

Abstract
Objectives
We aimed to explore the effects of pharmacologic inhibition of cannabinoid-1 (CB1) receptor in in vivo and in vitro models of doxorubicin (DOX)-induced cardiotoxicity.
Background
Doxorubicin is one of the most potent antitumor agents available; however, its clinical use is limited because of the risk of severe cardiotoxicity. Endocannabinoids mediate cardiodepressive effects through CB1 receptors in various pathophysiological conditions, and these effects can be reversed by CB1 antagonists.
Methods
Left ventricular function was measured by Millar pressure-volume system. Apoptosis markers, CB1/CB2 receptor expression, and endocannabinoid levels were determined by immunohistochemistry, Western blot, reverse transcription-polymerase chain reaction, real-time polymerase chain reaction, flow cytometry, fluorescent microscopy, and liquid chromatography/in-line mass spectrometry techniques.
Results
Five days after the administration of a single dose of DOX (20 mg/kg intraperitoneally) to mice, left ventricular systolic pressure, maximum first derivative of ventricular pressure with respect to time (+dP/dt), stroke work, ejection fraction, cardiac output, and load-independent indexes of contractility (end-systolic pressure–volume relation, preload-recruitable stroke work, dP/dt–end-diastolic volume relation) were significantly depressed, and the myocardial level of the endocannabinoid anandamide (but not CB1/CB2 receptor expression) was elevated compared with vehicle-treated control mice. Treatment with the CB1 antagonists rimonabant or AM281 markedly improved cardiac dysfunction and reduced DOX-induced apoptosis in the myocardium. Doxorubicin also decreased cell viability and induced apoptosis in the H9c2 myocardial cell line measured by flow cytometry and fluorescent microscopy, which were prevented by the preincubation of the cells with either CB1 antagonist, but not with CB1 and CB2 agonists and CB2 antagonists.
Conclusions
These data suggest that CB1 antagonists may represent a new cardioprotective strategy against DOX-induced cardiotoxicity.
Doxorubicin (DOX) (adriamycin) is one of the most potent broad-spectrum antitumor anthracycline antibiotics commonly used to treat a variety of cancers, including severe leukemias, lymphomas, and solid tumors (1–3). However, the clinical use of DOX is limited because of its serious cardiotoxicity, which often leads to irreversible degenerative cardiomyopathy and heart failure (3).
The mechanism of DOX-induced cardiotoxicity involves increased oxidative/nitrosative stress (4–7), matrix metalloproteinase activation (8,9), and alteration of cardiac energetics (10), which eventually lead to cell death by apoptosis or cell necrosis (11–15). However, the exact mechanisms have not been fully established, and optimal therapeutic approaches for cardioprotection remain undefined (3).
Endocannabinoids and their synthetic analogs exert powerful cardiodepressive effects mediated through cannabinoid-1 (CB1) receptors (16,17), which have recently been implicated in the mechanism of hypotension associated with hemorrhagic, endotoxic, and cardiogenic shock, and advanced liver cirrhosis, and these effects can be prevented or reversed by treatment with CB1 antagonists (16,17). Furthermore, the CB1 antagonist rimonabant is emerging as a novel therapeutic agent for obesity and related cardiometabolic risk factors in humans (17–20). Here, we explore the effects of 2 CB1 antagonists, rimonabant and AM281, on DOX-induced cardiac dysfunction and cardiotoxicity both in vivo and in vitro.
Materials and Methods
Animals
All protocols were approved by the Institutional Animal Care and Use Committee and were performed in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Male C57BL/6J mice weighing 25 to 35 g were administered a single dose of DOX HCl (Sigma/Aldrich, St. Louis, Missouri) at 20 mg/kg intraperitoneally, and used for functional measurements 5 days later when severe cardiac dysfunction is well established (6,14,15,21,22). Treatment with the CB1 antagonists AM281 or rimonabant (SR141716; 10 mg/kg intraperitoneally, respectively) started 1.5 h before the DOX injection and continued (10 mg/kg intraperitoneally/day) until the hemodynamic measurements were made.
Hemodynamic measurements in mice
Left ventricular performance was analyzed in mice anesthetized with 2% isoflurane by using 1-F microtip pressure-volume catheter (PVR 1045) and ARIA pressure–volume conductance system (Millar Instruments, Houston, Texas) coupled to a Powerlab/4SP A/D converter (AD Instruments, Mountain View, California) as described (6) (also see the Appendix).
Reagents, antibodies, and cell culture
Doxorubicin was purchased from Sigma Chemical. AM281, AM630, JWH133, and HU210 were purchased from Tocris (Baldwin, Missouri). SR144528 and rimonabant (SR141716) are from the National Institute on Drug Abuse Drug Supply Program (Research Triangle Park, North Carolina). Antibodies used are anti-actin mAb (Chemicon, Temecula, California), anti-caspase 3 mAb (Cell Signaling, Danvers, Massachusetts), and anti-active caspase-3 (Cell Signaling).
Rat embryonic ventricular myocardial H9c2 cells were obtained from American Type Culture Collection (Manassas, Virginia). Cells were cultured in DMEM (GIBCO, Invitrogen, Carlsbad, California) containing 10% fetal bovine serum (Invitrogen), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified atmosphere of 5% CO2 as described (23). Cells were always used at <80% of confluence.
Real-time polymerase chain reaction and reverse transcription-polymerase chain reaction analyses were performed from hearts and H9c2 cells for caspase-3, caspase-9, and CB1/2 receptor gene expression as detailed in the Appendix.
Western immunoblot analyses
Protein was extracted from tissue homogenates using radioimmunoprecipitation assay lysis buffer, containing protease inhibitor cocktail set III and phosphatase inhibitor cocktail set I (Calbiochem, EMD Biosciences, San Diego, California). Protein was measured by Dc protein assay kit (Bio-Rad, Hercules, California), and equal amounts (40 μg per lane) were fractionated on NuPAGE 4% to 12% Bis-Tris gel (Invitrogen) and transferred onto nitrocellulose membrane (Invitrogen) using a semidry transfer apparatus (Bio-Rad). The blots were detected with Supersignal West Pico chemiluminescent substrate (Pierce Biotechnology, Rockford, Illinois) and developed using Kodak Biomax film (PerkinElmer, Wellesley, Massachusetts). Immunoblots were scanned with Epson V750 Pro scanner and quantification after background correction was carried out by ImageQuant5.1 software (GE Healthcare, Piscataway, New Jersey); all values were normalized to beta-actin.
Cell viability assay
Cells were seeded at a density of 1 × 105 cells in 96 well plates, and cell viability was assessed via conventional XTT assays (Roche, Indianapolis, Indiana). After incubation, the cells were treated for 6 h with XTT solution at 37°C. The absorbance was measured at 570 nm using an enzyme-linked immunosorbent assay reader (SpectramaxPro, Molecular Devices, Union City, California).
