

Abstract
Xeroderma pigmentosum variant (XP-V) cells lack the damage-specific DNA polymerase η and have normal excision repair but show defective DNA replication after UV irradiation. Previous studies using cells transformed with SV40 or HPV16 (E6/E7) suggested that the S-phase response to UV damage is altered in XP-V cells with non-functional p53. To investigate the role of p53 directly we targeted p53 in normal and XP-V fibroblasts using short hairpin RNA. The shRNA reduced expression of p53, and the downstream cell cycle effector p21, in control and UV irradiated cells. Cells accumulated in late S phase after UV, but after down-regulation of p53 they accumulated earlier in S. Cells in which p53 was inhibited showed ongoing genomic instability at the replication fork. Cells exhibited high levels of UV induced S-phase γH2Ax phosphorylation representative of exposed single strand regions of DNA and foci of Mre11/Rad50/Nbs1 representative of double strand breaks. Cells also showed increased variability of genomic copy numbers after long-term inhibition of p53. Inhibition of p53 expression dominated the DNA damage response. Comparison with earlier results indicates that in virally transformed cells cellular targets other than p53 play important roles in the UV DNA damage response.
Keywords: DNA replication, ultraviolet, DNA polymerase η, p53, checkpoint, cell cycle, foci, DNA damage, genomic instability, γH2Ax
Introduction
The ultraviolet (UV) component of sunlight is the major carcinogen involved in the etiology of skin cancer [1, 2]. UV irradiation produces DNA photoproducts that are blocks to DNA replication by normal replicative DNA polymerases. A specialized, damage-specific, distributive polymerase, DNA polymerase η (Pol η) is required for replication past these photoproducts [3] In humans, inactivation of the Pol η gene results in sunlight sensitivity and causes the rare hereditary cancer-prone xeroderma pigmentosum variant syndrome (XP-V) [4, 5]. XP patients have a cancer risk 5,000 times greater than that of the general population, including basal cell carcinoma (BCC), squamous cell carcinoma (SCC), and melanoma [6]. Cells from XP-V individuals lack Pol η and have a reduced capacity to replicate UV-damaged DNA and show hypermutability after UV exposure [7-12].
TP53 is mutated in the majority of human cancers [13, 14], including skin cancers of the BCC and SCC type [1]. Mutations in TP53 are found with high frequency (>50%) in sporadic BCC, and at a slightly lower frequency (up to 45%) in sporadic SCC and these usually bear the UV signature CC to TT and C to T transitions indicative of ultraviolet radiation B (UV-B) exposure [1, 15]. The role of p53 in melanoma remains somewhat controversial, with some groups reporting mutation rates between 13-25% and others documenting much lower rates [1]. Since XP-V cells lack Pol η, they must employ other mechanisms aside from Pol η-dependent translesional synthesis to negotiate blocks to DNA replication, such as those posed by UV lesions in DNA. These alternative mechanisms involve the use of alternative polymerases such as Pol ι [16] or recombination between sister chomatids [17]. Several lines of evidence suggest that p53 plays a major role in mediating downstream events associated with S phase arrest from UV damage [18], especially in XP-V cells with protracted S phase arrest due to Pol η deficiency [19]. XP-V cells, in which p53 function was abrogated due to viral transformation by HPV16(E6) [20, 21] or SV40 [22], demonstrated a dramatic increase in sensitivity to the lethal and cytogenetic effects of UV light [23]. These p53-compromised cells employed Mre11 and Rad51-dependent recombination pathways to recover from UV damage [17, 24-28], implying that replication forks arrested by UV damage rapidly undergo a collapse leading to double strand breakage. These cells also show increased sister chromatid exchanges indicative of increased rates of recombination during the S phase [23].
To define the role of p53 in the UV response of XP-V cells without interference of oncogenic viral proteins, we chose to inhibit p53 with RNA interference using a sequence previously shown to be effective for p53 suppression in p53-normal primary dermal XP-V fibroblasts [29]. We demonstrate that the cellular DNA damage response to low-dose UV irradiation is dominated by loss of p53, that otherwise protects the DNA replication fork from breakdown, and masks the DNA replication deficiencies of the Pol η bypass polymerase.
Results
p53-suppressed XP-V and normal fibroblasts[30]
Primary dermal fibroblasts from an XP-V patient (GM3617) and a normal control (GM5659D) were immortalized with the catalytic subunit of telomerase (hTERT) which does not disrupt normal cell cycle checkpoints [30-32] [33] and cells were designated GM5659DT and GM3617T. We employed a gene knockdown approach using RNA interference directed against a target sequence of p53 [29]. We stably infected cells with vectors that synthesize short hairpin RNA (shRNA) directed against p53 under the control of the H1 promoter (designated pSuper.Retro.p53.Neo.Gfp vector or the empty vector pSuper.Retro.Neo.Gfp) to obtain persistent intracellular synthesis of siRNA. P53 and p21 gene expression was analyzed by Taqman (Figs 1A, B). GM5659DT with stable transfection of p53 shRNA showed p53 expression reduced to 9.1 % of vector-transfected GM5659DT (Fig 1 A). GM3617T with stable transfection of p53 shRNA showed p53 expression reduced to 8.4 % of vector-transfected GM3617DT (Fig. 1A). This shRNA was more effective at inhibiting the levels of p53 protein in XP-V (GM3617T) than in normal (5659DT) cultures (Fig. 1C). In wild-type cells, levels of damage-inducible p53 were reduced by 50% with shRNA (Fig. 1C). In XP-V cells, levels of damage-inducible p53 were reduced 20-fold with shRNA treatment (Fig. 1C). The results indicate that shRNA directed against p53 effectively reduced the levels of damage-inducible p53 in both cell types, although this effect was more pronounced in XP-V cells than in wild type cells. This difference may be idiosyncratic, and not necessarily related to the XP-V genotype.
