

Abstract
Interaction of the antigen-specific receptor of T lymphocytes with its antigenic ligand can lead either to cell activation or to a state of profound unresponsiveness (anergy). Although subtle changes in the nature of the ligand or of the antigen-presenting cell have been shown to affect the outcome of T cell receptor ligation, the mechanism by which the same receptor can induce alternative cellular responses is not completely understood. A model for explaining both positive (cell proliferation and cytokine production) and negative (anergy induction) signaling of T lymphocytes is described herein. This model relies on the autophosphorylative properties of the tyrosine kinases associated with the T cell receptor. One of its basic assumptions is that the kinase activity of these receptor-associated enzymes remains above background level after ligand removal and is responsible for cellular unresponsiveness. Using a simple Boolean formalism, we show how the timing of the binding and intracellular signal-transduction events can affect the properties of receptor signaling and determine the type of cellular response. The present approach integrates into a common framework a large body of experimental observations and allows specification of conditions leading to cellular activation or to anergy.
The initiation and regulation of an immune response to antigen are dependent on the activation of helper T lymphocytes by an appropriate antigen/major histocompatibility complex ligand (Ag/MHC). In the recent years, it has been observed that engagement of the T cell antigen receptor (TCR) by its cognate ligand triggers a series of biochemical events within the cell, which can lead either to cellular activation, i.e., lymphokine production and cell proliferation, or to the induction of a state of unresponsiveness termed anergy. This anergic state is characterized by a refractoriness to further antigenic stimulation despite normal TCR cell surface expression and normal response to exogenous growth factors. A variety of experimental models have led to the conclusion that T cell anergy can develop in response to a heterogeneous set of stimuli and under different experimental protocols, as summarized below.
A large body of data, obtained mostly in vitro, indicates that anergy may develop as a consequence of inadequate stimulation. Indeed, occupancy of the TCR by an appropriate ligand (an agonist Ag/MHC complex, anti-TCR-complex antibodies, or mitogenic lectins) in the absence of other antigen-presenting cell (APC)-derived signals (termed costimulatory signals) has been shown to induce T cell anergy rather than a functional, proliferative response (see ref. 1 for review). A state of T cell unresponsiveness can also be induced in mature T cells by analogs of immunogenic peptides (altered peptide ligands or APLs), which interact in an unproductive fashion with the TCR complex even when presented by adequate, costimulatory-expressing APCs (2). These ligands are thought to elicit a subset of normal intracellular activation events, leading to anergy rather than full activation (3, 4). Finally, anergy can be induced after stimulation of T cell clones by nominal (agonist) ligands presented by adequate accessory cells if interleukin 2 (IL-2)-induced proliferation is prevented by blocking antibodies to IL-2 or IL-2 receptors (5). Thus, a large body of data indicates that T cell anergy is induced in mature T cell populations when TCR stimulation is not followed by cell proliferation. In this context, clonal anergy develops after an abortive stimulation and therefore may represent a mechanism by which potentially harmful, autoreactive T lymphocytes that have escaped thymic deletion can be functionally inactivated (6).
More recently, numerous observations, mostly performed in vivo, have suggested an alternative way of anergy induction, in which a transient T cell activation precedes induction of unresponsiveness. Exposure of mature T cells in vivo to viral and bacterial superantigens leads to T cell activation (characterized by a costimulatory-dependent lymphokine production and entry into the cell cycle) followed by hyporesponsiveness (7). Similarly, administration of mitogenic anti-CD3 antibodies in vivo causes a potent inflammatory response secondary to the secretion of numerous T cell-derived lymphokines, followed by a state of T cell unresponsiveness characterized by defective TCR signaling (8). This form of “activation-induced anergy” may represent a mechanism for down-regulating excessive inflammatory responses that, although directed toward non-self-pathogens, are nevertheless a potential threat to the organism’s integrity.
The conditions determining the functional consequences of TCR engagement (memory-type response, induction of an anergic state or of a post-activation–refractory phase) are not completely understood. In this paper we present a model to account for both positive and negative signaling of T lymphocytes. Special emphasis is placed on specifying the conditions leading to alternative cellular behavior. Using a simple Boolean formalism developed by Thomas and coworkers (see e.g., refs. 9 and 10), we show how the timing of the signaling events may affect decision making at branching pathways and may determine the properties of receptor signaling and final state of the system. The goal of the present work is to integrate the different experimental observations described above into an unifying framework.