Flow cytometry
Early apoptosis and cytotoxity is determined by flow cytometry using propidium iodide and Annexin V staining (Molecular Probes, Invitrogen, Carlsbad, California) according to manufacturer’s recommendation. The cells were trypsinized for a very short time and collected via centrifugation at 1,000 μg for 5 min. The harvested cells were then washed with phosphate-buffered saline. The cells were resuspended at a density of 1 × 106 cells/ml in Hank’s balanced salt solution buffer containing calcium and magnesium. Flow-cytometry analyses included 10,000 events using a FACSCalibur (Becton Dickinson, San Jose, California). The data were acquired and analyzed using Cell Quest program (Becton Dickinson).
Fluorescence microscopy, DNA fragmentation assay, myocardial terminal deoxynucleotidyltransferase-mediated nick-end labeling (TUNEL) staining, and assay and caspase 3 activity were used according to manufacturer’s instructions as described in the Appendix.
Measurement of endocannabinoid levels
The levels of anandamide (AEA) and 2-arachidonoylglycerol (2-AG) were quantified by liquid chromatography/in-line mass spectrometry as detailed in the Appendix.
Statistical analysis
Results are reported as mean ± standard error of the mean. Probability values of p < 0.05 were considered significant. For detailed statistical analysis see the Appendix.
Results
CB1 antagonists improve DOX-induced cardiac dys-Function
Treatment of mice with DOX, 20 mg/kg intra-peritoneally, induced a significant decrease in heart rate, left ventricular systolic pressure, maximum first derivative of ventricular pressure with respect to time (+dP/dt), −dP/dt, stroke work, ejection fraction, cardiac output, and load-independent indexes of contractility (preload-recruitable stroke work, dP/dt–end-diastolic volume relation, and end-systolic pressure–volume relation, respectively), and an increase in left ventricular end-diastolic pressure and prolongation of relaxation time constants (τ Weiss and Glantz) (Figs. 1 and 2). Treatment with rimonabant or AM281 (10 mg/kg−1 intraperitoneally) immediately before and daily for 5 days after DOX treatment significantly attenuated the DOX-induced changes in ventricular function (Figs. 1 and 2). In vehicle-treated control animals, rimonabant or AM281 exerted no significant effects on the hemodynamic parameters studied (Fig. 1).
Figure 1. Effects of CB1 Antagonists on DOX-Induced Cardiac Dysfunction.
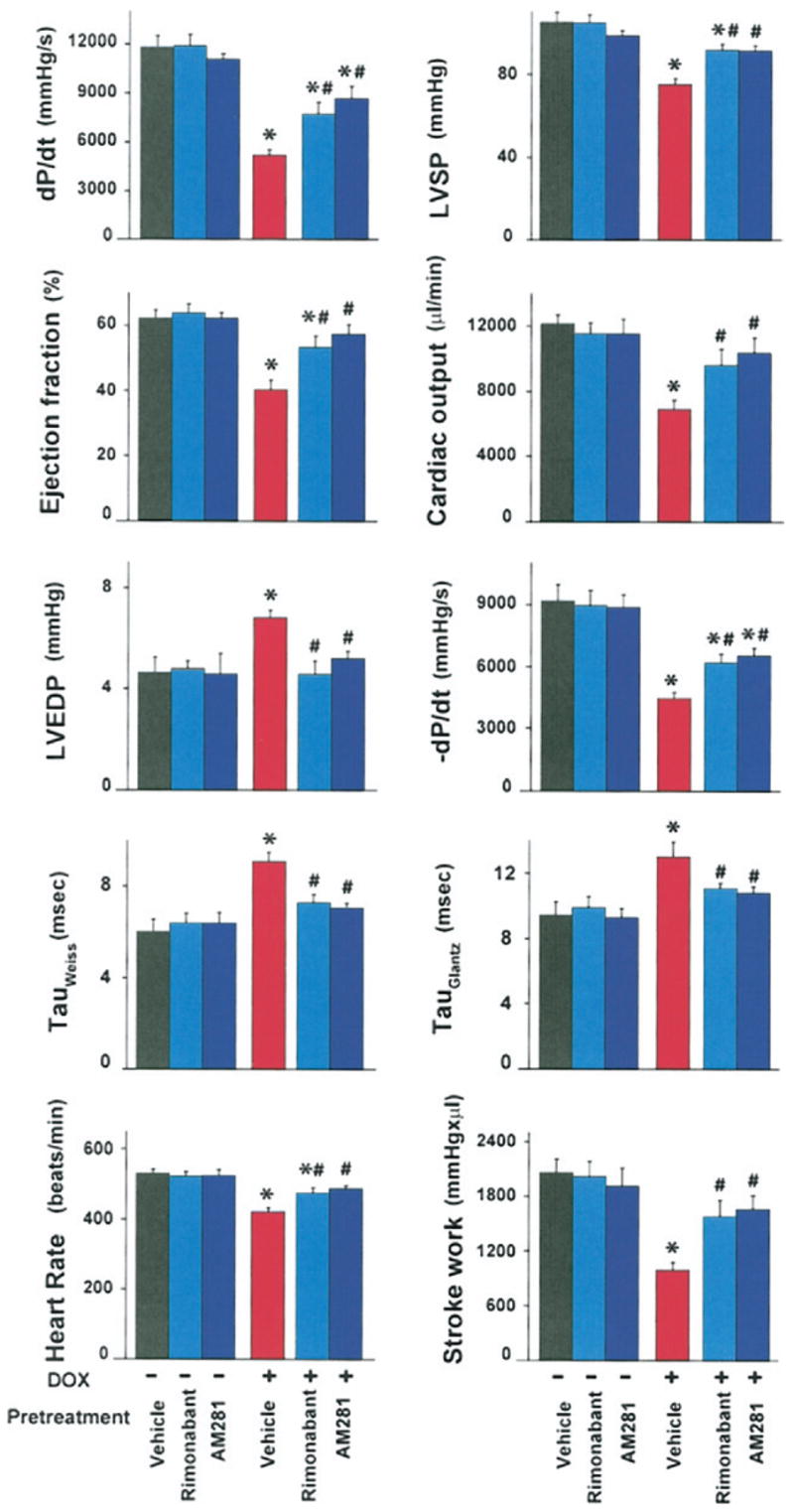
Effect of doxorubicin (DOX) on left ventricular systolic pressure (LVSP), left ventricular end-diastolic pressure (LVEDP), LV maximum first derivative of ventricular pressure with respect to time (+dP/dt), LV −dP/dt, heart rate, stroke work, ejection fraction, and cardiac output and tau (Weiss and Glantz) in mice. Mice were pretreated either with vehicle, rimonabant, or AM281, and treated with either vehicle or DOX, as indicated in the Methods section. Hemodynamic parameters were measured 5 days after DOX administration. Results are mean ± standard error of the mean of 7 to 13 experiments in each group. *p < 0.05 versus vehicle; #p < 0.05 versus DOX. CB1 = cannabinoid-1.
Figure 2. Effect of CB1 Antagonists on DOX-Induced Depression of Load-Independent Indexes of Cardiac Contractility.
