

Abstract
Protein kinase autophosphorylation of activation segment residues is a common regulatory mechanism in phosphorylation-dependent signalling cascades. However, the molecular mechanisms that guarantee specific and efficient phosphorylation of these sites have not been elucidated. Here, we report on three novel and diverse protein kinase structures that reveal an exchanged activation segment conformation. This dimeric arrangement results in an active kinase conformation in trans, with activation segment phosphorylation sites in close proximity to the active site of the interacting protomer. Analytical ultracentrifugation and chemical cross-linking confirmed the presence of dimers in solution. Consensus substrate sequences for each kinase showed that the identified activation segment autophosphorylation sites are non-consensus substrate sites. Based on the presented structural and functional data, a model for specific activation segment phosphorylation at non-consensus substrate sites is proposed that is likely to be common to other kinases from diverse subfamilies.
Keywords: activation segment, autophosphorylation, DAPK3, LOK, SLK
Introduction
Protein kinases are critical regulators for most cellular processes and misregulation of their activity is frequently implicated in disease development. Consequently, kinase activity is tightly controlled by a multitude of regulatory mechanisms. Many kinases are regulated by phosphorylation of one or more key residues within their activation segment, a region of typically 20–40 residues located within the large kinase lobe. This regulatory element consists of a magnesium-binding site (Asp-Phe-Gly (DFG) motif), a short β-strand (β9), the activation loop and the P+1 loop. The activation segment shows considerable conformational diversity, but structural comparisons have shown that two invariable anchor points, formed by the DFG motif and the central portion of the P+1 loop and helix αEF, lock both termini of this regulatory element in active kinases (Nolen et al, 2004).
For kinases that require phosphorylation for activity, the phosphate moiety at the primary phosphorylation site stabilizes the activation segment in a conformation suitable for substrate binding, whereas in the inactive unphosphorylated state, the activation segment is usually largely unstructured (Goldberg et al, 1996; Sicheri et al, 1997; Meng et al, 2002). The function of secondary phosphorylation sites in activation segments is less clear and may vary dramatically between different kinases. For instance, phosphorylation at a secondary site is required for the activity of extracellular signal-regulated kinase 2, whereas in glycogen synthase kinase 3 (GSK3) the catalytic activity increases only moderately. A further function of secondary phosphorylation sites in substrate recruitment has been suggested (Prowse and Lew, 2001; Dajani et al, 2003; Hu et al, 2003).
Many kinases autoactivate by phosphorylating their own activation segment. However, little is known about the underlying recognition events that make such phosphorylation specific and efficient. Especially unclear is the mechanism by which an unphosphorylated and thus, inactive kinase efficiently autophosphorylates its own activation segment. It is currently assumed that the activation segment of unphosphorylated kinases transiently adopts an active conformation, resulting in trans-phosphorylation of neighbouring kinase molecules. Intuitively, a high local concentration resulting from dimerization would make such autoactivation events more probable. Ligand-induced receptor dimerization has been suggested as a requirement for rapid autoactivation of receptor tyrosine kinases (Weiss and Schlessinger, 1998; Mellado et al, 2001). Alternatively, oligomerization as described for many cytoplasmic kinases, may also facilitate the necessary high local concentration of kinase domains. For example, calmodulin-dependent protein kinase 2 (CaMK2) forms dodecameric assemblies that autoactivate rapidly by trans-phosphorylation of neighbouring catalytic domains as a response to high Ca2+/calmodulin concentrations (Rosenberg et al, 2005, 2006).
However, many autophosphorylation sites in kinase activation segments are non-consensus substrate sites, suggesting that the mechanism of autophosphorylation is different from trans-phosphorylation of substrate molecules. Interestingly, an intramolecular transitional state has been suggested to explain autophosphorylation of a tyrosine residue within the activation segments of the S/T kinases dual-specificity Yak1-related kinase (DYRK) and GSK3 (Lochhead et al, 2005, 2006). The recently determined crystal structure of checkpoint kinase 2 (CHK2) revealed a domain-exchanged activation segment forming an active kinase in a trans configuration, suggesting for the first time a role of activation segment dimerization in CHK2 activation (Oliver et al, 2006, 2007).
Here, we report three high-resolution structures of catalytic domains from diverse kinase groups showing a similar dimeric arrangement. All three kinases are associated with important cellular functions. STE20-like kinase (SLK) is a microtubule-associated protein involved in the regulation of the cell cycle, cell adhesion and spreading (Burakov et al, 2005; O'Reilly et al, 2005), lymphocyte-originated kinase (LOK) has been described as an activator of Polo kinase (Walter et al, 2003) and death-associated protein kinase 3 (DAPK3) is a cytoskeleton-associated tumour suppressor involved in the regulation of autophagy and sensitization to pro-apoptotic signals (Shani et al, 2004).
Intriguingly, the three kinases have properties similar to CHK2, in that they autophosphorylate their respective activation segments despite the weak similarity to substrate recognition motifs. Analytical ultracentrifugation (AUC) demonstrated that all four catalytic domains self-associate in solution, whereas a site-directed mutant of SLK (Q185P), which is monomeric in solution, showed that dimerization is required for autophosphorylation. The presented structural and biophysical data suggest a model for effective phosphorylation of activation segment residues shared by largely diverse kinases.
Results
In this study, we used comparative structural analysis together with biophysical characterization to elucidate the mechanism of autophosphorylation of kinase activation segments. The STE20 family members SLK and LOK are large proteins (>1000 residues, Figure 1A) with long sequence stretches predicted to be unstructured. Optimal protein expression and stability was observed for constructs comprising a minimal kinase catalytic domain (residues 19–321, 17–317, 9–278 and 205–543, for SLK, LOK, DAPK3 and CHK2, respectively). All constructs were purified to homogeneity as confirmed by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) and mass spectrometry (MS). All kinases were catalytically active and autophosphorylated rapidly (data not shown). The SLK, LOK and DAPK3 kinase domains yielded crystals that diffracted to high resolution, whereas the structure of CHK2 has been reported previously (Oliver et al, 2006).
Figure 1.

Domain organization, structural overview and inhibitors used in this study. (A) Domain organization of the four kinases. The kinase domain is highlighted in red and has been marked with KD, coiled-coil domains are shown in blue. The FHA domain in CHK2 is shown in orange. (B) Schematic representation of the structure of SLK. The secondary structure elements of the diphosphorylated form of SLK in complex with K00546 are shown. The N-terminal lobe (green), C-terminal lobe (red) and activation segment region (blue) are highlighted along with the observed phosphorylation sites. (C) Chemical structures of inhibitors used for successful co-crystallization. K00225 (Pyridone 6), (2-(1,1-dimethylethyl)-9-fluoro-3,6-dihydro-7H-benz[h]-imidaz(4,5-f]isoquinolin-7-one); K00546, 5-amino-3-(4-sulphamoyl-phenylamino)-(1,2,4]triazole-1-carbothioic acid (2,6-difluoro-phenyl)-amide; K00606, (4-(4-(5-cyclopropyl-1H-pyrazol-3-ylamino)-quinazolin-2-ylamino)-phenyl)-acetonitrile; K00593 (SU11724), 3-(1-(3,5-dimethyl-4-(4-methyl-piperazine-1-carbonyl)-2H-pyrrol-2-yl)-meth-(Z)-ylidene)-2-oxo-2,3-dihydro-1H-indole-5-sulphonic acid (3-chloro-phenyl)-methyl-amide.