The Model
Our model is based on the following experimental observations and assumptions.
(i) An important feature of the early events of the TCR signaling cascade is the activation of protein tyrosine kinase (PTK) enzymes endowed with autocatalytic properties. Indeed, once the receptor-proximal src-family kinases Fyn and Lck and syk-related kinase ZAP-70 are activated, they can enhance further their own catalytic activity through autophosphorylation on a site of their activation segment (11). Recently, it has been suggested that phosphorylation of this catalytic site is dependent on the concentration of the kinases (12), consistent with a model in which TCR aggregation promotes intermolecular autophosphorylation. We have shown previously that this autocatalytic step may lead to a bistable behavior (13) characterized by the fact that, under identical conditions, the receptor-associated tyrosine kinases exist either in a state of low- or of high-phosphorylative activity, depending on the system’s antigenic history. As discussed in Kaufman et al. (13) this bistability allows for sustained PTK activity after removal of the stimulus. Thus, we assume here that after stimulation, the phosphorylative activity of the receptor-associated PTKs increases and may remain above background level after the ligands have been removed.
(ii) In addition to its positive role in T cell activation, we postulate that tyrosine phosphorylation also mediates a suppressive effect on the signaling events, which results in the inhibition of lymphokine secretion and cell proliferation. In particular, residual PTK activity after ligand dissociation may be responsible for a defective signal transduction capacity of the TCR system (see ref. 13).
(iii) Costimulation provided through an antigen-independent ligand/receptor interaction is required for optimal lymphokine production and subsequent cell proliferation. In the present model, costimulation does not act to suppress the negative signal but rather enhances and accelerates the positive signaling process (1, 14).
(iv) In agreement with the observation that signaling through the IL-2 receptor γ chain and/or cell proliferation reverses the anergic state (14, 15), we consider that positive signaling (i.e., IL-2 secretion and cell proliferation) inhibits, through a yet undefined mechanism, the persistent activity of the PTKs, thus reestablishing the signaling capacities of the TCR system.
The model shown in Fig. 1 consists of a series of events, each requiring a characteristic time to be realized. Binding of free ligand to the TCRs activates the receptor-associated tyrosine kinases. Receptor engagement together with PTK activation leads to positive signaling. Activated PTKs also trigger an inhibitory pathway, which negatively affects the positive response. Costimulation participates in signal generation. The positive action of the PTKs on themselves indicates that once receptor-associated PTK activity is established, it remains sustained even in the absence of ligand. This autocatalytic maintenance mechanism is suppressed by IL-2-linked signaling and proliferation. Except for the initial increase in PTK activity, the signal here includes both the early (calcium rise, protein kinase C, and Ras activation) and late transduction events (up-regulation of IL-2 receptors, IL-2 production, and cell proliferation). A distinction between different subsets of transduction events, while allowing to account for a wider variety of partial responses, does not change the basic properties of the model and will not be considered here.
Figure 1.

Schematic interaction diagram. f = free ligand; b = TCRs bound to ligand; k = receptor-associated PTK activity; x = tyrosine kinase-dependent inhibitory pathway; s = metabolic and mitogenic response. Positive and negative interactions are indicated by a plus and minus sign, respectively.
In our approach, for a cell with a large population of receptors, we assume that the number of receptors bound to ligand is sufficient to produce an observable response in optimal conditions. Moreover, each bound receptor contributes, on the average, in the same way to the global behavior. This means that for a qualitative description of the signaling process it is sufficient to consider the behavior of a single receptor.
We model the signaling that results from a single ligand-binding event per receptor. This situation is likely to occur at low ligand concentration and vast excess of receptors (i.e., the probability that a receptor binds more than once to a ligand is very small), which is often the case in physiological conditions. The effect of repeated binding of ligand to the same receptor during the course of the signaling process (e.g., at high ligand concentration) will be discussed briefly in the section titled Binding and Rebinding of Ligand.
Logical Description
Logical Variables and Logical Equations.