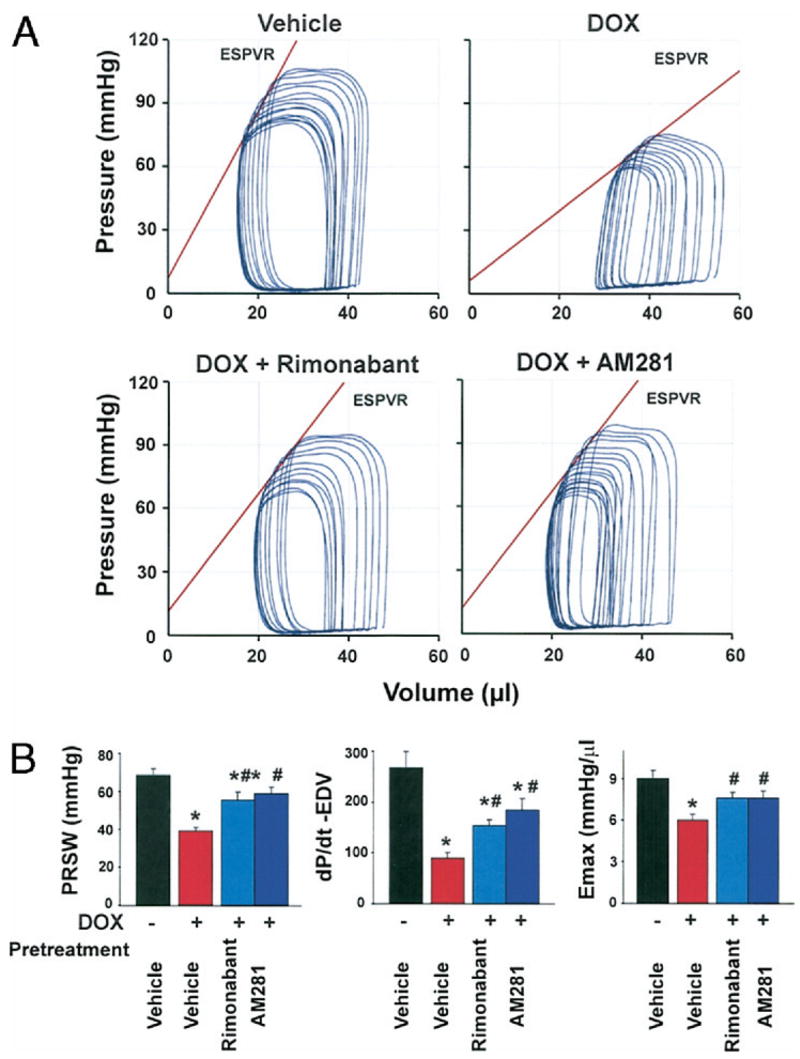
(A) Representative pressure–volume (P–V) loops obtained with a P–V conductance catheter system at different preloads after vena cava occlusion, showing differences in the end-systolic P–V relation (ESPVR) and between mice pretreated with vehicle, rimonabant, or AM281 and treated with vehicle or DOX. The less steep ESPVR in DOX-treated mice indicates decreased contractile function, which was improved by CB1 antagonists. (B) Effects of CB1 antagonists on DOX-induced depression of load-independent indexes of cardiac contractility. Results are mean ± standard error of the mean of 9 to 18 experiments in each group. *p < 0.05 versus vehicle; #p < 0.05 versus DOX. EDV = end-diastolic volume relation; ESPVR or Emax = end-systolic pressure–volume relation; PRSW = preload-recruitable stroke work; other abbreviations as in Figure 1.
CB1 antagonists protect against DOX-induced cell death in rat embryonic ventricular myocardial-derived H9c2 cells in vitro
Incubation of cells with 1 or 5 μmol/l DOX for 18 h resulted in significant decreases in cell viability, measured by a colorimetric XTT-based cell viability assay to 78.6 ± 1.6% or 74.4 ± 1.2%, respectively. Doxorubicin-induced cell death at 1 and 5 μmol/l was completely prevented by 2 h of preincubation (followed by continuous treatment during the 18 h DOX exposure) with 1 μmol/l AM281 (98.0 ± 4.5% and 95.3 ± 5.1%) or rimonabant (96.5 ±3.6% and 102.0 ± 4.3%, respectively) (Fig. 3A). Cannabinoid-1 receptor antagonists alone did not have any effect on cell viability (data not shown).
Figure 3. Effects of CB1 Antagonists on DOX-Induced Cell Death and Apoptosis In Vitro.
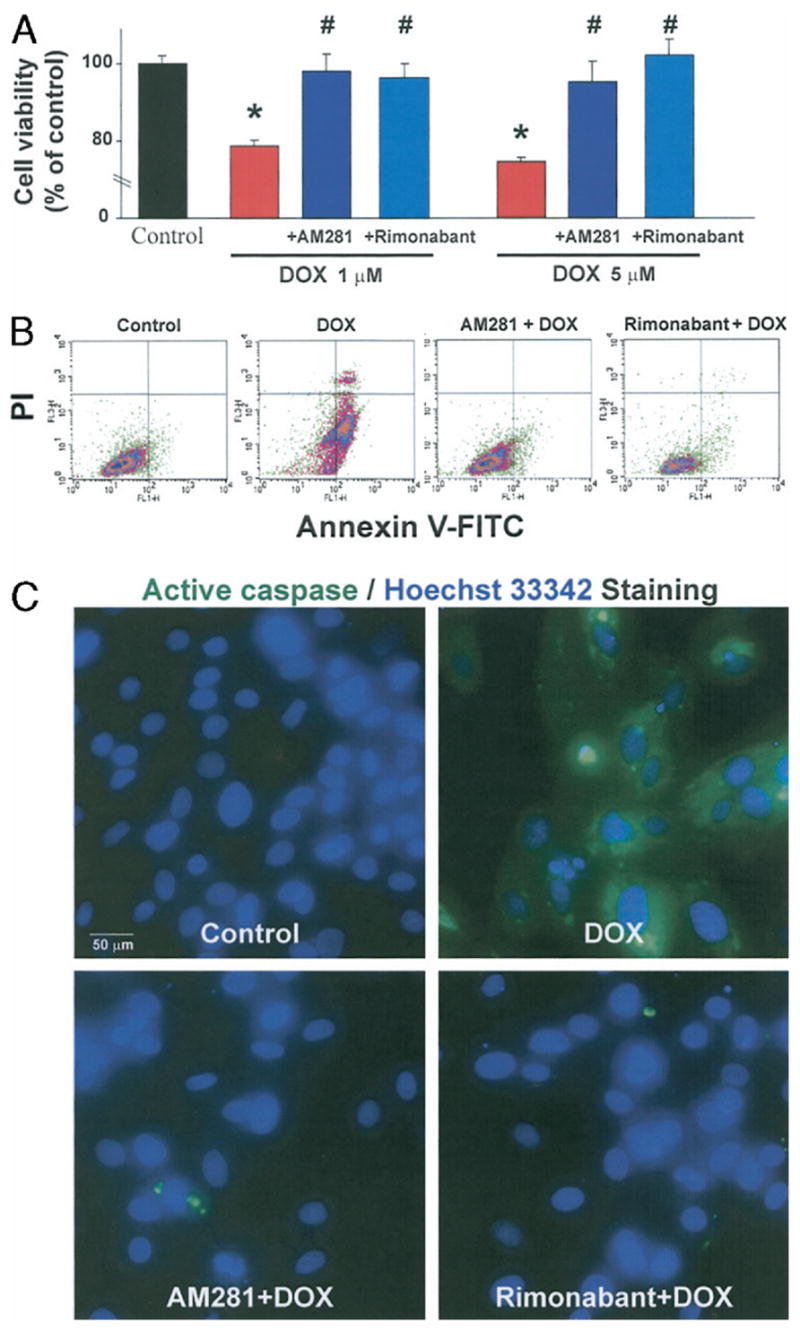
(A) Effects of CB1 antagonists on cell viability measured by XTT assays. AM281 and rimonabant (1 μmol/l) prevent cell death induced by 1 or 5 μmol/l of DOX. *p < 0.05 versus control group; #p < 0.05 versus DOX (n = 4). (B) Effects of CB1 antagonists on the early apoptosis marker fluorescent annexin V conjugate (Annexin V-FITC) and cell death detection dye propidium iodide (PI) measured by flow cytometric analysis of H9c2. Representative data from 3 experiments analyzed. (C) Effects of CB1 antagonists on active caspase expression (green) and nuclear staining pattern by Hoechst 33342 dye (blue). Please also note that some of the nuclei are fragmented in DOX-treated cells (indicative of late apoptosis), but not in CB1 antagonist-treated or control cells. Representative data from at least 15 experiments analyzed. Abbreviations as in Figure 1.
