinoid receptor type 1 decreases glutamatergic and GABAergic synaptic transmission in the lateral amygdala of the mouse. Learn Mem. 2003;10:116–28. doi: 10.1101/lm.53303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Hentges ST, Low MJ, Williams JT. Differential regulation of synaptic inputs by constitutively released endocannabinoids and exogenous cannabinoids. J Neurosci. 2005;25:9746–51. doi: 10.1523/JNEUROSCI.2769-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Di S, Boudaba C, Popescu IR, Weng FJ, Harris C, Marcheselli VL, et al. Activity-dependent release and actions of endocannabinoids in the rat hypothalamic supraoptic nucleus. J Physiol. 2005;569:751–60. doi: 10.1113/jphysiol.2005.097477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Szabo B, Wallmichrath I, Mathonia P, Pfreundtner C. Cannabinoids inhibit excitatory neurotransmission in the substantia nigra pars reticulata. Neuroscience. 2000;97:89–97. doi: 10.1016/s0306-4522(00)00036-1. [DOI] [PubMed] [Google Scholar]
- 478.Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci. 2004;24:53–62. doi: 10.1523/JNEUROSCI.4503-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Mendiguren A, Pineda J. CB1 cannabinoid receptors inhibit the glutamatergic component of KCl-evoked excitation of locus coeruleus neurons in rat brain slices. Neuropharmacology. 2007;52:617–25. doi: 10.1016/j.neuropharm.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 480.Levenes C, Daniel H, Soubrie P, Crepel F. Cannabinoids decrease excitatory synaptic transmission and impair long-term depression in rat cerebellar Purkinje cells. J Physiol. 1998;510(Pt 3):867–79. doi: 10.1111/j.1469-7793.1998.867bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Breivogel CS, Walker JM, Huang SM, Roy MB, Childers SR. Cannabinoid signaling in rat cerebellar granule cells: G-protein activation, inhibition of glutamate release and endogenous cannabinoids. Neuropharmacology. 2004;47:81–91. doi: 10.1016/j.neuropharm.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 482.Marcaggi P, Attwell D. Endocannabinoid signaling depends on the spatial pattern of synapse activation. Nat Neurosci. 2005;8:776–81. doi: 10.1038/nn1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Riedel G, Davies SN. Cannabinoid function in learning, memory and plasticity. Handb Exp Pharmacol. 2005:445–77. doi: 10.1007/3-540-26573-2_15. [DOI] [PubMed] [Google Scholar]
- 484.Hoffman AF, Oz M, Yang R, Lichtman AH, Lupica CR. Opposing actions of chronic Δ9-tetrahydrocannabinol and cannabinoid antagonists on hippocampal long-term potentiation. Learn Mem. 2007;14:63–74. doi: 10.1101/lm.439007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Ferraro L, Tomasini MC, Gessa GL, Bebe BW, Tanganelli S, Antonelli T. The cannabinoid receptor agonist WIN 55,212-2 regulates glutamate transmission in rat cerebral cortex: an in vivo and in vitro study. Cereb Cortex. 2001;11:728–33. doi: 10.1093/cercor/11.8.728. [DOI] [PubMed] [Google Scholar]
- 486.Pistis M, Ferraro L, Pira L, Flore G, Tanganelli S, Gessa GL, et al. Delta(9)-tetrahydrocannabinol decreases extracellular GABA and increases extracellular glutamate and dopamine levels in the rat prefrontal cortex: an in vivo microdialysis study. Brain Res. 2002;948:155–8. doi: 10.1016/s0006-8993(02)03055-x. [DOI] [PubMed] [Google Scholar]
- 487.Mato S, Robbe D, Puente N, Grandes P, Manzoni OJ. Presynaptic homeostatic plasticity rescues long-term depression after chronic Delta 9-tetrahydrocannabinol exposure. J Neurosci. 2005;25:11619–27. doi: 10.1523/JNEUROSCI.2294-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Tonini R, Ciardo S, Cerovic M, Rubino T, Parolaro D, Mazzanti M, et al. ERK-dependent modulation of cerebellar synaptic plasticity after chronic Δ9-tetrahydrocannabinol exposure. J Neurosci. 2006;26:5810–8. doi: 10.1523/JNEUROSCI.5469-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Nah SY, Saya D, Vogel Z. Cannabinoids inhibit agonist-stimulated formation of inositol phosphates in rat hippocampal cultures. Eur J Pharmacol. 1993;246:19–24. doi: 10.1016/0922-4106(93)90004-s. [DOI] [PubMed] [Google Scholar]
- 490.Doherty J, Dingledine R. Functional interactions between cannabinoid and metabotropic glutamate receptors in the central nervous system. Curr Opin Pharmacol. 2003;3:46–53. doi: 10.1016/s1471-4892(02)00014-0. [DOI] [PubMed] [Google Scholar]
- 491.Vaughan CW, Christie MJ. Retrograde signalling by endocannabinoids. Handb Exp Pharmacol. 2005:367–83. doi: 10.1007/3-540-26573-2_12. [DOI] [PubMed] [Google Scholar]
- 492.Justinova Z, Goldberg SR, Heishman SJ, Tanda G. Self-administration of cannabinoids by experimental animals and human marijuana smokers. Pharmacol Biochem Behav. 2005;81:285–99. doi: 10.1016/j.pbb.2005.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Lovinger DM, White G, Weight FF. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science. 1989;243:1721–1724. doi: 10.1126/science.2467382. [DOI] [PubMed] [Google Scholar]
- 494.Lima-Landman MT, Albuquerque EX. Ethanol potentiates and blocks NMDA-activated single-channel currents in rat hippocampal pyramidal cells. FEBS Lett. 1989;247:61–7. doi: 10.1016/0014-5793(89)81241-4. [DOI] [PubMed] [Google Scholar]
- 495.Hoffman PL, Rabe CS, Moses F, Tabakoff B. N-methyl-D-aspartate receptors and ethanol: inhibition of calcium flux and cyclic GMP production. J Neurochem. 1989;52:1937–40. doi: 10.1111/j.1471-4159.1989.tb07280.x. [DOI] [PubMed] [Google Scholar]
- 496.Dildy JE, Leslie SW. Ethanol inhibits NMDA-induced increases in free intracellular Ca2+ in dissociated brain cells. Brain Res. 1989;499:383–7. doi: 10.1016/0006-8993(89)90789-0. [DOI] [PubMed] [Google Scholar]
- 497.Lovinger DM, White G, Weight FF. Ethanol inhibition of neuronal glutamate receptor function. Ann Med. 1990;22:247–52. doi: 10.3109/07853899009148935. [DOI] [PubMed] [Google Scholar]
- 498.White G, Lovinger DM, Weight FF. Ethanol inhibits NMDA-activated current but does not alter GABA-activated current in an isolated adult mammalian neuron. Brain Res. 1990;507:332–6. doi: 10.1016/0006-8993(90)90292-j. [DOI] [PubMed] [Google Scholar]
- 499.Lovinger DM, White G, Weight FF. NMDA receptor-mediated synaptic excitation selectively inhibited by ethanol in hippocampal slice from adult rat. J Neurosci. 1990;10:1372–9. doi: 10.1523/JNEUROSCI.10-04-01372.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Kuner T, Schoepfer R, Korpi ER. Ethanol inhibits glutamate-induced currents in heteromeric NMDA receptor subtypes. Neuroreport. 1993;5:297–300. doi: 10.1097/00001756-199312000-00029. [DOI] [PubMed] [Google Scholar]
- 501.Masood K, Wu C, Brauneis U, Weight FF. Differential ethanol sensitivity of recombinant N-methyl-D-aspartate receptor subunits. Mol Pharmacol. 1994;45:324–329. [PubMed] [Google Scholar]
- 502.Chu B, Anantharam V, Treistman SN. Ethanol inhibition of recombinant heteromeric NMDA channels in the presence and absence of modulators. J Neurochem. 1995;65:140–148. doi: 10.1046/j.1471-4159.1995.65010140.x. [DOI] [PubMed] [Google Scholar]
- 503.Mirshahi T, Woodward JJ. Ethanol sensitivity of heteromeric NMDA receptors: effects of subunit assembly, glycine and NMDAR1 Mg(2+)-insensitive mutants. Neuropharmacology. 1995;34:347–355. doi: 10.1016/0028-3908(94)00155-l. [DOI] [PubMed] [Google Scholar]
- 504.Popp RL, Lickteig R, Browning MD, Lovinger DM. Ethanol sensitivity and subunit composition of NMDA receptors in cultured striatal neurons. Neuropharmacology. 1998;37:45–56. doi: 10.1016/s0028-3908(97)00186-x. [DOI] [PubMed] [Google Scholar]
- 505.Smothers CT, Clayton R, Blevins T, Woodward JJ. Ethanol sensitivity of recombinant human N-methyl-D-aspartate receptors. Neurochem Int. 2001;38:333–40. doi: 10.1016/s0197-0186(00)00094-2. [DOI] [PubMed] [Google Scholar]
- 506.Smothers CT, Woodward JJ. Effect of the NR3 subunit on ethanol inhibition of recombinant NMDA receptors. Brain Res. 2003;987:117–21. doi: 10.1016/s0006-8993(03)03315-8. [DOI] [PubMed] [Google Scholar]
- 507.Wirkner K, Eberts C, Poelchen W, Allgaier C, Illes P. Mechanism of inhibition by ethanol of NMDA and AMPA receptor channel functions in cultured rat cortical neurons. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:568–76. doi: 10.1007/s002100000262. [DOI] [PubMed] [Google Scholar]
- 508.Suvarna N, Borgland SL, Wang J, Phamluong K, Auberson YP, Bonci A, et al. Ethanol alters trafficking and functional N-methyl-D-aspartate receptor NR2 subunit ratio via H-Ras. J Biol Chem. 2005;280:31450–9. doi: 10.1074/jbc.M504120200. [DOI] [PubMed] [Google Scholar]
- 509.Wang J, Carnicella S, Phamluong K, Jeanblanc J, Ronesi JA, Chaudhri N, et al. Ethanol induces long-term facilitation of NR2B-NMDA receptor activity in the dorsal striatum: implications for alcohol drinking behavior. J Neurosci. 2007;27:3593–3602. doi: 10.1523/JNEUROSCI.4749-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Hoffman PL, Snell LD, Bhave SV, Tabakoff B. Ethanol inhibition of NMDA receptor function in primary cultures of rat cerebellar granule cells and cerebral cortical cells. Alcohol Alcohol Suppl. 1994;2:199–204. [PubMed] [Google Scholar]
- 511.Randoll LA, Wilson WR, Weaver MS, Spuhler-Phillips K, Leslie SW. N-methyl-D-aspartate-stimulated increases in intracellular calcium exhibit brain regional differences in sensitivity to inhibition by ethanol. Alcohol Clin Exp Res. 1996;20:197–200. doi: 10.1111/j.1530-0277.1996.tb01629.x. [DOI] [PubMed] [Google Scholar]
- 512.Nie Z, Madamba SG, Siggins GR. Ethanol inhibits glutamatergic neurotransmission in nucleus accumbens neurons by multiple mechanisms. J Pharmacol Exp Ther. 1994;271:1566–73. [PubMed] [Google Scholar]
- 513.Steffensen SC, Nie Z, Criado JR, Siggins GR. Ethanol inhibition of N-methyl-D-aspartate responses involves presynaptic γ-aminobutyric acidB receptors. J Pharmacol Exp Ther. 2000;294:637–647. [PubMed] [Google Scholar]
- 514.Grover CA, Wallace KA, Lindberg SA, Frye GD. Ethanol inhibition of NMDA currents in acutely dissociated medial septum/diagonal band neurons from ethanol dependent rats. Brain Res. 1998;782:43–52. doi: 10.1016/s0006-8993(97)01001-9. [DOI] [PubMed] [Google Scholar]
- 515.Criswell HE, Ming Z, Griffith BL, Breese GR. Comparison of effect of ethanol on N-methyl-D-aspartate- and GABA-gated currents from acutely dissociated neurons: absence of regional differences in sensitivity to ethanol. J Pharmacol Exp Ther. 2003;304:192–9. doi: 10.1124/jpet.102.041590. [DOI] [PubMed] [Google Scholar]
- 516.Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J Neurosci. 2004;24:1594–1603. doi: 10.1523/JNEUROSCI.5077-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Zhu W, Bie B, Pan ZZ. Involvement of non-NMDA glutamate receptors in central amygdala in synaptic actions of ethanol and ethanol-induced reward behavior. J Neurosci. 2007;27:289–98. doi: 10.1523/JNEUROSCI.3912-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Simson PE, Criswell HE, Breese GR. Inhibition of NMDA-evoked electrophysiological activity by ethanol in selected brain regions: evidence for ethanol-sensitive and ethanol-insensitive NMDA-evoked responses. Brain Res. 1993;607:9–16. doi: 10.1016/0006-8993(93)91483-9. [DOI] [PubMed] [Google Scholar]
- 519.Yang X, Criswell HE, Simson P, Moy S, Breese GR. Evidence for a selective effect of ethanol on N-methyl-d-aspartate responses: ethanol affects a subtype of the ifenprodil-sensitive N-methyl-d-aspartate receptors. J Pharmacol Exp Ther. 1996;278:114–24. [PubMed] [Google Scholar]
- 520.Peoples RW, White G, Lovinger DM, Weight FF. Ethanol inhibition of N-methyl-D-aspartate-activated current in mouse hippocampal neurones: whole-cell patch-clamp analysis. Br J Pharmacol. 1997;122:1035–42. doi: 10.1038/sj.bjp.0701483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Ariwodola OJ, Crowder TL, Grant KA, Daunais JB, Friedman DP, Weiner JL. Ethanol modulation of excitatory and inhibitory synaptic transmission in rat and monkey dentate granule neurons. Alcohol Clin Exp Res. 2003;27:1632–9. doi: 10.1097/01.ALC.0000089956.43262.17. [DOI] [PubMed] [Google Scholar]
- 522.Engberg G, Hajos M. Ethanol attenuates the response of locus coeruleus neurons to excitatory amino acid agonists in vivo. Naunyn Schmiedebergs Arch Pharmacol. 1992;345:222–6. doi: 10.1007/BF00165740. [DOI] [PubMed] [Google Scholar]
- 523.Frohlich R, Patzelt C, Illes P. Inhibition by ethanol of excitatory amino acid receptors and nicotinic acetylcholine receptors at rat locus coeruleus neurons. Naunyn Schmiedebergs Arch Pharmacol. 1994;350:626–31. doi: 10.1007/BF00169367. [DOI] [PubMed] [Google Scholar]
- 524.Nieber K, Poelchen W, Sieler D, Illes P. Inhibition by ethanol of excitatory amino acid receptors in rat locus coeruleus neurons in vitro. Naunyn Schmiedebergs Arch Pharmacol. 1998;357:299–308. doi: 10.1007/pl00005171. [DOI] [PubMed] [Google Scholar]
- 525.Allgaier C, Scheibler P, Muller D, Feuerstein TJ, Illes P. NMDA receptor characterization and subunit expression in rat cultured mesencephalic neurones. Br J Pharmacol. 1999;126:121–30. doi: 10.1038/sj.bjp.0702284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Stobbs SH, Ohran AJ, Lassen MB, Allison DW, Brown JE, Steffensen SC. Ethanol suppression of ventral tegmental area GABA neuron electrical transmission involves N-methyl-D-aspartate receptors. J Pharmacol Exp Ther. 2004;311:282–9. doi: 10.1124/jpet.104.071860. [DOI] [PubMed] [Google Scholar]