Figure 1.

Efficacy of retroviral shRNA directed against p53 in reducing gene expression and protein levels. (A) Effect on basal p53 expression as measured by Taqman in XP-V (GM3617T) and wild-type (GM5659DT) cells. (B) Effect on basal p21 expression as measured by Taqman in XP-V (GM3617T) and wild-type (GM5659DT) cells. (C) Western blots using antibody to ser-15 phospho-p53 showing the basal and damage-inducible p53 protein levels in XP-V (GM3617T) and wild-type (GM5659DT) cells. Bar graphs in the right panels are densitometry plots of band intensities in the adjacent Western blots, with basal p53 levels in vector-treated cells set to 100%. Loading controls, β actin, shown below each western. (D) Relative expression of p21, as a ratio to expression of GUS (see methods) as a function of UV irradiation and shRNA down-regulation of p53.
To examine the functionality of the p53 suppression, we measured the expression of the p53 target gene, p21. GM5659DT with stable transfection of p53 shRNA showed p21 expression reduced to 7.1 % of vector-transfected GM5659DT (Fig. 1B). GM3617T with stable transfection of p53 shRNA showed p21 expression reduced to 5.8 % of vector-transfected GM3617DT (Fig. 1B). At the gene expression level, the p53 shRNA was equally effective in normal and XPV cells. The induction of p21 by p53 after UV irradiation was also suppressed by shRNA (Fig 1 D). Normal cells showed higher base-line levels of p21 expression than XP-V cells, and both showed induction by UV. The relative level of induction as a ratio over isogenic control cells was greater in XP-V cells, but this owed more to the difference in base-line levels than the induced levels. The responses after shRNA were all suppressed, both control and UV irradiated, though as in the cells without shRNA the levels in XP-V cells were above those in normal cells.
UV-induced S-phase arrest
Changes in cell cycle distribution after UV were examined by FACS analysis (Fig. 2). Vector-infected normal and XP-V primary fibroblasts accumulated late in the S-phase arrest after UV damage, as has often been reported [19, 34] (Figs. 2B, F), relative to unirradiated controls (Figs 2A, E). Unirradiated cells containing shRNA appeared to have a slightly larger accumulation of cells in S-phase, (Figs. 2C, G) compared to vector-infected controls, for both normal and XP-V cells but this was not examined in detail (Figs. 2A, E). UV-irradiated cells containing shRNA exhibited an accumulation of cells earlier in the S phase (Figs. 2D, H) than occurred in vector-infected UV-irradiated cells (Figs. 2B, 2E) or unirradiated cells (Figs. 2C, G). This early S-phase arrest was similar in normal (Fig. 2D) and XP-V cells (Fig. 2H).
Figure 2.

Flow cytometric analysis of normal (GM5659DT) and XPV (GM3617T) cells irradiated with 0 (A,B,C,D) or 5.2 J/m2 of UV (C,D,G,H) and harvested 16 hours later and stained with propidium iodide. G1 and G2 peaks identified in upper left panel. A: GM5659DT cells infected with pUper.Retro.Neo.Gfp vector. E: unirradiated GM3617T cells infected with pSuper.Retro.Neo.Gfp vector. B: UV-irradiated GM5659DT cells infected with pSuper.Retro.Neo.Gfp vector. F: GM3617T cells infected with pSuper.Retro.Neo.Gfp vector, grown in medium after UV. C: UV-irradiated GM5659DT cells infected with pSuper.Retro.p53.Neo.Gfp or lower, unrradiated pSuper.Retro.p53.Neo.Gfp GM3617T cells. D: UV-irradiated GM5659DT cells infected with pSuper.Retro.p53.Neo.Gfp or H, UV-irradiated pSuper.Retro.p53.Neo.Gfp GM3617T cells.
UV survival
In a short-term assay for UV survival (Fig. 3), no appreciable differences were observed between XP-V and normal fibroblasts cells stably infected with pSuper.Retro.p53.Neo.Gfp or the pSuper.Retro.Neo.Gfp vector. Caffeine sensitized both normal and XP-V cells to cell killing by UV, but this sensitization did not discriminate normal from XP-V cells. We have found since that immortalization by hTERT can activate a caffeine-sensitive ATR pathway in late passage cells (de Feraudy and Cleaver unpublished observations), that can mask the characteristic XP-V sensitization that is small in primary fibroblasts but much larger in SV40 transformed cells [7, 35]. The shRNA appears to sensitize normal cells but not XP-V cells, suggesting thatloss of p53 increases lethal effects of UV damage in the presence of a functional Pol η but not in its absence.
