

Abstract
The oncoprotein c-Jun is a component of the activator protein-1 (AP-1) transcription factor complex, which is involved in cellular proliferation, transformation and death. The stabilization of c-Jun is critically important for its function. Phosphorylation of c-Jun by c-Jun N-terminal kinase 1 (JNK1) and extracellular signal-regulated protein kinases (ERKs) reduces c-Jun ubiquitination resulting in increased stabilization of c-Jun. In this report, we showed that C-terminal Src kinase (CSK) binds with and phosphorylates c-Jun at Y26 and Y170. Phosphorylation of c-Jun by CSK, in opposition to JNK1 and ERKs, promoted c-Jun degradation and reduced stability. By promoting c-Jun degradation, CSK helps to maintain a low steady-state level of c-Jun, thereby inhibiting AP-1 activity and cell transformation caused by c-Jun. These results indicated that this function of CSK controls cell proliferation under normal growth conditions and may have implications for CSK loss of function in carcinogenesis.
Keywords: phosphorylation, ubiquitination, transformation, AP-1
Introduction
The activator protein-1 (AP-1) transcription factor is involved in cellular proliferation, transformation and death (1). One of the mechanisms controlling the activation of AP-1 is the regulation of the activity of the various AP-1 protein components, particularly c-Jun. c-Jun is the most prevalent constituent of the AP-1 transcription factor complex, and its mRNA and protein expression are induced in response to various extracellular signals (2). However, following exposure to phorbol esters, growth factors and transforming oncogenes, post-translational modifications, especially phosphorylation, of c-Jun may be more important (3). Thus the identification of kinases responsible for c-Jun phosphorylation is essential. Currently, several kinases have been found that can phosphorylate c-Jun. These kinases include JNKs (4), p34cdc2, ERKs, PKC (5), CKII (6), GSK3 (7), DNA-PK (8) and c-Abl (9). Among these kinases, phosphorylation by JNKs is currently believed to be the most important positive regulator of c-Jun stability. However, identifying kinases that can phosphorylate c-Jun resulting in its down-regulation could be very helpful in understanding and suppressing the mechanism of neoplastic transformation that is caused by c-Jun.
The most well-characterized phosphorylation sites of c-Jun are Ser63 and Ser73 near the N-terminus (10). The phosphorylation of c-Jun by JNKs prevents the ubiquitin-dependent degradation of c-Jun, and this phosphorylation-dependent stabilization contributes to the efficient activation of c-Jun following exposure to growth factors, stress, or other stimulators of c-Jun activity (11). Currently, JNK2 is the only known kinase that functions as a negative regulator of the ubiquitin-dependent degradation of c-Jun under normal growth conditions (12). On the other hand, the positive regulation of the ubiquitin-dependent degradation of c-Jun may be critical in maintaining a low steady-state level of the c-Jun protein, which could result in a suppression of AP-1 activity and inhibition of c-Jun-caused cell transformation.
In this study, we identified CSK as a novel c-Jun kinase. Phosphorylation of c-Jun by CSK, in contrast to JNK1 and ERKs, was found to be a positive regulator of the ubiquitin-dependent degradation of c-Jun, thereby inhibiting c-Jun function under physiologic growth conditions. Our results indicated the existence of a new signaling route for maintaining a low steady-state level of c-Jun and thus, for controlling cell proliferation under normal growth conditions.
Materials and Methods
Plasmids and cell lines
72 kinases were isolated from the A549 cell line by RT-PCR and cloned into the pBIND vector and confirmed by sequencing. The MT35 (His6-tagged c-Jun) plasmids were kindly provided by Dr. Dirk Bohmann (11). csk-wt was kindly provided by Dr. Gordon Langsley (13). Site-directed mutagenesis was performed using the Quickchange kit (Stratagene, Cedar Creek, TX). Mutated c-jun plasmids were subcloned into pcDNA4-HismasA or into pGEX-5x-1 vectors. Expression of recombinant proteins was induced in Escherichia coli BL21 at 30 °C for 0.5–4 h by addition of 0.2 mM IPTG. GST-tagged proteins were affinity purified using glutathione–Sepharose 4B (Amersham Pharmacia Biotech, Piscataway, NJ). His-tagged protein were purified using Ni-NTA agarose (Qiagen, Valencia, CA) after transient transfection with Superfect (Qiagen,Valencia, CA).
Antibodies and co-immunoprecipitation
The c-Jun antibody was purchased from Santa Cruz Biotechnolgy, Inc (Santa Cruz, CA) and the phospho-tyrosine and CSK antibodies were purchased from Upstate Biotechnology, Inc. (Charlottesville, VA). Anti-phospho-Src-528 was purchased from Cell Signaling, Inc. (Beverly, MA). For co-immunoprecipitation, cell lysates were clarified by centrifugation at 4 °C after adding 1.0 μg of the control IgG and 20 μl agarose conjugated protein A-G beads. The supernatant fractions were transferred to new tubes and 2 μg of selected primary antibodies were added and incubated overnight. Agarose conjugated protein A-G beads (20 μl) were added and samples were incubated at 4 °C for 3 h. Samples were centrifuged and washed 3 times with RIPA buffer. The immunoprecipitates were collected by centrifugation and supernatant fractions were discarded. Pellets were resuspended in 40 μl of 2 x sample buffer and separated by 8% SDS-PAGE followed by western blotting.
In vitro kinase assay
Active CSK and 10 X kinase buffer were purchased from Upstate Biotechnology (Charlottesville, VA). c-Jun fusion proteins or peptides (1.5 μg) were mixed respectively together with CSK in 0.5 mM ATP and 1x kinase buffer, and then incubated at 30 °C for 15 min. Samples were separated by SDS-PAGE followed by autoradiography or Western blot analysis. The peptides were synthesized by Invitrogen (Carlsbad, CA).
In vitro ubiquitination assay
The Fraction II Hela Conjugation Kit was purchased from Boston Biochem, Cambridge, MA). In brief, 17 μl of Fraction II Hela (200 μg), 1.25 μl MG-132 (5 μM), 2 μl ubiquitin aldehyde were added in each reaction tube, vortexed and the inhibitor and Fraction II Hela were allowed to pre-incubate for 15 min at room temperature. Then 5 μl of the ubiquitin solutions were added into tubes for in vitro ubiquitination, but not into control tubes. The GST-c-Jun or GST-c-Jun mutant (c-Jun-YY26, 170FF) were added into control or experimental tubes, respectively, and the conjugation reaction was initiated with the addition of 5 μl of the energy regeneration solution (ERS). Samples were vortexed and incubated at 37 degrees centigrade for 1–2 hours. The samples were analyzed by Western blot using an ubiquitin antibody.
Pulse-chase analysis and cycloheximide chase analysis
CSK+/+ cells and CSK−/− cells were plated the day before the experiment. On the day of the experiment, the medium was aspirated from each plate of cells. Cells were washed 2 times in 5 ml warm PBS. Warm DMEM (1 ml, met/cys-free)/5% dialyzed fetal calf serum (FCS) was added and cells incubated for 30 min at 37 °C. Then 50 μl of Tran35S-label (at roughly 10 mCi/ml; equals 500 μCi total) were added to each plate. Cells were placed in a shoebox and incubated for 40 min at 37 °C. The radioactive media were carefully removed. Cells were washed in the plate with 2 ml warm PBS and then PBS was removed. Warm DMEM+FCS+2 mM methionine+2 mM cysteine (5 ml) was added to all plates except the zero (“0”) samples. Samples were collected at 0, 1.5, 3.0 and 4.5 h time points and c-Jun proteins were immunoprecipitated and separated on 10% SDS-polyacrylamide gels. The dried gels were exposed for autoradiography. For cycloheximide chase analysis, CSK+/+ cells and CSK−/− cells were incubated with 100 μg/ml of cycloheximide for various times. Cell lysates were prepared and the expression of c-Jun was analyzed by immunoblotting.
Anchorage-independent transformation assay
Cells (8,000/well) in a 6-well plate were exposed to EGF (10 ng/ml) in 1 ml of 0.33% BME agar containing 10% FBS over 3.5 ml of 0.5% BME agar containing 10% FBS medium. The cultures were maintained in a 37°C, 5% CO2 incubator for 10–14 days, and the induced cell colonies were scored by the methods described (14).