We define the state of the system in terms of four logical variables that take the logical values 0 or 1. Thus, b = 1 means receptor bound to ligand, otherwise b = 0; k = 1 means receptor-associated PTKs activated, otherwise k = 0; x = 1 means inhibitory pathway activated, otherwise x = 0; s = 1 means activatory pathway activated, otherwise s = 0. Costimulation is considered to accelerate the signaling process and is, at this stage of our analysis, accounted for in an implicit way as will appear below. Justification for treating the interactions as “on” or “off” comes from the observation that the effect of biological regulators often changes from inefficient to efficient over a rather small range of concentration and short period of time. In particular, such threshold phenomena have been described to be involved in T cell activation (16, 17).
To describe the dynamics of the system one has to consider transition rules that reflect the evolution of the state variables under the influence of the signaling interactions. Therefore, we associate with each variable b, k, x, and s a logical function B, K, X, and S, respectively, which is built to take the value 1 or 0 for the conditions in which its associated variable tends to or remains at 1 or 0. The logical functions thus give, at any time, the future value toward which the corresponding variable will tend as a function of the present state of the system. For our model we define the following set of logical equations:
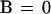 |
1 |
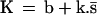 |
2 |
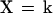 |
3 |
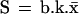 |
4 |
where we use the logical notations: z̄ = NOT z (logical complement); z.y = z AND y (logical product); z + y = z OR y (inclusive OR).
Eq. 1 describes the natural tendency of a bound TCR to dissociate from ligand, irrespective of the presence or absence of free ligand, and to remain unbound because no ligand rebinding is considered at this stage (B = 0 and b always tends toward or remains 0; see also section on binding and rebinding). Eq. 2 expresses that receptor-associated tyrosine kinase activity will increase above basal level if the receptors are bound to ligand (b = 1). It also involves a maintenance mechanism, through autophosphorylation, that persists in the absence of ligand-bound TCRs. This maintenance mechanism is suppressed by the occurrence of IL-2-linked signaling events and proliferation (s = 1). Thus, K = 1 (and k tends toward or remains at 1) if either or both b = 1, k.s̄ = 1; otherwise K = 0. Eq. 3 states that the receptor-associated tyrosine kinases activate, directly or indirectly, an inhibitory pathway. X = 1 if k = 1; otherwise X = 0. Finally, Eq. 4 means that positive signaling (i.e., cellular activation) will occur (S = 1) if the receptors are bound (b = 1) and the receptor-associated kinases are activated (k = 1) and if their positive action on cell activation is not inhibited (x = 0); otherwise S = 0.
There is a temporal relation between logical functions and associated variables: if the value of a logical function and its corresponding variable disagree, there is a commitment for the variable to align its value with the value of the function. This command will effectively be executed after a characteristic time delay, unless a counter order is received before the delay has elapsed. For example, the value of x will align on the value X = 1 after a time delay tx, provided there has not been a counter order, X = 0, before expiration of the delay. Similarly, if x = 1 and X = 0, then x will shift from x = 1 to x = 0 in a characteristic time tx̄. On and off delays for the other state variables are denoted similarly. Moreover, we adopt a fully asynchronous description in which all the time delays are different, except for accidental coincidence (9, 18).
State Table.
The state of the system is described by the combined logical values 0 or 1 of the state variables b, k, x, and s, presented in this defined order. Using Eqs. 1–4 one computes a state table that provides the value of the function vector BKXS for each of the 24 values of the state vector bkxs (Table 1).
Table 1.
| b | k | x | s | B | K | X | S |
|---|---|---|---|---|---|---|---|
| (0 | 0 | 0 | 0) | 0 | 0 | 0 | 0 |
| − | |||||||
| 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| − | − | ||||||
| 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| − | |||||||
| 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| (0 | 1 | 1 | 0) | 0 | 1 | 1 | 0 |
| − | − | ||||||
| 0 | 1 | 1 | 1 | 0 | 0 | 1 | 0 |
| − | + | − | |||||
| 0 | 1 | 0 | 1 | 0 | 0 | 1 | 0 |
| + | |||||||
| 0 | 1 | 0 | 0 | 0 | 1 | 1 | 0 |
| − | + | + | |||||
| 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 |
| − | + | ||||||
| 1 | 1 | 0 | 1 | 0 | 1 | 1 | 1 |
| − | − | ||||||
| 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 |
| − | |||||||
| 1 | 1 | 1 | 0 | 0 | 1 | 1 | 0 |
| − | + | − | |||||
| 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| − | + | − | − | ||||
| 1 | 0 | 1 | 1 | 0 | 1 | 0 | 0 |
| − | + | − | |||||
| 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 |
| − | + | ||||||
| 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
A plus or minus sign over the logical value of a state variable indicates that that variable is being driven upward (0 → 1) or downward (1 → 0) by the interactions. Parentheses or circles around a state indicate that the state is stable under the given input conditions. In such regime states each variable and its corresponding function have the same logical value.