CB1 antagonists, but not CB1 and CB2 agonists and CB2 antagonists, prevent DOX-induced apoptosis in vitro
The H9c2 cell line was subjected to flow cytometric analysis for apoptotic and total dead cells by Annexin V and propidium iodide staining. Early apoptotic marker Annexin V was significantly increased in cells exposed to 1 μmol/l DOX for 18 h but remained at control levels in cells pretreated with either 1 μmol/l AM281 or rimonabant starting from 2 h before DOX administration (Fig. 3B). The DOX-induced total cell death as indicated by positive propidium iodide staining (Fig. 3B) confirmed recent cell viability data, including the higher level of propidium iodide staining in apoptotic compared with normal cells (14). Activation of caspase is a key downstream event of apoptosis. We measured the active form of caspase by fluorescence microscopy. As an indication of caspase activity, green color is generated by cleavage of a generic caspase substrate (rhodamine 110, bis-[L-aspartic acid amide], trifluoroacetic acid salt) introduced into live cells. The green color was conspicuously present in all DOX-treated H9c2 cells (Fig. 3C). In contrast, the green fluorescence was completely absent in cells treated with 1 μmol/l AM281 or rimonabant. We simultaneously measured the late apoptotic marker represented as fragmented nuclei using Hoechst 33342 staining (Molecular Probes, Invitrogen). Few fragmented nuclei were observed in DOX-treated cells whereas no such nuclei were present in normal, AM281, or rimonabant pretreated samples. Thus, pretreatment with either AM281 or rimonabant protects H9c2 cells from DOX-induced apoptosis as shown by early-to-late apoptotic markers.
Pretreatment for 2 h (followed by continuous treatment during the 18 h DOX exposure) with CB2 agonist JWH133, CB1 agonist HU210, CB2 antagonists (AM630 and SR144528) (1 μmol/l each) did not have any protective effect against DOX-induced cell death and apoptosis (Fig. 4). Interestingly, CB1 agonist HU210 by itself significantly enhanced apoptosis (Fig. 4).
Figure 4. Effects of CB1 Agonist, CB2 Antagonists, and Agonists on DOX-Induced Apoptosis in H9c2 In Vitro.
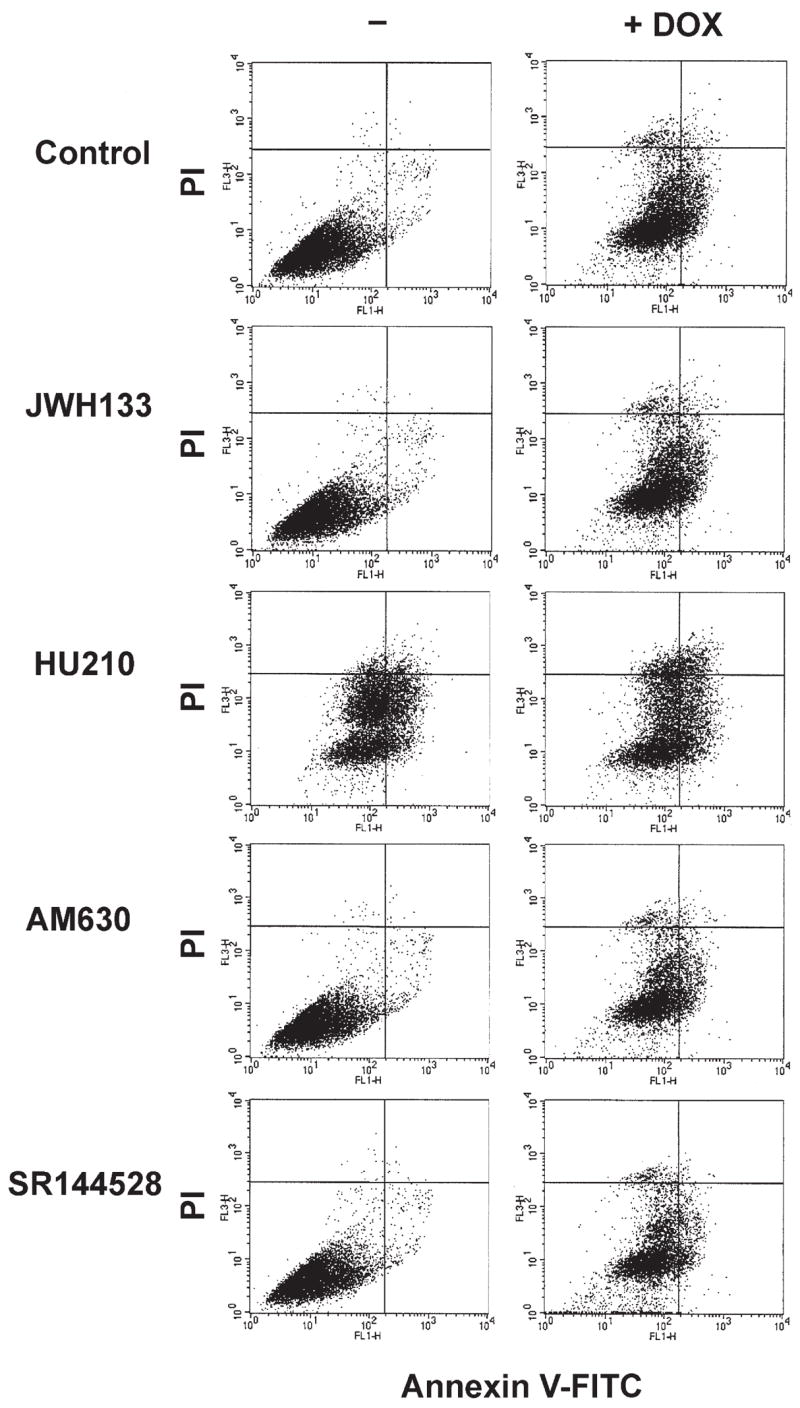
Effects of CB1 agonist, CB2 antagonists, and agonists on the early apoptosis marker Annexin V-FITC and cell death detection dye propidium iodide (PI) measured by flow cytometric analysis of H9c2. Representative data from 3 experiments analyzed. Abbreviations as in Figures 1 and 3.