Results
CSK phosphorylates c-Jun in vitro
To identify novel kinases that phosphorylate c-Jun, 72 kinases were cloned and each was individually inserted into the pBIND vector and c-jun was cloned into the pACT vector. Then pACT-c-jun and each individual pBIND-kinase were co-transfected into NIH3T3 cells. The interaction between c-Jun and each kinase was then tested by using the mammalian two-hybridization system (Promega). The luciferase (pG5lu) activity was found to be about 61 times higher in the pBIND-csk and pACT-c-jun co-transfection group (Fig. 1A, Lane 4) compared to the control group transfected with pG5lu alone (Fig. 1A, Lane 1). The luciferase activity of the positive co-transfection control group, pBIND-jnk1 and pACT-c-jun (Fig. 1A, Lane 5), was approximately 97 times higher than the pG5lu control (Fig. 1A, Lane 1). These results indicated that CSK could interact with c-Jun in NIH3T3 cells as determined by the mammalian two-hybridization system.
Figure 1.
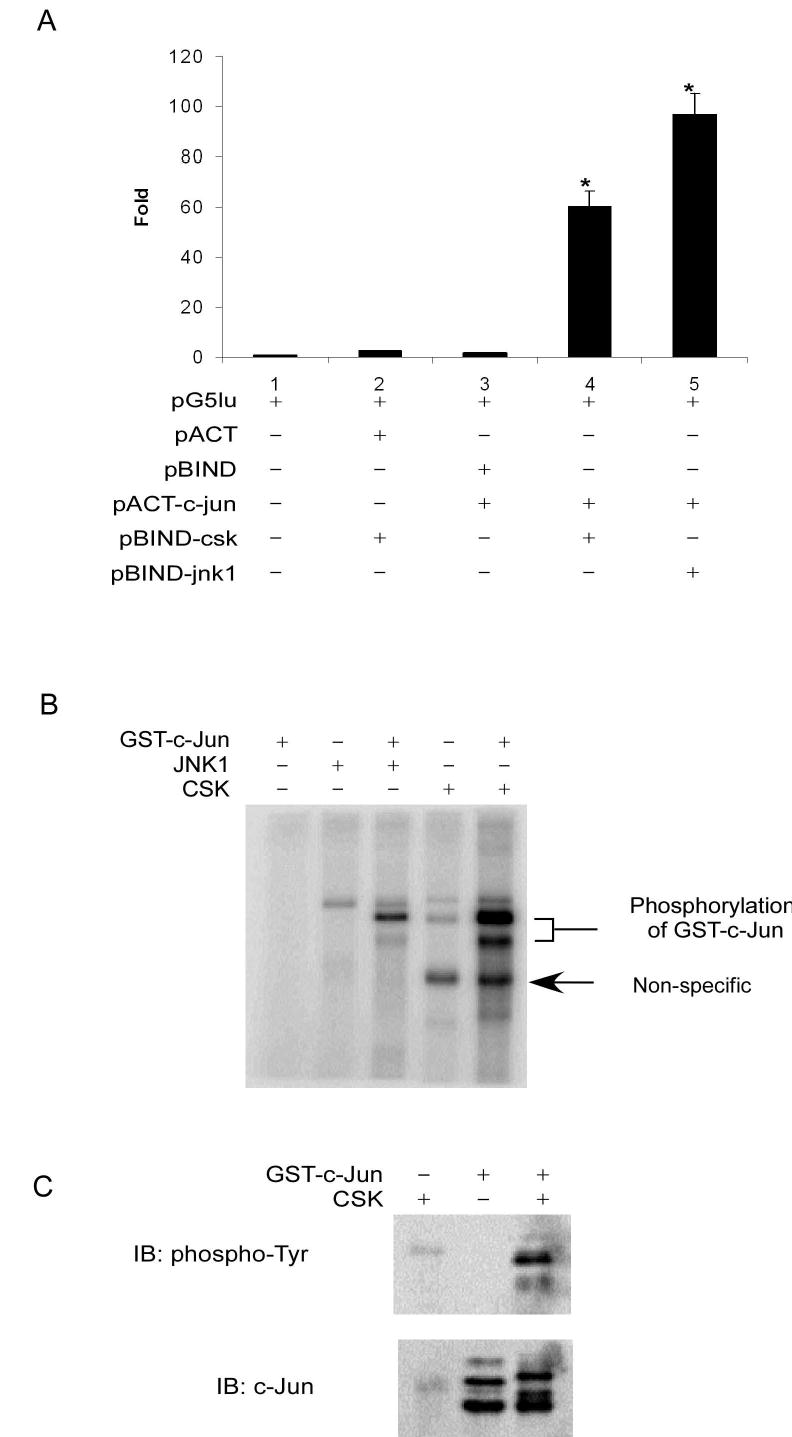
CSK binds with and phosphorylates c-Jun in vitro. A, the interaction between the two test proteins, as GAL4-CSK (pBIND-csk) and VP16-c-Jun (pACT-c-jun) fusion constructs (lane 4), resulted in an increase in luciferase expression compared to the negative controls (lanes 1, 2, and 3). JNK1 (pBIND-jnk1; lane 5) was used as a positive control. Data are represented as means ± S.D. of 3 independent experiments. Significant differences were evaluated using the Student’s t test (*, p < 0.01). B, CSK phosphorylates c-Jun in the in vitro kinase assay in the presence of 32P as visualized by autoradiography. C, CSK phosphorylates c-Jun at tyrosine sites as determined by an in vitro kinase assay with detection by Western blot.
To ascertain whether CSK can phosphorylate c-Jun after binding, in vitro kinase reactions, using active CSK with GST-c-Jun as a substrate, were performed with [γ-32P]ATP. Results indicated that CSK could phosphorylate c-Jun (Fig. 1B, Lane 5). Active JNK1 with GST-c-Jun as substrate served as the positive control (Lane 3). Overall these results indicated that CSK could phosphorylate c-Jun in vitro.
CSK is a protein tyrosine kinase and therefore the kinase reactions of CSK with GST-c-Jun as substrate were performed in the presence of unlabeled ATP. Then Western blot analysis, using a phospho-tyrosine antibody, was performed to determine whether CSK phosphorylated c-Jun at tyrosine sites. Results indicated that CSK phosphorylated c-Jun at tyrosine sites (Fig. 1C). These same samples were used in additional Western blots to determine whether CSK phosphorylates c-Jun at Ser63, Ser73, other serine or threonine sites. In this case, no signal was detected (data not shown) confirming a specificity of CSK for tyrosine phosphorylation of c-Jun in vitro.
CSK phosphorylates c-Jun in vivo
Next, using NIH3T3 cells, we tested whether c-Jun and CSK interacted with each other in vivo. In the mammalian two-hybridization system, c-Jun and CSK were both transported into the nucleus after translation because of the nuclear localization sequence present in the experimental GAL4 or VP16 tags. Therefore testing whether c-Jun and CSK have a similar localization in cells (in vivo) is important to eliminate the possibility of a false positive result. Localization experiments indicated that c-Jun and CSK were located both in the cytosol and the nucleus (Fig. 2A). The similar localization of c-Jun and CSK meant that the possibility exists for them to bind with each other in vivo. This possibility was confirmed with immunoprecipitation experiments in NIH3T3 cells using anti-c-Jun. CSK was detected by Western blot and the result confirmed that CSK interacted with c-Jun in NIH 3T3 cells (Fig. 2B).
Figure 2.