Note that when a state vector bkxs has more than one switch command, it is only if the corresponding time delays are strictly equal that the function vector BKXS coincides with the next state.
Transition Diagram.
From the state table one can derive all the possible temporal sequences of logical states, starting from any initial state.
To derive the signaling conditions for a single ligand-binding event per receptor, we start from the state (1000) where the receptor has just become bound by ligand. Depending on whether ligand dissociation or tyrosine kinase activation occurs first, two different pathways can be followed. If tyrosine kinase activity appears before unbinding of the receptors, again different branches can be followed depending on which next event is realized first, and so on. This defines a whole network of branching pathways (Fig. 2).
Figure 2.

Transition diagram showing the sequences of states and possible pathways. The total time to reach a given state, relative to time 0, is written next to each state in terms of the characteristic transition times (delays) of the state variables. An asterisk (∗) indicates the stages corresponding to cellular activation (s = 1).
Two groups of pathways may be distinguished. Pathways 1–3 reach a final stable state without the appearance of cellular activation. Pathways 4–12 all involve a stage of activation. Furthermore, the model predicts that two different stable final states can be reached after ligand dissociation:
(i) state (0000), where all the state variables are back to their initial zero value. This state corresponds to full immunocompetence.
(ii) state (0110), in which tyrosine kinase activity is on and inhibition is present. As shown in Fig. 3a, this state is characterized by an unresponsiveness to further stimulation and corresponds to the anergic state. Unresponsiveness, however, is reversed by external addition of the signaling component s (Fig. 3b), in agreement with the experimental observation that anergy is reversed after addition of exogenous IL-2.
Figure 3.

(a) Secondary response. Addition of free ligand to the anergic state fails to evoke a productive response (see Eq. 5 for ligand rebinding). After ligand dissociation and removal the system returns to the unresponsive state. (b) Reversal of anergy. Addition of the signaling component s to the anergic state restores the fully responsive state.
Each event in our logical network requires a characteristic time delay to be realized. We consider here that the effect of costimulation is to decrease the time delay ts for switching the positive signal on. Thus, we assume that ts < tx describes the situation in the presence of costimulation, whereas ts > tx corresponds to the absence of costimulation.
The state that will be reached after a branching point in the transition diagram is the one requiring the least time from the initial state where the receptor has just been bound (time t = 0 in Fig. 2). Note that the time to arrive at a given state in the network is a function of the time delays, calculated taking into account that a switch command may have been initiated at or several steps before the branching point. In this manner, the conditions for selecting alternative routes at each bifurcation are given by inequalities involving linear combinations of time delays. From the successive branching decisions, the simplest inequality conditions for traversing a given path can be derived. Using the tools of Boolean algebra and combinatorial logic, overall conditions then can be deduced for any particular cellular response or final situation. This type of analysis is described in ref. 9, and detailed calculations for the present study can be obtained upon request.
Timing-Dependent Signaling Properties
Fast Ligand Dissociation.
Path 1 is followed if ligand dissociation precedes kinase activation (tb̄ < tk) and corresponds to what is observed for “null” or inactive ligands. Dissociation is extremely rapid and no detectable changes are observed within the cells (4, 19).
Path 2 is followed if ligand dissociation is slower than kinase activation but faster than significant activation of the stimulatory and inhibitory pathways: tk < tb̄ < Min {tk + ts, tk + tx}. In the presence of residual kinase activity after ligand dissociation, the inhibitory component will switch on sooner or later and the system ends up in the anergic state. Upon restimulation with an activatory ligand in the presence of costimulation, productive signaling is prevented by the already activated inhibitory pathway.
This situation accounts for stimulation with APLs. These variants of immunogenic TCR ligands, characterized by strongly increased off-rates (19), generally lead only to unproductive initial phosphorylation reactions and drive the cells into the anergic state, even in the presence of the necessary costimulatory molecules (2, 3).