CB1 antagonists prevent DOX-induced apoptosis of cardiomyocytes in vivo
Myocardial caspase-3-dependent apoptosis was previously described in the mouse model of DOX-induced heart failure (14,24). We have observed significant level of cleaved caspase-3 in samples of heart tissue from DOX-treated mice. Two representative samples from each group are shown in Figure 5A. The total absence of cleaved caspase-3 in AM281 or rimonabant pretreated mice indicates the protective effect of CB1 antagonists against apoptosis.
Figure 5. Effects of CB1 Antagonists on DOX-Induced Apoptosis In Vivo.
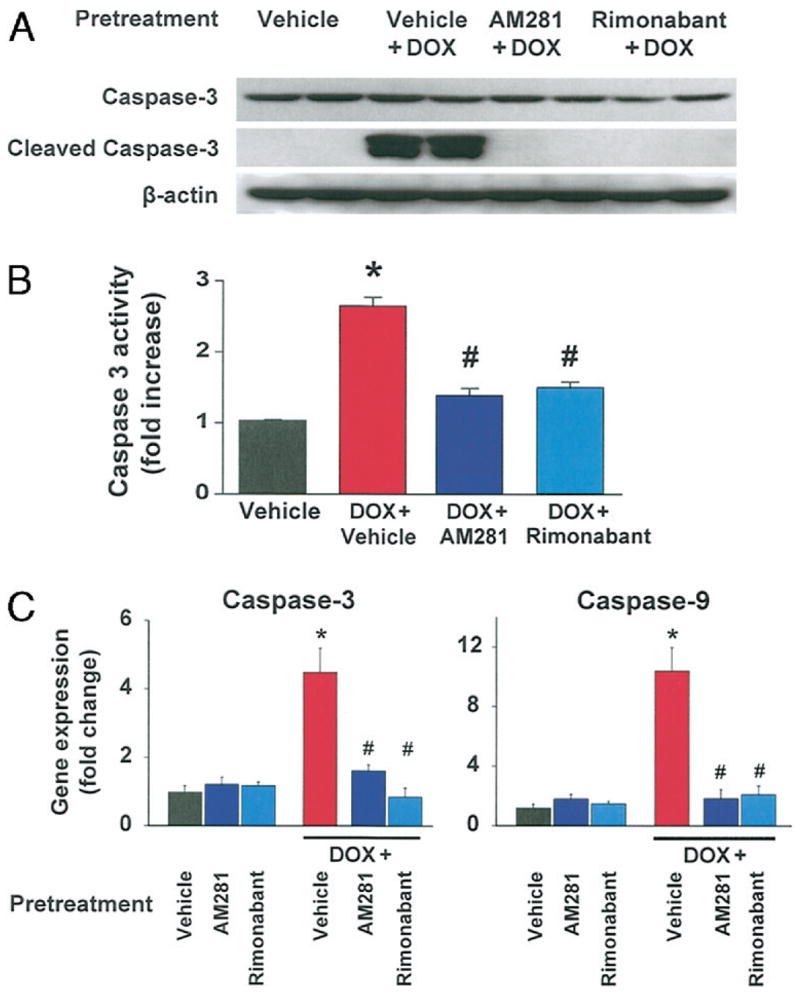
(A) Effects of CB1 antagonists on DOX-induced caspase-3 activation analyzed by Western blot from heart tissue homogenates. Treatment with rimonabant or AM281 reduced myocardial caspase-3 activation in DOX-treated mice. The blot was also probed for beta-actin level for loading control. Representative blot from at least 5 experiments. (B) Effects of CB1 antagonists on DOX-induced caspase-3 activity analyzed by colorimetric method from heart tissue homogenates. Treatment with rimonabant or AM281 reduced caspase-3 activity in DOX-treated mice. *p < 0.05 versus vehicle; #p < 0.05 versus DOX (n = 4 per group). (C) Effects of CB1 antagonists on DOX-induced caspase-3 and caspase-9 gene expression. Data were analyzed with 2 housekeeping genes, and data presented here were normalized with beta-actin. Treatment with rimonabant or AM281 reduced myocardial caspase-3 and caspase-9 gene expression in DOX-treated mice. *p < 0.05 versus vehicle; #p < 0.05 versus DOX (n = 6 to 15 per group). Abbreviations as in Figure 1.
As shown in Figure 5B, DOX treatment induced marked elevation in the caspase-3 activity in the myocardial tissues (~2.8-fold) compared with control mice. Rimonabant/AM281 pretreatment of mice prevented the DOX-induced increase in caspase-3 activity.
We also analyzed gene expression of 2 apoptotic markers, caspase-3 and caspase-9, from the same group of heart samples. In agreement with the increased protein expression, caspase-3 mRNA was also increased significantly by DOX treatment to 4.5 ± 0.7-fold over control group, and this increase was markedly attenuated by AM281 or rimonabant pretreatment (1.6 ± 0.2 and 0.9 ± 0.2, respectively) (Fig. 5C). Doxorubicin also induced a 10.4 ± 1.6-fold increase in caspase-9 gene expression compared with normal hearts, which was reduced by pretreatment with AM281 or rimonabant to a 1.8 ± 0.6 and 2.1 ± 0.6-fold change, respectively. In vehicle-treated control mice, AM281 or rimonabant treatment did not affect caspase-3 and caspase-9 gene expression.
CB1 receptor antagonists attenuate DOX-induced DNA fragmentation
Doxorubicin also increased myocardial TUNEL staining in the myocardium, which was largely attenuated by either rimonabant or AM281 pretreatment (Fig. 6A).
Figure 6. Effects of Rimonabant and AM281 on DOX-Induced Myocardial Apoptosis In Vivo Determined by TUNEL and DNA Fragmentation Assay.