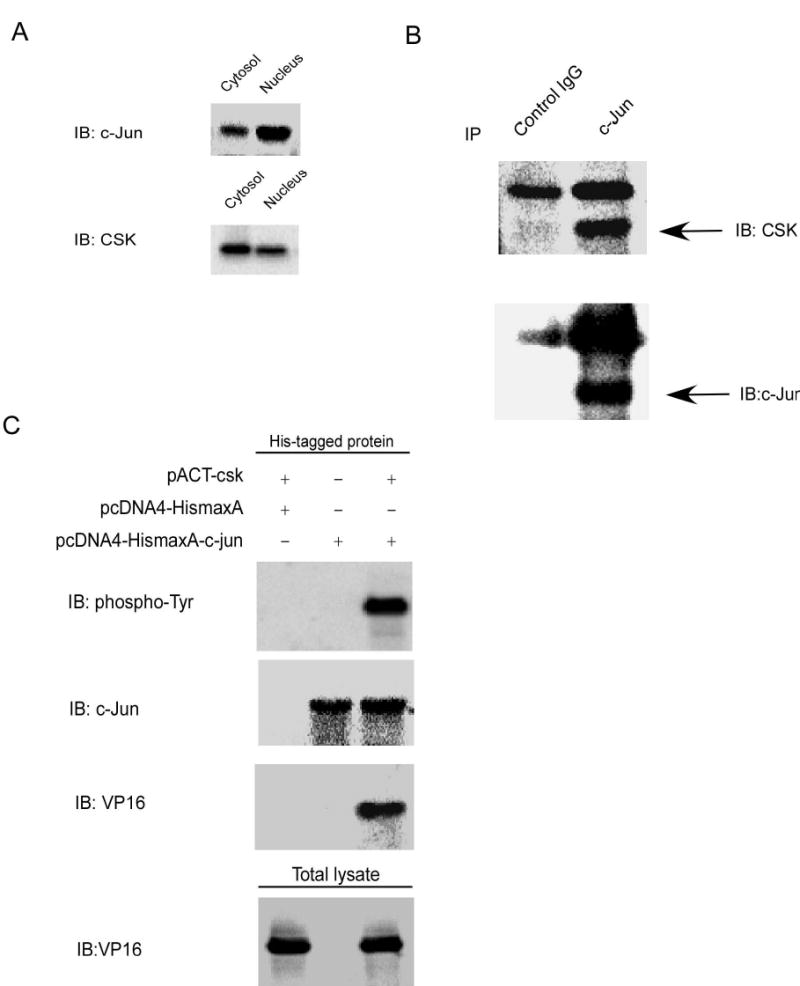
CSK binds with and phosphorylates c-Jun in vivo. A, c-Jun or CSK was expressed both in the nucleus and cytosol. B, CSK binds with c-Jun in vivo. CSK was co-immunoprecipitated from NIH3T3 cells with a c-Jun antibody. C, CSK binds with c-Jun and phosphorylates c-Jun at tyrosine sites. pACT-csk and pcDNA4-HismaxA-c-jun were transiently transfected into 293T cells, and 40 h later His-tagged proteins were purified followed by Western blot analysis.
To verify the in vitro experimental results, which showed that CSK could phosphorylate c-Jun at tyrosine sites, transient transfection experiments were used to test whether CSK could phosphorylate c-Jun in vivo. The pACT-csk and the pcDNA4-HismaxA vector or pcDNA4-HismaxA-c-jun were transiently transfected into HEK 293 cells. Forty hours after transfection, His-tagged proteins were purified by Ni-NTA magnetic beads, and protein expression was detected by Western blot using different antibodies. The results (Fig. 2C) confirmed that CSK could bind with c-Jun and phosphorylate it at tyrosine sites in vivo.
CSK phosphorylates c-Jun at Y26 and Y170
After confirming that CSK phosphorylates c-Jun at tyrosine sites in vitro and in vivo, the next important step is to identify the tyrosine sites of c-Jun that are phosphorylated by CSK. To address this question, we first identified potential tyrosine sites using NetPhos 2.0 (15). c-Jun contains five tyrosine sites (Fig. 3A) and the results of NetPhos 2.0 analysis showed that Y170, Y26, and Y10 are three sites that might be phosphorylated by CSK. We designed six peptides based on a computer analysis (Fig. 3A) to determine the tyrosine phosphorylation sites of c-Jun. Because the Y26 and Y28 sites of c-Jun are close together, we designed three peptides to distinguish between these possible phosphorylation sites. The in vitro kinase assays using different c-Jun peptides as substrates for CSK were carried out in the presence of [γ-32P]ATP. The results indicated that Y26 and Y170 were the two sites in c-Jun that were phosphorylated by CSK, whereas Y10, Y28 or Y190 in c-Jun cannot be phosphorylated by CSK (Fig. 3B). CSK is reported to be a highly selective tyrosine kinase in its phosphorylation of substrates (16). Therefore, we believed that our in vitro kinase assay results are reliable, but they still need to be confirmed using full-length c-Jun. We replaced each of the five tyrosine sites of c-Jun with phenylalanine and also made a double c-Jun mutant with Y26F and Y170F (YY26,170FF). The GST-mutant c-Jun proteins were expressed individually in Escherichia coli BL21 at 30 °C for 0.5–4 h by the addition of 0.2 mM IPTG. The in vitro kinase reactions of active CSK with GST-c-Jun or GST-c-Jun-mutants as substrates were carried out in the presence of [γ-32P]ATP. Samples were analyzed by SDS-PAGE and autoradiography. The results indicated that mutant Y170F or double mutant YY26,170FF c-Jun could not be phosphorylated by CSK, suggesting that Y170 of c-Jun is the most important site phosphorylated by CSK (Fig. 3C). The signal decreased slightly for the c-Jun mutant Y26F compared to GST-c-Jun wild type (Fig. 3C), but the result was not clear enough to conclude whether CSK could phosphorylate Y26 of c-Jun. Thus to determine whether Y26 of c-Jun could be phosphorylated by CSK, we used GST-c-Jun (1–89) (Cell Signaling, Danvers, MA) as the substrate to repeat the in vitro kinase assay, and the result indicated that CSK could indeed phosphorylate c-Jun at this tyrosine site (Fig. 3D) in vitro. The Y26 site is the only possible c-Jun site that can be phosphorylated between c-Jun residues 1 and 89 by CSK. Considering the results of the peptide site mapping and the in vitro kinase assay with GST-c-Jun-mutants; we believed that Y26 of c-Jun can also be phosphorylated by CSK. However, for reasons as yet unclear, the phosphorylation of Y26 was weaker than the phosphorylation of Y170 by CSK in vivo.
Figure 3.