Figure 3.

Survival of normal and XP-V cells after UV irradiation. , infected with pSuper.Retro.Neo.Gfp vector; , infected with pSuper.Retro.Neo.Gfp vector and grown in caffeine after UV; , infected with p53 shRNA-expressing pSuper.Retro.p53.Neo.Gfp; , infected with p53 shRNA-expressing pSuper.Retro.p53.Neo.Gfp and grown in caffeine after UV.
The S phase DNA damage response involving γH2Ax and Mre11
Phosphorylation of the minor histone H2Ax (γH2Ax) is a common marker of DNA damage. γH2Ax is formed as punctate foci at DNA double strand breaks caused by ionizing radiation or breaks at arrested replication forks. Following UV damage the predominant expression of γH2Ax is as nuclear-wide uniform intense staining in the S phase caused by single-stranded regions of DNA at arrested replication forks [36]. In a FACS scan of normal GM5659T control cells (Fig 4A) γH2Ax represented background levels of fluorescence, with a preponderance of G1 cells consistent with the cell cycle distribution (Fig 2A). After UV irradiation, an increased number of highly fluorescent cells were distributed in the S phase (Fig 4A). In GM5659T cells with shRNA and SV40 transformed normal cells there was a larger number of cells in the S and G2 phases cells. After UV irradiation the S phase distribution was greatly enhanced over that in irradiated GM5659T normal cells (Fig 4A). The level of γH2Ax was greatest in the shRNA treated cells indicating that depletion of p53 caused a greater replication arrest and consequent increased ATRIP/ATR signal transduction [37], even above that seen in SV40 transformed cells. This is consistent with replication arrest generating a stronger signal to the kinases that phosphorylate H2Ax in the absence of normal levels of p53.
Figure 4.
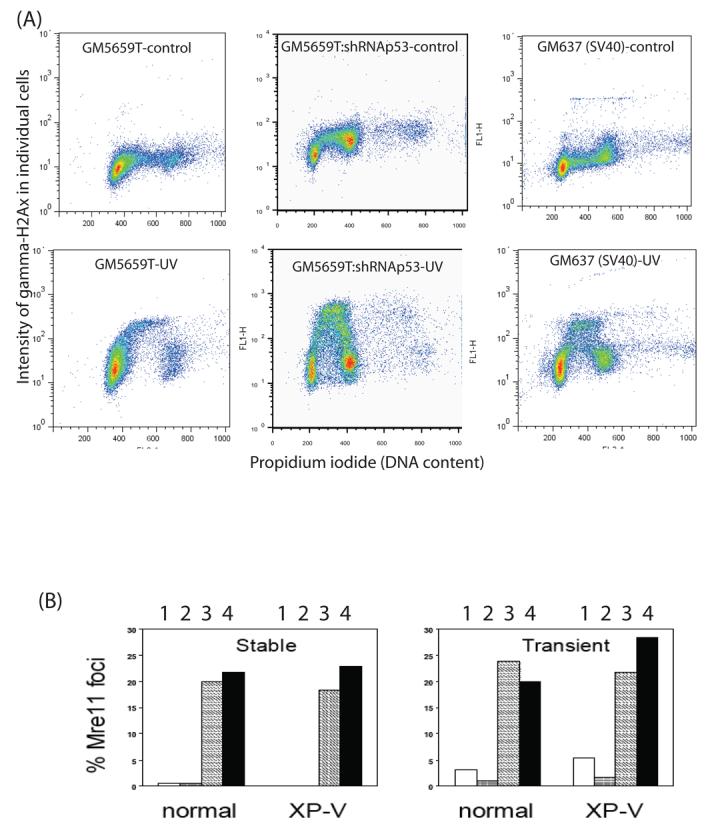
A. FACS scans of γH2Ax versus propidium iodide (DNA content) in GM5659T, GM5659T plus shRNA against p53, and GM637 (normal SV40 transformed) cells that were unirradiated or irradiated with 20 J/m2 and harvested 4 hr later. B. p53-dependence of Mre11 foci in XPV and normal cells. Histograms of the percentage of XP-V and normal cells showing Mre11 foci after stable (left panel) or transient (right panel) infection with pSuper.Retro.p53.Neo.Gfp or its vector control. Open bars (1), vector-infected no irradiation control; grey bars (2), vector-infected cells after 13 J/m2 UV; hatched bars (3), unirradiated cells infected with pSuper.Retro.p53.Neo.Gfp; black bars (4), cells infected with pSuper.Retro.p53.Neo.Gfp after 13 J/m2 UV.