Overall structures of SLK
Four crystal structures were determined for SLK, all of which were refined to acceptable R-values and geometry (Table I). Three structures contain ATP-mimetic inhibitors, whereas the fourth SLK structure comprises the apo-enzyme in its monophosphorylated (∼70% occupancy) state. All SLK structures adopt an active, closed conformation showing similar orientation of the kinase lobes, as well as similar overall positions of the main chain atoms (r.m.s.d., 0.15–0.32 Å; Figure 1B). Phosphorylation sites, detected by MS spectra, were clearly identified in electron density maps at T183 and S189. Neither site was completely phosphorylated, so occupancies of the two sites were estimated during refinement (Table I). The structure in the presence of the triazole-1-carbothioamide inhibitor K00546 (Figure 1C; Supplementary Figure S1) was determined in the unphosphorylated as well as the diphosphorylated state. This inhibitor binds to the SLK ATP-binding site forming three hydrogen bonds with the kinase hinge residues E109 and C111. The sulphamoyl moiety of K00546 also interacts with the main chain of L40 (Supplementary Figure S1A). The pyrazole-quinazoline inhibitor K00606 ((4-(4-(5-cyclopropyl-1H-pyrazol-3-ylamino)-quinazolin-2-ylamino)-phenyl)-acetonitrile) shows remarkable shape complementarities with the SLK active site and forms three hydrogen bonds with main chain atoms of the hinge region (Supplementary Figure S1B).
Table 1.
Structural refinement and data collection
| Data collection | SLK | SLK-monoP | SLK-Apo | SLK-diP | LOK | DAPK3 |
|---|---|---|---|---|---|---|
| PDB code | 2J51 | 2UV2 | 2JFM | 2JFL | 2J7T | 2J90 |
| Ligand | K00546 | K00606 | None | K00546 | K00593 | K00225 |
| Phosphorylation state observed | None | T183∼50% | T183∼70% | T183∼70%, S189 ∼30% | None | S50, T265 100% |
| Spacegroup | P6122 | P6122 | P6122 | P6122 | I222 | P43212 |
| Cell dimensions (Å) | a=b=101.2, c=177.2 | a=b=101.3, c=176.5 | a=b=102.1, c=177.0 | a=b=101.37, c=177.2 | a=49.2, b=112.9, c=133.8 | a=b=95.8, c=150.9 |
| α, β, γ | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 2.1 | 2.3 | 2.85 | 2.2 | 2.0 | 2.0 |
| Unique observations | 32094 | 24390 | 13398 | 27969 | 25648 | 48089 |
| Completenessa (%) | 100 (100) | 99.3 (95.2) | 100 (100) | 99.5 (97.3) | 99.9 (99.8) | 99.8 (100) |
| Redundancya | 8.9 (8.9) | 8.4 (6.0) | 4.4 (4.5) | 5.9 (4.1) | 5.3 (5.3) | 5.6 (5.6) |
| Rmergea | 0.08 (0.81) | 0.10 (0.86) | 0.08 (0.76) | 0.09 (0.71) | 0.09 (0.82) | 0.08 (0.67) |
| I/σIa | 16 (2.7) | 14.2 (2.1) | 12.5 (2.0) | 13 (2.0) | 11.4 (2.3) | 8.7 (2.0) |
| Refinement | ||||||
| Resolution (Å) | 60–2.1 | 60–2.3 | 30–2.85 | 50–2.2 | 55–2.0 | 50–2.0 |
| Reflections (Rfree) | 30437 (1589) | 23095 (1232) | 12654 (680) | 26523 (1377) | 24339 (1308) | 45599 (2433) |
| Rwork/Rfree (%) | 20/23.2 | 19.8/23.4 | 23.0/27.1 | 20.3/24.2 | 20.4/23.5 | 18.7/22.2 |
| Atoms (P/L/W/O)b | 2250/28/151/19 | 2249/29/148/26 | 2229/–/9/8 | 2274/28/146/24 | 2157/40/124/9 | 4242/46/329/19 |
| B-factors (P/L/W/O)b (Å2) | 51/51/46/58 | 55/51/48/61 | 89/–/64/113 | 58/58/52/68 | 43/38/38/57 | 41/36/45/45 |
| r.m.s.d bonds (Å) | 0.013 | 0.011 | 0.013 | 0.012 | 0.010 | 0.009 |
| r.m.s.d angles (deg) | 1.54 | 1.282 | 1.375 | 1.548 | 1.236 | 1.147 |
| Ramachandranc | ||||||
| Favoured (%) | 97.6 | 97.2 | 94.0 | 97.2 | 98.6 | 98.1 |
| Allowed (%) | 100 | 100 | 99.6 | 100 | 100 | 100 |
| aValues in brackets represent statistics for highest-resolution shells. | ||||||
| b(P/L/W/O): protein atoms/ligand atoms/water/other. Average B-factors include translation/libration/screw (TLS) contributions. | ||||||
| cMolprobity server analysis (http://molprobity.biochem.duke.edu/). | ||||||
Inhibitor-bound structure of LOK
The structure of unphosphorylated LOK was determined in complex with the pyrrole-indolinone inhibitor SU11274 (K00593, Figure 1C). It was well-ordered except for the loop segment connecting β3 strand with helix αC (residues 69–72) and the tip of the activation segment (residues 189–193). K00593 forms four hydrogen bonds with main chain LOK residues: two with the kinase hinge region, one with D175 and one with A117 (Supplementary Figure S1C).
Structure of inhibitor-bound DAPK3
The DAPK3 structure was determined in the presence of Pyridone 6 (K00225, Figure 1C), a potent, ATP-competitive inhibitor originally described to inhibit Janus protein tyrosine kinases (Pedranzini et al, 2006). The crystallized protein was diphosphorylated (>90%) as shown by MS. The phosphorylation sites at S50 and T265 were clearly identifiable in the electron density. T265 has been reported as an autophosphorylation site in vitro and in vivo, and is important for catalytic activity (Graves et al, 2005). T265 caps the N terminus of the DAPK3 C-terminal helix. Its phosphate moiety forms a hydrogen bond with the N267 backbone nitrogen, thereby presumably stabilizing this helix. The observed autophosphorylation of S50 has not been reported before. The region around S50 exhibits different conformations in each molecule of the DAPK3 dimer. In one molecule, the phosphate moiety forms an intramolecular hydrogen bond with R48 in helix αC, which may contribute to positioning and stabilization of this catalytically important helix. In the other molecule, the phosphate moiety is projected towards the dimer interface and forms intermolecular hydrogen bonds with R134 and K167. K00225 forms two hydrogen bonds with the DAPK3 hinge residues E94 and V96 (Supplementary Figure S1D).