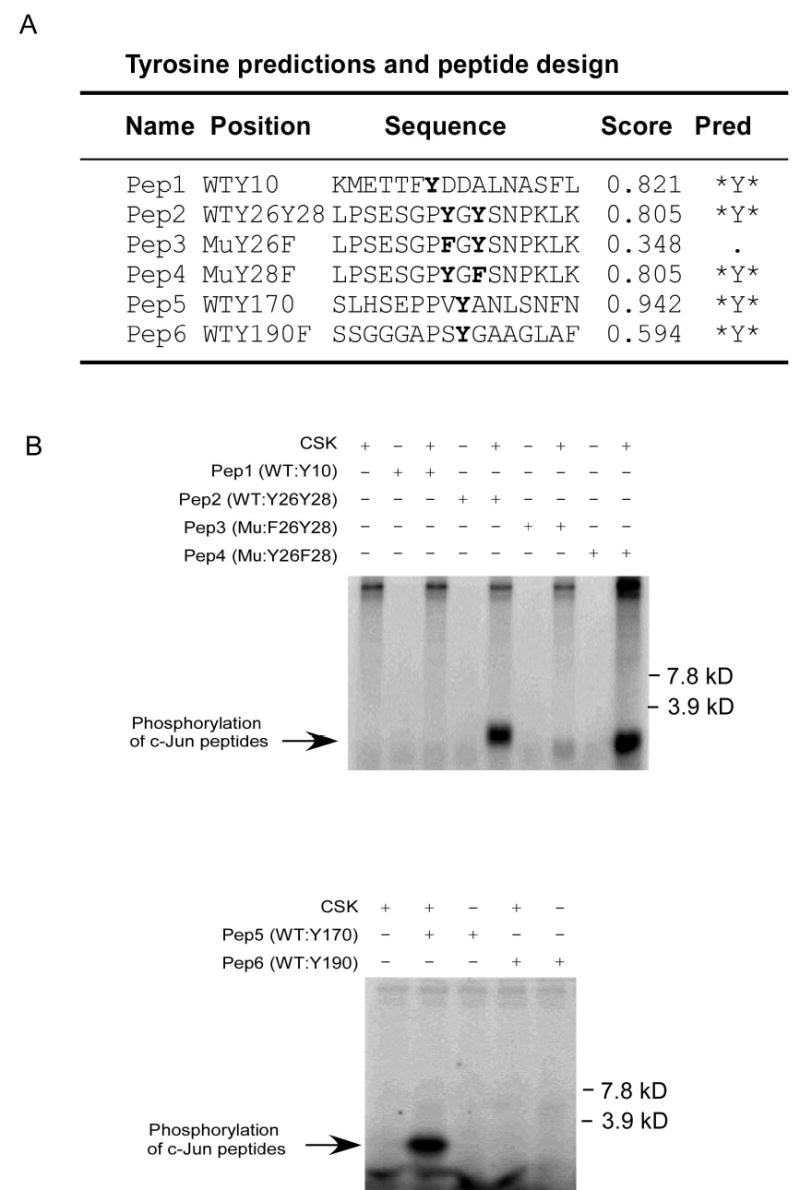
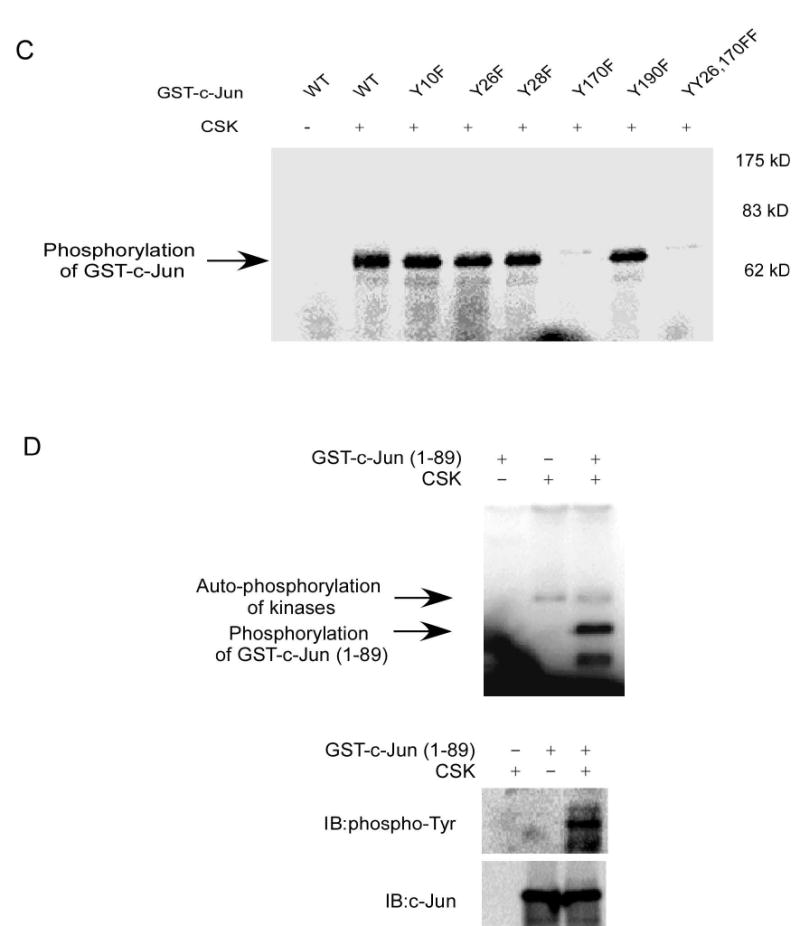
Mapping of the c-Jun tyrosine site(s) phosphorylated by CSK. A, tyrosine sites predicted by NetPhos 2.0 and peptide design following prediction. WT: wild type; Mu: mutant; Score: possibility that the site might be phosphorylated in vivo; Pred: prediction. B, peptide mapping of the tyrosine sites phosphorylated by CSK as determined by an in vitro kinase assay in the presence of 32P as visualized by autoradiography. C, confirmation of the peptide mapping results by using GST-c-Jun or a GST-c-Jun-mutant as substrate in the in vitro kinase assay in the presence of 32P as visualized by autoradiography. D, confirmation of Y26 in c-Jun as a phosphorylation site of CSK using GST-c-Jun (1–89) as a substrate in an in vitro kinase assay in the presence of 32P as visualized by autoradiography or by Western blot.
AP-1 activity and cell transformation are inhibited in CSK+/+ but not in CSK−/− cells
Next, we determined the biological consequence of the phosphorylation of c-Jun by CSK. c-Jun is a signal-transducing component of the AP-1 transcription factor complex, which is very important for cell transformation. In addition, c-Jun itself has transforming activity when overexpressed in chicken embryo fibroblasts (17). Therefore, we tested whether the phosphorylation of c-Jun by CSK had an effect on cell transformation through its effects on AP-1 activity. We examined the role of CSK in cell transformation using CSK+/+and CSK−/− (18) fibroblasts in the presence (10 ng/ml) or absence of EGF. Under normal growth conditions without EGF treatment, the CSK−/− cells generated more and bigger colonies, whereas CSK+/+ cells generated very few colonies (Fig. 4A and C). In addition, in untreated CSK−/− cells, AP-1 activity was much higher than that in CSK+/+ cells (Fig. 4B). These data strongly indicated that CSK−/− cells had a greater transforming ability and higher AP-1 activity. Combining these results with the finding that CSK phosphorylates c-Jun suggested that CSK had the ability to inhibit c-Jun function through its phosphorylation of c-Jun.
Figure 4.
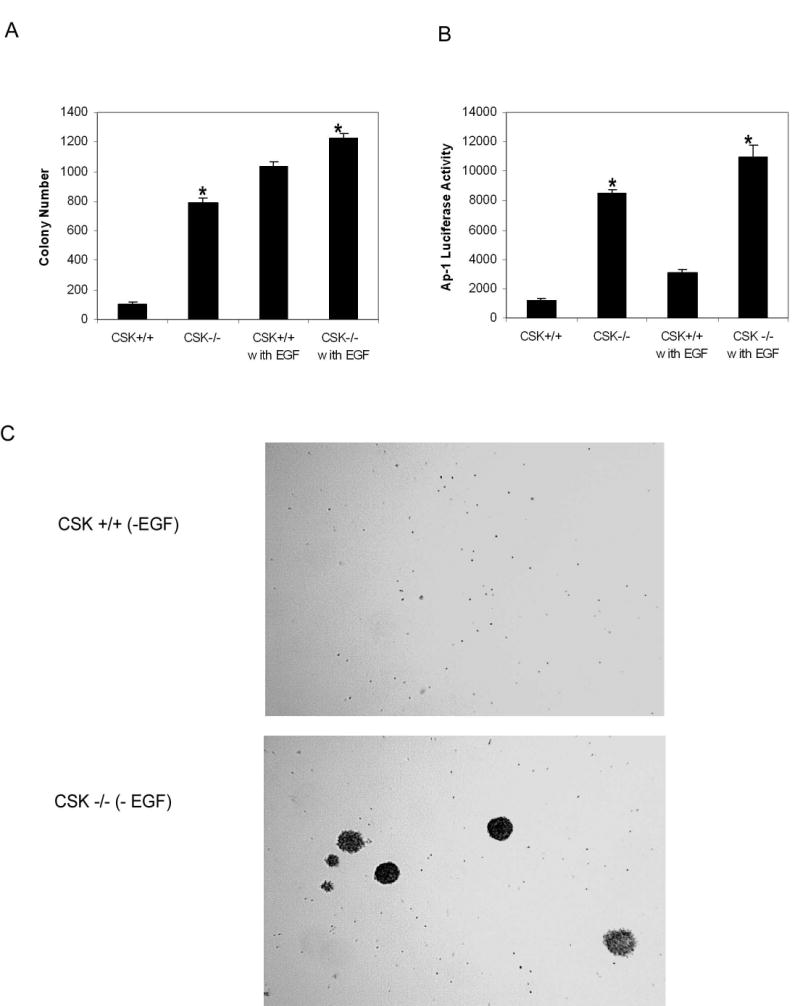
AP-1 activity and cell transformation are inhibited in CSK wildtype cells. A, CSK−/− cells had an increased transforming ability with or without EGF (10 ng/ml) induction. B, CSK−/− cells had an increased AP-1 activity with or without EGF (10 ng/ml) induction. Significant differences were evaluated using the Student’s t test (*, p < 0.01). C, photomicroscopy of anchorage-independent growth of CSK+/+ and CSK−/− cells without EGF treatment.