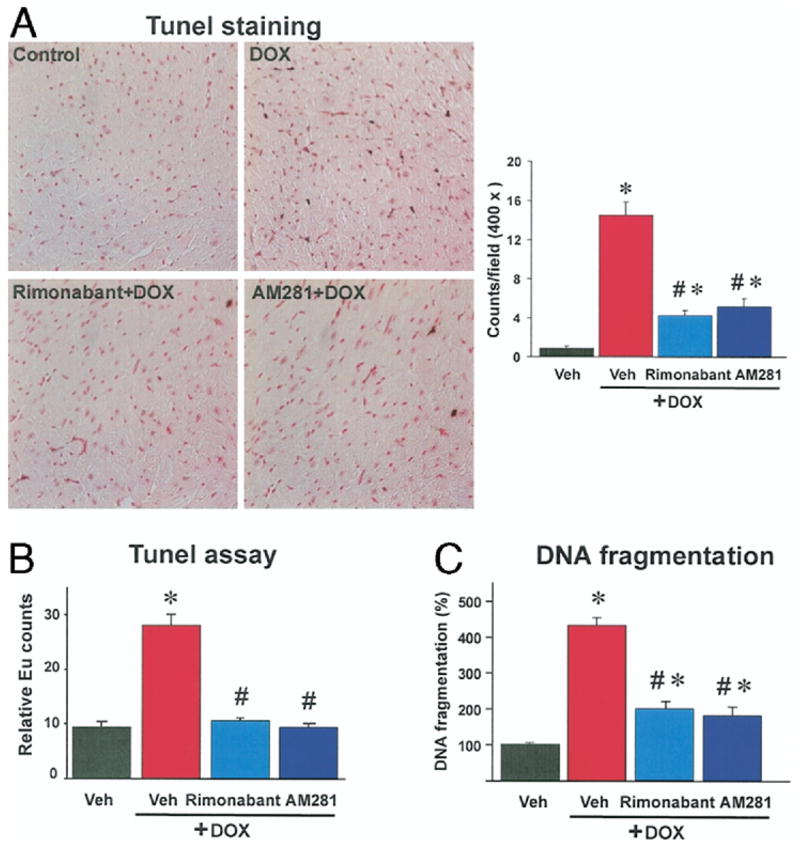
(A) Note the increased myocardial terminal deoxynucleotidyltransferase-mediated nick-end labeling (TUNEL) staining from doxorubicin (DOX)-treated mice (brown). Treatment with cannabinoid-1 antagonist rimonabant and AM281 reduced myocardial TUNEL staining in DOX-treated mice. Similar immunohistochemical profiles were seen in n = 3 hearts per group. Quantitative measurements were carried out in 20 fields per group. *p < 0.05 versus vehicle (Veh); #p < 0.05 versus DOX. (B) Effects of rimonabant and AM281 on DOX-induced myocardial apoptosis in vivo by TUNEL assay. *p < 0.05 versus vehicle; #p < 0.05 versus DOX (n = 4 per group). (C) Effects of rimonabant and AM281 on DOX-induced deoxyribonucleic acid (DNA) fragmentation in vivo. Changes are expressed in % of myocardial DNA fragmentation in vehicle-treated mice (100%). *p < 0.05 versus vehicle; #p < 0.05 versus DOX (n = 4 per group). Eu = Europium.
To strengthen the preceding conclusion, we used 2 additional quantitative methods. Doxorubicin-induced DNA fragmentations in the myocardial tissue was increased 2.9- and 4.5-fold compared with sham control measured by quantitative TUNEL and DNA fragmentation assays, and was largely attenuated by pretreatment with CB1 antagonists (Figs. 6B and 6C).
Effects of DOX on myocardial CB1 and CB2 receptors
Myocardial CB1 receptor protein levels were unchanged in DOX-treated mice as documented by using Western blots (Fig. 7A). The specificity of the antibodies used was confirmed by the absence of a specific band using myocardial tissue from a CB1 knockout mouse. Myocardial CB1 or CB2 receptor gene expression was not affected by DOX treatment in mice as verified using reverse transcription-polymerase chain reaction (Fig. 7B) as well as quantitative real-time polymerase chain reaction where the CB1 or CB2 receptor gene expressions were normalized to beta-actin gene expression (Fig. 7C). Similar results were obtained in cardiomyocytes (Fig. 7D, lower part).
Figure 7. Effects of DOX on Myocardial Endocannabinoid Content and CB 1/2 Receptor Expression.
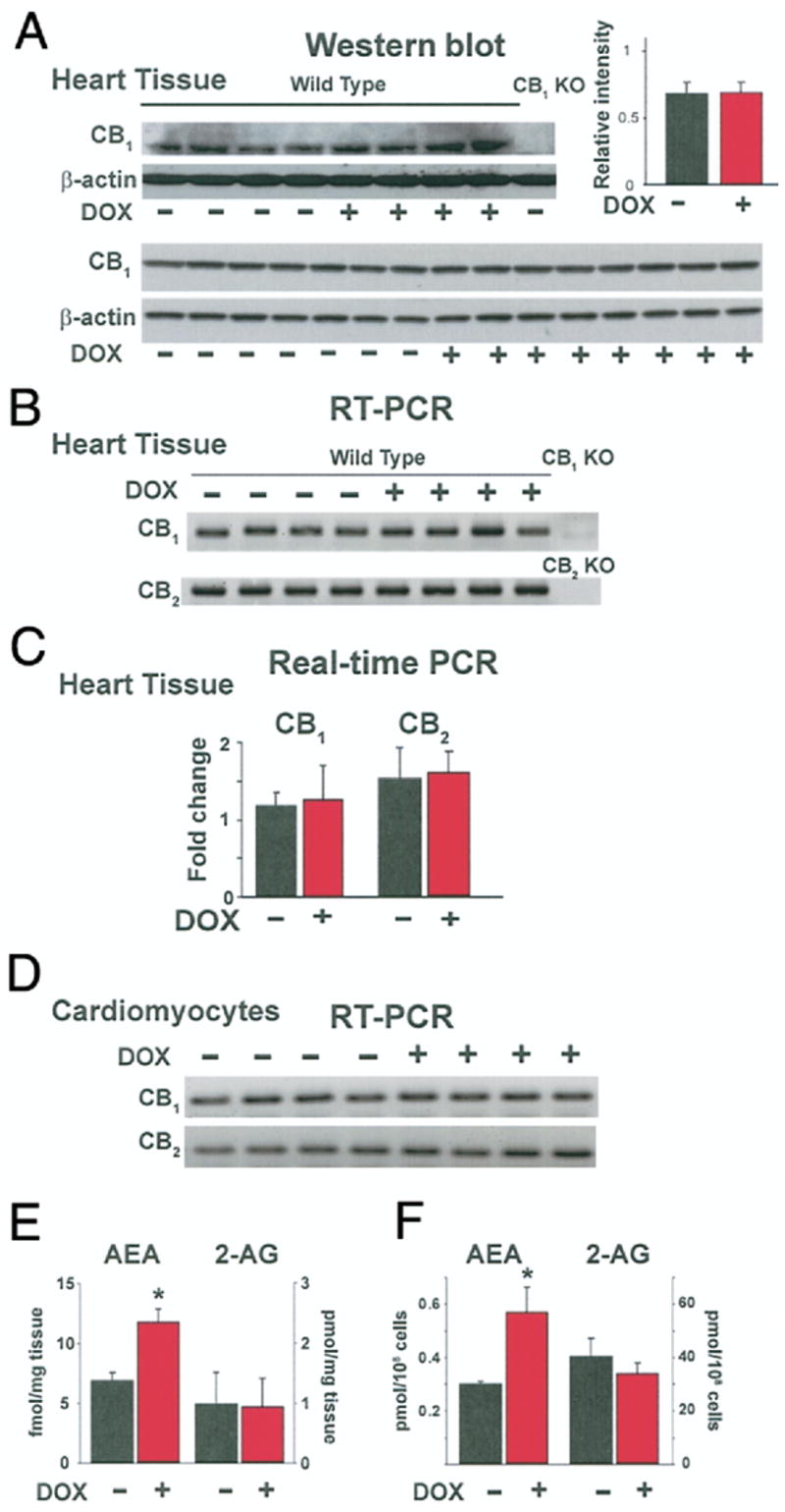
(A) Evidence of CB1 receptor protein expression in total lysate of DOX-treated mouse heart tissue homogenate by Western blot analysis. (B and D) Evidence of CB1 and CB2 receptor gene expression by semiquantitative reverse transcription-polymerase chain reaction (RT-PCR) from complementary deoxyribonucleic acid of both untreated and DOX-treated mouse heart samples (B) and cardiomyocytes (D). (C) Evidence of CB1 receptor and CB2 receptor gene expression in heart tissue by quantitative real-time PCR after normalization to beta-actin (n = 6 to 15). (E) Effect of DOX on anandamide (AEA) and 2-arachidonylglycerol (2-AG) production in vivo by liquid chromatography mass spectrometry (LCMS) analysis of heart tissue samples. *p < 0.05 versus vehicle (n = 6). (F) Effect of DOX (1 μM for 2 h) on AEA and 2-AG production in vitro by LCMS analysis of H9c2 cells. Increased AEA levels were observed in doxorubicin treated samples. *p < 0.05 versus vehicle (n = 3). KO = knockout; other abbreviations as in Figure 1.