- 527.Engblom AC, Courtney MJ, Kukkonen JP, Akerman KE. Ethanol specifically inhibits NMDA receptors with affinity for ifenprodil in the low micromolar range in cultured cerebellar granule cells. J Neurochem. 1997;69:2162–8. doi: 10.1046/j.1471-4159.1997.69052162.x. [DOI] [PubMed] [Google Scholar]
- 528.Blitzer RD, Gil O, Landau EM. Long-term potentiation in rat hippocampus is inhibited by low concentrations of ethanol. Brain Res. 1990;537:203–8. doi: 10.1016/0006-8993(90)90359-j. [DOI] [PubMed] [Google Scholar]
- 529.Morrisett RA, Swartzwelder HS. Attenuation of hippocampal long-term potentiation by ethanol: a patch-clamp analysis of glutamatergic and GABAergic mechanisms. J Neurosci. 1993;13:2264–72. doi: 10.1523/JNEUROSCI.13-05-02264.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Givens B, McMahon K. Ethanol suppresses the induction of long-term potentiation in vivo. Brain Res. 1995;688:27–33. doi: 10.1016/0006-8993(95)00499-g. [DOI] [PubMed] [Google Scholar]
- 531.Pyapali GK, Turner DA, Wilson WA, Swartzwelder HS. Age and dose-dependent effects of ethanol on the induction of hippocampal long-term potentiation. Alcohol. 1999;19:107–11. doi: 10.1016/s0741-8329(99)00021-x. [DOI] [PubMed] [Google Scholar]
- 532.Yin HH, Park BS, Adermark L, Lovinger DM. Ethanol reverses the direction of long-term synaptic plasticity in the dorsomedial striatum. Eur J Neurosci. 2007;25:3226–32. doi: 10.1111/j.1460-9568.2007.05606.x. [DOI] [PubMed] [Google Scholar]
- 533.Weitlauf C, Egli RE, Grueter BA, Winder DG. High-frequency stimulation induces ethanol-sensitive long-term potentiation at glutamatergic synapses in the dorsolateral bed nucleus of the stria terminalis. J Neurosci. 2004;24:5741–7. doi: 10.1523/JNEUROSCI.1181-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Hendricson AW, Miao CL, Lippmann MJ, Morrisett RA. Ifenprodil and ethanol enhance NMDA receptor-dependent long-term depression. J Pharmacol Exp Ther. 2002;301:938–44. doi: 10.1124/jpet.301.3.938. [DOI] [PubMed] [Google Scholar]
- 535.Jin C, Woodward JJ. Effects of 8 different NR1 splice variants on the ethanol inhibition of recombinant NMDA receptors. Alcohol Clin Exp Res. 2006;30:673–9. doi: 10.1111/j.1530-0277.2006.00079.x. [DOI] [PubMed] [Google Scholar]
- 536.Rabe CS, Tabakoff B. Glycine site-directed agonists reverse the actions of ethanol at the N-methyl-D-aspartate receptor. Mol Pharmacol. 1990;38:753–7. [PubMed] [Google Scholar]
- 537.Martin D, Morrisett RA, Bian XP, Wilson WA, Swartzwelder HS. Ethanol inhibition of NMDA mediated depolarizations is increased in the presence of Mg2+ Brain Res. 1991;546:227–34. doi: 10.1016/0006-8993(91)91486-k. [DOI] [PubMed] [Google Scholar]
- 538.Dildy-Mayfield JE, Leslie SW. Mechanism of inhibition of N-methyl-D-aspartate-stimulated increases in free intracellular Ca2+ concentration by ethanol. J Neurochem. 1991;56:1536–43. doi: 10.1111/j.1471-4159.1991.tb02048.x. [DOI] [PubMed] [Google Scholar]
- 539.Morrisett RA, Martin D, Oetting TA, Lewis DV, Wilson WA, Swartzwelder HS. Ethanol and magnesium ions inhibit N-methyl-D-aspartate-mediated synaptic potentials in an interactive manner. Neuropharmacology. 1991;30:1173–8. doi: 10.1016/0028-3908(91)90162-5. [DOI] [PubMed] [Google Scholar]
- 540.Popp RL, Lickteig RL, Lovinger DM. Factors that enhance ethanol inhibition of N-methyl-D-aspartate receptors in cerebellar granule cells. J Pharmacol Exp Ther. 1999;289:1564–1574. [PubMed] [Google Scholar]
- 541.Mirshahi T, Anders DL, Ronald KM, Woodward JJ. Intracellular calcium enhances the ethanol sensitivity of NMDA receptors through an interaction with the C0 domain of the NR1 subunit. J Neurochem. 1998;71:1095–107. doi: 10.1046/j.1471-4159.1998.71031095.x. [DOI] [PubMed] [Google Scholar]
- 542.Miyakawa T, Yagi T, Kitazawa H, Yasuda M, Kawai N, Tsuboi K, et al. Fyn-kinase as a determinant of ethanol sensitivity: relation to NMDA-receptor function. Science. 1997;278:698–701. doi: 10.1126/science.278.5338.698. [DOI] [PubMed] [Google Scholar]
- 543.Anders DL, Blevins T, Sutton G, Swope S, Chandler LJ, Woodward JJ. Fyn tyrosine kinase reduces the ethanol inhibition of recombinant NR1/NR2A but not NR1/NR2B NMDA receptors expressed in HEK 293 cells. J Neurochem. 1999;72:1389–1393. doi: 10.1046/j.1471-4159.1999.721389.x. [DOI] [PubMed] [Google Scholar]
- 544.Yaka R, Phamluong K, Ron D. Scaffolding of Fyn kinase to the NMDA receptor determines brain region sensitivity to ethanol. J Neurosci. 2003;23:3623–32. doi: 10.1523/JNEUROSCI.23-09-03623.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Ferrani-Kile K, Randall PK, Leslie SW. Acute ethanol affects phosphorylation state of the NMDA receptor complex: implication of tyrosine phosphatases and protein kinase A. Mol Brain Res. 2003;115:78–86. doi: 10.1016/s0169-328x(03)00186-4. [DOI] [PubMed] [Google Scholar]
- 546.Xu M, Woodward JJ. Ethanol inhibition of NMDA receptors under conditions of altered protein kinase A activity. J Neurochem. 2006;96:1760–7. doi: 10.1111/j.1471-4159.2006.03703.x. [DOI] [PubMed] [Google Scholar]
- 547.Snell LD, Tabakoff B, Hoffman PL. Involvement of protein kinase C in ethanol-induced inhibition of NMDA receptor function in cerebellar granule cells. Alcohol Clin Exp Res. 1994;18:81–85. doi: 10.1111/j.1530-0277.1994.tb00884.x. [DOI] [PubMed] [Google Scholar]
- 548.Maldve RE, Zhang TA, Ferrani-Kile K, Schreiber SS, Lippmann MJ, Snyder GL, et al. DARPP-32 and regulation of the ethanol sensitivity of NMDA receptors in the nucleus accumbens. Nat Neurosci. 2002;5:641–648. doi: 10.1038/nn877. [DOI] [PubMed] [Google Scholar]
- 549.Wilson WR, Bosy TZ, Ruth JA. NMDA agonists and antagonists alter the hypnotic response to ethanol in LS and SS mice. Alcohol. 1990;7:389–95. doi: 10.1016/0741-8329(90)90021-4. [DOI] [PubMed] [Google Scholar]
- 550.Danysz W, Dyr W, Jankowska E, Glazewski S, Kostowski W. The involvement of NMDA receptors in acute and chronic effects of ethanol. Alcohol Clin Exp Res. 1992;16:499–504. doi: 10.1111/j.1530-0277.1992.tb01407.x. [DOI] [PubMed] [Google Scholar]
- 551.Beleslin DB, Djokanovic, Jovanovic Micic D, Samardzic R. Opposite effects of GABAA and NMDA receptor antagonists on ethanol-induced behavioral sleep in rats. Alcohol. 1997;14:167–73. doi: 10.1016/s0741-8329(96)00140-1. [DOI] [PubMed] [Google Scholar]
- 552.Silveri MM, Spear LP. The effects of NMDA and GABAA pharmacological manipulations on ethanol sensitivity in immature and mature animals. Alcohol Clin Exp Res. 2002;26:449–56. [PubMed] [Google Scholar]
- 553.Robledo P, Kaneko W, Ehlers CL. Combined effects of ethanol and MK 801 on locomotor activity in the rat. Pharmacol Biochem Behav. 1991;39:513–516. doi: 10.1016/0091-3057(91)90218-q. [DOI] [PubMed] [Google Scholar]
- 554.Bienkowski P, Koros E, Piasecki J, Kostowski W. Interactions of ethanol with nicotine, dizocilpine, CGP 40116, and 1-(m-chlorophenyl)-biguanide in rats. Pharmacol Biochem Behav. 1997;58:1159–65. doi: 10.1016/s0091-3057(97)00317-1. [DOI] [PubMed] [Google Scholar]
- 555.Shen EH, Phillips TJ. MK-801 potentiates ethanol’s effects on locomotor activity in mice. Pharmacol Biochem Behav. 1998;59:135–43. doi: 10.1016/s0091-3057(97)00389-4. [DOI] [PubMed] [Google Scholar]
- 556.Grant KA, Kinsley JS, Tabakoff B, Barrett JE, Balster RL. Ethanol-like discriminative stimulus effects of noncompetitive N-methyl-D-aspartate antagonists. Behav Pharmacol. 1991;2:87–95. [PubMed] [Google Scholar]
- 557.Colombo G, Grant KA. NMDA receptor complex antagonists have ethanol-like discriminative stimulus effects. Ann N Y Acad Sci. 1992;654:421–3. doi: 10.1111/j.1749-6632.1992.tb25986.x. [DOI] [PubMed] [Google Scholar]
- 558.Balster RL, Grech DM, Bobelis DJ. Drug discrimination analysis of ethanol as an N-methyl-D-aspartate receptor antagonist. Eur J Pharmacol. 1992;222:39–42. doi: 10.1016/0014-2999(92)90460-l. [DOI] [PubMed] [Google Scholar]
- 559.Grant K, Colombo G. Discriminative stimulus effects of ethanol: effect of training dose on the substitution on N-methyl-D-aspartate antagonists. J Pharmacol Exp Ther. 1993;264:1241–1247. [PubMed] [Google Scholar]
- 560.Schechter MD, Meehan SM, Gordon TL, McBurney DM. The NMDA receptor antagonist MK-801 produces ethanol-like discrimination in the rat. Alcohol. 1993;10:197–201. doi: 10.1016/0741-8329(93)90035-m. [DOI] [PubMed] [Google Scholar]
- 561.Sanger DJ. Substitution by NMDA antagonists and other drugs in rats trained to discriminate ethanol. Behav Pharmacol. 1993;4:523–528. [PubMed] [Google Scholar]
- 562.Shelton KL, Balster RL. Ethanol drug discrimination in rats: substitution with GABA agonists and NMDA antagonists. Behav Pharmacol. 1994;5:441–451. [PubMed] [Google Scholar]
- 563.Bienkowski P, Stefanski R, Kostowski W. Competitive NMDA receptor antagonist, CGP 40116, substitutes for the discriminative stimulus effects of ethanol. Eur J Pharmacol. 1996;314:277–80. doi: 10.1016/s0014-2999(96)00658-9. [DOI] [PubMed] [Google Scholar]
- 564.Kotlinska J, Liljequist S. The NMDA/glycine receptor antagonist, L-701,324, produces discriminative stimuli similar to those of ethanol. Eur J Pharmacol. 1997;332:1–8. doi: 10.1016/s0014-2999(97)01069-8. [DOI] [PubMed] [Google Scholar]
- 565.Hundt W, Danysz W, Holter SM, Spanagel R. Ethanol and N-methyl-D-aspartate receptor complex interactions: A detailed drug discrimination study in the rat. Psychopharmacology. 1998;135:44–51. doi: 10.1007/s002130050484. [DOI] [PubMed] [Google Scholar]
- 566.Krystal JH, Petrakis IL, Webb E, Cooney NL, Karper LP, Namanworth S, et al. Dose-related ethanol-like effects of the NMDA antagonist, ketamine, in recently detoxified alcoholics. Arch Gen Psychiatry. 1998;55:354–60. doi: 10.1001/archpsyc.55.4.354. [DOI] [PubMed] [Google Scholar]
- 567.Bienkowski P, Kostowski W. Discrimination of ethanol in rats: effects of nicotine, diazepam, CGP 40116, and 1-(m-chlorophenyl)-biguanide. Pharmacol Biochem Behav. 1998;60:61–9. doi: 10.1016/s0091-3057(97)00469-3. [DOI] [PubMed] [Google Scholar]
- 568.Hodge CW, Cox AA. The discriminative stimulus effects of ethanol are mediated by NMDA and GABAA receptors in specific limbic brain regions. Psychopharmacology. 1998;139:95–107. doi: 10.1007/s002130050694. [DOI] [PubMed] [Google Scholar]
- 569.Hölter SM, Danysz W, Spanagel R. Novel uncompetitive N-methyl-D-aspartate (NMDA)-receptor antagonist MRZ 2/579 suppresses ethanol intake in long-term ethanol-experienced rats and generalizes to ethanol cue in drug discrimination procedure. J Pharmacol Exp Ther. 2000;292:545–552. [PubMed] [Google Scholar]
- 570.Soyka M, Bondy B, Eisenburg B, Schutz CG. NMDA receptor challenge with dextromethorphan -subjective response, neuroendocrinological findings and possible clinical implications. J Neural Transm. 2000;107:701–14. doi: 10.1007/s007020070071. [DOI] [PubMed] [Google Scholar]
- 571.Stolerman IP, Olufsen K. Generalisation of ethanol with drug mixtures containing a positive modulator of the GABAA receptor and an NMDA antagonist. Neuropharmacology. 2001;40:123–30. doi: 10.1016/s0028-3908(00)00100-3. [DOI] [PubMed] [Google Scholar]
- 572.Hodge CW, Cox AA, Bratt AM, Camarini R, Iller K, Kelley SP, et al. The discriminative stimulus properties of self-administered ethanol are mediated by GABAA and NMDA receptors in rats. Psychopharmacology. 2001;154:13–22. doi: 10.1007/s002130000619. [DOI] [PubMed] [Google Scholar]
- 573.Shelton KL, Grant KA. Discriminative stimulus effects of ethanol in C57BL/6J and DBA/2J inbred mice. Alcohol Clin Exp Res. 2002;26:747–57. [PubMed] [Google Scholar]
- 574.Vivian JA, Waters CA, Szeliga KT, Jordan K, Grant KA. Characterization of the discriminative stimulus effects of N-methyl-D-aspartate ligands under different ethanol training conditions in the cynomolgus monkey (Macaca fascicularis) Psychopharmacology. 2002;162:273–81. doi: 10.1007/s00213-002-1086-2. [DOI] [PubMed] [Google Scholar]
- 575.Shelton KL. Substitution profiles of N-methyl-D-aspartate antagonists in ethanol-discriminating inbred mice. Alcohol. 2004;34:165–75. doi: 10.1016/j.alcohol.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 576.Trevisan L, Fitzgerald LW, Brose N, Gasic GP, Heinemann SF, Duman RS, et al. Chronic ingestion of ethanol up-regulates NMDAR1 receptor subunit immunoreactivity in rat hippocampus. J Neurochem. 1994;62:1635–8. doi: 10.1046/j.1471-4159.1994.62041635.x. [DOI] [PubMed] [Google Scholar]
- 577.Follesa P, Ticku MK. Chronic ethanol treatment differentially regulates NMDA receptor subunit mRNA expression in rat brain. Mol Brain Res. 1995;29:99–106. doi: 10.1016/0169-328x(94)00235-7. [DOI] [PubMed] [Google Scholar]
- 578.Hu XJ, Follesa P, Ticku MK. Chronic ethanol treatment produces a selective upregulation of the NMDA receptor subunit gene expression in mammalian cultured cortical neurons. Mol Brain Res. 1996;36:211–218. doi: 10.1016/0169-328x(95)00223-f. [DOI] [PubMed] [Google Scholar]
- 579.Morrow AL, Devaud LL, Bucci D, Smith FD. GABAA and NMDA receptor subunit mRNA expression in ethanol dependent rats. Alcohol Alcohol. 1994;2:89–95. [PubMed] [Google Scholar]
- 580.Follesa P, Ticku MK. Chronic ethanol-mediated up-regulation of the N-methyl-D-aspartate receptor polypeptide subunits in mouse cortical neurons in culture. J Biol Chem. 1996;271:13297–13299. doi: 10.1074/jbc.271.23.13297. [DOI] [PubMed] [Google Scholar]