The Mre11-Rad50-Nbs1 (MRN) complex acts as a double strand break sensor and is involved in repair of double strand breaks (DSBs) by nonhomologous end joining (NHEJ) [38, 39] and relocates into foci after UV irradiation [25, 27]. XP-V and normal fibroblasts the were infected either transiently or stably with the control pSuper.Retro.p53.Neo.Gfp showed very few (1-5%) cells positive for Mre11 foci (Fig. 4 B). In contrast, the frequency of cells containing nuclear Mre11 foci was in the range of 20-25% in all cells infected with pSuper.Retro.p53.Neo.Gfp, both normal and XP-V cells irrespective of UV irradiation. Increased Mre11 foci were also seen in transiently infected cells after p53 suppression and again the effect was similar in normal and XP-V cells irrespective of irradiation.
Genomic instability after p53 suppression
After growing the pSuper.Retro.p53.Neo.Gfp-infected cells for 5 passages, we observed that the cellular morphology of some of the cells became less elongated, reminiscent of an epithelial morphology, and the cells began to grow in dense clusters. In contrast, vector-infected control cells maintained the normal elongated morphology characteristic of primary fibroblasts (Fig. 5, A, C). By passage 15, the pSuper.Retrop53.Neo.Gfp-infected cultures were dominated by the new morphology (Fig. 5, B, D), and by passage 20, the pSuper.Retro-p53-infected cultures no longer grew in clusters, but contained cells with epithelial morphology that grew at an even, open density. In contrast, vector-infected control cells continued to maintain the normal elongated morphology characteristic of primary fibroblasts.
Figure 5.

Chromosome gains and losses in normal and XP-V fibroblasts with or without p53 inhibition grown continuously for 20 passages. The genes are ordered by chromosome location from left to right and plotted against amplitude of copy number change. A, normal fibroblasts stably infected with the pRetro vector; B, vector- infected XP-V fibroblasts; C, normal fibroblasts stably infected with pRetro-p53; D, XP-V fibroblasts stably infected with pRetro-p53; E, SV40-transformed normal fibroblasts (GM637); F, SV40-transformed XP-V fibroblasts (XP30RO).
Genomic instability was investigated with genome-wide array comparative genomic hybridization (CGH) [40] to conduct a high resolution evaluation of the effect of p53 inhibition in normal and XPV fibroblasts at passage 20. Gains and losses of chromosomal regions were essentially absent in vector-infected normal (Fig. 5 E) and XP-V (Fig. 5 I) fibroblasts, with the exception of small losses in chromosomes 6 and 9 in the normal human fibroblasts, which was not observed when replicating the experiment. In contrast, genomic instability was clearly evident in pSuper.Retrop53.Neo.Gfp-infected normal and XP-V fibroblasts, with gains and losses both commonly observed (Fig. 5, F,J). For comparison, genomic instability was also examined in E6/E7 transformed (Fig. 5 G, K) and SV40-transformed versions of the same normal and XP-V fibroblasts (Fig. 4, H,L). Virally transformed cells also exhibited gains and losses of chromosomal regions.
Discussion
In XP-V cells that lack the translesional DNA Pol η, UV damage to DNA causes an asymmetric replication fork arrest due to a replication block on the leading strand of DNA [41-46]. These structures trigger the activation of intra-S-phase checkpoints [44]. These arrested replication forks contain long (several kB) regions of single-stranded DNA, which are covered by RPA which in turn binds p53, ATRIP/ATR and other proteins [42, 47-49]. Competition between p53 and these other factors at the ssDNA regions plays a complex role the intra-S-phase checkpoint [18], producing varied results depending on the cell types and transformed status.
We therefore examined the role of p53 directly by downregulating its expression with shRNA. Previous experiments in XP-V cells showed greatly increased UV sensitivity after transformation with HPV16 (E6) that inhibits p53 but not the retinoblastoma cell cycle regulator gene (Rb). This sensitivity was not observed after transformation of XP-V cells with HPV16 (E7) that inhibits Rb but not p53 [23, 24]. Similarly, transformation of XP-V cells with SV40, which compromises p53 function through binding to the viral antigen Large T, also increases the sensitivity of XP-V cells to the lethal and cytogenetic effects of UV light, especially when sensitized by caffeine [23, 35, 50]. In contrast, we observed smaller increases in post-UV sensitivity after p53 inhibition (Fig 3), suggesting that loss of p53 alone does not play a major role in the lethal effects of UV even though ii does increase genomic instability (Figs 4,5).
We observed that inhibition of p53 brought the UV-induced S-phase arrest earlier in the S phase (Fig 2) and increased the S phase γH2Ax intensity (Fig 4A) and the frequency of cells with Mre11 (Fig 4B). This indicates that p53 is a key player in protecting UV-damaged replication forks from generating or undergoing damaging downstream events. The dominance of p53 suppression over Pol η deficiency after UV indicates that p53 acts upstream or before Pol η bypass replication, acting earlier in the S phase. Reduction of p53 by shRNA had a similar effect as viral transformation in allowing increased signaling to γH2Ax formation in the S phase (Fig 4 A). No differences, however, in S-phase arrest associated with p53-deficiency were reported after symmetric replication fork stalling with hydroxyurea [51], suggesting that arrested forks generated by UV damage represent particularly fragile structures in which p53 has a major protective role prior to Pol η bypass.