Absence of Costimulation.
If ts > tx and tb̄ > tk + tx, path 3 will be followed. Here, the receptor-associated kinases are activated but inhibition precedes significant signal transmission, so that there is no positive signaling. After ligand dissociation, the system ends up again in the unresponsive state and restimulation in optimal conditions does not lead to cellular activation. Path 3 thus may account for stimulation with an activatory ligand in the absence of costimulatory signals (1).
Positive Signaling.
The general conditions for positive signaling are: ts < tx and tb̄ > tk + ts. Positive signaling should be faster than significant inhibition, and the ligand residence time must exceed the time required for activation of the kinases and signal transmission. These timing conditions correspond to paths 4 to 12 and account for signaling upon stimulation with an activatory ligand in the presence of costimulation. Along paths 8–12 (tb̄ < tk + tx), the time length of the activation phase essentially is determined by the ligand residence time on the receptors, whereas along paths 4–7 (tb̄ > tk + tx), it is mainly determined by the time lag between positive signaling and inhibition.
After activation the system will end up in one of two stable steady states, corresponding either to recovery of responsiveness (0000) or to anergy (0110). Independently of the precise pathway that is followed, the necessary and sufficient conditions to reach each of these two final states can be determined by using the tools of combinatorial logic. These additional conditions follow.
(i) For positive signaling with recovery of responsiveness:
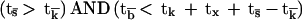 |
which means that the activatory phase will be followed by recovery of immunocompetence both if the ligand does not bind too strongly and positive signaling does not decay before inactivation of the kinases. This situation requires that the ligand residence time be in an optimal range and is related to a memory-type response.
(ii) For positive signaling followed by anergy:
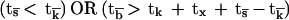 |
which means that the activatory phase will be followed by anergy if either or both the positive signal decays rapidly or ligand dissociation is very slow. This situation corresponds to “activation-induced anergy.”
It should be stressed that, in addition to the ligand residence time on the receptors, which is inversely proportional to the kinetic dissociation rate, the conditions to follow different paths involve time delays that will be determined by the cell type and nature of costimulatory functions.
Binding and Rebinding of Ligand.
To investigate the effect on the signaling process of multiple binding of ligand to the same receptor, we generalize Eq. 1 and describe receptor/ligand association and dissociation by the logical equation (see also ref. 10):
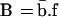 |
5 |
where f is an input variable that stands for free ligand available (f = 1) or not (f = 0).
This equation states that in the presence of free ligand (f = 1), an unbound receptor (b = 0) will become bound in a characteristic time tb inversely related to the free ligand concentration, whereas bound receptors (b = 1) will dissociate from ligand at a rate 1/tb̄ dependent on the kinetic dissociation constant. In the absence of free ligand, f = 0; therefore, B = 0 and b will tend to or remain at 0.
With Eq. 5 for ligand binding together with Eqs. 2–4, one can again compute the temporal state transitions, upon addition of free ligand to the virgin state (0000).
If the time scale of reassociation is slow compared with the downstream events (e.g., at low ligand concentration), the results are identical to those described above. Essentially the same scenarios also are found for rapid rebinding of ligand (e.g., at high ligand concentration). However, one additional feature appears for fast-dissociating ligands, characterized by tk < tb̄ < Min {tk + ts, tk + tx}, which may now, upon rebinding to a partially activated system, induce a full activatory signal, providing there is costimulation (ts < tx) and ts < tb̄ < tk + tx − (tb + ts) with tb < tx − ts (M.K., unpublished results).
The model thus predicts that certain ligands that do not activate and induce unresponsiveness at a low concentration may become activatory at high concentrations (4, 20).
Discussion
Anergy has been defined as a cellular state in which a lymphocyte fails to respond optimally (in terms of both cytokine production and proliferation) when stimulated through its antigen-specific receptor. Recently, it has been recognized that this unresponsive state can be achieved by multiple means (see Introduction). The goal of the present work is to propose a common mechanism underlying all known forms of T cell unresponsiveness. Our logical description is based on the approach developed by Thomas and D’Ari, which emphasizes the importance of feedback loops and asynchronous switching for the dynamic properties of regulatory networks (9). This formalism already has been applied to other biological examples such as the decision between lysis and lysogenization in bacteriophage λ (21), the choice between memory and paralysis in the humoral immune response (22), or the influence of hormone residence time on the insulin receptor for metabolic versus mitogenic signaling (10).