- 581.Snell LD, Nunley KR, Lickteig RL, Browning MD, Tabakoff B, Hoffman PL. Regional and subunit specific changes in NMDA receptor mRNA and immunoreactivity in mouse brain following chronic ethanol ingestion. Mol Brain Res. 1996;40:71–78. doi: 10.1016/0169-328x(96)00038-1. [DOI] [PubMed] [Google Scholar]
- 582.Chen X, Michaelis ML, Michaelis EK. Effects of chronic ethanol treatment on the expression of calcium transport carriers and NMDA/glutamate receptor proteins in brain synaptic membranes. J Neurochem. 1997;69:1559–1569. doi: 10.1046/j.1471-4159.1997.69041559.x. [DOI] [PubMed] [Google Scholar]
- 583.Kumari M, Ticku MK. Ethanol and regulation of the NMDA receptor subunits in fetal cortical neurons. J Neurochem. 1998;70:1467–1473. doi: 10.1046/j.1471-4159.1998.70041467.x. [DOI] [PubMed] [Google Scholar]
- 584.Kalluri HS, Mehta AK, Ticku MK. Up-regulation of NMDA receptor subunits in rat brain following chronic ethanol treatment. Mol Brain Res. 1998;58:221–4. doi: 10.1016/s0169-328x(98)00112-0. [DOI] [PubMed] [Google Scholar]
- 585.Chandler LJ, Norwood D, Sutton G. Chronic ethanol upregulates NMDA and AMPA, but not kainate receptor subunit proteins in rat primary cortical cultures. Alcohol Clin Exp Res. 1999;23:363–70. [PubMed] [Google Scholar]
- 586.Hardy PA, Chen W, Wilce PA. Chronic ethanol exposure and withdrawal influence NMDA receptor subunit and splice variant mRNA expression in the rat cerebral cortex. Brain Res. 1999;819:33–39. doi: 10.1016/s0006-8993(98)01340-7. [DOI] [PubMed] [Google Scholar]
- 587.Chen X, Moore-Nichols D, Nguyen H, Michaelis EK. Calcium influx through NMDA receptors, chronic receptor inhibition by ethanol and 2-amino-5-phosponopentanoic acid, and receptor protein expression. J Neurochem. 1999;72:1969–80. doi: 10.1046/j.1471-4159.1999.0721969.x. [DOI] [PubMed] [Google Scholar]
- 588.Devaud LL, Morrow AL. Gender-selective effects of ethanol dependence on NMDA receptor subunit expression in cerebral cortex, hippocampus and hypothalamus. Eur J Pharmacol. 1999;369:331–4. doi: 10.1016/s0014-2999(99)00103-x. [DOI] [PubMed] [Google Scholar]
- 589.Narita M, Soma M, Narita M, Mizoguchi H, Tseng LF, Suzuki T. Implications of the NR2B subunit-containing NMDA receptor localized in mouse limbic forebrain in ethanol dependence. Eur J Pharmacol. 2000;401:191–195. doi: 10.1016/s0014-2999(00)00428-3. [DOI] [PubMed] [Google Scholar]
- 590.Kumari M. Differential effects of chronic ethanol treatment on N-methyl-D-aspartate R1 splice variants in fetal cortical neurons. J Biol Chem. 2001;276:29764–71. doi: 10.1074/jbc.M100317200. [DOI] [PubMed] [Google Scholar]
- 591.Bao X, Hui D, Naassila M, Michaelis EK. Chronic ethanol exposure increases gene transcription of subunits of an N-methyl-D-aspartate receptor-like complex in cortical neurons in culture. Neurosci Lett. 2001;315:5–8. doi: 10.1016/s0304-3940(01)02317-5. [DOI] [PubMed] [Google Scholar]
- 592.Nagy J, Kolok S, Dezso P, Boros A, Szombathelyi Z. Differential alterations in the expression of NMDA receptor subunits following chronic ethanol treatment in primary cultures of rat cortical and hippocampal neurones. Neurochem Int. 2003;42:35–43. doi: 10.1016/s0197-0186(02)00062-1. [DOI] [PubMed] [Google Scholar]
- 593.Henniger MS, Wotjak CT, Holter SM. Long-term voluntary ethanol drinking increases expression of NMDA receptor 2B subunits in rat frontal cortex. Eur J Pharmacol. 2003;470:33–6. doi: 10.1016/s0014-2999(03)01787-4. [DOI] [PubMed] [Google Scholar]
- 594.Devaud LL, Alele P. Differential effects of chronic ethanol administration and withdrawal on gamma-aminobutyric acid type A and NMDA receptor subunit proteins in male and female rat brain. Alcohol Clin Exp Res. 2004;28:957–65. doi: 10.1097/01.alc.0000128225.83916.40. [DOI] [PubMed] [Google Scholar]
- 595.Nelson TE, Ur CL, Gruol DL. Chronic intermittent ethanol exposure enhances NMDA-receptor-mediated synaptic responses and NMDA receptor expression in hippocampal CA1 region. Brain Res. 2005;1048:69–79. doi: 10.1016/j.brainres.2005.04.041. [DOI] [PubMed] [Google Scholar]
- 596.Sheela Rani CS, Ticku MK. Comparison of chronic ethanol and chronic intermittent ethanol treatments on the expression of GABA(A) and NMDA receptor subunits. Alcohol. 2006;38:89–97. doi: 10.1016/j.alcohol.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 597.Sircar R, Sircar D. Repeated ethanol treatment in adolescent rats alters cortical NMDA receptor. Alcohol. 2006;39:51–8. doi: 10.1016/j.alcohol.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 598.Marutha Ravindran CR, Ticku MK. Changes in methylation pattern of NMDA receptor NR2B gene in cortical neurons after chronic ethanol treatment in mice. Mol Brain Res. 2004;121:19–27. doi: 10.1016/j.molbrainres.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 599.Marutha Ravindran CR, Ticku MK. Role of CpG islands in the up-regulation of NMDA receptor NR2B gene expression following chronic ethanol treatment of cultured cortical neurons of mice. Neurochem Int. 2005;46:313–27. doi: 10.1016/j.neuint.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 600.Ortiz J, Fitzgerald LW, Charlton M, Lane S, Trevisan L, Guitart X, et al. Biochemical actions of chronic ethanol exposure in the mesolimbic dopamine system. Synapse. 1995;21:289–298. doi: 10.1002/syn.890210403. [DOI] [PubMed] [Google Scholar]
- 601.Floyd DW, Jung KY, McCool BA. Chronic ethanol ingestion facilitates N-methyl-D-aspartate receptor function and expression in rat lateral/basolateral amygdala neurons. J Pharmacol Exp Ther. 2003;307:1020–9. doi: 10.1124/jpet.103.057505. [DOI] [PubMed] [Google Scholar]
- 602.Grant KA, Valverius P, Hudspith M, Tabakoff B. Ethanol withdrawal seizures and the NMDA receptor complex. Eur J Pharmacol. 1990;176:289–96. doi: 10.1016/0014-2999(90)90022-x. [DOI] [PubMed] [Google Scholar]
- 603.Iorio KR, Reinlib L, Tabakoff B, Hoffman PL. Chronic exposure of cerebellar granule cells to ethanol results in increased N-methyl-D-aspartate receptor function. Mol Pharmacol. 1992;41:1142–8. [PubMed] [Google Scholar]
- 604.Sanna E, Serra M, Cossu A, Colombo G, Follesa P, Cuccheddu T, et al. Chronic ethanol intoxication induces differential effects on GABAA and NMDA receptor function in the rat brain. Alcohol Clin Exp Res. 1993;17:115–123. doi: 10.1111/j.1530-0277.1993.tb00735.x. [DOI] [PubMed] [Google Scholar]
- 605.Ahern KB, Lustig HS, Greenberg DA. Enhancement of NMDA toxicity and calcium responses by chronic exposure of cultured cortical neurons to ethanol. Neurosci Lett. 1994;165:211–4. doi: 10.1016/0304-3940(94)90747-1. [DOI] [PubMed] [Google Scholar]
- 606.Molleman A, Little HJ. Increases in non-N-methyl-D-aspartate glutamatergic transmission, but no change in gamma-aminobutyric acidB transmission, in CA1 neurons during withdrawal from in vivo chronic ethanol treatment. J Pharmacol Exp Ther. 1995;274:1035–41. [PubMed] [Google Scholar]
- 607.Blevins T, Mirshahi T, Woodward JJ. Increased agonist and antagonist sensitivity of N-methyl-D-aspartate stimulated calcium flux in cultured neurons following chronic ethanol exposure. Neurosci Lett. 1995;200:214–8. doi: 10.1016/0304-3940(95)12086-j. [DOI] [PubMed] [Google Scholar]
- 608.Ibbotson T, Field MJ, Boden PR. Effect of chronic ethanol treatment in vivo on excitability in mouse cortical neurones in vitro. Br J Pharmacol. 1997;122:956–62. doi: 10.1038/sj.bjp.0701471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609.Carpenter-Hyland EP, Woodward JJ, Chandler LJ. Chronic ethanol induces synaptic but not extrasynaptic targeting of NMDA receptors. J Neurosci. 2004;24:7859–68. doi: 10.1523/JNEUROSCI.1902-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610.Tsai G, Coyle JT. The role of glutamatergic neurotransmission in the pathophysiology of alcoholism. Annu Rev Med. 1998;49:173–184. doi: 10.1146/annurev.med.49.1.173. [DOI] [PubMed] [Google Scholar]
- 611.Tsai G, Gastfriend DR, Coyle JT. The glutamatergic basis of human alcoholism. Am J Psychiatry. 1995;152:332–340. doi: 10.1176/ajp.152.3.332. [DOI] [PubMed] [Google Scholar]
- 612.Grant KA, Snell LD, Rogawski MA, Thurkauf A, Tabakoff B. Comparison of the effects of the uncompetitive N-methyl-D-aspartate antagonist (+-)-5-aminocarbonyl-10,11-dihydro-5H-dibenzo[a,d] cyclohepten-5,10-imine (ADCI) with its structural analogs dizocilpine (MK-801) and carbamazepine on ethanol withdrawal seizures. J Pharmacol Exp Ther. 1992;260:1017–22. [PubMed] [Google Scholar]
- 613.Morrisett RA, Rezvani AH, Overstreet D, Janowsky DS, Wilson WA, Swartzwelder HS. MK-801 potently inhibits alcohol withdrawal seizures in rats. Eur J Pharmacol. 1990;176:103–5. doi: 10.1016/0014-2999(90)90138-v. [DOI] [PubMed] [Google Scholar]
- 614.Liljequist S. The competitive NMDA receptor antagonist, CGP 39551, inhibits ethanol withdrawal seizures. Eur J Pharmacol. 1991;192:197–8. doi: 10.1016/0014-2999(91)90092-5. [DOI] [PubMed] [Google Scholar]
- 615.Erden BF, Ozdemirci S, Yildiran G, Utkan T, Gacar N, Ulak G. Dextromethorphan attenuates ethanol withdrawal syndrome in rats. Pharmacol Biochem Behav. 1999;62:537–41. doi: 10.1016/s0091-3057(98)00175-0. [DOI] [PubMed] [Google Scholar]
- 616.Malinowska B, Napiórkowska-Pawlak D, Pawlak R, Buczko W, Göthert M. Ifenprodil influences changes in mouse behaviour related to acute and chronic ethanol administration. Eur J Pharmacol. 1999;377:13–19. doi: 10.1016/s0014-2999(99)00393-3. [DOI] [PubMed] [Google Scholar]
- 617.Bienkowski P, Krzascik P, Koros E, Kostowski W, Scinska A, Danysz W. Effects of a novel uncompetitive NMDA receptor antagonist, MRZ 2/579 on ethanol self-administration and ethanol withdrawal seizures in the rat. Eur J Pharmacol. 2001;413:81–89. doi: 10.1016/s0014-2999(01)00743-9. [DOI] [PubMed] [Google Scholar]
- 618.Kotlinska J. NMDA antagonists inhibit the development of ethanol dependence in rats. Pol J Pharmacol. 2001;53:47–50. [PubMed] [Google Scholar]
- 619.Veatch LM, Becker HC. Lorazepam and MK-801 effects on behavioral and electrographic indices of alcohol withdrawal sensitization. Brain Res. 2005;1065:92–106. doi: 10.1016/j.brainres.2005.10.047. [DOI] [PubMed] [Google Scholar]
- 620.Roberto M, Bajo M, Crawford E, Madamba SG, Siggins GR. Chronic ethanol exposure and protracted abstinence alter NMDA receptors in central amygdala. Neuropsychopharmacology. 2006;31:988–96. doi: 10.1038/sj.npp.1300840. [DOI] [PubMed] [Google Scholar]
- 621.Dildy-Mayfield JE, Harris RA. Comparison of ethanol sensitivity of rat brain kainate, DL-alpha-amino-3-hydroxy-5-methyl-4-isoxalone proprionic acid and N-methyl-D-aspartate receptors expressed in Xenopus oocytes. J Pharmacol Exp Ther. 1992;262:487–94. [PubMed] [Google Scholar]
- 622.Lovinger DM. High ethanol sensitivity of recombinant AMPA-type glutamate receptors expressed in mammalian cells. Neurosci Lett. 1993;159:83–7. doi: 10.1016/0304-3940(93)90804-t. [DOI] [PubMed] [Google Scholar]
- 623.Dildy-Mayfield JE, Harris RA. Ethanol inhibits kainate responses of glutamate receptors expressed in xenopus oocytes: role of calcium and protein kinase C. J Neurosci. 1995;15:3162–3171. doi: 10.1523/JNEUROSCI.15-04-03162.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Martin D, Tayyeb MI, Swartzwelder HS. Ethanol inhibition of AMPA and kainate receptor-mediated depolarizations of hippocampal area CA1. Alcohol Clin Exp Res. 1995;19:1312–6. doi: 10.1111/j.1530-0277.1995.tb01617.x. [DOI] [PubMed] [Google Scholar]
- 625.Valenzuela CF, Bhave S, Hoffman P, Harris RA. Acute effects of ethanol on pharmacologically isolated kainate receptors in cerebellar granule neurons: comparison with NMDA and AMPA receptors. J Neurochem. 1998;71:1777–80. doi: 10.1046/j.1471-4159.1998.71041777.x. [DOI] [PubMed] [Google Scholar]
- 626.Valenzuela CF, Cardoso RA. Acute effects of ethanol on kainate receptors with different subunit compositions. J Pharmacol Exp Ther. 1999;288:1199–1206. [PubMed] [Google Scholar]
- 627.Lu SM, Yeh HH. Ethanol modulates AMPA-induced current responses of primary somatosensory cortical neurons. Neurochem Int. 1999;35:175–83. doi: 10.1016/s0197-0186(99)00059-5. [DOI] [PubMed] [Google Scholar]
- 628.Weiner JL, Dunwiddie TV, Valenzuela CF. Ethanol inhibition of synaptically evoked kainate responses in rat hippocampal CA3 pyramidal neurons. Mol Pharmacol. 1999;56:85–90. doi: 10.1124/mol.56.1.85. [DOI] [PubMed] [Google Scholar]
- 629.Wang MY, Rampil IJ, Kendig JJ. Ethanol directly depresses AMPA and NMDA glutamate currents in spinal cord motor neurons independent of actions on GABAA or glycine receptors. J Pharmacol Exp Ther. 1999;290:362–7. [PubMed] [Google Scholar]
- 630.Costa ET, Soto EE, Cardoso RA, Olivera DS, Valenzuela CF. Acute effects of ethanol on kainate receptors in cultured hippocampal neurons. Alcohol Clin Exp Res. 2000;24:220–5. [PubMed] [Google Scholar]
- 631.Frye GD, Fincher A. Sustained ethanol inhibition of native AMPA receptors on medial septum/diagonal band (MS/DB) neurons. Br J Pharmacol. 2000;129:87–94. doi: 10.1038/sj.bjp.0703039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Fink K, Meder W, Dooley DJ, Gothert M. Inhibition of neuronal Ca2+ influx by gabapentin and subsequent reduction of neurotransmitter release from rat neocortical slices. Br J Pharmacol. 2000;130:900–6. doi: 10.1038/sj.bjp.0703380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Akinshola BE. Straight-chain alcohols exhibit a cutoff in potency for the inhibition of recombinant glutamate receptor subunits. Br J Pharmacol. 2001;133:651–8. doi: 10.1038/sj.bjp.0704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634.Crowder TL, Ariwodola OJ, Weiner JL. Ethanol antagonizes kainate receptor-mediated inhibition of evoked GABAA inhibitory postsynaptic currents in the rat hippocampal CA1 region. J Pharmacol Exp Ther. 2002;303:937–44. doi: 10.1124/jpet.102.038471. [DOI] [PubMed] [Google Scholar]