CSK, but not c-Src, regulates c-Jun total protein levels under normal culture conditions
In our experiments, we noticed that under normal cell culture conditions, c-Jun total protein level was higher (~1.7 fold) in CSK−/− cells compared to CSK+/+ cells. In addition, the phosphorylation of c-Jun at Ser63 and Ser73 was fairly low. On the other hand, phosphorylation of Src at Tyr528 detected at the same time as c-Jun on the same membrane was much stronger in CSK+/+ cells compared to CSK−/− cells (Fig. 5A). In cells treated with EGF, the total c-Jun level was also higher in CSK−/− cells than in CSK+/+ cells, but the basal or EGF-induced phosphorylation of c-Jun at Ser63 and Ser73 in CSK−/− cells showed no dramatic difference between cell lines. CSK was reported to negatively regulate Src function by phosphorylating Src at Tyr528 (19). Thus the higher level of total c-Jun might be caused by a higher activity of Src in CSK−/− cells because of the absence of the inhibitory function of CSK. Thus we examined whether Src had an effect on the total c-Jun protein level and phosphorylation at Ser63 and Ser73 of c-Jun in Src-deficient SYF cells and SYF cells overexpressing c-Src (20). The results indicated that the total levels of c-Jun under normal culture conditions were not different between the two cell lines and the phosphorylation of c-Jun at Ser63 and Ser73 without stimulation was barely detectable. Following treatment with EGF, the total level of c-Jun and the phosphorylation of c-Jun at Ser63 and Ser73 was slightly higher in SYF cells expressing c-Src than in SYF (−Src) cells (Fig. 5B). This suggested that c-Src might be involved in the higher level of c-Jun expression stimulated by EGF treatment but not under normal culture conditions without added EGF. Further, EGF strongly induced c-Jun phosphorylation at Ser63 and Ser73 in CSK−/− cells or SYF (−Src) cells, indicating that phosphorylation of c-Jun at Ser63 and Ser73 occurred independently of CSK or Src kinase activity. The level of phosphorylated tyrosines of c-Jun under normal cell culture conditions was similar in SYF (−Src) cells and SYF (+Src) cells (Fig. 5C). These results suggested that Src was neither related to the total c-Jun protein level nor to the level of phosphorylated tyrosines of c-Jun under normal cell culture conditions.
Figure 5.
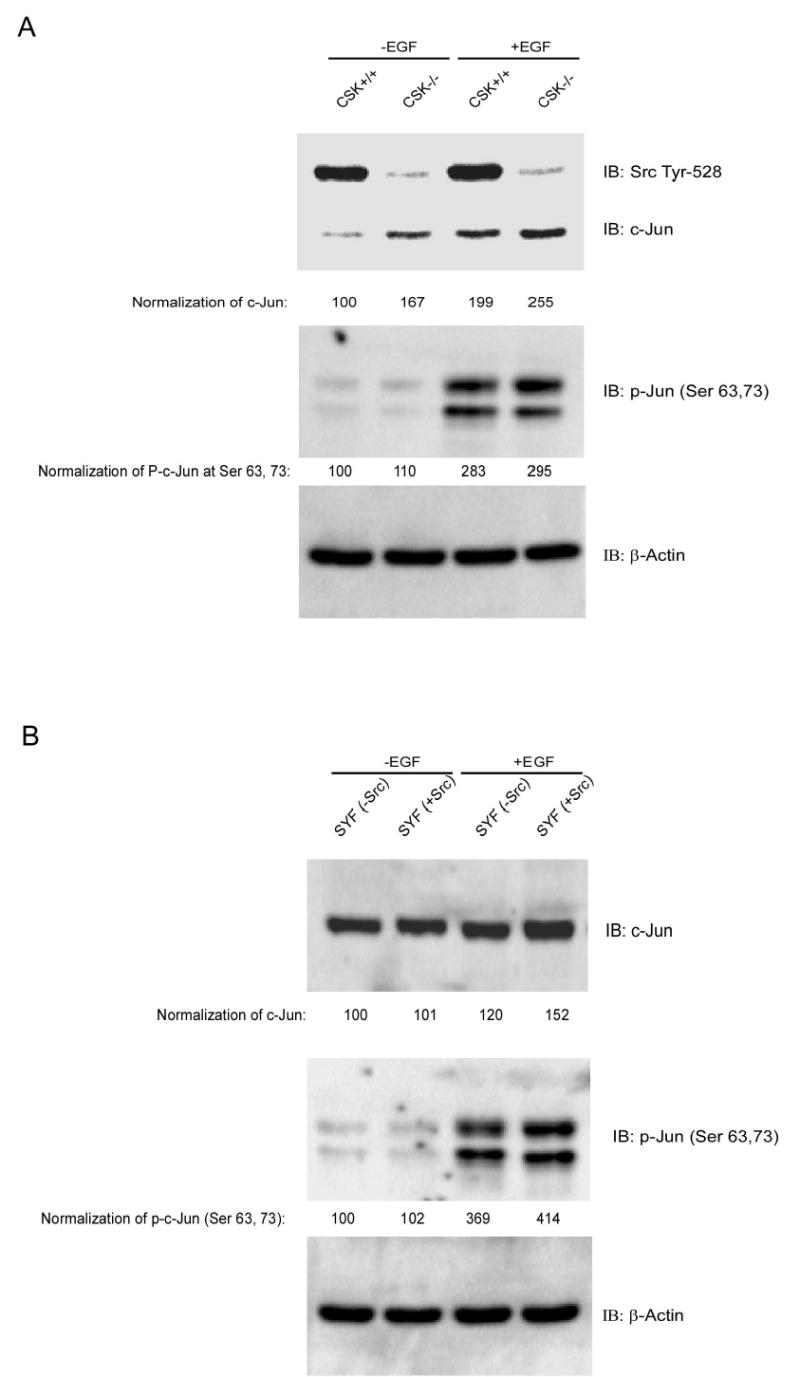
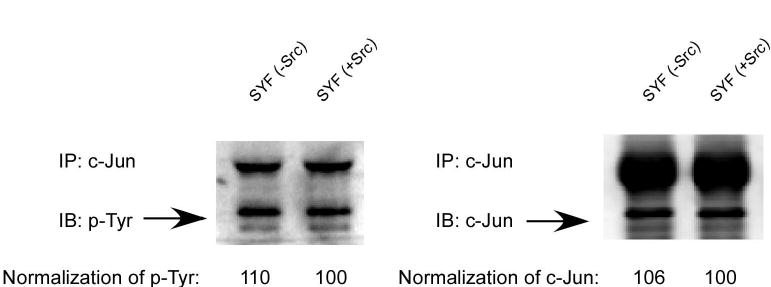
CSK, but not c-Src, is associated with c-Jun total protein level under normal culture conditions. A, total level of c-Jun under normal cell culture conditions is higher in CSK−/− cells than in CSK+/+ cells with or without EGF stimulation. No dramatic difference was observed in c-Jun (Ser63, 73) phosphorylation in wildtype or knockout CSK cells. B, total level of c-Jun expression under normal cell culture conditions is similar in SYF (−Src) cells and SYF (+Src) cells with or without EGF stimulation. c-Jun phosphorylation at serine 63 or 73 in SYF (−Src) cells is slightly weaker than in SYF (+Src) cells. C, the level of phosphoylated tyrosine of c-Jun under normal cell culture conditions is similar in SYF (−Src) cells and SYF (+Src) cells.
Under normal culture conditions, the phosphorylation level of c-Jun at Ser63 and Ser73 in either CSK+/+ or CSK−/− cells was barely detectable and therefore, presumably, the activity of JNKs or ERKs, which phosphorylate c-Jun at Ser 63 and 73, was also weak. Under EGF stimulation conditions, CSK activity is inhibited (21) and ERK or JNK is activated and c-Jun is phosphorylated at Ser 63 and 73. The mechanism of c-Jun regulation by ERKs or JNKs under EGF stimulation is well characterized and CSK activity is inhibited with EGF stimulation. Therefore in our experiments we focused only on the regulation of c-Jun under normal culture conditions.