In previous studies, it was observed that XP-V cells with impaired p53 employed an MRN-dependent recombination pathway in response to UV damage [17, 25, 27, 28], implying DSBs are generated during replication arrest [49, 52-54]. We observed that p53 suppression leads to a similar increase in the frequency of normal and XP-V cells with Mre11 foci, even without UV irradiation. Studies treating p53-normal and SV40 transformed cells with UV demonstrated that the stability of stalled replication forks depends on p53, such that DSBs accumulate after UV in SV40-transformed cells but not normal cells [49]. The same study also examined p53-normal and SV40-transformed XP-V cells and found that the effect of p53 was even more pronounced in XP-V cells; SV40-transformed XP-V cells converted all their UV-induced ssDNA regions into DSBs [49]. Our results indicate that p53 inactivation is a stronger factor for generation of S phase genomic instability (γH2Ax and Mre11) than Pol η deficiency alone. P53 deficiency alone does not elevate sister chromatid exchange frequencies (SCE) [55] but increases SCE frequencies dramatically at low UV doses [23]. Previously, virally transformed cells showed increased frequencies of cells with Mre11 foci only after UV irradiation, and XP-V cells were consistently higher than control. The similar increases in Mre11 foci-positive cells in both control and UV irradiated normal and XP-V cells after shRNA (Fig 4 B) reveals a dominant role for p53 levels in maintaining replication fork stability not seen in virally transformed cells.
Gross chromosomal rearrangements, amplifications and deletions are a common event in human cancer [56]. XP-V cells immortalized with hTERT show high frequencies of DNA breaks but few frank chromosomal anomalies [43]. These observations suggest that checkpoint function protects the broken DNA from generating major chromosomal changes. We observed multiple small gains and losses in chromosomal regions in hTERT-immortalized XP-V cells after down-regulation of p53, as measured by comparative genomic hybridization. The overall pattern emerging from this analysis is that of many small chromosomal changes, rather than gains or losses of entire chromosomes, as has been observed for certain skin tumors such as melanoma [57]. This effect of genomic instability was seen in both XP-V and normal fibroblasts, suggesting that p53 is a major driver for chromosomal instability that dominates that of Pol η deficiency alone.
The present results provide a basis for understanding previous reports on the DNA damage response of SV40 and HPV16(E6/E7) transformed XP-V cells [23-27]. Clearly, there are additional phenotypes in the virally transformed cells not evident in cells treated with shRNA against p53 (Table 1). Each of the three means of inactivating p53 are distinct, although all appear to destabilize the S phase in UV damaged cells and contribute to higher levels of DNA breakage (Fig 4) and increased importance of the ATR signal transduction pathway. XP-V cells transformed with SV40 uniquely show increased UV sensitivity when ATR is inhibited by caffeine [7, 35, 43]; this may be due to the elevated levels of p53 in equilibrium with large T that mimic high levels of damage and induce an apoptotic response [28]. XP-V cells transformed by HPV16(E6/E7) are highly UV sensitive without further sensitization [23], suggesting that there are additional targets for the E6 ubiquitin ligase that play supporting roles in the UV response. These could include the ATR pathway or homologous recombination, both of which are important in recovery from DNA replication arrest and lead to increased cell killing when blocked [58-60].
Table 1.
Phenotypes of human XP-V cells in which p53 is inactivated by different means and possible additional p53-dependent or independent mechanisms involved in viral transformed cells (see [23] and this study).
| Cell type |
P53 status | UV sensitivity |
Caffeine sensitization |
Apoptosis (A) or senescence (S) |
Additional possible mechanisms |
|---|---|---|---|---|---|
| Normal | Normal | Normal | Normal | S | --------------- |
| SV40 | Large T complex |
+ | +++ | A | Enhanced UV apoptosis by p53 |
| HPV16 (E6/E7) |
Degraded | +++ | No further increase in sensitivity |
S | ATR or HR targeted by E6 ubiquitination (?) |
| shRNA | Reduced expression |
+ | Same as in normal cells |
S | ------------------- |
Materials and methods
Expression constructs
pSuper.Retro.Neo.Gfp, a retroviral vector system for expression of short interfering RNA and puromycin resistance (OligoEngine, WA), was digested with BglII/Hind. An effective siRNA target sequence for the silencing of p53 has been previously described (GACUCCAGUGGUAAUCUAC) [29]. The ends of this sequence were modified for the pSuper.Retro.Neo.Gfp vector, and two oligonucleotides, encoding a sense and antisense sequence for the expression of an siRNA directed against p53, were annealed: p53_FWD 5'-GAT CCC GAC TCC AGTG GTA ATC TAC TTC AAG AGA GTA GAT TAC CAC TGG AGT CTT TTT GGA AA-3' and p53_REV 5'-AGC TTT TCC AAA AAG ACT CCA GTG GTA ATC TAC TCT CTT GAA GTA GAT TAC CAC TGG AGT CGG G-3' and ligated into the above vector. Successful clones were verified by sequencing and were designated pSuper.Retro.p53.Neo.Gfp.