- 635.Hsiao SH, Frye GD. AMPA receptors on developing medial septum/diagonal band neurons are sensitive to early postnatal binge-like ethanol exposure. Dev Brain Res. 2003;142:89–99. doi: 10.1016/s0165-3806(03)00034-8. [DOI] [PubMed] [Google Scholar]
- 636.Fischer W, Franke H, Illes P. Effects of acute ethanol on the Ca2+ response to AMPA in cultured rat cortical GABAergic nonpyramidal neurons. Alcohol Alcohol. 2003;38:394–9. doi: 10.1093/alcalc/agg108. [DOI] [PubMed] [Google Scholar]
- 637.Carta M, Ariwodola OJ, Weiner JL, Valenzuela CF. Alcohol potently inhibits the kainate receptor-dependent excitatory drive of hippocampal interneurons. Proc Natl Acad Sci U S A. 2003;100:6813–8. doi: 10.1073/pnas.1137276100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638.Moykkynen T, Korpi ER, Lovinger DM. Ethanol inhibits alpha-amino-3-hydyroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor function in central nervous system neurons by stabilizing desensitization. J Pharmacol Exp Ther. 2003;306:546–55. doi: 10.1124/jpet.103.050666. [DOI] [PubMed] [Google Scholar]
- 639.Akinshola BE, Yasuda RP, Peoples RW, Taylor RE. Ethanol sensitivity of recombinant homomeric and heteromeric AMPA receptor subunits expressed in Xenopus oocytes. Alcohol Clin Exp Res. 2003;27:1876–83. doi: 10.1097/01.ALC.0000098874.65490.52. [DOI] [PubMed] [Google Scholar]
- 640.Bruckner MK, Rossner S, Arendt T. Differential changes in the expression of AMPA receptors genes in rat brain after chronic exposure to ethanol: an in situ hybridization study. J Hirnforsch. 1997;38:369–76. [PubMed] [Google Scholar]
- 641.Netzeband JG, Trotter C, Caguioa JN, Gruol DL. Chronic ethanol exposure enhances AMPA-elicited Ca2+ signals in the somatic and dendritic regions of cerebellar Purkinje neurons. Neurochem Int. 1999;35:163–74. doi: 10.1016/s0197-0186(99)00058-3. [DOI] [PubMed] [Google Scholar]
- 642.Netzeband JG, Trotter C, Parsons KL, Gruol DL. Chronic ethanol treatment alters AMPA-induced calcium signals in developing Purkinje neurons. Brain Res. 1999;826:270–80. doi: 10.1016/s0006-8993(99)01309-8. [DOI] [PubMed] [Google Scholar]
- 643.Minami K, Gereau RWt, Minami M, Heinemann SF, Harris RA. Effects of ethanol and anesthetics on type 1 and 5 metabotropic glutamate receptors expressed in Xenopus laevis oocytes. Mol Pharmacol. 1998;53:148–56. doi: 10.1124/mol.53.1.148. [DOI] [PubMed] [Google Scholar]
- 644.Smith TL. Selective effects of ethanol exposure on metabotropic glutamate receptor and guanine nucleotide stimulated phospholipase C activity in primary cultures of astrocytes. Alcohol. 1994;11:405–9. doi: 10.1016/0741-8329(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 645.Simonyi A, Christian MR, Sun AY, Sun GY. Chronic ethanol-induced subtype- and subregion-specific decrease in the mRNA expression of metabotropic glutamate receptors in rat hippocampus. Alcohol Clin Exp Res. 2004;28:1419–23. doi: 10.1097/01.alc.0000139825.35438.a4. [DOI] [PubMed] [Google Scholar]
- 646.Shelton KL, Balster RL. Effects of gamma-aminobutyric acid agonists and N-methyl-D-aspartate antagonists on a multiple schedule of ethanol and saccharin self-administration in rats. J Pharmacol Exp Ther. 1997;280:1250–1260. [PubMed] [Google Scholar]
- 647.Piasecki J, Koros E, Dyr W, Kostowski W, Danysz W, Bienkowski P. Ethanol-reinforced behaviour in the rat: effects of uncompetitive NMDA receptor antagonist, memantine. Eur J Pharmacol. 1998;354:135–43. doi: 10.1016/s0014-2999(98)00442-7. [DOI] [PubMed] [Google Scholar]
- 648.Bienkowski P, Koros E, Kostowski W, Danysz W. Effects of N-methyl-D-aspartate receptor antagonists on reinforced and nonreinforced responding for ethanol in rats. Alcohol. 1999;18:131–137. doi: 10.1016/s0741-8329(98)00075-5. [DOI] [PubMed] [Google Scholar]
- 649.McMillen BA, Joyner PW, Parmar CA, Tyer WE, Williams HL. Effects of NMDA glutamate receptor antagonist drugs on the volitional consumption of ethanol by a genetic drinking rat. Brain Res Bull. 2004;64:279–84. doi: 10.1016/j.brainresbull.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 650.Kotlinska J, Biala G, Rafalski P, Bochenski M, Danysz W. Effect of neramexane on ethanol dependence and reinforcement. Eur J Pharmacol. 2004;503:95–8. doi: 10.1016/j.ejphar.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 651.Escher T, Call SB, Blaha CD, Mittleman G. Behavioral effects of aminoadamantane class NMDA receptor antagonists on schedule-induced alcohol and self-administration of water in mice. Psychopharmacology. 2006;187:424–34. doi: 10.1007/s00213-006-0465-5. [DOI] [PubMed] [Google Scholar]
- 652.Lin N, Hubbard JI. An NMDA receptor antagonist reduces ethanol preference in untrained but not trained rats. Brain Res Bull. 1995;36:421–4. doi: 10.1016/0361-9230(94)00215-m. [DOI] [PubMed] [Google Scholar]
- 653.Rassnick S, Pulvirenti L, Koob GF. Oral ethanol self-administration in rats is reduced by the administration of dopamine and glutamate receptor antagonists into the nucleus accumbens. Psychopharmacology. 1992;109:92–98. doi: 10.1007/BF02245485. [DOI] [PubMed] [Google Scholar]
- 654.Vengeliene V, Bachteler D, Danysz W, Spanagel R. The role of the NMDA receptor in alcohol relapse: a pharmacological mapping study using the alcohol deprivation effect. Neuropharmacology. 2005;48:822–9. doi: 10.1016/j.neuropharm.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 655.Backstrom P, Hyytia P. Ionotropic glutamate receptor antagonists modulate cue-induced reinstatement of ethanol-seeking behavior. Alcohol Clin Exp Res. 2004;28:558–65. doi: 10.1097/01.alc.0000122101.13164.21. [DOI] [PubMed] [Google Scholar]
- 656.Bachteler D, Economidou D, Danysz W, Ciccocioppo R, Spanagel R. The effects of acamprosate and neramexane on cue-induced reinstatement of ethanol-seeking behavior in rat. Neuropsychopharmacology. 2005;30:1104–1110. doi: 10.1038/sj.npp.1300657. [DOI] [PubMed] [Google Scholar]
- 657.Biala G, Kotlinska J. Blockade of the acquisition of ethanol-induced conditioned place preference by N-methyl-D-aspartate receptor antagonists. Alcohol Alcohol. 1999;34:175–182. doi: 10.1093/alcalc/34.2.175. [DOI] [PubMed] [Google Scholar]
- 658.Boyce-Rustay JM, Cunningham CL. The role of NMDA receptor binding sites in ethanol place conditioning. Behav Neurosci. 2004;118:822–34. doi: 10.1037/0735-7044.118.4.822. [DOI] [PubMed] [Google Scholar]
- 659.Broadbent J, Weitemier AZ. Dizocilpine (MK-801) prevents the development of sensitization to ethanol in DBA/2J mice. Alcohol Alcohol. 1999;34:283–8. doi: 10.1093/alcalc/34.3.283. [DOI] [PubMed] [Google Scholar]
- 660.Camarini R, Frussa-Filho R, Montiero MG, Calil HM. MK-801 blocks the development of behavioral sensitization to ethanol. Alcohol Clin Exp Res. 2000;24:285–290. [PubMed] [Google Scholar]
- 661.Broadbent J, Kampmueller KM, Koonse SA. Expression of behavioral sensitization to ethanol by DBA/2J mice: the role of NMDA and non-NMDA glutamate receptors. Psychopharmacology. 2003;167:225–34. doi: 10.1007/s00213-003-1404-3. [DOI] [PubMed] [Google Scholar]
- 662.Meyer PJ, Phillips TJ. Bivalent effects of MK-801 on ethanol-induced sensitization do not parallel its effects on ethanol-induced tolerance. Behav Neurosci. 2003;117:641–9. doi: 10.1037/0735-7044.117.3.641. [DOI] [PubMed] [Google Scholar]
- 663.Kotlinska J, Bochenski M, Danysz W. N-methyl-D-aspartate and group I metabotropic glutamate receptors are involved in the expression of ethanol-induced sensitization in mice. Behav Pharmacol. 2006;17:1–8. doi: 10.1097/01.fbp.0000181600.95405.c7. [DOI] [PubMed] [Google Scholar]
- 664.Vosler PS, Bombace JC, Kosten TA. A discriminative two-lever test of dizocilpine’s ability to reinstate ethanol-seeking behavior. Life Sci. 2001;69:591–8. doi: 10.1016/s0024-3205(01)01150-x. [DOI] [PubMed] [Google Scholar]
- 665.Stephens DN, Brown G. Disruption of operant oral self-administration of ethanol, sucrose, and saccharin by the AMPA/kainate antagonist, NBQX, but not the AMPA antagonist, GYKI 52466. Alcohol Clin Exp Res. 1999;23:1914–1920. [PubMed] [Google Scholar]
- 666.Sanchis-Segura C, Borchardt T, Vengeliene V, Zghoul T, Bachteler D, Gass P, et al. Involvement of AMPA receptor GluR-C subunit in alcohol-seeking behavior and relapse. J Neurosci. 2006;26:1231–1238. doi: 10.1523/JNEUROSCI.4237-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 667.Lominac KD, Kapasova Z, Hannun RA, Patterson C, Middaugh LD, Szumlinski KK. Behavioral and neurochemical interactions between group I mGluR antagonists and ethanol: potential insight into their anti-addictive properties. Drug Alcohol Depend. 2006;85:142–156. doi: 10.1016/j.drugalcdep.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 668.Hodge CW, Miles MF, Sharko AC, Stevenson RA, Hillmann JR, Lepoutre V, et al. The mGluR5 antagonist MPEP selectively inhibits the onset and maintenance of ethanol self-administration in C57BL/6J mice. Psychopharmacology. 2006;183:429–38. doi: 10.1007/s00213-005-0217-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669.Backstrom P, Hyytia P. Suppression of alcohol self-administration and cue-induced reinstatement of alcohol seeking by the mGlu2/3 receptor agonist LY379268 and the mGlu8 receptor agonist (S)-3,4-DCPG. Eur J Pharmacol. 2005;528:110–118. doi: 10.1016/j.ejphar.2005.10.051. [DOI] [PubMed] [Google Scholar]
- 670.Zhao Y, Dayas CV, Aujla H, Baptista MAS, Martin-Fardon R, Weiss F. Activation of group II metabotropic glutamate receptors attenuates both stress and cue-induced ethanol-seeking and modulates c-fos expression in the hippocampus and amygdala. J Neurosci. 2006;26:9967–9974. doi: 10.1523/JNEUROSCI.2384-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 671.Backstrom P, Bachteler D, Koch S, Hyytia P, Spanagel R. mGluR5 antagonist MPEP reduces ethanol-seeking and relapse behavior. Neuropsychopharmacology. 2004;29:921–928. doi: 10.1038/sj.npp.1300381. [DOI] [PubMed] [Google Scholar]
- 672.McMillen BA, Crawford MS, Kulers CM, Williams HL. Effects of a metabotropic, mglu5, glutamate receptor antagonist on ethanol consumption by genetic drinking rats. Alcohol Alcohol. 2005;40:494–7. doi: 10.1093/alcalc/agh200. [DOI] [PubMed] [Google Scholar]
- 673.Cowen MS, Djouma E, Lawrence AJ. The metabotropic glutamate 5 receptor antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl)-pyridine reduces ethanol self-administration in multiple strains of alcohol-preferring rats and regulates olfactory glutamatergic systems. J Pharmacol Exp Ther. 2005;315:590–600. doi: 10.1124/jpet.105.090449. [DOI] [PubMed] [Google Scholar]
- 674.Olive MF, McGeehan AJ, Kinder JR, McMahon T, Hodge CW, Janak PH, et al. The mGluR5 antagonist 6-methyl-2-(phenylethynyl)pyridine decreases ethanol consumption via a protein kinase Cε-dependent mechanism. Mol Pharmacol. 2005;67:349–55. doi: 10.1124/mol.104.003319. [DOI] [PubMed] [Google Scholar]
- 675.Schroeder JP, Overstreet DH, Hodge CW. The mGluR5 antagonist MPEP decreases operant ethanol self-administration during maintenance and after repeated alcohol deprivations in alcohol-preferring (P) rats. Psychopharmacology. 2005;179:262–70. doi: 10.1007/s00213-005-2175-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676.Cowen MS, Krstew E, Lawrence AJ. Assessing appetitive and consummatory phases of ethanol self-administration in C57BL/6J mice under operant conditions: regulation by mGlu5 receptor antagonism. Psychopharmacology. 2007;190:21–9. doi: 10.1007/s00213-006-0583-0. [DOI] [PubMed] [Google Scholar]
- 677.Besheer J, Stevenson RA, Hodge CW. mGlu5 receptors are involved in the discriminative stimulus effects of self-administered ethanol in rats. Eur J Pharmacol. 2006;551:71–75. doi: 10.1016/j.ejphar.2006.08.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 678.Crabbe JC, Phillips TJ, Harris RA, Arends MA, Koob GF. Alcohol-related genes: contributions from studies with genetically engineered mice. Addict Biol. 2006;11:195–269. doi: 10.1111/j.1369-1600.2006.00038.x. [DOI] [PubMed] [Google Scholar]
- 679.Kiefer F, Jahn H, Koester A, Montkowski A, Reinscheid RK, Wiedemann K. Involvement of NMDA receptors in alcohol-mediated behavior: mice with reduced affinity of the NMDA R1 glycine binding site display an attenuated sensitivity to ethanol. Biol Psychiatry. 2003;53:345–51. doi: 10.1016/s0006-3223(02)01486-5. [DOI] [PubMed] [Google Scholar]
- 680.Gordey M, Mekmanee L, Mody I. Altered effects of ethanol in NR2A(ΔC/ΔC) mice expressing C-terminally truncated NR2A subunit of NMDA receptor. Neuroscience. 2001;105:987–97. doi: 10.1016/s0306-4522(01)00234-2. [DOI] [PubMed] [Google Scholar]
- 681.Boyce-Rustay JM, Holmes A. Functional roles of NMDA receptor NR2A and NR2B subunits in the acute intoxicating effects of ethanol in mice. Synapse. 2005;56:222–5. doi: 10.1002/syn.20143. [DOI] [PubMed] [Google Scholar]