For our simple model, the logical analysis provides conditions for the selection of specific cellular responses and final situations, which are expressed in terms of inequalities among the time delays required for activation or deactivation of the components of the model. These conditions link the properties of the ligand/TCR interactions, cellular characteristics and experimental conditions for determining the outcome of cell stimulation.
Central to our model is the dual role of receptor-associated tyrosine kinases in the regulation of cellular competence. As discussed in a previous work (13), the autophosphorylative activity of these kinases is compatible with a bistable behavior, thereby allowing these enzymes to persist either in a resting or in an active state (depending on previous antigenic experience) in the absence of physiological ligands. We hypothesize that the residual tyrosine kinase activity that persists after ligand removal is responsible for actively maintaining an inhibitory pathway at work, thus leading to anergy. PTK-dependent negative signaling also contributes to the termination of a response.
The assumption that tyrosine kinase activity does not rapidly return to basal levels upon ligand removal and correlates with a defective transduction of activation signals recently has received experimental support. The kinase activity of p59fyn was found constitutively elevated in anergic human and murine Th1 clones (23–26). In a particular study, anergic human Th2 clones displayed elevated tyrosine kinase activity possibly associated with an enhanced p56lck phosphorylated status (27). Moreover, several in vivo models, including unresponsive T cells from lpr (28) and gld (29) mice or from HIV-infected patients (30), also suggest a possible link between constitutive kinase activity and T cell anergy.
We originally proposed that down-regulation of TCR-transducing properties in anergic T cells expressing elevated levels of kinase activity could result from the recruitment of an inhibitory molecule to the TCR-associated transducing complex, thus able to block and/or disrupt a crucial receptor–target interaction needed for proper signaling (see ref. 13). This hypothesis is in good agreement with recent observations performed in a human T cell clone rendered anergic after inadequate stimulation. In this experimental model, anergy was found to correlate with the recruitment to the TCR complex of constitutively tyrosine-phosphorylated proteins able to antagonize Ras function, thus blocking an obligatory signaling step leading to IL-2 gene transcription (26). An alternative mechanism by which hyperphosphorylation of selected substrates may lead to unresponsiveness has been suggested recently by the finding that the phosphorylation status of a given protein may affect its intracellular localization (31). According to this hypothesis, phosphorylation may render key substrates inaccessible to important downstream enzymes or proteins, thus leading to improper signaling. Collectively, these experimental observations provide possible mechanisms by which, as postulated in the present model, increased tyrosine kinase activity can lead to T cell unresponsiveness.
Several experimental (19, 32) and theoretical papers (33, 34) recently have stressed the influence of ligand-dissociation rate on T cell activation. In particular, the kinetic discrimination model proposed by Rabinowitz et al. (34) relates T cell activation, as a function of ligand-dissociation rate, to a ratio of complete and incomplete signals, thus allowing for a discrimination between agonist and antagonist ligands in terms of their rate of dissociation. The purpose of our study is to extend these ideas to provide a link between the activating properties of a given ligand/costimulation combination and anergy induction. As noted earlier, the selection of specific responses in our model is dictated by the kinetic characteristics of TCR occupation by a given ligand as well as by the time delay required to generate a negative signal (tx) versus the time delay required to achieve positive signaling (ts). On this basis, our theoretical approach defines conditions for three different cases of anergy induction.
(i) In agreement with numerous experimental studies, stimulation of a T lymphocyte by an appropriate ligand (adequate concentration and dissociation rate) in the absence of APC-derived costimulation will be sufficient to induce kinase activity and negative signaling, but will fail to generate a complete activation signal. As a consequence, these cells will not respond properly and will be induced into an anergic state (1). (ii) Stimulation of a lymphocyte by an APL (characterized by a high dissociation rate) or by suboptimal doses of an agonist ligand (unable to occupy the TCR complex for the time period required to generate positive signaling) will induce anergy even in the presence of costimulation, as described recently (35). (iii) Our model also predicts that although excessive stimulation can lead to cell proliferation and cytokine secretion, it will also lead inevitably to a state of unresponsiveness. This particular situation (referred to as post-activation anergy) is expected to occur as a consequence of prolonged TCR stimulation causing continuous stimulation of the kinase-dependent, negative signal. Experimentally, this may correspond to stimulation of T cells by high-affinity TCR ligands (characterized by a slow off-rate), such as anti-TCR-complex antibodies. We indeed have shown that stimulation of T cells by soluble anti-CD3 antibodies in the presence of the required accessory cells induces a costimulatory-dependent activation phase, followed by anergy (8). It also has been demonstrated recently that mature T cells exposed in vivo to high and persistent doses of autoantigen undergo an initial phase of expansion before becoming anergic (36).