Activation segment exchange
The most striking feature of each of the structures is an exchanged activation segment that interacts with either a symmetry-related molecule or a second molecule in the asymmetric unit (Figure 2; Supplementary Figure S2; Table II). The domain exchange encompasses the entire activation segment, that is, it starts with the DFG motif and ends with the αEF/αF loop. All activation segments were well-defined by electron density (Supplementary Figure S3). A comparison with monomeric kinase domain structures shows that domain-exchanged activation segments are hinged in the loop region between helices αF and αEF and after the short strand β9, whereas conformation of the DFG motif is not affected (Figure 3). In LOK, the short strand β9 is replaced with a helical insert termed helix αAL (AL, activation loop). Short helical inserts have also been reported in activation segments of inactive cyclin-dependent kinase 2 (CDK2) and NIMA (never in mitosis gene a)-related expressed kinase 2 (NEK2), indicating a general propensity of some kinases to form helices in this region (Brown et al, 1999; Rellos et al, 2007). Interestingly, SLK and CHK2 also contain short helices C-terminal to the DFG motif, while in DAPK3 helix αAL is distorted and reduced to half a turn. Generally, αAL is characterized by high temperature factors, suggesting that αAL is relatively flexible (Supplementary Figure S4). It may therefore, function as an adaptor to compensate for differences in sequence lengths in activation segments enabling key target residues to be positioned proximal to catalytic residues in the catalytic domain. A second common feature of all exchanged activation segments is the presence of an aromatic residue at the tip of the activation segment, that binds to a deep cleft formed by αF and αG of the interacting protomer. The short helix αEF, C-terminal to the APE motif, is conserved in all activation segments. In LOK, it is extended by a second helical turn, while in CHK2 a short distorted helical segment is added. A comparison of the overall structures of all four activation segments is shown in Supplementary Figure S4.
Figure 2.

Domain-exchanged kinase dimers. The activation segment regions from each monomer extend to form an extensive intermolecular interface. The arrangements of monomers within each dimer are distinct and the tip of each activation segment binds in a narrow cleft formed between helix αF and αG in the C-terminal lobe of the kinase. For each dimer, two views are shown separated by a 45° and a molecular surface (gold) is shown for one monomer, while the other monomer is shown in ribbon form (dark blue). Disordered regions are shown as dotted lines and bound inhibitors are depicted in space-filling form. Each dimer is shown in the same reference frame as that of SLK.
Table 2.
Dimer interfaces
| SLK | SLK-diP | LOKa | DAPK3a | CHK2 | |
|---|---|---|---|---|---|
| PDB code | 2J51 | 2JFL | 2J7T | 2J90 | 2CN5 |
| Dimer type (N/C)b | C | C | C | N | C |
| Interface-accessible surface area per monomer (ASA; Å2)c | 1735 | 2032 | 1730 | 1940 | 1335 |
| % ASAc | 11.5 | 13 | 11.5 | 12.9 | 8.5 |
| Interface residues (atoms)c | 48 (181) | 55 (212) | 46 (176) | 56 (210) | 39 (139) |
| % Polar atoms in interfaced | 32 | 34 | 28 | 32 | 30 |
| % Nonpolar atoms in interfaced | 68 | 66 | 72 | 68 | 70 |
| Length/breadth of interface (Å)d | 61/32 | 61/28 | 47/22 | 52/33 | 52/23 |
| Salt bridgesc | 2 | 2 | 4 | 3 | 2 |
| Direct hydrogen bondsc | 18 | 29 | 23 | 21 | 14 |
| aLOK and DAPK3 (molA) have incomplete activation loop regions due to disorder and the extent of the dimer interface is therefore likely to be under-represented by the values shown. | |||||
| bDimer type defined as noncrystallographic or crystallographic. | |||||
| cCalculated using MSD-PISA server (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html). | |||||
| dCalculated using protein–protein interaction server (http://www.biochem.ucl.ac.uk/bsm/PP/server/). | |||||
Figure 3.

Conformation of activation segments. (A) Activation segment nomenclature. Schematic representation of the typical architecture of a kinase activation segment based on the active mono-phosphorylated STE kinase PAK4 (PDB: 2CDZ). The activation segment region runs from the magnesium-binding DFG motif (orange) to the αF helix (grey). The β9 strand (green), the activation segment (magenta), the P+1 loop (turquoise), APE (αEF) helix (blue) and αEF/αF loop (yellow) are highlighted. (B) Comparison of the activation segments of SLK (green), LOK (orange), DAPK3A (cyan) and CHK2 (magenta; PDB: 2CN5) after superimposition. The activation segments adopt a spectrum of orientations that are dictated by the relative orientation of each monomer with each respective dimer. The disordered region of LOK is indicated by a dotted line. For DAPK3, the activation segment from molecule B is shown as the corresponding region in molecule A is disordered. Inset: overall superposition of kinases indicating area of interest shown in main panel. (C) Structure-based sequence alignment of activation segment regions. The sequences of the activation segments of SLK, LOK, DAPK3, CHK2 and PAK4 are shown. Known phosphorylation sites are highlighted in orange. Secondary structure elements are shown for each kinase below the alignment and coloured using the same scheme as in panel B. The structural elements of PAK4 (PDB: 2CDZ), in which the activation segment adopts a classical intramolecular orientation, are shown for reference.
The intermolecular interface
In all four catalytic domain structures, the exchanged activation segment forms an extended dimer interface. A large accessible surface area (ASA) that is predominantly hydrophobic in character, ranging from 1730 Å2 for LOK (11.5% ASA) to 2030 Å2 for the diphosphorylated SLK (13% ASA), is buried in each of the dimer interfaces (Supplementary Figure S5). The extent of the interface compares favourably with the previously observed intermolecular CHK2 dimer (1330 Å2 (8.5% ASA); Table II) and with the properties of other bona fide protein–protein complexes (Ponstingl et al, 2000). In addition, the interfaces encompass a surface area that is more than fivefold larger than any interface involved in packing contacts observed in crystal contact regions of any of the kinases studied here. Nonetheless, it could not be ruled out that the observed dimeric structures of catalytic domains were related to crystal packing effects or binding of the cocrystallized inhibitor. However, additional structures of SLK and LOK rendered the possibility of artificial dimerization due to crystal packing or/and ligand binding to be unlikely: both the apo-structure of SLK and a complex with an inhibitor of the pyrazole-quinazoline class (K00546) were identical to the structure of the triazole-1-carbothioamide (K00606) complex apart from local differences in side chain conformations. Moreover, the conformation of the activation segment in LOK that was crystallized in a monoclinic (P21) form was identical to that observed in the I222 form presented here (data not shown).
Domain-exchanged kinase catalytic domains are dimeric in solution
Self-association of catalytic domains was investigated by AUC and cross-linking studies. Sedimentation velocity experiments revealed the presence of catalytic domain dimers in solution at physiological pH and salt concentrations for all four kinases (Figure 4). The dimeric state represented between 5 and 15% of the total protein mass at 30 μM. A dissociation constant (Kd) of 140±40.0 μM was estimated for unphosphorylated LOK from sedimentation equilibrium experiments. The affinity of the DAPK3 protomers was somewhat weaker (Kd=290±30 μM). Cross-linking experiments confirmed the presence of dimers for all four kinases (Figure 4B). In contrast, no dimerization was observed using either method for serine/threonine kinase 38 (STK38) or for casein kinase 1γ.