DOX increased endocannabinoid AEA content both in vivo and in vitro
Doxorubicin treatment resulted in increased AEA but not 2-AG content in the myocardium (Fig. 7E). Similarly, elevated AEA levels were detected in DOX-treated H9c2 cells as compared with the vehicle-treated control cells (Fig. 7F).
Discussion
The natural ligands of CB1 receptors are the endocannabinoids AEA and 2-AG, both of which are present in the myocardium, along with CB1 receptors (16,17). Numerous previous experimental studies have demonstrated that activation of the endocannabinoid system may contribute to the hypotension and compromised cardiovascular function in a variety of pathophysiological states (e.g., hemorrhagic, endotoxic, and cardiogenic shock, advanced liver cirrhosis, and cirrhotic cardiomyopathy) through the activation of myocardial and vascular CB1 receptors. Importantly, in all these conditions, the cardiovascular depressive effects could be prevented or reversed by various CB1 antagonists (16,17).
Our present findings provide evidence that endocannabinoid AEA is overproduced in a well-established model of DOX-induced acute heart failure, and CB1 antagonists improve compromised contractile function. Furthermore, we show that CB1 antagonists exert powerful cytoprotective effect in cardiomyocytes against DOX-induced cardiotoxicity both in vivo and in vitro by reducing apoptosis, offering a novel approach for the prevention of this devastating complication of chemotherapy.
There is limited and conflicting information about the role of cannabinoid receptor activation in cell-protective mechanisms against ischemia/reperfusion (I/R) damage in the heart (16,17,25). A major limitation of these studies is the use of buffer-perfused isolated heart preparations, in which the effects of endocannabinoids and synthetic agonists on immune cells, which are pivotal in reperfusion damage, cannot be studied, as well as the use of nonselective cannabinoid ligands. In a more relevant in vivo model of myocardial I/R injury induced by coronary occlusion/reocclusion in anesthetized mice, the published evidence points to the protective role of CB2 but not CB1 receptor activation (CB2 activation on immune cells was responsible for the reduced leukocyte-dependent myocardial damage associated with reperfusion) (26). The presence of CB2 receptors in the myocardium and their function has never been convincingly demonstrated so far (selective CB2 receptor agonists do not decrease blood pressure and myocardial contractility in vivo, in contrast to the well-known hypotensive and cardiodepressive effects of CB1 agonists). Two studies using rat models of acute and chronic myocardial infarction have demonstrated that endocannabinoids contribute to the hypotension and cardiodepression associated with acute cardiogenic shock, which could be attenuated by CB1 antagonists (27,28). Cannabinoid-1 receptor antagonist AM251 was suggested to promote remodeling in the later chronic heart failure study, but it tended to improve survival. In contrast, the treatment with CB1 agonist HU210 was shown to prevent endothelial dysfunction, but it increased left ventricular end-diastolic pressure (28). However, the major limitation of the previous study (28) is the use of high dose of HU210, which was previously reported to cause severe psychotropic side effects and hypothermia, and the use of suboptimal doses of CB1 antagonist AM251 (17). Furthermore, HU210 was previously demonstrated to exert antiarrhythmic, anti-inflammatory properties, and to decrease myocardial damage in perfused hearts by mechanisms not related to CB1 activation, complicating the interpretation of these results (17).
In our present study, we used a well-established mouse model of DOX-induced acute heart failure in which the cardiac dysfunction has been characterized by using either echocardiography (14,21), tissue Doppler imaging (22), or pressure–volume systems (6,15). Importantly, in this model, a direct correlation between the degree of myocardial apoptosis and the severity of DOX-induced heart failure has been established (22). Apoptotic cell death is a key component in DOX-induced cardiotoxicity as established by numerous studies (11,13,14,24,29), and also indicated by the present findings.
Here we demonstrate that pharmacologic inhibition of CB1 receptors with rimonabant and AM281 conferred marked protection against DOX-induced cardiac dysfunction and cell death both in vivo and in vitro. The protective effect against DOX-induced cell death was not observed with CB1 and CB2 agonists and CB2 antagonists in vitro. Thus, CB1 antagonists may protect against DOX-induced cardiotoxicity by exerting potent cytoprotective effects on the one hand, and by antagonizing the cardiodepression elicited by endocannabinoid AEA on the other.
The observed protective effect of CB1 antagonists and the DOX-induced increase in myocardial AEA content may, therefore, suggest that DOX-induced cardiotoxicity is associated with and mediated, at least in part, by activation of the endocannabinoid system. Collectively, the present findings suggest that CB1 antagonists may represent a new cardioprotective strategy against DOX-induced cardiotoxicity and perhaps other cardiovascular pathologies associated with increased cell death. Further studies are warranted to investigate the underlying signaling mechanisms of these beneficial effects.
Supplemental Materials
Acknowledgments
Dr. Pacher would like to dedicate this study to his beloved mother Bolfert Iren who died from the cardiovascular complications of chemotherapy. The authors are indebted to Millar Instruments for the excellent customer support and generous gift of PVR 1045 P-V catheter.
This study was supported by the Intramural Research Program of NIH/NIAAA (to Dr. Pacher).
Abbreviations and Acronyms
- AEA
anandamide
- AG
arachidonoylglycerol
- CB1/CB2
cannabinoid-1/-2
- DOX
doxorubicin/adriamycin
- I/R
ischemia/reperfusion
- TUNEL
terminal deoxynucleotidyltransferase-mediated nick-end labeling
APPENDIX
For expanded Materials and Methods, please see the online version of this article.