An additional prediction of our model is that anergy is not determined solely by the nature of APC-derived signals (including Ag/MHC complexes and costimulatory functions) but will also depend on the rate of the intracellular biochemical processes and, hence, on factors such as the density, spatial distribution, and stability of some key components. The importance of the cellular characteristics in anergy induction has been illustrated recently by the finding that naive and activated T cells expressing identical TCRs are not equally sensitive to anergy induction when stimulated by the same ligand presented by chemically treated, costimulatory-deficient APCs (37).
In conclusion, although our simplified description does not fully characterize all the processes involved in T cell signaling, it accounts for a large body of experimental data and allows to integrate most experimental observations related to activation and anergy into a common framework.
Acknowledgments
We thank R. Thomas and J. Urbain for helpful discussions and J. F. Lahou for help in the realization of the artwork. This work has been supported by the Belgian program on interuniversity attraction poles, initiated by the Belgian State, Prime Minister’s Office Policy Programming. The scientific responsibility is assumed by its authors.
ABBREVIATIONS
- Ag
antigen
- MHC
major histocompatibility complex
- TCR
T cell receptor
- APC
antigen-presenting cell
- APL
altered peptide ligand
- IL
interleukin
- PTK
protein tyrosine kinase
References
- 1.Schwartz R H. J Exp Med. 1996;184:1–8. doi: 10.1084/jem.184.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sloan-Lancaster J, Evavold B D, Allen P M. Nature (London) 1993;363:156–159. doi: 10.1038/363156a0. [DOI] [PubMed] [Google Scholar]
- 3.Jameson S C, Bevan M J. Immunity. 1995;2:1–11. doi: 10.1016/1074-7613(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 4.Rabinowitz J D, Beeson C, Wülfing C, Tate K, Allen P M, Davis M M, McConnell H M. Immunity. 1996;5:125–135. doi: 10.1016/s1074-7613(00)80489-6. [DOI] [PubMed] [Google Scholar]
- 5.DeSilva D R, Urdahl K B, Jenkins M. J Immunol. 1991;147:3261–3267. [PubMed] [Google Scholar]
- 6.Quill H. J Immunol. 1996;156:1325–1327. [PubMed] [Google Scholar]
- 7.MacDonald H R, Lees R K, Baschieri S, Herrmann T, Lussow A R. Immunol Rev. 1993;133:105–117. doi: 10.1111/j.1600-065x.1993.tb01512.x. [DOI] [PubMed] [Google Scholar]
- 8.Andris F, Van Mechelen M, De Mattia F, Baus E, Urbain J, Leo O. Eur J Immunol. 1996;26:1187–1195. doi: 10.1002/eji.1830260534. [DOI] [PubMed] [Google Scholar]
- 9.Thomas R, D’Ari R. Biological Feedback. Boca Raton, FL: CRC Press; 1990. [Google Scholar]
- 10.Shymko R M, De Meyts P, Thomas R. Biochem J. 1997;326:463–469. doi: 10.1042/bj3260463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian D, Weiss A. Curr Opin Cell Biol. 1997;9:205–212. doi: 10.1016/s0955-0674(97)80064-6. [DOI] [PubMed] [Google Scholar]
- 12.Mayer B J. Curr Biol. 1997;7:295–298. doi: 10.1016/s0960-9822(06)00141-2. [DOI] [PubMed] [Google Scholar]
- 13.Kaufman M, Andris F, Leo O. Int Immunol. 1996;8:613–624. doi: 10.1093/intimm/8.4.613. [DOI] [PubMed] [Google Scholar]
- 14.Beverly B, Kang S-M, Lenardo M J, Schwartz R H. Int Immunol. 1992;4:661–671. doi: 10.1093/intimm/4.6.661. [DOI] [PubMed] [Google Scholar]