Figure 4.

Kinase self-association in solution. (A) Size distribution of the sedimentation coefficient for 30 μM unphosphorylated LOK observed by an AUC velocity experiment. The first peak corresponds to the monomer and the second peak to the LOK dimer. A dimer concentration of approximately 17% was estimated. (B) Detection of dimers by cross-linking. The sizes for the dimers of CHK2 (lane 4), DAPK3 (lane 5) and LOK (lane 6) are indicated by an arrow. No band corresponding to dimers was observed for CSNK1G (lane 2) and STK38 (lane 3). Lane 1, molecular weight marker. (C) ESI-MS spectrum of wild-type SLK (black), wild-type SLK after autophosphorylation (blue), T183A (red) and T183A mutant after autophosphorylation (green) under identical conditions (48 h at 4°C). The expected position of the T183 autophosphorylation peak is indicated by an arrow. (D) Size distribution of the sedimentation coefficient for 30 μM SLK mutant Q185P (black line) and the corresponding wild-type protein (red line). Dimerization was only detected in the wild-type protein as indicated by a second peak at 3.4 S.
Dimerization is necessary for activation segment autophosphorylation
To address the role of dimerization in activation loop phosphorylation, we mutated residues in the dimer interface with the goal of obtaining monomeric protein. Mutations of the conserved tryptophan residue (SLK mutants: W196A, W196R) resulted in insoluble protein. Comparison with the monomeric structure of p21-activated kinase 4 (PAK4, Protein Data Bank (PDB) code: 2BVA) revealed that this residue inserts deeply into a hydrophobic pocket in the lower kinase lobe, suggesting that the generated mutants would also be destabilized in the monomeric state. Mutation of the less conserved tyrosine residue in SLK (Y195A) resulted in a stable construct that, however, still dimerized (data not shown).
Comparison with PAK4 and other group II PAKs that are monomeric in solution identified a proline residue in the position corresponding to Q185 in SLK. Proline in that position was expected to disrupt the first activation segment helix (αAL), impairing dimer formation without perturbing the monomeric state of the kinase. Indeed, the SLK mutant Q185P had similar stability as wild-type SLK (data not shown) and was monomeric in solution. Most importantly, the mutant did not autophosphorylate to a significant level, demonstrating that dimerization is necessary for SLK autophosphorylation (Figure 4 C and D).
Kinases with domain-exchanged activation segments are in an active conformation
The kinase domains described here are in a closed active conformation and contain a correctly positioned αC helix as indicated by the presence of a salt bridge formed between the conserved αC glutamate (E78 in SLK) and the active site lysine (E63 in SLK) (Figure 5A). In SLK, the catalytically important αC helix is further stabilized by a hydrogen bond between the activation segment residues K179 and D81 and by hydrophobic interactions with the N terminus, which forms an additional β-strand (β0). Although LOK also exhibits a closed active conformation, the canonical salt bridge is disrupted by the inhibitor K00593, which intercalates between the active site lysine (K65) and αC glutamate (E81), resulting in a partial disordering of the lysine side chain.
Figure 5.

Comparison of active site and phosphorylated activation segment regions. (A) Superimposition of the active site regions for SLK (green), LOK (orange) and DAPK3 (cyan). Critical residues for kinase function are shown in ball and stick representation and the major secondary structure elements are labelled. (B, C) Active site stabilization via activation segment phosphorylation. (B) Details of the key interactions formed by the activating phosphate moiety in the STE kinase PAK4 (PDB id: 2CDZ) are depicted. The phosphate group on S474 is coloured red/orange and the activation segment is coloured according to the scheme in Figure 3A. (C) Details of key interactions formed by the two phosphate moieties in the dimer interface of the STE kinase SLK. The core of the kinase is shown along with the domain-exchanged activation segment from the adjacent molecule (residues indicated by *). The colour scheme is as in (B). Hydrogen bonds are indicated by dotted lines. The catalytic base (Asp155) is coloured with green carbons and labelled in red.
We were interested to determine whether the observed conformation and dimerization can also occur between phosphorylated catalytic domains and determined the structure of both di- and unphosphorylated SLKs in the presence of the same ligand. Superimposition of the main chains of both structures revealed that the overall structures were identical (r.m.s.d. 0.15 Å), strongly suggesting that phosphorylation at either site within the activation segment does not inhibit activation segment exchange. Phosphorylation increased the number of direct hydrogen bonds formed in the dimer interface from 18 to 29, as well as the surface area buried in the interface (Table II). This is mainly a consequence of ordering of the R186 and R187 side chains flanking the phosphothreonine at position T183. However, AUC experiments showed that the dimer concentration for unphosphorylated SLK was twofold higher than for the phosphorylated catalytic domain (data not shown).
The role of phosphate moieties in activated kinases is to stabilize the activation segment in a conformation suitable for substrate binding. Typical interactions stabilizing the activation segment of the monomeric kinase PAK4 are shown in Figure 5B. In SLK, phosphorylation of T183 results in formation of a hydrogen bond network involving the activation segment residues R186 and R187 (Figure 5C), as well as two hydrogen bonds to main chain atoms located on both termini of the activation segment (R187 and S177). In the absence of phosphorylation at T183, the side chains of R186 and R187 are disordered. The second phosphorylation site, identified at S189, is orientated towards the solvent and is unlikely to contribute to the stability of the activation segment in its dimeric state. However, comparison with known activation sites in STE20 kinases (e.g., PAK1, PAK4) suggested that S189 is the relevant residue for kinase activity in the monomeric state. We mutated the SLK residues T183 and S189 to alanine and determined the autophosphorylation activity of the dephosphorylated protein in vitro. The S189A mutant autophosphorylated to a similar level as wild type SLK, confirming that phosphorylation at this site is not relevant for phosphorylation on T183. Interestingly, the T183A mutant showed significantly (∼fivefold) lower autophosphorylation levels under identical conditions (Supplementary Figure S6), thus supporting its crucial role in activation segment phosphorylation.
Sequence requirements of active site-mediated phosphorylation
We determined the sequence specificity of the kinase catalytic domains using degenerate peptide libraries (Figure 6). The peptide array data revealed a particularly strong preference of SLK for tyrosine in position −2, bulky hydrophobic or aromatic residues in position +1, and for either arginine or lysine in position +2, thus providing the consensus sequence X-X-X-Y-X-T*-Φ-R/K-X-X-X (*, site for phosphorylation; Φ, hydrophobic or aromatic). As the LOK substrate site is closely related to that of SLK, similar sequence specificity is assumed for LOK.
Figure 6.

Substrate specificities of SLK, CHK2 and DAPK3. Biotinylated peptides bearing the indicated residue at the indicated position relative to a central S/T phosphor-acceptor site were subjected to phosphorylation using radiolabelled ATP. Aliquots of each reaction were subsequently spotted onto a streptavidin membrane, which was washed, dried and exposed to a phosphor screen.