Cell culture
The XPV GM3617 fibroblasts (also designated XP30R0) and the wild-type control 5659D were obtained from The Human Genetic Mutant Cell Repository, Camden, NJ. The XPV fibroblast GM3617 contains a mutation in the XPV gene, which results in a chain termination of DNA Pol η. To immortalize these cells with telomerase we used a retrovirus to express the catalytic subunit of telomerase (hTERT)(kindly provided by Russel Pieper, UCSF). Stably infected cells were selected with blastacidin (200μM), and designated GM3617T and GM5659DT. SV40 transformed normal cells (GM637) and XP-V cells (derived from GM3617 cells REFERENCE) were also used in some experiments for comparison. Cells were grown in Eagle's MEM, supplemented with 2 mM glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% FBS (Life Technologies, Gaithersburg, MD).
Phi-NX packaging cells (a kind gift of Dr. Garry Nolan, Stanford) were transfected with pSuper.Retro.Neo-GFP or pSuper.Retro.Neo-GFP.p53 using standard calcium-phosphate techniques. Twenty-four hours later, the media overlying the Phi-NX cells was transferred to 80% confluent fibroblasts. After an additional 48 hours, neomycin (600 ug/mL) selection was initiated to obtain stable pooled cultures.
Expression Analysis
For p53 expression analysis, RNA from 250,000 cells was isolated using the PicoPure kit (Arcturus, Mountain View, CA) and for p21 expression RNA was isolated using the RNEasy kit (Qiagen Inc., Valencia, CA) and quantitated using a Nanodrop spectrophotometer (Nanodrop Technologies, Wilmington, DE). cDNA was synthesized from 100 ng of total RNA using an iScript kit (BioRad Laboratories, Hercules, CA) according to the manufacturer's instructions. P53 expression was normalized to that of the control gene Snrpd3. TaqMan probes for TP53 (HS00153340) and the control gene Snrpd3 (Hs00188207) were from Applied Biosystems (Foster City, CA). p21 expression was normalized to that of the control gene glucuronidase β (GUS). Sequences for the GUS primers and probe were: forward primer: CTC ATT TGG AAT TTT GCC GAT T, reverse primer: CCG AGT GAA GAT CCC CTT TTT A and probe: FAM/TGA ACA GTC ACC GAC GAG AGT GCT GG/BHQ1. Sequences for the p21 primers and probe were: forward primer: TGG AGA CTC TCA GGG TCG AAA, reverse primer: AGG ACT GCA GGC TTC CTG TG, and probe: FAM/CAG ACC AGC ATG ACA GAT TTC TAC CAC TCC A/BHQ1. All 6 oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA). Expression data was acquired on an ABI 7900 HT Sequence Detection System (Applied Biosystems, Foster City, CA) according to the manufacturer's recommendations. For data analysis, basal level of p53 or p21 expression in vector-treated cells was set to 100%.
Immunoblotting
Cells that had been stably infected with pSuper.Retro.p53.Neo.Gfp or the pSuper.Retro.Neo.Gfp vector were irradiated with 20 J/m2 of UV or sham-irradiated and harvested 2 hours later. A nuclear lysate was extracted using previously described methods [19]. Western blotting was carried out according to standard techniques, with uniformity of protein loading assessed by Ponceau staining. Nitrocellulose membranes were blocked overnight in 5% skim milk in TBST and then were probed with the phospho-specific rabbit polyclonal antibody Ser-15 p53 (9284, Cell Signaling, Beverly, MA) at a 1:1,000 dilution for 1 hour at room temperature. After 3 washes in TBST, the membrane was incubated with HRP-conjugated secondary antibody (goat anti-rabbit, Upstate, Waltham, MA) at a dilution of 1:10,000 for 1 hour at room temperature. After 5 washes in TBST, detection was conducted with enhanced chemiluminescence (Visualizer, Upstate, Waltham, MA) and exposure to ECL-reactive film (Amersham) with development in an automatic photodeveloper (Eastman Kodak). The membrane was stripped with Restore stripping buffer (Pierce, Rockford, IL) at 37 °C for 30 minutes and rinsed with TBST. The membrane was exposed to chemiluminescence detection as above to confirm that stripping was complete. The membrane was blocked again, and then incubated as above, but with monoclonal p53 antibody DO-1 (Santa Cruz Biotechnology), which recognizes total p53, at a dilution of 1:500. The remainder of the blotting procedure was conducted as described above. To verify similar protein loading between lanes, the membrane was stripped again, and incubated with monoclonal anti-beta-actin Clone AC15 (Sigma, St. Louis, MO) at a dilution of 1:10,000.
Cell cycle analysis
Cells that had been stably infected with pSuper.Retro.p53.Neo.Gfp or the pSuper.Retro.Neo.Gfp vector were irradiated with 5.2 J/m2 of UV or sham-irradiated and harvested 16 hours later. Fixed cells were processed and analyzed as described previously, using a FACSCalibur instrument (BD Biosciences) [19].
Cell survival
For cell survival, a rapid well assay was used involving measuring the growth of cells for 7 days in 24-well plates, as described previously [61]. In some experiments, cells were grown in caffeine (1 mM) after irradiation, which is known to sensitize XPV cells to UV light [7, 50].