- 682.Boyce-Rustay JM, Holmes A. Ethanol-related behaviors in mice lacking the NMDA receptor NR2A subunit. Psychopharmacology. 2006;187:455–66. doi: 10.1007/s00213-006-0448-6. [DOI] [PubMed] [Google Scholar]
- 683.Sato Y, Seo N, Kobayashi E. Ethanol-induced hypnotic tolerance is absent in N-methyl-D-aspartate receptor ε1 subunit knockout mice. Anesth Analg. 2006;103:117–20. doi: 10.1213/01.ane.0000220944.27963.b1. [DOI] [PubMed] [Google Scholar]
- 684.Cowen MS, Schroff KC, Gass P, Sprengel R, Spanagel R. Neurobehavioral effects of alcohol in AMPA receptor subunit (GluR1) deficient mice. Neuropharmacology. 2003;45:325–33. doi: 10.1016/s0028-3908(03)00174-6. [DOI] [PubMed] [Google Scholar]
- 685.Blednov YA, Walker D, Osterndorf-Kahanek E, Harris RA. Mice lacking metabotropic glutamate receptor 4 do not show the motor stimulatory effect of ethanol. Alcohol. 2004;34:251–9. doi: 10.1016/j.alcohol.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 686.Boehm SL, 2nd, Peden L, Chang R, Harris RA, Blednov YA. Deletion of the fyn-kinase gene alters behavioral sensitivity to ethanol. Alcohol Clin Exp Res. 2003;27:1033–40. doi: 10.1097/01.ALC.0000075822.80583.71. [DOI] [PubMed] [Google Scholar]
- 687.Yaka R, Tang KC, Camarini R, Janak PH, Ron D. Fyn kinase and NR2B-containing NMDA receptors regulate acute ethanol sensitivity but not ethanol intake or conditioned reward. Alcohol Clin Exp Res. 2003;27:1736–42. doi: 10.1097/01.ALC.0000095924.87729.D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 688.Cowen MS, Schumann G, Yagi T, Spanagel R. Role of Fyn tyrosine kinase in ethanol consumption by mice. Alcohol Clin Exp Res. 2003;27:1213–9. doi: 10.1097/01.ALC.0000081630.14159.02. [DOI] [PubMed] [Google Scholar]
- 689.Szumlinski KK, Lominac KD, Oleson EB, Walker JK, Mason A, Dehoff MH, et al. Homer2 is necessary for EtOH-induced neuroplasticity. J Neurosci. 2005;25:7054–61. doi: 10.1523/JNEUROSCI.1529-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 690.Moghaddam B, Bolinao ML. Biphasic effect of ethanol on extracellular accumulation of glutamate in the hippocampus and nucleus accumbens. Neurosci Lett. 1994;178:99–102. doi: 10.1016/0304-3940(94)90299-2. [DOI] [PubMed] [Google Scholar]
- 691.Quertemont E, de Neuville J, De Witte P. Changes in the amygdala amino acid microdialysate after conditioning with a cue associated with ethanol. Psychopharmacology. 1998;139:71–78. doi: 10.1007/s002130050691. [DOI] [PubMed] [Google Scholar]
- 692.Selim M, Bradberry CW. Effect of ethanol on extracellular 5-HT and glutamate in the nucleus accumbens and prefrontal cortex: comparison between the Lewis and Fischer 344 rat strains. Brain Res. 1996;716:157–164. doi: 10.1016/0006-8993(95)01385-7. [DOI] [PubMed] [Google Scholar]
- 693.Szumlinski KK, Diab ME, Friedman R, Henze LM, Lominac KD, Bowers MS. Accumbens neurochemical adaptations produced by binge-like alcohol consumption. Psychopharmacology. 2007;190:415–31. doi: 10.1007/s00213-006-0641-7. [DOI] [PubMed] [Google Scholar]
- 694.Yan Q-S, Reith MEA, Yan SG, Jobe PC. Effects of systemic ethanol on basal and stimulated glutamate release in the nucleus accumbens of freely moving Sprague-Dawley rats: a microdialysis study. Neurosci Lett. 1998;258:29–32. doi: 10.1016/s0304-3940(98)00840-4. [DOI] [PubMed] [Google Scholar]
- 695.Piepponen TP, Kiianmaa K, Ahtee L. Effects of ethanol on the accumbal output of dopamine, GABA and glutamate in alcohol-tolerant and alcohol-nontolerant rats. Pharmacol Biochem Behav. 2002;74:21–30. doi: 10.1016/s0091-3057(02)00937-1. [DOI] [PubMed] [Google Scholar]
- 696.Smith A, Watson CJ, Frantz KJ, Eppler B, Kennedy RT, Peris J. Differential increase in taurine levels by low-dose ethanol in the dorsal and ventral striatum revealed by microdialysis with on-line capillary electrophoresis. Alcohol Clin Exp Res. 2004;28:1028–38. doi: 10.1097/01.alc.0000131979.78003.34. [DOI] [PubMed] [Google Scholar]
- 697.Zuo GC, Yang JY, Hao Y, Dong YX, Wu CF. Ethanol and acetaldehyde induce similar changes in extracellular levels of glutamate, taurine and GABA in rat anterior cingulate cortex. Toxicol Lett. 2007;169:253–8. doi: 10.1016/j.toxlet.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 698.Smith TL. Regulation of glutamate uptake in astrocytes continuously exposed to ethanol. Life Sci. 1997;61:2499–505. doi: 10.1016/s0024-3205(97)00985-5. [DOI] [PubMed] [Google Scholar]
- 699.Othman T, Sinclair CJ, Haughey N, Geiger JD, Parkinson FE. Ethanol alters glutamate but not adenosine uptake in rat astrocytes: evidence for protein kinase C involvement. Neurochem Res. 2002;27:289–96. doi: 10.1023/a:1014955111742. [DOI] [PubMed] [Google Scholar]
- 700.Melendez RI, Hicks MP, Cagle SS, Kalivas PW. Ethanol exposure decreases glutamate uptake in the nucleus accumbens. Alcohol Clin Exp Res. 2005;29:326–33. doi: 10.1097/01.alc.0000156086.65665.4d. [DOI] [PubMed] [Google Scholar]
- 701.Dahchour A, Quertemont E, De Witte P. Taurine increases in the nucleus accumbens microdialysate after acute ethanol administration to naive and chronically alcoholised rats. Brain Res. 1996;735:9–19. doi: 10.1016/0006-8993(96)00537-9. [DOI] [PubMed] [Google Scholar]
- 702.Dahchour A, Quertemont E, De Witte P. Acute ethanol increases taurine but neither glutamate nor GABA in the nucleus accumbens of male rats: a microdialysis study. Alcohol Alcohol. 1994;29:485–487. [PubMed] [Google Scholar]
- 703.De Witte P, Dahchour A, Quertemont E. Acute and chronic alcohol injections increase taurine in the nucleus accumbens. Alcohol Alcohol Suppl. 1994;2:229–233. [PubMed] [Google Scholar]
- 704.Quertemont E, Linotte S, De Witte P. Differential taurine responsiveness to ethanol in high- and low-alcohol sensitive rats: a brain microdialysis study. Eur J Pharmacol. 2002;444:143–150. doi: 10.1016/s0014-2999(02)01648-5. [DOI] [PubMed] [Google Scholar]
- 705.Dahchour A, Hoffman A, Deitrich R, De Witte P. Effects of ethanol on extracellular amino acid levels in high- and low-alcohol sensitive rats: a microdialysis study. Alcohol Alcohol. 2000;35:548–553. doi: 10.1093/alcalc/35.6.548. [DOI] [PubMed] [Google Scholar]
- 706.Rossetti ZL, Carboni S. Ethanol withdrawal is associated with increased extracellular glutamate in the rat striatum. Eur J Pharmacol. 1995;283:177–183. doi: 10.1016/0014-2999(95)00344-k. [DOI] [PubMed] [Google Scholar]
- 707.Rossetti ZL, Carboni S, Fadda F. Glutamate-induced increase of extracellular glutamate through N-methyl-D-aspartate receptors in ethanol withdrawal. Neuroscience. 1999;93:1135–1140. doi: 10.1016/s0306-4522(99)00250-x. [DOI] [PubMed] [Google Scholar]
- 708.Dahchour A, De Witte P. Taurine blocks the glutamate increase in the nucleus accumbens microdialysate of ethanol-dependent rats. Pharmacol Biochem Behav. 2000;65:345–350. doi: 10.1016/s0091-3057(99)00197-5. [DOI] [PubMed] [Google Scholar]
- 709.Dahchour A, De Witte P. Excitatory and inhibitory amino acid changes during repeated episodes of ethanol withdrawal: an in vivo microdialysis study. Eur J Pharmacol. 2003;459:171–178. doi: 10.1016/s0014-2999(02)02851-0. [DOI] [PubMed] [Google Scholar]
- 710.Dahchour A, De Witte P. Effect of repeated ethanol withdrawal on glutamate microdialysate in the hippocampus. Alcohol Clin Exp Res. 1999;23:1698–1703. doi: 10.1111/j.1530-0277.1999.tb04063.x. [DOI] [PubMed] [Google Scholar]
- 711.Becker HC. Alcohol withdrawal: neuroadaptation and sensitization. CNS Spectrums. 1999;4:38–65. [Google Scholar]
- 712.Dinwiddie SH. Abuse of inhalants: a review. Addiction. 1994;89:925–39. doi: 10.1111/j.1360-0443.1994.tb03348.x. [DOI] [PubMed] [Google Scholar]
- 713.Brouette T, Anton R. Clinical review of inhalants. Am J Addict. 2001;10:79–94. doi: 10.1080/105504901750160529. [DOI] [PubMed] [Google Scholar]
- 714.Anderson CE, Loomis GA. Recognition and prevention of inhalant abuse. Am Fam Physician. 2003;68:869–74. [PubMed] [Google Scholar]
- 715.Balster RL. Neural basis of inhalant abuse. Drug Alcohol Depend. 1998;51:207–214. doi: 10.1016/s0376-8716(98)00078-7. [DOI] [PubMed] [Google Scholar]
- 716.Riegel AC, French ED. Abused inhalants and central reward pathways: electrophysiological and behavioral studies in the rat. Ann N Y Acad Sci. 2002;965:281–91. [PubMed] [Google Scholar]
- 717.Riegel AC, Zapata A, Shippenberg TS, French ED. The abused inhalant toluene increases dopamine release in the nucleus accumbens by directly stimulating ventral tegmental area neurons. Neuropsychopharmacology. 2007;32:1558–69. doi: 10.1038/sj.npp.1301273. [DOI] [PubMed] [Google Scholar]
- 718.Cruz SL, Mirshahi T, Thomas B, Balster RL, Woodward JJ. Effects of the abused solvent toluene on recombinant N-methyl-D-aspartate and non-N-methyl-D-aspartate receptors expressed in Xenopus oocytes. J Pharmacol Exp Ther. 1998;286:334–40. [PubMed] [Google Scholar]
- 719.Del Re AM, Dopico AM, Woodward JJ. Effects of the abused inhalant toluene on ethanol-sensitive potassium channels expressed in oocytes. Brain Res. 2006;1087:75–82. doi: 10.1016/j.brainres.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 720.Bale AS, Tu Y, Carpenter-Hyland EP, Chandler LJ, Woodward JJ. Alterations in glutamatergic and gabaergic ion channel activity in hippocampal neurons following exposure to the abused inhalant toluene. Neuroscience. 2005;130:197–206. doi: 10.1016/j.neuroscience.2004.08.040. [DOI] [PubMed] [Google Scholar]
- 721.Bjornaes S, Naalsund LU. Biochemical changes in different brain areas after toluene inhalation. Toxicology. 1988;49:367–74. doi: 10.1016/0300-483x(88)90020-0. [DOI] [PubMed] [Google Scholar]
- 722.Williams JM, Stafford D, Steketee JD. Effects of repeated inhalation of toluene on ionotropic GABAA and glutamate receptor subunit levels in rat brain. Neurochem Int. 2005;46:1–10. doi: 10.1016/j.neuint.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 723.Win-Shwe TT, Mitsushima D, Nakajima D, Ahmed S, Yamamoto S, Tsukahara S, et al. Toluene induces rapid and reversible rise of hippocampal glutamate and taurine neurotransmitter levels in mice. Toxicol Lett. 2007;168:75–82. doi: 10.1016/j.toxlet.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 724.Winder DG, Egli RE, Schramm NL, Matthews RT. Synaptic plasticity in drug reward circuitry. Curr Mol Med. 2002;2:667–76. doi: 10.2174/1566524023361961. [DOI] [PubMed] [Google Scholar]
- 725.Thomas MJ, Malenka RC. Synaptic plasticity in the mesolimbic dopamine system. Philos Trans R Soc Lond B Biol Sci. 2003;358:815–9. doi: 10.1098/rstb.2002.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 726.Jay TM. Dopamine: a potential substrate for synaptic plasticity and memory mechanisms. Prog Neurobiol. 2003;69:375–90. doi: 10.1016/s0301-0082(03)00085-6. [DOI] [PubMed] [Google Scholar]
- 727.Malenka RC. Synaptic plasticity and AMPA receptor trafficking. Ann N Y Acad Sci. 2003;1003:1–11. doi: 10.1196/annals.1300.001. [DOI] [PubMed] [Google Scholar]
- 728.Kauer JA. Learning mechanisms in addiction: synaptic plasticity in the ventral tegmental area as a result of exposure to drugs of abuse. Annu Rev Physiol. 2004;66:447–75. doi: 10.1146/annurev.physiol.66.032102.112534. [DOI] [PubMed] [Google Scholar]
- 729.Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–79. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 730.Jones S, Bonci A. Synaptic plasticity and drug addiction. Curr Opin Pharmacol. 2005;5:20–5. doi: 10.1016/j.coph.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 731.Nicola SM, Kombian SB, Malenka RC. Psychostimulants depress excitatory synaptic transmission in the nucleus accumbens via presynaptic D1-like dopamine receptors. J Neurosci. 1996;16:1591–604. doi: 10.1523/JNEUROSCI.16-05-01591.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 732.Nicola SM, Malenka RC. Dopamine depresses excitatory and inhibitory synaptic transmission by distinct mechanisms in the nucleus accumbens. J Neurosci. 1997;17:5697–710. doi: 10.1523/JNEUROSCI.17-15-05697.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 733.Jones S, Kauer JA. Amphetamine depresses excitatory synaptic transmission via serotonin receptors in the ventral tegmental area. J Neurosci. 1999;19:9780–9787. doi: 10.1523/JNEUROSCI.19-22-09780.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 734.Jones S, Kornblum JL, Kauer JA. Amphetamine blocks long-term synaptic depression in the ventral tegmental area. J Neurosci. 2000;20:5575–80. doi: 10.1523/JNEUROSCI.20-15-05575.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 735.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–26. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 736.Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–79. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- 737.Esteban JA. AMPA receptor trafficking: a road map for synaptic plasticity. Mol Interv. 2003;3:375–85. doi: 10.1124/mi.3.7.375. [DOI] [PubMed] [Google Scholar]
- 738.Cull-Candy S, Kelly L, Farrant M. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol. 2006;16:288–97. doi: 10.1016/j.conb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 739.Sprengel R. Role of AMPA receptors in synaptic plasticity. Cell Tissue Res. 2006;326:447–55. doi: 10.1007/s00441-006-0275-4. [DOI] [PubMed] [Google Scholar]
- 740.Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–31. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 741.Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature. 2007;446:1086–90. doi: 10.1038/nature05726. [DOI] [PubMed] [Google Scholar]