CHK2 showed a very strong preference for arginine in position −3, excluding most other residues in that position. This feature is similar to consensus sequences determined for other members of the CaMK family, and it is also shared by DAPK3, which additionally showed a very strong selectivity for arginine in position −2. Importantly, for none of the four studied kinases the determined consensus sequences matched the activation segment phosphorylation sites (Figure 3C), but corresponded well with sequences of known substrates for the respective kinases.
Discussion
This study reports the previously unknown structures for the three human protein kinases DAPK3, LOK and SLK. The structural data revealed that the activation segment, a key regulatory element of kinase function, may exchange with the activation segment of an adjacent kinase molecule forming an active kinase in trans conformation. Structural comparison, mutagenesis and biophysical characterization suggest a model for activation segment autophosphorylation, which may be common to kinases from diverse families.
Flexibility of the activation segment in the inactive state
The high degree of flexibility in a region that constitutes the substrate-binding site in inactive kinases raises the question of how activation segments are specifically recognized. The segment exchange observed in the four catalytic domain structures leads to ordering of the activation segment in the nonphosphorylated state. This conformation is stabilized by a large number of interactions at the dimer interface. Residues targeted for phosphorylation are generally in close proximity (between 9 and 12 Å) to the catalytic aspartate in nonphosphorylated segments, and only a small local reorganization would be sufficient to arrange these residues in positions competent for phosphoryl transfer. Dimerization has also been described for the dual-specificity kinases MEK1 and MEK2. However, no activation segment exchange has been reported for the published structures (Ohren et al, 2004).
The main function of activation segment phosphorylation in SLK and LOK is autoactivation. However, DAPK3 has been reported not to require activation segment phosphorylation for catalytic activity. This is supported by our observation that DAPK3 is autophosphorylated at two sites (S50 and T265) in the absence of a phosphorylation within the activation segment. Autophosphorylation at the activation segment residue T180 has been suggested to be of functional importance for DAPK3 activity, because a T180A mutant is resistant to activation and suppresses DAPK3 function in cells (Graves et al, 2005). Interestingly, the DAPK3 phosphorylation site at T180 is analogous to the second autophosphorylation site in CHK2 at T387, and the T387A mutant of CHK2 is unable to trigger the G1 checkpoint, suggesting that phosphorylation at this site is critical for CHK2 activity (Lee and Chung, 2001).
Active conformation of domain-exchanged kinases
The proposed mechanism for autophosphorylation requires that the activation segment-exchanged kinase dimers retain an active conformation. Comparison of active kinase structures shows that helix αC is positioned such that a salt bridge between the active site lysine and the conserved αC glutamate can form, provided this interaction is not perturbed by interaction with an inhibitor as was observed in the structure of the LOK/K00593 complex.
Comparison between the activation segment sequence of SLK and other STE kinases suggests that S189, and not T183, is the primary phosphorylation site. In the structure of diphosphorylated dimeric SLK, T183 forms a hydrogen bond network with the activation segment residues R187 and R186, whereas phosphorylation of S189 did not result in formation of new polar interactions in dimeric SLK. The length of the activation segment is invariant among the PAK and SLK/LOK families, suggesting that S189 may form similar interactions in monomeric SLK/LOK, as observed for the analogous residue in PAK4 (S474) (Eswaran et al, 2007). This suggests that the role of T183 phosphorylation is to promote activation segment phosphorylation at S189 positioned in close proximity to the catalytic aspartate (D155) of the interacting protomer (Figure 5C). This interpretation is supported strongly by the impairment of autophosphorylation observed for the T183A mutant. Multiple activation segment phosphorylation sites have been reported for a number of kinases. From our results, it could be deduced that these sites play functional roles in activation segment phosphorylation and kinase activation, but may be less relevant for the catalytic activity once the kinase is activated.
Activation segment phosphorylation on non-consensus sites
A common feature of the discussed kinases is that all activation segment autophosphorylation sites identified in this study or reported in the literature do not match the sequence requirements for consensus substrate binding or phosphorylation. Furthermore, it was shown recently that full-length, activated CHK2 does not recognize an isolated CHK2 kinase domain as a bona fide substrate despite rapid autophosphorylation (Oliver et al, 2006). Thus, the mechanism for the recognition of autophosphorylation residues differs from substrate recognition and likely requires dimerization of the catalytic domain as observed in the crystal structure.
In addition, improper activation across kinase cascades would also be prevented. For example, CHK1 shares a large number of substrates with CHK2. Yet, in the absence of dimerization and activation segment exchange, CHK1 cannot phosphorylate activation segment residues and activate CHK2, as they do not resemble CHK1 consensus substrates.
Requirement of additional domains that stabilize dimer formation
AUC demonstrated that dimerization is also observed in solution. However, the determined dissociation constants suggest that the studied catalytic domains require additional mechanisms to stabilize dimers and increase the effective concentrations of the kinase domains in vivo to achieve efficient dimerization. Accordingly, all kinases studied here possess additional dimerization domains adjacent to the kinase domain. In CHK2, dimerization is regulated by phosphorylation of the FHA domain residue T68 by the DNA damage sensor ATM (Ahn et al, 2002; Xu et al, 2006). Regulation of dimerization by post-translational modification offers an attractive model of how kinase activity may be controlled by dimerization. Several recent studies also strongly support the idea that DAPK3 dimerization is regulated by phosphorylation. Mutation of the leucine zipper present in the DAPK3 C terminus results in loss of self-association, autophosphorylation and a decrease in the ability of the enzyme to induce cell death upon overexpression (Kawai et al, 1998; Graves et al, 2005). Interestingly, the DAPK kinase domain itself is sufficient for self-association and this oligomerization might be relevant not only for autoactivation, but may also regulate crosstalk with other DAPK family members. Recently, it has been shown that DAPK3 and DAPK1, which share about 80% sequence identity, associate in cells and that the DAPK3 catalytic domain is sufficient for self- as well as hetero-association (Shani et al, 2004). The DAPK3 structure indicates that dimerization may be mediated by phosphorylation at S50, which is part of a hydrogen bond network at the dimer interface involving K167, R135 and E70 of the interacting kinase domain (Supplementary Figure S7B). Interestingly, S50 is located in the DAPK family-specific basic loop region that links the strand β3 with helix αC, and interaction studies showed that mutation of conserved arginine residues in this region reduces dimerization of DAPK3 (Shani et al, 2004). To study the effect of S50 phosphorylation on dimerization, we coexpressed DAPK3 with λ-phosphatase and obtained a homogeneous, unphosphorylated protein sample. Indeed, AUC velocity experiments showed that the unphosphorylated DAPK3 kinase domain is entirely monomeric in solution (Supplementary Figure S7A). Furthermore, unphosphorylated DAPK3 crystallizes as a monomer (unpublished data). Based on these data, we propose a stepwise activation of DAPK3 comprising (a) autophosphorylation at consensus sites, (b) dimerization of the catalytic domain stabilized by phosphorylation at S50 and activation segment exchange, and (c) phosphorylation of the activation segment non-consensus site T180 (Supplementary Figure S7C).