CSK-mediated phosphorylation promotes c-Jun degradation
The ubiquitin-dependent proteolytic pathway plays an important role in protein degradation, and the activity of many short-lived regulatory proteins comprising numerous transcription factors is controlled by proteolysis through the ubiquitin pathway. The phosphorylation-dependent ubiquitination of c-Jun plays an important role in its function (11, 22, 23). Based on our results thus far, c-Jun may likely be targeted for degradation after phosphorylation by CSK. To test this hypothesis, GST wild type c-Jun and the GST c-Jun double mutant (c-Jun-YY26, 170FF) proteins were used for an in vitro ubiquitination assay. Results (Fig. 6A) indicated that GST wild type c-Jun had a higher level of ubiquitination compared to the GST c-Jun double mutant (c-Jun-YY26, 170FF). To further confirm this result, c-Jun was immunoprecipitated from CSK+/+ cells and CSK−/− cells and used for Western blot analysis to detect c-Jun, ubiquitin, and phosphorylation of tyrosines. Results indicated a much higher level of phosphorylated tyrosines (3.3 fold) and a lower lever of total c-Jun (~0.7 fold) protein present in CSK+/+ cells compared to CSK−/− cells (Fig. 6B). In addition, ubiquitination of c-Jun was greater in CSK+/+ cells and loss of functional CSK inhibited c-Jun ubiquitination (Fig. 6B). One possible reason for the persistent tyrosine phosphorylation signal for c-Jun in CSK−/− cells is most likely due to the presence of other c-Jun tyrosine kinases, such as c-Abl, in these cells (9). These results suggested that CSK-mediated c-Jun phosphorylation promotes ubiquitin-dependent degradation of c-Jun. Next, the stability of c-Jun was analyzed in CSK+/+ cells and CSK−/− cells by immunoblotting after inhibition of protein synthesis with cycloheximide. The c-Jun protein was much more stable in CSK−/− cells than in CSK+/+ cells (Fig. 6C). A pulse-chase experiment was performed to determine the half-life of c-Jun in CSK+/+ cells and CSK−/− cells and the results indicated that c-Jun had a longer half-life in CSK−/− cells than in CSK+/+ cells (Fig. 6D). Our results overall indicated that c-Jun is phosphorylated by CSK in CSK+/+ cells under normal growth conditions and is more likely to be degraded through the ubiquitination pathway (Fig. 6), resulting in a lower total level of c-Jun protein in CSK+/+ cells (Fig. 5A and 6). In contrast, c-Jun cannot be phosphorylated by CSK in CSK−/− cells and is thus less likely to be degraded through the ubiquitination pathway resulting in a higher total level of c-Jun in these cells. The decreased c-Jun protein level associated with CSK phosphorylation is related to c-Jun stability, and therefore is also related to AP-1 activity and cell transformation. Our data therefore suggested that a lower phosphorylation level of c-Jun by CSK is responsible for higher AP-1 activity and cell transformation in CSK−/− cells. However, stimulation with EGF can down regulate the CSK protein and its subsequent activity (21), resulting in increased AP-1 activation and cell transformation caused by c-Jun.
Figure 6.
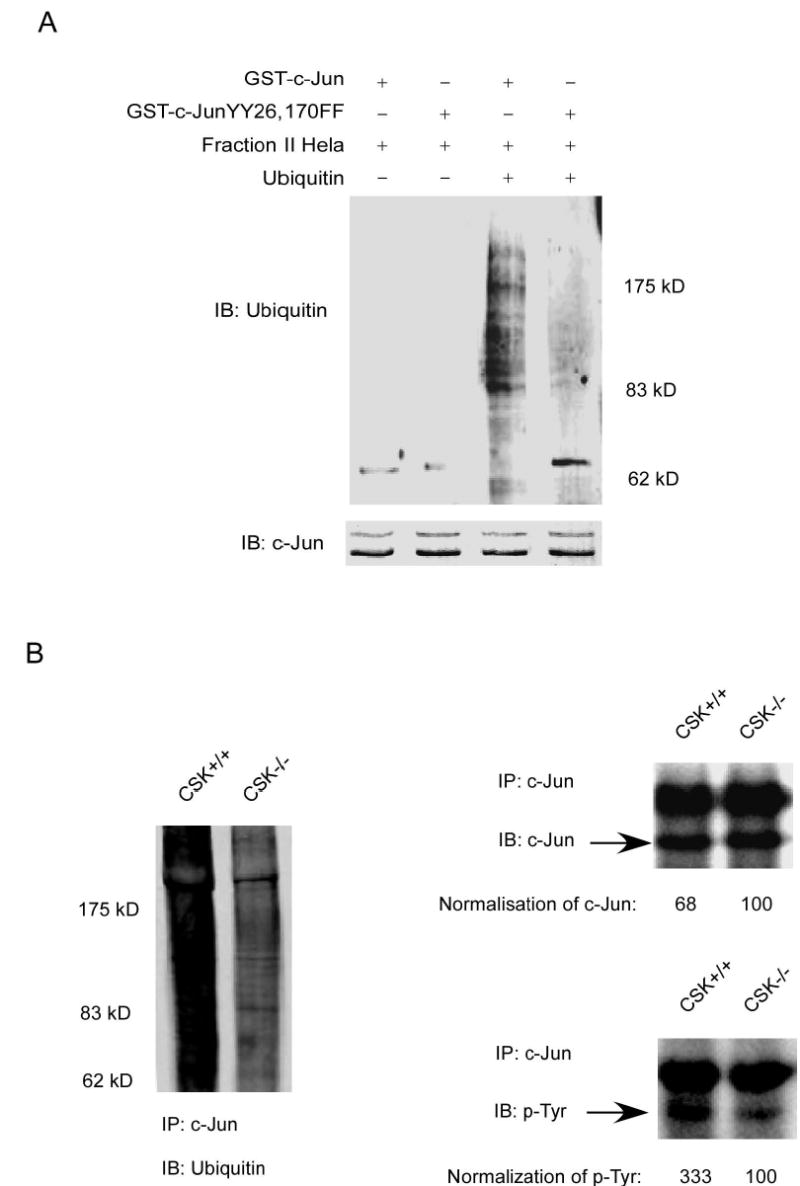
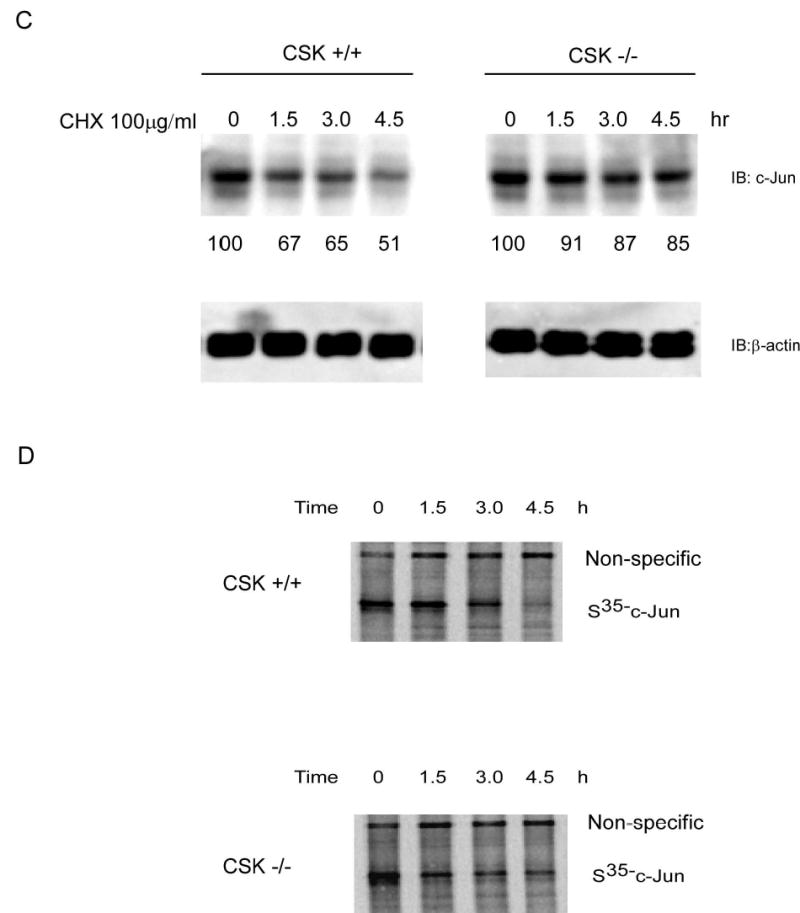
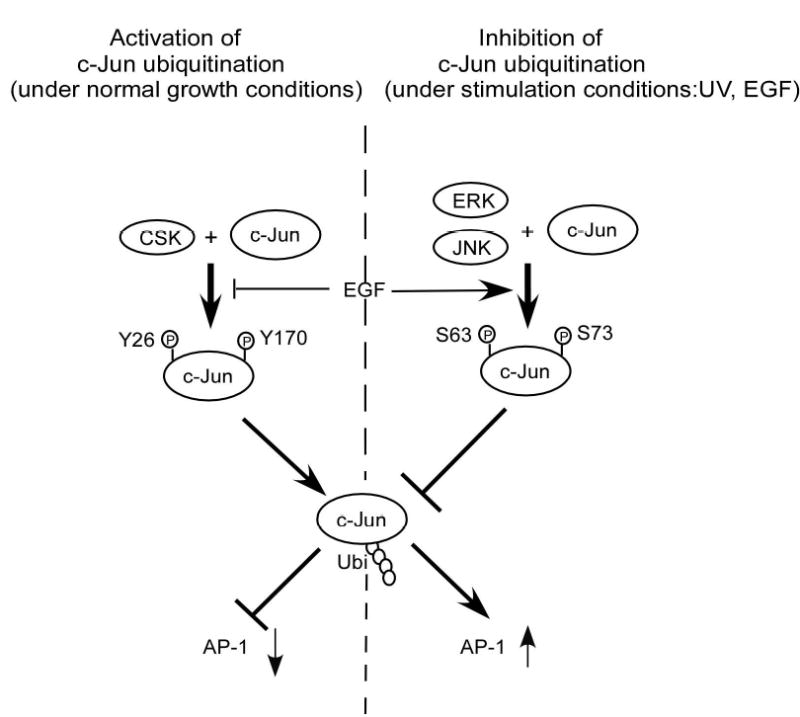
CSK-mediated c-Jun phosphorylation promotes c-Jun degradation. A, in vitro ubiquitination assay for GST wild type c-Jun or GST c-Jun double mutant (YY26, 170FF). B, the level of phosphorylated tyrosine was much higher in CSK+/+ cells than in CSK−/− cells and the total amount of c-Jun protein was also higher in CSK−/− cells than in CSK+/+ cells. Ubiquitination of c-Jun was stronger in CSK+/+ cells than in CSK−/− cells. C, stability of c-Jun was analyzed in CSK+/+ cells or CSK−/− cells at indicated time points by immunoblotting after addition of cycloheximide. D, stability of c-Jun in CSK+/+ cells or CSK−/− cells was analyzed by pulse-chase experiments. E, schematic illustration of the regulation of c-Jun ubiquitination. Under normal growth conditions, CSK phosphorylates c-Jun, which promotes ubiquitin-dependent degradation of c-Jun, resulting in a decrease in AP-1 activity. In contrast, with EGF stimulation, CSK is inhibited and c-Jun is activated by phosphorylation by MAP kinases, which inhibits ubiquitin-dependent degradation of c-Jun, resulting in an increased AP-1 activity.
Discussion