References
- 1.Young RC, Ozols RF, Myers CE. The anthracycline antineoplastic drugs. N Engl J Med. 1981;305:139–53. doi: 10.1056/NEJM198107163050305. [DOI] [PubMed] [Google Scholar]
- 2.Hortobagyi GN. Anthracyclines in the treatment of cancer. An overview Drugs. 1997;54(Suppl 4):1–7. doi: 10.2165/00003495-199700544-00003. [DOI] [PubMed] [Google Scholar]
- 3.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–5. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 4.Myers CE, McGuire WP, Liss RH, Ifrim I, Grotzinger K, Young RC. Adriamycin: the role of lipid peroxidation in cardiac toxicity and tumor response. Science. 1977;197:165–7. doi: 10.1126/science.877547. [DOI] [PubMed] [Google Scholar]
- 5.Doroshow JH, Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem. 1986;261:3068–74. [PubMed] [Google Scholar]
- 6.Pacher P, Liaudet L, Bai P, et al. Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation. 2003;107:896–904. doi: 10.1161/01.cir.0000048192.52098.dd. [DOI] [PubMed] [Google Scholar]
- 7.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bai P, Mabley JG, Liaudet L, Virag L, Szabo C, Pacher P. Matrix metalloproteinase activation is an early event in doxorubicin-induced cardiotoxicity. Oncol Rep. 2004;11:505–8. [PubMed] [Google Scholar]
- 9.Kizaki K, Ito R, Okada M, et al. Enhanced gene expression of myocardial matrix metalloproteinases 2 and 9 after acute treatment with doxorubicin in mice. Pharmacol Res. 2006;53:341–6. doi: 10.1016/j.phrs.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Tokarska-Schlattner M, Zaugg M, Zuppinger C, Wallimann T, Schlattner U. New insights into doxorubicin-induced cardiotoxicity: the critical role of cellular energetics. J Mol Cell Cardiol. 2006;41:389–405. doi: 10.1016/j.yjmcc.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 11.Kumar D, Kirshenbaum LA, Li T, Danelisen I, Singal PK. Apoptosis in adriamycin cardiomyopathy and its modulation by probucol. Anti-oxid Redox Signal. 2001;3:135–45. doi: 10.1089/152308601750100641. [DOI] [PubMed] [Google Scholar]
- 12.Wu S, Ko YS, Teng MS, et al. Adriamycin-induced cardiomyocyte and endothelial cell apoptosis: in vitro and in vivo studies. J Mol Cell Cardiol. 2002;34:1595–607. doi: 10.1006/jmcc.2002.2110. [DOI] [PubMed] [Google Scholar]
- 13.Ueno M, Kakinuma Y, Yuhki K, et al. Doxorubicin induces apoptosis by activation of caspase-3 in cultured cardiomyocytes in vitro and rat cardiac ventricles in vivo. J Pharmacol Sci. 2006;101:151–8. doi: 10.1254/jphs.fp0050980. [DOI] [PubMed] [Google Scholar]
- 14.Li K, Sung RY, Huang WZ, et al. Thrombopoietin protects against in vitro and in vivo cardiotoxicity induced by doxorubicin. Circulation. 2006;113:2211–20. doi: 10.1161/CIRCULATIONAHA.105.560250. [DOI] [PubMed] [Google Scholar]
- 15.Pacher P, Liaudet L, Bai P, et al. Activation of poly(ADP-ribose) polymerase contributes to development of doxorubicin-induced heart failure. J Pharmacol Exp Ther. 2002;300:862–7. doi: 10.1124/jpet.300.3.862. [DOI] [PubMed] [Google Scholar]
- 16.Pacher P, Batkai S, Kunos G. Cardiovascular pharmacology of cannabinoids. Handb Exp Pharmacol. 2005;168:599–625. doi: 10.1007/3-540-26573-2_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Despres JP, Golay A, Sjostrom L. Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N Engl J Med. 2005;353:2121–34. doi: 10.1056/NEJMoa044537. [DOI] [PubMed] [Google Scholar]
- 19.Pi-Sunyer FX, Aronne LJ, Heshmati HM, Devin J, Rosenstock J. Effect of rimonabant, a cannabinoid-1 receptor blocker, on weight and cardiometabolic risk factors in overweight or obese patients: RIO-North America: a randomized controlled trial. JAMA. 2006;295:761–75. doi: 10.1001/jama.295.7.761. [DOI] [PubMed] [Google Scholar]
- 20.Gelfand EV, Cannon CP. Rimonabant: a cannabinoid receptor type 1 blocker for management of multiple cardiometabolic risk factors. J Am Coll Cardiol. 2006;47:1919–26. doi: 10.1016/j.jacc.2005.12.067. [DOI] [PubMed] [Google Scholar]
- 21.Delgado RM, 3rd, Nawar MA, Zewail AM, et al. Cyclooxygenase-2 inhibitor treatment improves left ventricular function and mortality in a murine model of doxorubicin-induced heart failure. Circulation. 2004;109:1428–33. doi: 10.1161/01.CIR.0000121354.34067.48. [DOI] [PubMed] [Google Scholar]
- 22.Neilan TG, Jassal DS, Perez-Sanz TM, et al. Tissue Doppler imaging predicts left ventricular dysfunction and mortality in a murine model of cardiac injury. Eur Heart J. 2006;27:1868–75. doi: 10.1093/eurheartj/ehl013. [DOI] [PubMed] [Google Scholar]
- 23.Pacher P, Thomas AP, Hajnoczky G. Ca2+ marks: miniature calcium signals in single mitochondria driven by ryanodine receptors. Proc Natl Acad Sci U S A. 2002;99:2380–5. doi: 10.1073/pnas.032423699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nozaki N, Shishido T, Takeishi Y, Kubota I. Modulation of doxorubicin-induced cardiac dysfunction in toll-like receptor-2-knockout mice. Circulation. 2004;110:2869–74. doi: 10.1161/01.CIR.0000146889.46519.27. [DOI] [PubMed] [Google Scholar]
- 25.Lamontagne D, Lepicier P, Lagneux C, Bouchard JF. The endogenous cardiac cannabinoid system: a new protective mechanism against myocardial ischemia. Arch Mal Coeur Vaiss. 2006;99:242–6. [PubMed] [Google Scholar]
- 26.Di Filippo C, Rossi F, Rossi S, D’Amico M. Cannabinoid CB2 receptor activation reduces mouse myocardial ischemia-reperfusion injury: involvement of cytokine/chemokines and PMN. J Leukoc Biol. 2004;75:453–9. doi: 10.1189/jlb.0703303. [DOI] [PubMed] [Google Scholar]
- 27.Wagner JA, Hu K, Bauersachs J, et al. Endogenous cannabinoids mediate hypotension after experimental myocardial infarction. J Am Coll Cardiol. 2001;38:2048–54. doi: 10.1016/s0735-1097(01)01671-0. [DOI] [PubMed] [Google Scholar]
- 28.Wagner JA, Hu K, Karcher J, et al. CB(1) cannabinoid receptor antagonism promotes remodeling and cannabinoid treatment prevents endothelial dysfunction and hypotension in rats with myocardial infarction. Br J Pharmacol. 2003;138:1251–8. doi: 10.1038/sj.bjp.0705156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Childs AC, Phaneuf SL, Dirks AJ, Phillips T, Leeuwenburgh C. Doxorubicin treatment in vivo causes cytochrome C release and cardiomyocyte apoptosis, as well as increased mitochondrial efficiency, superoxide dismutase activity, and Bcl-2:Bax ratio. Cancer Res. 2002;62:4592–8. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.