- 15.Boussiotis V A, Barber D L, Nakarai T, Freeman G J, Gribben J G, Bernstein G M, D’Anrea A D, Ritz J, Nadler L M. Science. 1994;266:1039–1041. doi: 10.1126/science.7973657. [DOI] [PubMed] [Google Scholar]
- 16.Fiering S, Northrop J P, Nolan G P, Mattila P S, Crabtree G R, Herzenberg L A. Genes Dev. 1990;4:1823–1834. doi: 10.1101/gad.4.10.1823. [DOI] [PubMed] [Google Scholar]
- 17.Viola A, Lanzavecchia A. Science. 1996;273:104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 18.Thomas R. J Theor Biol. 1991;153:1–23. [Google Scholar]
- 19.Lyons D S, Lieberman S A, Hampl J, Boniface J J, Chien Y-H, Berg L J, Davis M M. Immunity. 1996;5:53–61. doi: 10.1016/s1074-7613(00)80309-x. [DOI] [PubMed] [Google Scholar]
- 20.Bertoletti A, Sette A, Chisari F V, Penna A, Levrero M, De Carli M, Fiaccadori F, Ferrari C. Nature (London) 1994;369:407–410. doi: 10.1038/369407a0. [DOI] [PubMed] [Google Scholar]
- 21.Thomas R. Lecture Notes Biomath. 1979;29:354–401. [Google Scholar]
- 22.Kaufman M, Urbain J, Thomas R. J Theor Biol. 1985;114:507–534. doi: 10.1016/s0022-5193(85)80042-4. [DOI] [PubMed] [Google Scholar]
- 23.Quill H, Riley M P, Cho E A, Casnellie J E, Reed J C, Torigoe T. J Immunol. 1992;149:2887–2893. [PubMed] [Google Scholar]
- 24.Gajewski T F, Quian D, Fields P, Fitch F W. Proc Natl Acad Sci USA. 1994;91:38–42. doi: 10.1073/pnas.91.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gajewski T F, Fields P, Fitch F W. Eur J Immunol. 1995;25:1836–1842. doi: 10.1002/eji.1830250707. [DOI] [PubMed] [Google Scholar]
- 26.Boussiotis V A, Freeman G J, Berezovskaya A, Barber D L, Nadler L M. Science. 1997;278:124–128. doi: 10.1126/science.278.5335.124. [DOI] [PubMed] [Google Scholar]
- 27.Faith A, Akdis C A, Akdis M, Simon H-U, Blaser K. J Immunol. 1997;159:53–60. [PubMed] [Google Scholar]
- 28.Samelson L E, Davidson W F, Morse H C, Klausner R D. Nature (London) 1986;324:674–676. doi: 10.1038/324674a0. [DOI] [PubMed] [Google Scholar]
- 29.Katagiri T, Urakawa K, Yamanashi Y, Semba K, Takahashi T, Toyoshima K, Yamamoto T, Kano K. Proc Natl Acad Sci USA. 1989;86:10064–10068. doi: 10.1073/pnas.86.24.10064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phipps D J, Yousefi S, Branch D R. Blood. 1997;90:3603–3612. [PubMed] [Google Scholar]
- 31.Wartmann M, Hofer P, Turowski P, Saltiel A R, Hynes N E. J Biol Chem. 1997;272:3915–3923. doi: 10.1074/jbc.272.7.3915. [DOI] [PubMed] [Google Scholar]
- 32.Valitutti S, Lanzavecchia A. Immunol Today. 1997;18:299–304. [PubMed] [Google Scholar]
- 33.McKeithan T W. Proc Natl Acad Sci USA. 1995;92:5042–5046. doi: 10.1073/pnas.92.11.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rabinowitz J D, Beeson C, Lyons D S, Davis M M, McConnell H M. Proc Natl Acad Sci USA. 1996;93:1401–1405. doi: 10.1073/pnas.93.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryan K R, Evavold B D. J Exp Med. 1998;187:89–96. doi: 10.1084/jem.187.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lanoue A, Bona C, von Boehmer H, Sarukhan A. J Exp Med. 1997;185:405–414. doi: 10.1084/jem.185.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hayashi R J, Loh D Y, Kanagawa O, Wang F. J Immunol. 1998;160:33–38. [PubMed] [Google Scholar]