- 742.Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- 743.Todtenkopf MS, Parsegian A, Naydenov A, Neve RL, Konradi C, Carlezon WA., Jr Brain reward regulated by AMPA receptor subunits in nucleus accumbens shell. J Neurosci. 2006;26:11665–11669. doi: 10.1523/JNEUROSCI.3070-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 744.Li Y, Kauer JA. Repeated exposure to amphetamine disrupts dopaminergic modulation of excitatory synaptic plasticity and neurotransmission in nucleus accumbens. Synapse. 2004;51:1–10. doi: 10.1002/syn.10270. [DOI] [PubMed] [Google Scholar]
- 745.Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A. Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci. 2006;9:868–9. doi: 10.1038/nn1713. [DOI] [PubMed] [Google Scholar]
- 746.Sesack SR, Carr DB, Omelchenko N, Pinto A. Anatomical substrates for glutamate-dopamine interactions: evidence for specificity of connections and extrasynaptic actions. Ann N Y Acad Sci. 2003;1003:36–52. doi: 10.1196/annals.1300.066. [DOI] [PubMed] [Google Scholar]
- 747.Bonci A, Bernardi G, Grillner P, Mercuri NB. The dopamine-containing neuron: maestro or simple musician in the orchestra of addiction? Trends Pharmacol Sci. 2003;24:172–177. doi: 10.1016/S0165-6147(03)00068-3. [DOI] [PubMed] [Google Scholar]
- 748.Chao SZ, Lu W, Lee HK, Huganir RL, Wolf ME. D1 dopamine receptor stimulation increases GluR1 phosphorylation in postnatal nucleus accumbens cultures. J Neurochem. 2002;81:984–92. doi: 10.1046/j.1471-4159.2002.00877.x. [DOI] [PubMed] [Google Scholar]
- 749.Chao SZ, Ariano MA, Peterson DA, Wolf ME. D1 dopamine receptor stimulation increases GluR1 surface expression in nucleus accumbens neurons. J Neurochem. 2002;83:704–12. doi: 10.1046/j.1471-4159.2002.01164.x. [DOI] [PubMed] [Google Scholar]
- 750.Mangiavacchi S, Wolf ME. D1 dopamine receptor stimulation increases the rate of AMPA receptor insertion onto the surface of cultured nucleus accumbens neurons through a pathway dependent on protein kinase A. J Neurochem. 2004;88:1261–71. doi: 10.1046/j.1471-4159.2003.02248.x. [DOI] [PubMed] [Google Scholar]
- 751.Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci. 2005;25:7342–51. doi: 10.1523/JNEUROSCI.4603-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 752.Gao C, Sun X, Wolf ME. Activation of D1 dopamine receptors increases surface expression of AMPA receptors and facilitates their synaptic incorporation in cultured hippocampal neurons. J Neurochem. 2006;98:1664–77. doi: 10.1111/j.1471-4159.2006.03999.x. [DOI] [PubMed] [Google Scholar]
- 753.O’Brien CP, Childress AR, McLellan AT, Ehrman R. A learning model of addiction. Res Publ Assoc Res Nerv Ment Dis. 1992;70:157–77. [PubMed] [Google Scholar]
- 754.Robbins TW, Everitt BJ. Limbic-striatal memory systems and drug addiction. Neurobiol Learn Mem. 2002;78:625–636. doi: 10.1006/nlme.2002.4103. [DOI] [PubMed] [Google Scholar]
- 755.White NM. Addictive drugs as reinforcers: multiple partial actions on memory systems. Addiction. 1996;91:921–49. [PubMed] [Google Scholar]
- 756.Di Chiara G. A motivational learning hypothesis of the role of mesolimbic dopamine in compulsive drug use. J Psychopharmacol. 1998;12:54–67. doi: 10.1177/026988119801200108. [DOI] [PubMed] [Google Scholar]
- 757.Di Chiara G. Drug addiction as dopamine-dependent associative learning disorder. Eur J Pharmacol. 1999;375:13–30. doi: 10.1016/s0014-2999(99)00372-6. [DOI] [PubMed] [Google Scholar]
- 758.Volkow ND, Fowler JS, Wang G-J, Goldstein RZ. Role of dopamine, the frontal cortex and memory circuits in drug addiction: insights from imaging studies. Neurobiol Learn Mem. 2002;78:610–624. doi: 10.1006/nlme.2002.4099. [DOI] [PubMed] [Google Scholar]
- 759.Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–94. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- 760.Hyman SE. Addiction: a disease of learning and memory. Am J Psychiatry. 2005;162:1414–22. doi: 10.1176/appi.ajp.162.8.1414. [DOI] [PubMed] [Google Scholar]
- 761.Childress AR, McLellan AT, Ehrman R, O’Brien CP. Classically conditioned responses in opioid and cocaine dependence: a role in relapse? NIDA Res Monogr. 1988;84:25–43. [PubMed] [Google Scholar]
- 762.O’Brien CP, Childress AR, Ehrman R, Robbins SJ. Conditioning factors in drug abuse: can they explain compulsion? J Psychopharmacol. 1998;12:15–22. doi: 10.1177/026988119801200103. [DOI] [PubMed] [Google Scholar]
- 763.Childress AR, Hole AV, Ehrman RN, Robbins SJ, McLellan AT, O’Brien CP. Cue reactivity and cue reactivity interventions in drug dependence. NIDA Res Monogr. 1993;137:73–95. [PubMed] [Google Scholar]
- 764.Conklin CA, Tiffany ST. Applying extinction research and theory to cue-exposure addiction treatments. Addiction. 2002;97:155–67. doi: 10.1046/j.1360-0443.2002.00014.x. [DOI] [PubMed] [Google Scholar]
- 765.Bouton ME. Context, ambiguity, and unlearning: sources of relapse after behavioral extinction. Biol Psychiatry. 2002;52:976–86. doi: 10.1016/s0006-3223(02)01546-9. [DOI] [PubMed] [Google Scholar]
- 766.Bouton ME. Context and behavioral processes in extinction. Learn Mem. 2004;11:485–94. doi: 10.1101/lm.78804. [DOI] [PubMed] [Google Scholar]
- 767.Berke JD, Hyman SE. Addiction, dopamine and the molecular mechanisms of memory. Neuron. 2000;25:515–532. doi: 10.1016/s0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- 768.Hyman SE, Malenka RC. Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Neurosci Rev. 2001;2:695–703. doi: 10.1038/35094560. [DOI] [PubMed] [Google Scholar]
- 769.Nestler EJ. Is there a common molecular pathway for addiction? Nat Neurosci. 2005;8:1445–9. doi: 10.1038/nn1578. [DOI] [PubMed] [Google Scholar]
- 770.Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- 771.Davis M, Myers KM, Chhatwal J, Ressler KJ. Pharmacological treatments that facilitate extinction of fear: relevance to psychotherapy. NeuroRx. 2006;3:82–96. doi: 10.1016/j.nurx.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 772.Davis M, Ressler K, Rothbaum BO, Richardson R. Effects of D-cycloserine on extinction: translation from preclinical to clinical work. Biol Psychiatry. 2006;60:369–75. doi: 10.1016/j.biopsych.2006.03.084. [DOI] [PubMed] [Google Scholar]
- 773.Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci. 2002;22:2343–51. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 774.Lee JLC, Milton AL, Everitt BJ. Reconsolidation and extinction of conditioned fear: inhibition and potentiation. J Neurosci. 2006;26:10051–10056. doi: 10.1523/JNEUROSCI.2466-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 775.Mao SC, Hsiao YH, Gean PW. Extinction training in conjunction with a partial agonist of the glycine site on the NMDA receptor erases memory trace. J Neurosci. 2006;26:8892–9. doi: 10.1523/JNEUROSCI.0365-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 776.Botreau F, Paolone G, Stewart J. d-Cycloserine facilitates extinction of a cocaine-induced conditioned place preference. Behav Brain Res. 2006;172:173–8. doi: 10.1016/j.bbr.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 777.Kelley JB, Anderson KL, Itzhak Y. Long-term memory of cocaine-associated context: disruption and reinstatement. Neuroreport. 2007;18:777–780. doi: 10.1097/WNR.0b013e3280c1e2e7. [DOI] [PubMed] [Google Scholar]
- 778.Bouton ME, Swartzentruber D. Sources of relapse after extinction in Pavlovian and instrumental learning. Clin Psychol Rev. 1991;11:123–140. [Google Scholar]
- 779.Rescorla RA. Preservation of Pavlovian associations through extinction. Q J Exp Psychol. 1996;49B:245–258. [Google Scholar]
- 780.Schmidt EF, Sutton MA, Schad CA, Karanian DA, Brodkin ES, Self DW. Extinction training regulates tyrosine hydroxylase during withdrawal from cocaine self-administration. J Neurosci. 2001;21:RC137. doi: 10.1523/JNEUROSCI.21-07-j0003.2001. 1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 781.Self DW, Choi KH, Simmons D, Walker JR, Smagula CS. Extinction training regulates neuroadaptive responses to withdrawal from chronic cocaine self-administration. Learn Mem. 2004;11:648–57. doi: 10.1101/lm.81404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 782.Sutton MA, Schmidt EF, Choi K-H, Schad CA, Whisler K, Simmons D, et al. Extinction-induced upregulation in AMPA receptors reduces cocaine-seeking behaviour. Nature. 2003;421:70–75. doi: 10.1038/nature01249. [DOI] [PubMed] [Google Scholar]
- 783.Miller CA, Marshall JF. Molecular substrates for retrieval and reconsolidation of cocaine-associated contextual memory. Neuron. 2005;47:873–84. doi: 10.1016/j.neuron.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 784.Zavala AR, Biswas S, Harlan RE, Neisewander JL. Fos and glutamate AMPA receptor subunit coexpression associated with cue-elicited cocaine-seeking behavior in abstinent rats. Neuroscience. 2007;145:438–452. doi: 10.1016/j.neuroscience.2006.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 785.Boismare F, Daoust M, Moore N, Saligaut C, Lhuintre JP, Chretien P, et al. A homotaurine derivative reduces the voluntary intake of ethanol by rats: are cerebral GABA receptors involved? Pharmacol Biochem Behav. 1984;21:787–789. doi: 10.1016/s0091-3057(84)80020-9. [DOI] [PubMed] [Google Scholar]
- 786.Lhuintre JP, Daoust M, Moore ND, Chretien P, Saligaut C, Tran G, et al. Ability of calcium bis acetyl homotaurine, a GABA agonist, to prevent relapse in weaned alcoholics. Lancet. 1985;1(8436):1014–1016. doi: 10.1016/s0140-6736(85)91615-0. [DOI] [PubMed] [Google Scholar]
- 787.Carmen B, Angeles M, Ana M, Maria AJ. Efficacy and safety of naltrexone and acamprosate in the treatment of alcohol dependence: a systematic review. Addiction. 2004;99:811–28. doi: 10.1111/j.1360-0443.2004.00763.x. [DOI] [PubMed] [Google Scholar]
- 788.Mann K, Lehert P, Morgan MY. The efficacy of acamprosate in the maintenance of abstinence in alcohol-dependent individuals: results of a meta-analysis. Alcohol Clin Exp Res. 2004;28:51–63. doi: 10.1097/01.ALC.0000108656.81563.05. [DOI] [PubMed] [Google Scholar]
- 789.Boothby LA, Doering PL. Acamprosate for the treatment of alcohol dependence. Clin Ther. 2005;27:695–714. doi: 10.1016/j.clinthera.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 790.Mason BJ. Acamprosate in the treatment of alcohol dependence. Expert Opin Pharmacother. 2005;6:2103–15. doi: 10.1517/14656566.6.12.2103. [DOI] [PubMed] [Google Scholar]
- 791.Rosenthal RN. Current and future drug therapies for alcohol dependence. J Clin Psychopharmacol. 2006;26(Suppl 1):S20–9. [Google Scholar]
- 792.Soyka M, Roesner S. New pharmacological approaches for the treatment of alcoholism. Expert Opin Pharmacother. 2006;7:2341–53. doi: 10.1517/14656566.7.17.2341. [DOI] [PubMed] [Google Scholar]
- 793.Anton RF, O’Malley SS, Ciraulo DA, Cisler RA, Couper D, Donovan DM, et al. Combined pharmacotherapies and behavioral interventions for alcohol dependence - the COMBINE study: a randomized controlled trial. JAMA. 2006;295:2003–17. doi: 10.1001/jama.295.17.2003. [DOI] [PubMed] [Google Scholar]
- 794.Daoust M, Lhuintre JP, Saligaut C, Chretien P, Moore N, Boismare F. Calcium bis acetyl homotaurine: un nouvel agoniste gabaergique? J Pharmacol. 1985;16:521. [Google Scholar]
- 795.Madamba SG, Schweitzer P, Zieglgänsberger W, Siggins GR. Acamprosate (calcium acetylhomotaurinate) enhances the N-methyl-D-aspartate component of excitatory neurotransmission in rat hippocampal CA1 neurons in vitro. Alcohol Clin Exp Res. 1996;20:651–658. doi: 10.1111/j.1530-0277.1996.tb01667.x. [DOI] [PubMed] [Google Scholar]
- 796.Berton F, Francesconi WG, Madamba SG, Zieglgänsberger W, Siggins GR. Acamprosate enhances N-methyl-D-apartate receptor-mediated neurotransmission but inhibits presynaptic GABAB receptors in nucleus accumbens neurons. Alcohol Clin Exp Res. 1998;22:183–191. [PubMed] [Google Scholar]
- 797.Dahchour A, Durbin P, De Witte P. Ethanol and acamprosate increase the extracellular taurine in the nucleus accumbens: a microdialysis study. Alcohol Alcohol. 1995;30:483. [Google Scholar]
- 798.Dahchour A, De Witte P. Ethanol and amino acids in the central nervous system: assessment of the pharmacological actions of acamprosate. Prog Neurobiol. 2000;60:343–362. doi: 10.1016/s0301-0082(99)00031-3. [DOI] [PubMed] [Google Scholar]
- 799.Zeise ML, Kasparow S, Capogna M, Zieglgänsberger W. Calciumdiacetylhomotaurinate (CA-AOTA) decreases the action of excitatory amino acids in the rat neocortex in vitro. Prog Clin Biol Res. 1990;351:237–42. [PubMed] [Google Scholar]
- 800.Zeise ML, Kasparov S, Capogna M, Zieglgänsberger W. Acamprosate (calciumacetylhomotaurinate) decreases postsynaptic potentials in the rat neocortex: possible involvement of excitatory amino acid receptors. Eur J Pharmacol. 1993;231:47–52. doi: 10.1016/0014-2999(93)90682-8. [DOI] [PubMed] [Google Scholar]
- 801.Rammes G, Mahal B, Putzke J, Parsons C, Spielmanns P, Pestel E, et al. The anti-craving compound acamprosate acts as a weak NMDA-receptor antagonist, but modulates NMDA-receptor subunit expression similar to memantine and MK-801. Neuropharmacology. 2001;40:749–760. doi: 10.1016/s0028-3908(01)00008-9. [DOI] [PubMed] [Google Scholar]
- 802.Putzke J, Spanagel R, Tolle TR, Zieglgansberger W. The novel anti-craving drug acamprosate alters the expression of NMDA1 receptor splice variant mRNAs in the rat brain. J Neural Transm. 1996;103:XLV–XLVI. [Google Scholar]