CSK is a protein tyrosine kinase, which selectively phosphorylates Src and negatively regulates Src activity (18, 19, 24). CSK negatively regulates cell proliferation during development in Drosophila by inhibiting Src activity (25). It also negatively regulates organ growth and cell proliferation through inhibition of the Src, Jun N-terminal kinase, and STAT pathways (26). Reduced CSK activities have been found in hepatocellular carcinoma (27) and in colorectal carcinoma (28). Overexpression of the csk gene suppresses tumor metastasis (29, 30) and CSK also inhibits transcriptional activation of AP-1 (13). CSK inhibits cell transformation through negative regulation of Src activity. In this report, we found that under normal culture conditions, CSK inhibits AP-1 activity and cell transformation caused directly by c-Jun. This function of CSK is not dependent on the inhibition of Src kinase.
We found that CSK phosphorylates c-Jun directly. The phosphorylation of c-Jun by CSK promotes ubiquitin-dependent degradation of c-Jun, thereby inhibiting AP-1 activity and cell transformation independent of Src inhibition. Our data can be summarized in a model (Fig. 6E). Under normal growth conditions, CSK phosphorylates c-Jun at Y26 and Y170 and the phosphorylated c-Jun is more likely to be degraded through the ubiquitination pathway. On the other hand, JNKs, ERKs or Src are not activated and therefore c-Jun is not phosphorylated at Ser63 or Ser73. AP-1 activity decreases because of this negative regulation of c-Jun by CSK. By this mechanism, c-Jun is degraded and maintained at a low steady-state level resulting in low AP-1 activity and proper control of cell proliferation under normal growth conditions. Conversely, under stimulation conditions, CSK activity is inhibited (21) and the phosphorylation of c-Jun at Y26 and Y170 is also suppressed. This results in a decrease in the degradation of c-Jun and an increase in total c-Jun protein level. At the same time, JNKs or ERKs are activated and the phosphorylation of c-Jun at Ser63 and Ser73 increases. c-Jun is more stable, total c-Jun protein level increases, and AP-1 activity is higher (Fig. 6E).
The selective degradation of many short-lived proteins in eukaryotic cells is carried out by the ubiquitin system. Ubiquitin-mediated degradation of regulatory proteins plays an important role in the control of numerous processes (31). The liability of signal-dependent transcription factors is crucially important, because changes in their stabilities could have a significant impact on activities of downstream target genes. c-Jun is activated after phosphorylation at Ser63 and Ser73 by MAPKs. The activation of c-Jun after phosphorylation is accompanied by a reduction in c-Jun ubiquitination (11). The δ domain of c-Jun is also suggested to mediate the ubiquitin-dependent c-Jun degradation (22). MAPKs inhibit c-Jun ubiquitination after exposure of cells to growth factors or stress. However, under normal growth conditions, CSK is responsible for c-Jun ubiquitin-dependent degradation and is more important in regulating c-Jun stability while MAPKs are not activated. The function of CSK is suppressed after exposure of cells to growth factors or stress, and the MAPKs are activated and responsible for c-Jun stability. Thus, CSK and MAPK act in opposition to regulate c-Jun ubiquitin-dependent degradation under different circumstances, and by this mechanism, CSK and MAPKs facilitate cellular responses to environmental changes.
c-Jun is suggested to contain an activator domain (A1) that is negatively regulated by a cell type-specific inhibitor (32). The HDAC3-containing repressor complex was identified (33) and a de-repression model, which regulates c-Jun transcription activity, was provided (34). The Y26 and Y170 phosphorylation sites are located in the A1 activation domain in c-Jun. c-Jun has been reported to be phosphorylated at Y170 by c-Abl, and this process is inhibited when JNK is activated (9). Our data strongly suggested that CSK is one of the repressors that can bind with c-Jun at the A1 activation domain, and negatively regulate c-Jun function through ubiquitin-dependent degradation thereby regulating AP-1 activity. The repression caused by CSK can be relieved by stimulation, such as EGF treatment. A low level of CSK has been observed in carcinogenesis, and our model suggests a new mechanism by which cell transformation can be caused by loss of functional CSK. The loss of CSK results in an increased AP-1 activity by allowing c-Jun to escape the ubiquitin-dependent degradation that would normally follow its phosphorylation by CSK.
Acknowledgments
We thank Dr. Dirk Bohmann and Dr. Gordon Langsley for kindly providing the plasmids and cell lines. We appreciate the contributions and helpful discussions of various members in the Zigang Dong Lab. We also thank Ms. Andria Hansen for secretarial assistance.
Footnotes
Note: The University of Minnesota is an equal opportunity educator and employer.