As shown for SLK, activation segment phosphorylation weakens self-association. However, in the crystal structure, diphosphorylated SLK was still found dimeric. It is tempting to speculate that dimers of activated kinases may serve as a specificity filter that would allow only specific substrates with high affinity to the substrate-binding site to bind. Thus, they may lead to dissociation of the dimer and may be phosphorylated by the kinase. Such a model is supported by a recent report, on the dimeric state of the kinase domain of PAK2, where it was shown that the kinase dissociates into monomers upon addition of a high-affinity substrate (Pirruccello et al, 2006).
Implications for development of specific inhibitors
Targeting the inactive conformation of kinases has resulted in the development of a number of highly successful and specific kinases inhibitors. All kinases examined here represent potential targets for inhibitor development for cancer therapy: the DAPK family has been linked to misregulation of oncogenes (Gozuacik and Kimchi, 2006). A mutation in LOK (K277E) has been implicated in the development of testicular germ-cell tumours, and its role as an activator of polo-like kinases suggests that LOK-selective inhibitors may be beneficial for the treatment of certain tumour types (Bignell et al, 2006). The observed dimeric nature of the studied kinases may give rise to novel types of inhibitors. Activation segment exchange creates a pocket in the dimer interface adjacent to the ATP-binding site. This pocket is lined by the two strands β3 and β5, αC as well as by residues of the interacting protomer activation segment (Supplementary Figure S8). Several high-affinity type I inhibitors have been cocrystallized with the kinases discussed here, constituting a suitable starting point for further design of such inhibitors.
Materials and methods
Cloning, expression and purification
Catalytic domain residues were cloned into the vector pNIC28-Bsa4, by ligation-independent cloning (Stols et al, 2002). The vector includes a TEV-cleavable (*) N-terminal His6 tag (MHHHHHHSSGVDLGTENLYFQ*SM). Proteins were expressed in Escherichia coli and purified using immobilized Ni-affinity chromatography followed by proteolytic cleavage of the His6 tag and size-exclusion chromatography. Recombinant proteins were 95% pure as judged by SDS–PAGE and the correct molecular weight was confirmed using electrospray (ESI) MS. Unphosphorylated variants of SLK and LOK were obtained by treating the purified protein for 12 h with GST-lambda phosphatase. Phosphorylated SLK was obtained by incubating the recombinant protein with 5 mM ATP and 10 mM MgCl2 for 12 h. Autophosphorylation was carried out using 1.5 mM ATP, 3 mM MgCl2, 3 mM MnCl2 at 4°C for 48 h. Mutants were generated using the QuickChange kit (Stratagene) and were verified by DNA sequencing.
Crystallization
All crystals were obtained at 4°C using the sitting-drop vapour diffusion method.
SLK. Crystals of unphosphorylated and diphosphorylated SLK were obtained by mixing 100 nl of protein (10–12 mg/ml containing 1 mM Cdk1/2 Inhibitor III (K00546; Calbiochem)) with 50 nl of a well solution containing 16–18% PEG3350, 0.15 M KSCN, 10% ethylene glycol, 0.1 M bis-Tris propane, pH 6.5. Crystals of the K00606 complex were obtained under the same conditions using predominantly monophosphorylated protein (10 mg/ml) containing 1 mM of the inhibitor. Crystals of apo-SLK were obtained by mixing protein (10.5 mg/ml) with reservoir solution (20% PEG3350, 0.2 M KSCN, 10% ethylene glycol, 0.1 M bis-Tris propane, pH 7.5 at a ratio of 2:1). Prior to vitrification in liquid nitrogen, all crystals were briefly soaked in mother liquor supplemented with 15% ethylene glycol.
LOK. Crystals were obtained by mixing 100 nl of protein solution (12.8 mg/ml containing 1 mM K00593/SU11274 (3-(1-(3,5-dimethyl-4-(4-methyl-piperazine-1-carbonyl)-2H-pyrrol-2-yl)-meth-(Z)-ylidene)-2-oxo-2,3-dihydro-1H-indole-5-sulphonic acid (3-chloro-phenyl)-methyl-amide)) with 50 nl of a well solution containing 45% PEG300, 0.24 M calcium acetate, 0.1 M sodium cacodylate, pH 6.5). Crystals were vitirified directly in liquid nitrogen prior to data collection.
DAPK3. Crystals were grown from a drop consisting of 150 nl protein (10.3 mg/ml) containing 1 mM K00225/Pyridone 6 (2-(1,1-dimethylethyl)-9-fluoro-3,6-dihydro-7H-benz[h]-imidaz(4,5-f)isoquinolin-7-one) and 50 nl well solution. The drop was equilibrated against well solution containing 30% PEG1000, 0.1 M succinic acid-phosphate-glycine (SPG) buffer pH 8.0. Prior to flash freezing, the crystals were briefly soaked in mother liquor supplemented with 20% ethylene glycol.
Data collection and processing
Diffraction data were collected from flash-frozen crystals at 100 K on beamline X10SA at the Swiss Light Source (SLS). Images were recorded on a MAR CCD detector (Mar Research), indexed and integrated using MOSFLM and scaled using SCALA in the CCP4 suite of programs. Data collection statistics and cell parameters are listed in Table I.
Structure solution and refinement
SLK, LOK. The structure of the SLK/K00546 co-complex was solved by molecular replacement using PHASER (Storoni et al, 2004) with the coordinates of human TAO2 kinase (PDB code: 1U5R) that had been sequence-truncated using CHAINSAW (Schwarzenbacher et al, 2004) as a search model. The refined SLK/K00546 complex was then used as a starting model, after rigid-body refinement in REFMAC5 (Murshudov et al, 1997), for all other SLK complexes. The LOK structure was subsequently solved using the SLK/K00546 structure.
DAPK3. The structure was solved by molecular replacement using PHASER with a composite model comprising coordinates from DAPK3 (PDB code: 1YRP) and DAPK1 (PDB code 1JKT/1WVX) as a search model. Initial automated model building was carried out using ARP/WARP (Morris et al, 2003). In all cases, iterative rounds of restrained refinement using REFMAC5 (Murshudov et al, 1997) with appropriate TLS restraints, interspersed with manual rebuilding using Coot (Emsley and Cowtan, 2004). Final models were validated using MOLPROBITY (Davis et al, 2004, 2007).
Identification of inhibitors
Thermal denaturation experiments were carried out using a real-time PCR (Mx3005p, Stratagene) as described previously (Vedadi et al, 2006; Niesen et al, 2007). K00546 and K00589 were purchased from Calbiochem. K00606 was a generous gift from Professor Kevan Shokat (University of California).