Flow cytometry
Detection of cellular γH2AX was carried out using the H2AX phosphorylation assay kit for flow cytometry (Upstate Biotechnology). The assay was carried out according to the manufacturers instructions with one major modification, the FITC labeled antibody was incubated overnight at 40C instead of 20 min on ice. Cells were suspended in flow buffer (Ca2+ and Mg2+ free 1x PBS with 1%BSA containing 10 mg/ml propidium iodide (PI) and 100 mg/ml RNaseA) and analyzed using a Becton Dickinson FACS Caliber Flow Cytometer (Becton Dikinson) equipped with Cell Quest software. Data was analyzed using both Cell Quest and FloJo software (Becton Dickinson and Tree Star Inc).
Immunohistochemistry
Cells that had been stably infected with pSuper.Retro.p53.Neo.Gfp or the pSuper.Retro.Neo.Gfp vector were grown on chamber slides (Nunc) and 24 hours after plating were irradiated with 13 J/m2 of UV or sham-irradiated and harvested 4 hours later. Cells were fixed and processed for Mre11 foci according to previously published methods [25]. Briefly, cells were fixed with methanol at −20 °C, kept at −80 °C, then permeabilized with ice-cold acetone:methanol (1:1) for 20 minutes and blocked for 1 hour at 37 °C in 10% fetal bovine serurm/1% bovine serum albumin in PBS and stained with rabbit polyclonal anti-Mre11 (Novus Biochemicals) at 1:200 for 1 hour at 37 °C in antibody dilution buffer with background reducing components (Dako cytomation), washed three times in PBS and incubated with secondary antibody, goat-anti-rabbit conjugated to Alexa Fluor 488 (Molecular Probes) at 1:1,000 for 1 hour at 37 °C. Slides were washed with PBS three times for 5 minutes each and mounted with ProLong Antifade (Molecular Probes). Staining was visualized using a Zeiss Axiocam microscope and Mre11-positive cells were counted as those with greater than three distinctly visible nuclear foci. Counts were done on three separate experiments and verified by two independent observers.
Comparative Genomic Hybridization
Genomic DNA was isolated from two 100mm2 dishes of cultures grown continuously for 20 passages from the lines GM3617T/pSuper.Retro.Neo.Gfp, GM3617T/pSuper.Retro.p53.Neo.Gfp, GM5659DT/pSuper.Retro.Neo.Gfp, and GM5659DT/pSuper.Retro.p53.Neo.Gfp cells. Cells were trypsinized and resuspended in 3 mL of Tris-EDTA buffer, to which was added 100 μL of 20% SDS and 20 μL of 200 mg/mL DNAse-free proteinase K (Roche Biochemicals) and incubated overnight at 55° C. Subsequently, 1 mL of saturated NaCl solution was added, followed by 10 mL of ethanol. The DNA precipitated and was collected by centrifugation at 2,000 X g for 5 minutes, washed with 70% ethanol, and dissolved in water. The DNA was further purified by two extractions with phenol:chloroform:isoamyl alcohol (25:24:1, Invitrogen) followed by one extraction with chloroform:isoamyl alcohol (24:1, Invitrogen) and dissolved in water. The DNA concentration was measured using a fluorimeter (Versa Fluor, BioRad) and Hoecst dye. 2 μg of DNA was hybridized onto a BAC array by the staff at the Microarray Core, Comprehensive Cancer Center, University of California, San Francisco, according to published methods [40]. Data were analyzed using SPOT [62] and SPROC software.
Acknowledgements
Comparative Genomic Hybridization was conducted by the Microarray Core, Comprehensive Cancer Center, University of California, San Francisco. The work described here was supported by the following grants: a National Cancer Institute Ruth Kirstensen Postdoctoral Fellowship 1 F32 CA099499-01A1 (to RRL) and the National Institutes of Environmental Health Sciences grant 1 RO1 ES 8061 (to JEC) and the National Institutes of Neurological Disorders and Stroke 1R01NS052781 (to JEC). Additional support for JEC came from the Cancer Center Support grant P30 CA82103 (PI: McCormick), and a program project grant P01 AR050440-01 (PI: Epstein). We are also grateful to the XP Society, Poughkeepsie, NY, for their continued support and encouragement. We are also grateful to Dr. Russell Pieper, Department of Neurosurgery, for the gift of retroviral hTERT used in developing the cell lines for this study, and to Dr. Charles Limoli, Department of Radiation Oncology, for assistance and advice in immunofluorescence.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tsai KY, Tsao H. The genetics of skin cancer. Amer J Medical Genetics. 2004;131C:82–92. doi: 10.1002/ajmg.c.30037. [DOI] [PubMed] [Google Scholar]
- 2.Cleaver JE, Mitchell DL. Ultraviolet Radiation Carcinogenesis. In: Bast RJ, Kufe DW, Pollock RE, Weichselbaum RW, Holland JF, Frei E III, editors. Cancer Medicine. 7th edition Vol. 1. 2005. [Google Scholar]