Grant support: This work was supported in part by The Hormel Foundation and National Institutes of Health grants CA77646, CA88961, CA111356, and CA27502.
References
- 1.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–6. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 2.Lamph WW, Wamsley P, Sassone-Corsi P, Verma IM. Induction of proto-oncogene JUN/AP-1 by serum and TPA. Nature. 1988;334:629–31. doi: 10.1038/334629a0. [DOI] [PubMed] [Google Scholar]
- 3.Binetruy B, Smeal T, Karin M. Ha-Ras augments c-Jun activity and stimulates phosphorylation of its activation domain. Nature. 1991;351:122–7. doi: 10.1038/351122a0. [DOI] [PubMed] [Google Scholar]
- 4.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–48. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 5.Baker SJ, Kerppola TK, Luk D, et al. Jun is phosphorylated by several protein kinases at the same sites that are modified in serum-stimulated fibroblasts. Mol Cell Biol. 1992;12:4694–705. doi: 10.1128/mcb.12.10.4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin A, Frost J, Deng T, et al. Casein kinase II is a negative regulator of c-Jun DNA binding and AP-1 activity. Cell. 1992;70:777–89. doi: 10.1016/0092-8674(92)90311-y. [DOI] [PubMed] [Google Scholar]
- 7.Boyle WJ, Smeal T, Defize LH, et al. Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell. 1991;64:573–84. doi: 10.1016/0092-8674(91)90241-p. [DOI] [PubMed] [Google Scholar]
- 8.Bannister AJ, Gottlieb TM, Kouzarides T, Jackson SP. c-Jun is phosphorylated by the DNA-dependent protein kinase in vitro; definition of the minimal kinase recognition motif. Nucleic Acids Res. 1993;21:1289–95. doi: 10.1093/nar/21.5.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barila D, Mangano R, Gonfloni S, et al. A nuclear tyrosine phosphorylation circuit: c-Jun as an activator and substrate of c-Abl and JNK. Embo J. 2000;19:273–81. doi: 10.1093/emboj/19.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Derijard B, Hibi M, Wu IH, et al. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–37. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 11.Musti AM, Treier M, Bohmann D. Reduced ubiquitin-dependent degradation of c-Jun after phosphorylation by MAP kinases. Science. 1997;275:400–2. doi: 10.1126/science.275.5298.400. [DOI] [PubMed] [Google Scholar]
- 12.Sabapathy K, Hochedlinger K, Nam SY, Bauer A, Karin M, Wagner EF. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol Cell. 2004;15:713–25. doi: 10.1016/j.molcel.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 13.Baumgartner M, Angelisova P, Setterblad N, et al. Constitutive exclusion of Csk from Hck-positive membrane microdomains permits Src kinase-dependent proliferation of Theileria-transformed B lymphocytes. Blood. 2003;101:1874–81. doi: 10.1182/blood-2002-02-0456. [DOI] [PubMed] [Google Scholar]
- 14.Colburn NH, Wendel EJ, Abruzzo G. Dissociation of mitogenesis and late-stage promotion of tumor cell phenotype by phorbol esters: mitogen-resistant variants are sensitive to promotion. Proc Natl Acad Sci U S A. 1981;78:6912–6. doi: 10.1073/pnas.78.11.6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–62. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 16.Sondhi D, Xu W, Songyang Z, Eck MJ, Cole PA. Peptide and protein phosphorylation by protein tyrosine kinase CSK: insights into specificity and mechanism. Biochemistry. 1998;37:165–72. doi: 10.1021/bi9722960. [DOI] [PubMed] [Google Scholar]
- 17.Bos TJ, Monteclaro FS, Mitsunobu F, et al. Efficient transformation of chicken embryo fibroblasts by c-Jun requires structural modification in coding and noncoding sequences. Genes Dev. 1990;4:1677–87. doi: 10.1101/gad.4.10.1677. [DOI] [PubMed] [Google Scholar]
- 18.Imamoto A, Soriano P. Disruption of the csk gene, encoding a negative regulator of Src family tyrosine kinases, leads to neural tube defects and embryonic lethality in mice. Cell. 1993;73:1117–24. doi: 10.1016/0092-8674(93)90641-3. [DOI] [PubMed] [Google Scholar]
- 19.Nada S, Okada M, MacAuley A, Cooper JA, Nakagawa H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature. 1991;351:69–72. doi: 10.1038/351069a0. [DOI] [PubMed] [Google Scholar]
- 20.Klinghoffer RA, Sachsenmaier C, Cooper JA, Soriano P. Src family kinases are required for integrin but not PDGFR signal transduction. Embo J. 1999;18:2459–71. doi: 10.1093/emboj/18.9.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ren Y, Meng S, Mei L, Zhao ZJ, Jove R, Wu J. Roles of Gab1 and SHP2 in paxillin tyrosine dephosphorylation and Src activation in response to epidermal growth factor. J Biol Chem. 2004;279:8497–505. doi: 10.1074/jbc.M312575200. [DOI] [PubMed] [Google Scholar]
- 22.Treier M, Staszewski LM, Bohmann D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell. 1994;78:787–98. doi: 10.1016/s0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- 23.Musti AM, Treier M, Peverali FA, Bohmann D. Differential regulation of c-Jun and JunD by ubiquitin-dependent protein degradation. Biol Chem. 1996;377:619–24. doi: 10.1515/bchm3.1996.377.10.619. [DOI] [PubMed] [Google Scholar]
- 24.Thomas SM, Soriano P, Imamoto A. Specific and redundant roles of Src and Fyn in organizing the cytoskeleton. Nature. 1995;376:267–71. doi: 10.1038/376267a0. [DOI] [PubMed] [Google Scholar]
- 25.Pedraza LG, Stewart RA, Li DM, Xu T. Drosophila Src-family kinases function with Csk to regulate cell proliferation and apoptosis. Oncogene. 2004;23:4754–62. doi: 10.1038/sj.onc.1207635. [DOI] [PubMed] [Google Scholar]
- 26.Read RD, Bach EA, Cagan RL. Drosophila C-terminal Src kinase negatively regulates organ growth and cell proliferation through inhibition of the Src, Jun N-terminal kinase, and STAT pathways. Mol Cell Biol. 2004;24:6676–89. doi: 10.1128/MCB.24.15.6676-6689.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masaki T, Okada M, Tokuda M, et al. Reduced C-terminal Src kinase (Csk) activities in hepatocellular carcinoma. Hepatology. 1999;29:379–84. doi: 10.1002/hep.510290239. [DOI] [PubMed] [Google Scholar]
- 28.Cam WR, Masaki T, Shiratori Y, et al. Reduced C-terminal Src kinase activity is correlated inversely with pp60(c-src) activity in colorectal carcinoma. Cancer. 2001;92:61–70. doi: 10.1002/1097-0142(20010701)92:1<61::aid-cncr1292>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 29.Nakagawa T, Tanaka S, Suzuki H, et al. Overexpression of the csk gene suppresses tumor metastasis in vivo. Int J Cancer. 2000;88:384–91. [PubMed] [Google Scholar]
- 30.Rengifo-Cam W, Konishi A, Morishita N, et al. Csk defines the ability of integrin-mediated cell adhesion and migration in human colon cancer cells: implication for a potential role in cancer metastasis. Oncogene. 2004;23:289–97. doi: 10.1038/sj.onc.1207041. [DOI] [PubMed] [Google Scholar]
- 31.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 32.Baichwal VR, Tjian R. Control of c-Jun activity by interaction of a cell-specific inhibitor with regulatory domain delta: differences between v- and c-Jun. Cell. 1990;63:815–25. doi: 10.1016/0092-8674(90)90147-7. [DOI] [PubMed] [Google Scholar]
- 33.Weiss C, Schneider S, Wagner EF, Zhang X, Seto E, Bohmann D. JNK phosphorylation relieves HDAC3-dependent suppression of the transcriptional activity of c-Jun. Embo J. 2003;22:3686–95. doi: 10.1093/emboj/cdg364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiss C, Bohmann D. Deregulated repression of c-Jun provides a potential link to its role in tumorigenesis. Cell Cycle. 2004;3:111–3. [PubMed] [Google Scholar]