- 803.Allgaier C, Franke H, Sobottka H, Scheibler P. Acamprosate inhibits Ca2+ influx mediated by NMDA receptors and voltage-sensitive Ca2+ channels in cultured rat mesencephalic neurones. Naunyn-Schmiedeberg Arch Pharmacol. 2000;362:440–443. doi: 10.1007/s002100000285. [DOI] [PubMed] [Google Scholar]
- 804.Popp RL, Lovinger DM. Interaction of acamprosate with ethanol and spermine on NMDA receptors in primary cultured neurons. Eur J Pharmacol. 2000;394:221–231. doi: 10.1016/s0014-2999(00)00195-3. [DOI] [PubMed] [Google Scholar]
- 805.De Witte P, Littleton J, Parot P, Koob G. Neuroprotective and abstinence-promoting effects of acamprosate : elucidating the mechanism of action. CNS Drugs. 2005;19:517–37. doi: 10.2165/00023210-200519060-00004. [DOI] [PubMed] [Google Scholar]
- 806.Naassila M, Hammoumi S, Legrand E, Durbin P, Daoust M. Mechanism of action of acamprosate. Part I. Characterization of spermidine-sensitive acamprosate binding site in rat brain. Alcohol Clin Exp Res. 1998;22:802–809. [PubMed] [Google Scholar]
- 807.al Qatari M, Bouchenafa O, Littleton J. Mechanism of action of acamprosate. Part II. Ethanol dependence modifies effects of acamprosate on NMDA receptor binding in membranes from rat cerebral cortex. Alcohol Clin Exp Res. 1998;22:810–814. [PubMed] [Google Scholar]
- 808.Harris BR, Prendergast MA, Gibson DA, Rogers DT, Blanchard JA, Holley RC, et al. Acamprosate inhibits the binding of neurotoxic effects on trans-ACPD, suggesting a novel site of action at metabotropic glutamate receptors. Alcoholism Clinical and Experimental Research. 2002;26:1779–1793. doi: 10.1097/01.ALC.0000042011.99580.98. [DOI] [PubMed] [Google Scholar]
- 809.Zieglgansberger W, Rammes G, Spanagel R, Danysz W, Parsons C. Mechanisms of action of acamprosate focusing on the glutamatergic system. In: Herman BH, editor. Glutamate and Addiction. Humana Press; Totowa, NJ: 2003. pp. 399–406. [Google Scholar]
- 810.Harris BR, Gibson DA, Prendergast MA, Blanchard JA, Holley RC, Hart SR, et al. The neurotoxicity induced by ethanol withdrawal in mature organotypic hippocampal slices might involve cross-talk between metabotropic glutamate type 5 receptors and N-methyl-D-aspartate receptors. Alcohol Clin Exp Res. 2003;27:1724–35. doi: 10.1097/01.ALC.0000093601.33119.E3. [DOI] [PubMed] [Google Scholar]
- 811.Spanagel R, Pendyala G, Abarca C, Zghoul T, Sanchis-Segura C, Magnone MC, et al. The clock gene Per2 influences the glutamatergic system and modulates alcohol consumption. Nat Med. 2005;11:35–42. doi: 10.1038/nm1163. [DOI] [PubMed] [Google Scholar]
- 812.Dahchour A, De Witte P. Effects of acamprosate on excitatory amino acids during multiple ethanol withdrawal periods. Alcohol Clin Exp Res. 2003;27:465–470. doi: 10.1097/01.ALC.0000056617.68874.18. [DOI] [PubMed] [Google Scholar]
- 813.Mcgeehan AJ, Olive MF. The anti-relapse compound acamprosate inhibits the development of a conditioned place preference to ethanol and cocaine but not morphine. Br J Pharmacol. 2003;138:9–12. doi: 10.1038/sj.bjp.0705059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 814.Spanagel R, Sillaber I, Zieglgansberger W, Corrigall WA, Stewart J, Shaham Y. Acamprosate suppresses the expression of morphine-induced sensitization in rats but does not affect heroin self-administration or relapse induced by heroin or stress. Psychopharmacology. 1998;139:391–401. doi: 10.1007/s002130050730. [DOI] [PubMed] [Google Scholar]
- 815.Mcgeehan AJ, Olive MF. Attenuation of cocaine-induced reinstatement of cocaine conditioned place preference by acamprosate. Behav Pharmacol. 2006;17:363–367. doi: 10.1097/01.fbp.0000224384.01863.5f. [DOI] [PubMed] [Google Scholar]
- 816.Bowers MS, Chen BT, Chou JK, Osborne MPH, Gass JT, See RE, et al. Acamprosate attenuates cocaine and cue-induced reinstatement of cocaine-seeking behavior in rats. doi: 10.1007/s00213-007-0904-y. Submitted. [DOI] [PubMed] [Google Scholar]
- 817.Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22:9134–41. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 818.Larowe SD, Mardikian P, Malcolm R, Myrick H, Kalivas PW, McFarland K, et al. Safety and tolerability of N-acetylcysteine in cocaine-dependent individuals. Am J Addict. 2006;15:105–110. doi: 10.1080/10550490500419169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 819.Mardikian PN, Larowe SD, Hedden S, Kalivas PW, Malcolm RJ. An open-label trial of N-acetylcysteine for the treatment of cocaine dependence: a pilot study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:389–394. doi: 10.1016/j.pnpbp.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 820.Ballon JS, Feifel D. A systematic review of modafinil: potential clinical uses and mechanisms of action. J Clin Psychiatry. 2006;67:554–66. doi: 10.4088/jcp.v67n0406. [DOI] [PubMed] [Google Scholar]
- 821.Ferraro L, Antonelli T, O’Connor WT, Tanganelli S, Rambert F, Fuxe K. The antinarcoleptic drug modafinil increases glutamate release in thalamic areas and hippocampus. NeuroReport. 1997;8:2883–2887. doi: 10.1097/00001756-199709080-00016. [DOI] [PubMed] [Google Scholar]
- 822.Ferraro L, Antonelli T, Tanganelli S, O’Connor WT, Perez de la Mora M, Mendez-Franco J, et al. The vigilance promoting drug modafinil increases extracellular glutamate levels in the medial preoptic area and the posterior hypothalamus of the conscious rat: prevention by local GABAA receptor blockade. Neuropsychopharmacology. 1999;20:346–56. doi: 10.1016/S0893-133X(98)00085-2. [DOI] [PubMed] [Google Scholar]
- 823.Ferraro L, Antonelli T, O’Connor WT, Tanganelli S, Rambert FA, Fuxe K. The effects of modafinil on striatal, pallidal and nigral GABA and glutamate release in the conscious rat: evidence for a preferential inhibition of striato-pallidal GABA transmission. Neurosci Lett. 1998;253:135–8. doi: 10.1016/s0304-3940(98)00629-6. [DOI] [PubMed] [Google Scholar]
- 824.Perez de la Mora M, Aguilar-Garcia A, Ramon-Frias T, Ramirez-Ramirez R, Mendez-Franco J, Rambert F, et al. Effects of the vigilance promoting drug modafinil on the synthesis of GABA and glutamate in slices of rat hypothalamus. Neurosci Lett. 1999;259:181–5. doi: 10.1016/s0304-3940(98)00905-7. [DOI] [PubMed] [Google Scholar]
- 825.Dackis CA, Lynch KG, Yu E, Samaha FF, Kampman KM, Cornish JW, et al. Modafinil and cocaine: a double-blind, placebo-controlled drug interaction study. Drug Alcohol Depend. 2003;70:29–37. doi: 10.1016/s0376-8716(02)00335-6. [DOI] [PubMed] [Google Scholar]
- 826.Malcolm R, Swayngim K, Donovan JL, DeVane CL, Elkashef A, Chiang N, et al. Modafinil and cocaine interactions. Am J Drug Alcohol Abuse. 2006;32:577–87. doi: 10.1080/00952990600920425. [DOI] [PubMed] [Google Scholar]
- 827.Dackis CA, Kampman KM, Lynch KG, Pettinati HM, O’Brien CP. A double-blind, placebo-controlled trial of modafinil for cocaine dependence. Neuropsychopharmacology. 2005;30:205–211. doi: 10.1038/sj.npp.1300600. [DOI] [PubMed] [Google Scholar]
- 828.Donovan JL, DeVane CL, Malcolm RJ, Mojsiak J, Chiang CN, Elkashef A, et al. Modafinil influences the pharmacokinetics of intravenous cocaine in healthy cocaine-dependent volunteers. Clin Pharmacokinet. 2005;44:753–65. doi: 10.2165/00003088-200544070-00006. [DOI] [PubMed] [Google Scholar]
- 829.Landmark CJ. Targets for antiepileptic drugs in the synapse. Med Sci Monit. 2007;13:RA1–7. [PubMed] [Google Scholar]
- 830.Rogawski MA, Loscher W. The neurobiology of antiepileptic drugs. Nat Rev Neurosci. 2004;5:553–64. doi: 10.1038/nrn1430. [DOI] [PubMed] [Google Scholar]
- 831.Dickenson AH, Ghandehari J. Anti-convulsants and anti-depressants. Handb Exp Pharmacol. 2007:145–77. doi: 10.1007/978-3-540-33823-9_6. [DOI] [PubMed] [Google Scholar]
- 832.Gryder DS, Rogawski MA. Selective antagonism of GluR5 kainate-receptor-mediated synaptic currents by topiramate in rat basolateral amygdala neurons. J Neurosci. 2003;23:7069–74. doi: 10.1523/JNEUROSCI.23-18-07069.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 833.Kaminski RM, Banerjee M, Rogawski MA. Topiramate selectively protects against seizures induced by ATPA, a GluR5 kainate receptor agonist. Neuropharmacology. 2004;46:1097–104. doi: 10.1016/j.neuropharm.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 834.Zullino DF, Khazaal Y, Hattenschwiler J, Borgeat F, Besson J. Anticonvulsant drugs in the treatment of substance withdrawal. Drugs Today (Barc) 2004;40:603–19. doi: 10.1358/dot.2004.40.7.850478. [DOI] [PubMed] [Google Scholar]
- 835.Krupitsky EM, Rudenko AA, Burakov AM, Slavina TY, Grinenko AA, Pittman B, et al. Antiglutamatergic strategies for ethanol detoxification: comparison with placebo and diazepam. Alcohol Clin Exp Res. 2007;31:604–11. doi: 10.1111/j.1530-0277.2007.00344.x. [DOI] [PubMed] [Google Scholar]
- 836.Komanduri R. Two cases of alcohol craving curbed by topiramate. J Clin Psychiatry. 2003;64:612. doi: 10.4088/jcp.v64n0518d. [DOI] [PubMed] [Google Scholar]
- 837.Rubio G, Ponce G, Jimenez-Arriero MA, Palomo T, Manzanares J, Ferre F. Effects of topiramate in the treatment of alcohol dependence. Pharmacopsychiatry. 2004;37:37–40. doi: 10.1055/s-2004-815473. [DOI] [PubMed] [Google Scholar]
- 838.Johnson BA, Ait-Daoud N, Akhtar FZ, Ma JZ. Oral topiramate reduces the consequences of drinking and improves the quality of life of alcohol-dependent individuals: a randomized controlled trial. Arch Gen Psychiatry. 2004;61:905–12. doi: 10.1001/archpsyc.61.9.905. [DOI] [PubMed] [Google Scholar]
- 839.Anderson N, Oliver MN. Oral topiramate effective for alcoholism. J Fam Pract. 2003;52:682–3. 687. [PubMed] [Google Scholar]
- 840.Kampman KM, Pettinati H, Lynch KG, Dackis C, Sparkman T, Weigley C, et al. A pilot trial of topiramate for the treatment of cocaine dependence. Drug Alcohol Depend. 2004;75:233–40. doi: 10.1016/j.drugalcdep.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 841.Johnson BA, Ait-Daoud N, Akhtar FZ, Javors MA. Use of oral topiramate to promote smoking abstinence among alcohol-dependent smokers: a randomized controlled trial. Arch Intern Med. 2005;165:1600–5. doi: 10.1001/archinte.165.14.1600. [DOI] [PubMed] [Google Scholar]
- 842.Khazaal Y, Cornuz J, Bilancioni R, Zullino DF. Topiramate for smoking cessation. Psychiatry Clin Neurosci. 2006;60:384–8. doi: 10.1111/j.1440-1819.2006.01518.x. [DOI] [PubMed] [Google Scholar]
- 843.Akhondzadeh S, Hampa AD. Topiramate prevents ecstasy consumption: a case report. Fundam Clin Pharmacol. 2005;19:601–2. doi: 10.1111/j.1472-8206.2005.00355.x. [DOI] [PubMed] [Google Scholar]
- 844.Leach MJ, Marden CM, Miller AA. Pharmacological studies on lamotrigine, a novel potential antiepileptic drug: II. Neurochemical studies on the mechanism of action. Epilepsia. 1986;27:490–7. doi: 10.1111/j.1528-1157.1986.tb03573.x. [DOI] [PubMed] [Google Scholar]
- 845.Lees G, Leach MJ. Studies on the mechanism of action of the novel anticonvulsant lamotrigine (Lamictal) using primary neurological cultures from rat cortex. Brain Res. 1993;612:190–9. doi: 10.1016/0006-8993(93)91660-k. [DOI] [PubMed] [Google Scholar]
- 846.Teoh H, Fowler LJ, Bowery NG. Effect of lamotrigine on the electrically-evoked release of endogenous amino acids from slices of dorsal horn of the rat spinal cord. Neuropharmacology. 1995;34:1273–8. doi: 10.1016/0028-3908(95)00104-e. [DOI] [PubMed] [Google Scholar]
- 847.Waldmeier PC, Baumann PA, Wicki P, Feldtrauer JJ, Stierlin C, Schmutz M. Similar potency of carbamazepine, oxcarbazepine, and lamotrigine in inhibiting the release of glutamate and other neurotransmitters. Neurology. 1995;45:1907–13. doi: 10.1212/wnl.45.10.1907. [DOI] [PubMed] [Google Scholar]
- 848.Waldmeier PC, Martin P, Stocklin K, Portet C, Schmutz M. Effect of carbamazepine, oxcarbazepine and lamotrigine on the increase in extracellular glutamate elicited by veratridine in rat cortex and striatum. Naunyn Schmiedebergs Arch Pharmacol. 1996;354:164–72. doi: 10.1007/BF00178716. [DOI] [PubMed] [Google Scholar]
- 849.Lingamaneni R, Hemmings HC., Jr Effects of anticonvulsants on veratridine- and KCl-evoked glutamate release from rat cortical synaptosomes. Neurosci Lett. 1999;276:127–30. doi: 10.1016/s0304-3940(99)00810-1. [DOI] [PubMed] [Google Scholar]
- 850.Cunningham MO, Jones RS. The anticonvulsant, lamotrigine decreases spontaneous glutamate release but increases spontaneous GABA release in the rat entorhinal cortex in vitro. Neuropharmacology. 2000;39:2139–46. doi: 10.1016/s0028-3908(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 851.Wang SJ, Sihra TS, Gean PW. Lamotrigine inhibition of glutamate release from isolated cerebrocortical nerve terminals (synaptosomes) by suppression of voltage-activated calcium channel activity. Neuroreport. 2001;12:2255–8. doi: 10.1097/00001756-200107200-00042. [DOI] [PubMed] [Google Scholar]
- 852.Ahmad S, Fowler LJ, Whitton PS. Effects of acute and chronic lamotrigine treatment on basal and stimulated extracellular amino acids in the hippocampus of freely moving rats. Brain Res. 2004;1029:41–7. doi: 10.1016/j.brainres.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 853.Sitges M, Chiu LM, Guarneros A, Nekrassov V. Effects of carbamazepine, phenytoin, lamotrigine, oxcarbazepine, topiramate and vinpocetine on Na+ channel-mediated release of [3H]glutamate in hippocampal nerve endings. Neuropharmacology. 2007;52:598–605. doi: 10.1016/j.neuropharm.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 854.Margolin A, Avants SK, DePhilippis D, Kosten TR. A preliminary investigation of lamotrigine for cocaine abuse in HIV-seropositive patients. Am J Drug Alcohol Abuse. 1998;24:85–101. doi: 10.3109/00952999809001700. [DOI] [PubMed] [Google Scholar]