Analytical ultracentrifugation
Sedimentation velocity experiments were carried out on a Beckman XL-I Analytical Ultracentrifuge equipped with a Ti-50 rotor. Protein samples were studied at a concentration of 15, 30 and 60 μM in 10 mM HEPES, pH 7.4, 150 mM NaCl, employing a rotor speed of 50 000 rpm. Radial absorbance scans were collected in 1 min intervals. Data were analysed using SEDFIT (Brown and Schuck, 2006) to calculate c(s) distributions. The software package SEDNTERP (http://www.jphilo.mailway.com) was used to normalize the obtained sedimentation coefficient values to the corresponding values in water at 20°C, 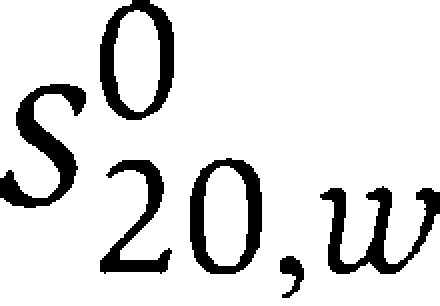 . Sedimentation equilibrium experiments were performed at 4°C and a protein concentration of 15, 30 and 60 μM. The dissociation constant, Kd, was calculated from the fitted apparent association constant, Ka,obs, according to the equation
. Sedimentation equilibrium experiments were performed at 4°C and a protein concentration of 15, 30 and 60 μM. The dissociation constant, Kd, was calculated from the fitted apparent association constant, Ka,obs, according to the equation 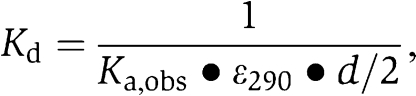 where d is the optical pathlength and ɛ290 the extinction coefficient at 290 nm.
where d is the optical pathlength and ɛ290 the extinction coefficient at 290 nm.
Determination of peptide phosphorylation specificity
Phosphorylation motifs for SLK, CHK2 and DAPK3 were determined using a positional scanning peptide library approach essentially as described previously (Hutti et al, 2004). Reactions were carried out in multiwell plates in 50 mM HEPES, pH 7.4, 10 mM MnCl2, 1 mM DTT, 0.1% Tween 20, 100 μM ATP (including 0.3 μCi/μl γ-[33P]-ATP), 50 μM peptide substrate and 50 μg/ml kinase for 2–4 h at 30°C. Peptide substrates had the general sequence YAXXXXX-S/T-XXXXAGKK(biotin), where S/T represents an even mixture of serine and threonine, K(biotin) is ɛ-(biotinamidocaproyl)lysine and X is a equimolar mixture of the 17 amino acids excluding cysteine, serine and threonine. Each well contained a distinct peptide in which one of the X positions was replaced with one of the 20 residues (one of the unmodified proteogenic amino acids excluding S and T). In addition, three additional wells were included that contained either no peptide, the peptide YAXXXXX-S-XXXXAGKK(biotin) or the peptide YAXXXXX-T-XXXXAGKK(biotin), to test phosphoacceptor residue preference. At the end of the incubation time, aliquots of each reaction were spotted onto streptavidin membrane, which was processed as described previously (Hutti et al, 2004).
Structure analysis
To facilitate analysis, all coordinates were superimposed onto a common frame using the SLKA/K00546 complex as a reference using THESEUS-3D (Theobald and Wuttke, 2006). The dimer interface was characterized using the MSD-PISA server (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html). Figures 1B, 2, 3A and B, 5A–C and 6 were created using PYMOL (DeLano, 2002). Atomic coordinates and structure factors have been deposited in the PDB, accession codes 2J51, 2UV2, 2JFL, 2JFM, 2J7T and 2J90.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Acknowledgments
We thank Dr Oleg Fedorov for inhibitor screening support. The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research and the Wellcome Trust. LHP and AWO gratefully acknowledge the support of a Programme Grant from Cancer Research UK. BET and SAP are supported by the National Institutes of Health (GM079498).
References
- Ahn JY, Li X, Davis HL, Canman CE (2002) Phosphorylation of threonine 68 promotes oligomerization and autophosphorylation of the Chk2 protein kinase via the forkhead-associated domain. J Biol Chem 277: 19389–19395 [DOI] [PubMed] [Google Scholar]
- Bignell G, Smith R, Hunter C, Stephens P, Davies H, Greenman C, Teague J, Butler A, Edkins S, Stevens C, O'Meara S, Parker A, Avis T, Barthorpe S, Brackenbury L, Buck G, Clements J, Cole J, Dicks E, Edwards K et al. (2006) Sequence analysis of the protein kinase gene family in human testicular germ-cell tumors of adolescents and adults. Genes Chromosomes Cancer 45: 42–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NR, Noble ME, Lawrie AM, Morris MC, Tunnah P, Divita G, Johnson LN, Endicott JA (1999) Effects of phosphorylation of threonine 160 on cyclin-dependent kinase 2 structure and activity. J Biol Chem 274: 8746–8756 [DOI] [PubMed] [Google Scholar]
- Brown PH, Schuck P (2006) Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys J 90: 4651–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burakov AV, Kovalenko OV, Potekhina ES, Nadezhdina ES, Zinovkina LA (2005) LOSK (SLK) protein kinase activity is necessary for microtubule organization in the interphase cell centrosome. Dokl Biol Sci 403: 317–319 [DOI] [PubMed] [Google Scholar]
- Dajani R, Fraser E, Roe SM, Yeo M, Good VM, Thompson V, Dale TC, Pearl LH (2003) Structural basis for recruitment of glycogen synthase kinase 3beta to the axin-APC scaffold complex. EMBO J 22: 494–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Bryan Arendall W III, Snoeyink J, Richardson JS, Richardson DC (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 89: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis IW, Murray LW, Richardson JS, Richardson DC (2004) MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res 32: W615–W619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMOL Molecular Graphics System. Paolo Alto, CA, USA: DeLano Scientific [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Eswaran J, Lee WH, Debreczeni JE, Filippakopoulos P, Turnbull A, Fedorov O, Deacon SW, Peterson JR, Knapp S (2007) Crystal Structures of the p21-activated kinases PAK4, PAK5, and PAK6 reveal catalytic domain plasticity of active group II PAKs. Structure 15: 201–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg J, Nairn AC, Kuriyan J (1996) Structural basis for the autoinhibition of calcium/calmodulin-dependent protein kinase I. Cell 84: 875–887 [DOI] [PubMed] [Google Scholar]
- Gozuacik D, Kimchi A (2006) DAPk protein family and cancer. Autophagy 2: 74–79 [DOI] [PubMed] [Google Scholar]
- Graves PR, Winkfield KM, Haystead TA (2005) Regulation of zipper-interacting protein kinase activity in vitro and in vivo by multisite phosphorylation. J Biol Chem 280: 9363–9374 [DOI] [PubMed] [Google Scholar]
- Hu J, Liu J, Ghirlando R, Saltiel AR, Hubbard SR (2003) Structural basis for recruitment of the adaptor protein APS to the activated insulin receptor. Mol Cell 12: 1379–1389 [DOI] [PubMed] [Google Scholar]
- Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A, Cantley LC, Turk BE (2004) A rapid method for determining protein kinase phosphorylation specificity. Nat Methods 1: 27–29 [DOI] [PubMed] [Google Scholar]
- Kawai T, Matsumoto M, Takeda K, Sanjo H, Akira S (1998) ZIP kinase, a novel serine/threonine kinase which mediates apoptosis. Mol Cell Biol 18: 1642–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Chung JH (2001) The hCds1 (Chk2)-FHA domain is essential for a chain of phosphorylation events on hCds1 that is induced by ionizing radiation. J Biol Chem 276: 30537–30541 [DOI] [PubMed] [Google Scholar]
- Lochhead PA, Kinstrie R, Sibbet G, Rawjee T, Morrice N, Cleghon V (2006) A chaperone-dependent GSK3beta transitional intermediate mediates activation-loop autophosphorylation. Mol Cell 24: 627–633 [DOI] [PubMed] [Google Scholar]
- Lochhead PA, Sibbet G, Morrice N, Cleghon V (2005) Activation-loop autophosphorylation is mediated by a novel transitional intermediate form of DYRKs. Cell 121: 925–936 [DOI] [PubMed] [Google Scholar]
- Mellado M, Rodriguez-Frade JM, Manes S, Martinez AC (2001) Chemokine signaling and functional responses: the role of receptor dimerization and TK pathway activation. Annu Rev Immunol 19: 397–421 [DOI] [PubMed] [Google Scholar]
- Meng W, Swenson LL, Fitzgibbon MJ, Hayakawa K, Ter Haar E, Behrens AE, Fulghum JR, Lippke JA (2002) Structure of mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 suggests a bifunctional switch that couples kinase activation with nuclear export. J Biol Chem 277: 37401–37405 [DOI] [PubMed] [Google Scholar]
- Morris RJ, Perrakis A, Lamzin VS (2003) ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol 374: 229–244 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Niesen FH, Berglund H, Vedadi M (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2: 2212–2221 [DOI] [PubMed] [Google Scholar]
- Nolen B, Taylor S, Ghosh G (2004) Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell 15: 661–675 [DOI] [PubMed] [Google Scholar]
- O'Reilly PG, Wagner S, Franks DJ, Cailliau K, Browaeys E, Dissous C, Sabourin LA (2005) The Ste20-like kinase SLK is required for cell cycle progression through G2. J Biol Chem 280: 42383–42390 [DOI] [PubMed] [Google Scholar]
- Ohren JF, Chen H, Pavlovsky A, Whitehead C, Zhang E, Kuffa P, Yan C, McConnell P, Spessard C, Banotai C, Mueller WT, Delaney A, Omer C, Sebolt-Leopold J, Dudley DT, Leung IK, Flamme C, Warmus J, Kaufman M, Barrett S et al. (2004) Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol 11: 1192–1197 [DOI] [PubMed] [Google Scholar]
- Oliver AW, Knapp S, Pearl LH (2007) Activation segment exchange: a common mechanism of kinase autophosphorylation? Trends Biochem Sci 32: 351–356 [DOI] [PubMed] [Google Scholar]
- Oliver AW, Paul A, Boxall KJ, Barrie SE, Aherne GW, Garrett MD, Mittnacht S, Pearl LH (2006) Trans-activation of the DNA-damage signalling protein kinase Chk2 by T-loop exchange. EMBO J 25: 3179–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedranzini L, Dechow T, Berishaj M, Comenzo R, Zhou P, Azare J, Bornmann W, Bromberg J (2006) Pyridone 6, a pan-janus-activated kinase inhibitor, induces growth inhibition of multiple myeloma cells. Cancer Res 66: 9714–9721 [DOI] [PubMed] [Google Scholar]
- Pirruccello M, Sondermann H, Pelton JG, Pellicena P, Hoelz A, Chernoff J, Wemmer DE, Kuriyan J (2006) A dimeric kinase assembly underlying autophosphorylation in the p21 activated kinases. J Mol Biol 361: 312–326 [DOI] [PubMed] [Google Scholar]
- Ponstingl H, Henrick K, Thornton JM (2000) Discriminating between homodimeric and monomeric proteins in the crystalline state. Proteins 41: 47–57 [DOI] [PubMed] [Google Scholar]
- Prowse CN, Lew J (2001) Mechanism of activation of ERK2 by dual phosphorylation. J Biol Chem 276: 99–103 [DOI] [PubMed] [Google Scholar]
- Rellos P, Ivins FJ, Baxter JE, Pike A, Nott TJ, Parkinson DM, Das S, Howell S, Fedorov O, Shen QY, Fry AM, Knapp S, Smerdon SJ (2007) Structure and regulation of the human Nek2 centrosomal kinase. J Biol Chem 282: 6833–6842 [DOI] [PubMed] [Google Scholar]
- Rosenberg OS, Deindl S, Comolli LR, Hoelz A, Downing KH, Nairn AC, Kuriyan J (2006) Oligomerization states of the association domain and the holoenyzme of Ca2+/CaM kinase II. FEBS J 273: 682–694 [DOI] [PubMed] [Google Scholar]
- Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J (2005) Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell 123: 849–860 [DOI] [PubMed] [Google Scholar]
- Schwarzenbacher R, Godzik A, Grzechnik SK, Jaroszewski L (2004) The importance of alignment accuracy for molecular replacement. Acta Crystallogr D 60: 1229–1236 [DOI] [PubMed] [Google Scholar]
- Shani G, Marash L, Gozuacik D, Bialik S, Teitelbaum L, Shohat G, Kimchi A (2004) Death-associated protein kinase phosphorylates ZIP kinase, forming a unique kinase hierarchy to activate its cell death functions. Mol Cell Biol 24: 8611–8626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicheri F, Moarefi I, Kuriyan J (1997) Crystal structure of the Src family tyrosine kinase Hck. Nature 385: 602–609 [DOI] [PubMed] [Google Scholar]
- Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI (2002) A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif 25: 8–15 [DOI] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ (2004) Likelihood-enhanced fast rotation functions. Acta Crystallogr D 60: 432–438 [DOI] [PubMed] [Google Scholar]
- Theobald DL, Wuttke DS (2006) THESEUS: maximum likelihood superpositioning and analysis of macromolecular structures. Bioinformatics 22: 2171–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedadi M, Niesen FH, Allali-Hassani A, Fedorov OY, Finerty PJ Jr, Wasney GA, Yeung R, Arrowsmith C, Ball LJ, Berglund H, Hui R, Marsden BD, Nordlund P, Sundstrom M, Weigelt J, Edwards AM (2006) Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Natl Acad Sci USA 103: 15835–15840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter SA, Cutler RE Jr, Martinez R, Gishizky M, Hill RJ (2003) Stk10, a new member of the polo-like kinase kinase family highly expressed in hematopoietic tissue. J Biol Chem 278: 18221–18228 [DOI] [PubMed] [Google Scholar]
- Weiss A, Schlessinger J (1998) Switching signals on or off by receptor dimerization. Cell 94: 277–280 [DOI] [PubMed] [Google Scholar]
- Xu YJ, Davenport M, Kelly TJ (2006) Two-stage mechanism for activation of the DNA replication checkpoint kinase Cds1 in fission yeast. Genes Dev 20: 990–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8