- 855.Brown ES, Nejtek VA, Perantie DC, Orsulak PJ, Bobadilla L. Lamotrigine in patients with bipolar disorder and cocaine dependence. J Clin Psychiatry. 2003;64:197–201. doi: 10.4088/jcp.v64n0213. [DOI] [PubMed] [Google Scholar]
- 856.Berger SP, Winhusen TM, Somoza EC, Harrer JM, Mezinskis JP, Leiderman DB, et al. A medication screening trial evaluation of reserpine, gabapentin and lamotrigine pharmacotherapy of cocaine dependence. Addiction. 2005;100(Suppl 1):58–67. doi: 10.1111/j.1360-0443.2005.00983.x. [DOI] [PubMed] [Google Scholar]
- 857.Brown ES, Perantie DC, Dhanani N, Beard L, Orsulak P, Rush AJ. Lamotrigine for bipolar disorder and comorbid cocaine dependence: a replication and extension study. J Affect Disord. 2006;93:219–22. doi: 10.1016/j.jad.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 858.Winther LC, Saleem R, McCance-Katz EF, Rosen MI, Hameedi FA, Pearsall HR, et al. Effects of lamotrigine on behavioral and cardiovascular responses to cocaine in human subjects. Am J Drug Alcohol Abuse. 2000;26:47–59. doi: 10.1081/ada-100100590. [DOI] [PubMed] [Google Scholar]
- 859.Rubio G, Lopez-Munoz F, Alamo C. Effects of lamotrigine in patients with bipolar disorder and alcohol dependence. Bipolar Disord. 2006;8:289–93. doi: 10.1111/j.1399-5618.2006.00292.x. [DOI] [PubMed] [Google Scholar]
- 860.Shen YC. Treatment of inhalant dependence with lamotrigine. Prog Neuropsychopharmacol Biol Psychiatry. 2006;31:769–771. doi: 10.1016/j.pnpbp.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 861.Shimoyama M, Shimoyama N, Hori Y. Gabapentin affects glutamatergic excitatory neurotransmission in the rat dorsal horn. Pain. 2000;85:405–14. doi: 10.1016/S0304-3959(99)00283-3. [DOI] [PubMed] [Google Scholar]
- 862.Dooley DJ, Mieske CA, Borosky SA. Inhibition of K+-evoked glutamate release from rat neocortical and hippocampal slices by gabapentin. Neurosci Lett. 2000;280:107–10. doi: 10.1016/s0304-3940(00)00769-2. [DOI] [PubMed] [Google Scholar]
- 863.Maneuf YP, McKnight AT. Block by gabapentin of the facilitation of glutamate release from rat trigeminal nucleus following activation of protein kinase C or adenylyl cyclase. Br J Pharmacol. 2001;134:237–40. doi: 10.1038/sj.bjp.0704227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 864.Maneuf YP, Blake R, Andrews NA, McKnight AT. Reduction by gabapentin of K+-evoked release of [3H]-glutamate from the caudal trigeminal nucleus of the streptozotocin-treated rat. Br J Pharmacol. 2004;141:574–9. doi: 10.1038/sj.bjp.0705579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 865.Cunningham MO, Woodhall GL, Thompson SE, Dooley DJ, Jones RS. Dual effects of gabapentin and pregabalin on glutamate release at rat entorhinal synapses in vitro. Eur J Neurosci. 2004;20:1566–76. doi: 10.1111/j.1460-9568.2004.03625.x. [DOI] [PubMed] [Google Scholar]
- 866.Coderre TJ, Kumar N, Lefebvre CD, Yu JS. A comparison of the glutamate release inhibition and anti-allodynic effects of gabapentin, lamotrigine, and riluzole in a model of neuropathic pain. J Neurochem. 2007;100:1289–99. doi: 10.1111/j.1471-4159.2006.04304.x. [DOI] [PubMed] [Google Scholar]
- 867.Myrick H, Malcolm R, Brady KT. Gabapentin treatment of alcohol withdrawal. Am J Psychiatry. 1998;155:1632. [PubMed] [Google Scholar]
- 868.Bonnet U, Banger M, Leweke FM, Maschke M, Kowalski T, Gastpar M. Treatment of alcohol withdrawal syndrome with gabapentin. Pharmacopsychiatry. 1999;32:107–9. doi: 10.1055/s-2007-979203. [DOI] [PubMed] [Google Scholar]
- 869.Bozikas V, Petrikis P, Gamvrula K, Savvidou I, Karavatos A. Treatment of alcohol withdrawal with gabapentin. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26:197–9. doi: 10.1016/s0278-5846(01)00234-2. [DOI] [PubMed] [Google Scholar]
- 870.Voris J, Smith NL, Rao SM, Thorne DL, Flowers QJ. Gabapentin for the treatment of ethanol withdrawal. Subst Abus. 2003;24:129–32. doi: 10.1080/08897070309511541. [DOI] [PubMed] [Google Scholar]
- 871.Martinez-Raga J, Sabater A, Perez-Galvez B, Castellano M, Cervera G. Add-on gabapentin in the treatment of opiate withdrawal. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:599–601. doi: 10.1016/j.pnpbp.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 872.Rustembegovic A, Sofic E, Tahirovic I, Kundurovic Z. A study of gabapentin in the treatment of tonic-clonic seizures of alcohol withdrawal syndrome. Med Arh. 2004;58:5–6. [PubMed] [Google Scholar]
- 873.Mariani JJ, Rosenthal RN, Tross S, Singh P, Anand OP. A randomized, open-label, controlled trial of gabapentin and phenobarbital in the treatment of alcohol withdrawal. Am J Addict. 2006;15:76–84. doi: 10.1080/10550490500419110. [DOI] [PubMed] [Google Scholar]
- 874.Watson WP, Robinson E, Little HJ. The novel anticonvulsant, gabapentin, protects against both convulsant and anxiogenic aspects of the ethanol withdrawal syndrome. Neuropharmacology. 1997;36:1369–75. doi: 10.1016/s0028-3908(97)00118-4. [DOI] [PubMed] [Google Scholar]
- 875.Bailey CP, Molleman A, Little HJ. Comparison of the effects of drugs on hyperexcitability induced in hippocampal slices by withdrawal from chronic ethanol consumption. Br J Pharmacol. 1998;123:215–22. doi: 10.1038/sj.bjp.0701596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 876.Bisaga A, Aharonovich E, Garawi F, Levin FR, Rubin E, Raby WN, et al. A randomized placebo-controlled trial of gabapentin for cocaine dependence. Drug Alcohol Depend. 2006;81:267–74. doi: 10.1016/j.drugalcdep.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 877.Gonzalez G, Desai R, Sofuoglu M, Poling J, Oliveto A, Gonsai K, et al. Clinical efficacy of gabapentin versus tiagabine for reducing cocaine use among cocaine dependent methadone-treated patients. Drug Alcohol Depend. 2007;87:1–9. doi: 10.1016/j.drugalcdep.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 878.Raby WN. Gabapentin therapy for cocaine cravings. Am J Psychiatry. 2000;157:2058–9. doi: 10.1176/appi.ajp.157.12.2058-a. [DOI] [PubMed] [Google Scholar]
- 879.Myrick H, Henderson S, Brady KT, Malcolm R. Gabapentin in the treatment of cocaine dependence: a case series. J Clin Psychiatry. 2001;62:19–23. doi: 10.4088/jcp.v62n0105. [DOI] [PubMed] [Google Scholar]
- 880.Raby WN, Coomaraswamy S. Gabapentin reduces cocaine use among addicts from a community clinic sample. J Clin Psychiatry. 2004;65:84–6. doi: 10.4088/jcp.v65n0114. [DOI] [PubMed] [Google Scholar]
- 881.Haney M, Hart C, Collins ED, Foltin RW. Smoked cocaine discrimination in humans: effects of gabapentin. Drug Alcohol Depend. 2005;80:53–61. doi: 10.1016/j.drugalcdep.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 882.Heinzerling KG, Shoptaw S, Peck JA, Yang X, Liu J, Roll J, et al. Randomized, placebo-controlled trial of baclofen and gabapentin for the treatment of methamphetamine dependence. Drug Alcohol Depend. 2006;85:177–84. doi: 10.1016/j.drugalcdep.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 883.White WD, Crockford D, Patten S, El-Guebaly N. A randomized, open-label pilot comparison of gabapentin and bupropion SR for smoking cessation. Nicotine Tob Res. 2005;7:809–13. doi: 10.1080/14622200500259887. [DOI] [PubMed] [Google Scholar]
- 884.Bisaga A, Evans SM. The acute effects of gabapentin in combination with alcohol in heavy drinkers. Drug Alcohol Depend. 2006;83:25–32. doi: 10.1016/j.drugalcdep.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 885.Myrick H, Anton R, Voronin K, Wang W, Henderson S. A double-blind evaluation of gabapentin on alcohol effects and drinking in a clinical laboratory paradigm. Alcohol Clin Exp Res. 2007;31:221–7. doi: 10.1111/j.1530-0277.2006.00299.x. [DOI] [PubMed] [Google Scholar]
- 886.Vosburg SK, Hart CL, Haney M, Foltin RW. An evaluation of the reinforcing effects of memantine in cocaine-dependent humans. Drug Alcohol Depend. 2005;79:257–60. doi: 10.1016/j.drugalcdep.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 887.Bisaga A, Comer SD, Ward AS, Popik P, Kleber HD, Fischman MW. The NMDA antagonist memantine attenuates the expression of opioid physical dependence in humans. Psychopharmacology (Berl) 2001;157:1–10. doi: 10.1007/s002130100739. [DOI] [PubMed] [Google Scholar]
- 888.Bisaga A, Evans SM. Acute effects of memantine in combination with alcohol in moderate drinkers. Psychopharmacology. 2004;172:16–24. doi: 10.1007/s00213-003-1617-5. [DOI] [PubMed] [Google Scholar]
- 889.Krupitsky EM, Neznanova O, Masalov D, Burakov AM, Didenko T, Romanova T, et al. Effect of memantine on cue-induced alcohol craving in recovering alcohol-dependent patients. Am J Psychiatry. 2007;164:519–23. doi: 10.1176/ajp.2007.164.3.519. [DOI] [PubMed] [Google Scholar]
- 890.Arias AJ, Feinn R, Covault J, Karnzler HR. Memantine for alcohol dependence: an open-label pilot study. Addict Disord Treat. 2007;6:77–83. [Google Scholar]
- 891.Evans SM, Levin FR, Brooks DJ, Garawi F. A pilot double-blind treatment trial of memantine for alcohol dependence. Alcohol Clin Exp Res. 2007;31:775–82. doi: 10.1111/j.1530-0277.2007.00360.x. [DOI] [PubMed] [Google Scholar]
- 892.Collins ED, Ward AS, McDowell DM, Foltin RW, Fischman MW. The effects of memantine on the subjective, reinforcing and cardiovascular effects of cocaine in humans. Behav Pharmacol. 1998;9:587–98. doi: 10.1097/00008877-199811000-00014. [DOI] [PubMed] [Google Scholar]
- 893.Collins ED, Vosberg SK, Ward AS, Haney M, Foltin RW. The effects of acute pretreatment with high-dose memantine on the cardiovascular and behavioral effects of cocaine in humans. Exp Clin Psychopharmacol. 2007;15:228–37. doi: 10.1037/1064-1297.15.3.228. [DOI] [PubMed] [Google Scholar]
- 894.Sander T, Ostapowicz A, Samochowiec J, Smolka M, Winterer G, Schmidt LG. Genetic variation of the glutamate transporter EAAT2 gene and vulnerability to alcohol dependence. Psychiatr Genet. 2000;10:103–7. doi: 10.1097/00041444-200010030-00001. [DOI] [PubMed] [Google Scholar]
- 895.Foley PF, Loh EW, Innes DJ, Williams SM, Tannenberg AE, Harper CG, et al. Association studies of neurotransmitter gene polymorphisms in alcoholic Caucasians. Ann N Y Acad Sci. 2004;1025:39–46. doi: 10.1196/annals.1316.005. [DOI] [PubMed] [Google Scholar]
- 896.Petrakis IL, Limoncelli D, Gueorguieva R, Jatlow P, Boutros NN, Trevisan L, et al. Altered NMDA glutamate receptor antagonist response in individuals with a family vulnerability to alcoholism. Am J Psychiatry. 2004;161:1776–82. doi: 10.1176/ajp.161.10.1776. [DOI] [PubMed] [Google Scholar]
- 897.Wernicke C, Samochowiec J, Schmidt LG, Winterer G, Smolka M, Kucharska-Mazur J, et al. Polymorphisms in the N-methyl-D-aspartate receptor 1 and 2B subunits are associated with alcoholism-related traits. Biol Psychiatry. 2003;54:922–8. doi: 10.1016/s0006-3223(03)00072-6. [DOI] [PubMed] [Google Scholar]
- 898.Rujescu D, Soyka M, Dahmen N, Preuss U, Hartmann AM, Giegling I, et al. GRIN1 locus may modify the susceptibility to seizures during alcohol withdrawal. Am J Med Genet B Neuropsychiatr Genet. 2005;133:85–7. doi: 10.1002/ajmg.b.30112. [DOI] [PubMed] [Google Scholar]
- 899.Preuss UW, Zill P, Koller G, Bondy B, Hesselbrock V, Soyka M. Ionotropic glutamate receptor gene GRIK3 Ser310Ala functional polymorphism is related to delirium tremens in alcoholics. Pharmacogenomics J. 2006;6:34–41. doi: 10.1038/sj.tpj.6500343. [DOI] [PubMed] [Google Scholar]
- 900.Samochowiec J, Grzywacz A, Kucharska-Mazur J, Samochowiec A, Horodnicki J, Pelka-Wysiecka J, et al. Family-based and case-control association studies of glutamate receptor GRIK3 Ser310Ala polymorphism in Polish patients and families with alcohol dependence. Neurosci Lett. 2006;396:159–62. doi: 10.1016/j.neulet.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 901.Schumann G, Rujescu D, Szegedi A, Singer P, Wiemann S, Wellek S, et al. No association of alcohol dependence with a NMDA-receptor 2B gene variant. Mol Psychiatry. 2003;8:11–2. doi: 10.1038/sj.mp.4001245. [DOI] [PubMed] [Google Scholar]
- 902.Sansig G, Bushell TJ, Clarke VR, Rozov A, Burnashev N, Portet C, et al. Increased seizure susceptibility in mice lacking metabotropic glutamate receptor 7. J Neurosci. 2001;21:8734–45. doi: 10.1523/JNEUROSCI.21-22-08734.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 903.Preuss UW, Koller G, Bahlmann M, Zill P, Soyka M, Bondy B. No association between metabotropic glutamate receptors 7 and 8 (mGlur7 and mGlur8) gene polymorphisms and withdrawal seizures and delirium tremens in alcohol-dependent individuals. Alcohol Alcohol. 2002;37:174–8. doi: 10.1093/alcalc/37.2.174. [DOI] [PubMed] [Google Scholar]
- 904.Kalivas PW. Glutamate systems in cocaine addiction. Curr Opin Pharmacol. 2004;4:23–29. doi: 10.1016/j.coph.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 905.Dahl JP, Kampman KM, Oslin DW, Weller AE, Lohoff FW, Ferraro TN, et al. Association of a polymorphism in the Homer1 gene with cocaine dependence in an African American population. Psychiatr Genet. 2005;15:277–83. doi: 10.1097/00041444-200512000-00010. [DOI] [PubMed] [Google Scholar]
- 906.Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic Press; 2001. [Google Scholar]