- 3.Ohmori H, Friedberg EC, Fuchs RPP, Goodman MF, Hanaoka F, Hinkle D, Kunkel TA, Lawrence CW, Livneh Z, Nohmi T, Prakash L, Prakash S, Todo T, Walker GC, Wang Z, Woodgate R. The Y-Family of DNA Polymerases. Molecular Cell. 2001;8:7–8. doi: 10.1016/s1097-2765(01)00278-7. [DOI] [PubMed] [Google Scholar]
- 4.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokol M, Yuasa M, Araki M, Iwa S, Takio K, Hanoaka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 5.Johnson RE, Kondratick CM, Prakash S, Prakash L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 1999;264:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 6.Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum Cutaneous, ocilar, and neurologic abnormalities in 830 published cases. Archiv Dermatol. 1975;123:241–250. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- 7.Arlett CF, Harcourt SA, Broughton BC. The influence of caffeine on cell survival in excision-proficient and excision-deficient xeroderma pigmentosum and normal cell strains following ultraviolet light. Mutation Research. 1975;33:341–346. doi: 10.1016/0027-5107(75)90209-2. [DOI] [PubMed] [Google Scholar]
- 8.Maher VM, Oulette LM, Curren RD, McCormick JJ. Frequency of ultraviolet light-induced mutations is higher in xeroderma pigmentosum variant cells than in normal human cells. Nature. 1976;261:593–595. doi: 10.1038/261593a0. [DOI] [PubMed] [Google Scholar]
- 9.McGregor WG, Wei D, Maher VM, McCormick JJ. Abnormal, error-prone bypass of photoproducts by xeroderma pigmentosum variant cell extracts results in extreme strand bias for the kinds of mutations induced by ultraviolet light. Molecular Cell Biology. 1999;19:147–154. doi: 10.1128/mcb.19.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang YC, Maher VM, McCormick JJ. Xeroderma pigmentosum variant cells are less likely than normal cells to incorporate dAMP opposite photoproducts during replication of UV-irradiated plasmids. Proceedings of National Academy of Sciences USA. 1991;88:7810–7814. doi: 10.1073/pnas.88.17.7810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang YC, Maher VM, Mitchell DL, McCormick JJ. Evidence from mutation spectra that the UV hypermutability of xeroderma pigmentosum variant cells reflects abnormal error-prone replication on a template containing photoproducts. Molecular Cellular Biology. 1993;13:4276–4283. doi: 10.1128/mcb.13.7.4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waters HL, Seetharam S, Seidman MM, Kraemer KH. Ultraviolet hypermutability of a shuttle vector propagated in xeroderma pigmentosum variant cells. J. of Investigative Dermatology. 1993;101:744–748. doi: 10.1111/1523-1747.ep12371686. [DOI] [PubMed] [Google Scholar]
- 13.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutation in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 14.Levine AJ, Momand J, Finlay CA. The p53 tumor supporessor gene. nature. 1991;351:453–456. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 15.Ananthaswamy HN, Ouhtit A, Evans RL, Gorny A, Khaskina P, Sands AT, Conti CJ. Persistence of p53 mutations and resistance of keratinocytes to apoptosis are associated with the increased susceptibility of mice lacking the XPC gene to UV carcinogenesis. Oncogene. 1999;18:7395–7398. doi: 10.1038/sj.onc.1203147. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Woodgate R, McManus TP, Mead S, McCormick JJ, Maher VM. Evidence that in xeroderma pigmentosum variant cells, which lack DNA polymerase eta, DNA polymerase iota causes the very high frequency and unique spectrum of UV-induced mutations. Cabcer res. 2007;67:3018–3026. doi: 10.1158/0008-5472.CAN-06-3073. [DOI] [PubMed] [Google Scholar]
- 17.Limoli CL, Giedzinski E, Cleaver JE. Alternative recombination pathways in UV-irradiated XP Variant cells. Oncogene. 2005;24:3708–3714. doi: 10.1038/sj.onc.1208515. [DOI] [PubMed] [Google Scholar]
- 18.Sengupta S, Harris CC. p53: traffic cop at the crossroads of DNA repair and recombination. Net Rev Mol Cell Biol. 2005;6:44–55. doi: 10.1038/nrm1546. [DOI] [PubMed] [Google Scholar]
- 19.Laposa RR, Feeney L, Cleaver JE. Recapitulation of the cellular xeroderma pigmentosum-variant phenotype using short interfering RNA for DNA polymerase H. Cancer Res. 2003;63:3909–3912. [PubMed] [Google Scholar]
- 20.Thomas M, Massimi P, Jenkins J, Banks L. HPV-18 E6 mediated inhibition of p53 DNA binding activity is independent of E6 induced degradation. Oncogene. 1995;10:261–268. [PubMed] [Google Scholar]
- 21.Thomas M, Massimi P, Banks L. HPV-18 E6 inhibits DNA binding activity regardless of the oligomeric state of p53 or the exact p53 recognition sequence. Oncogene. 1996;13:471–480. [PubMed] [Google Scholar]
- 22.Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Research. 1994;54:4855–4878. [PubMed] [Google Scholar]
- 23.Cleaver JE, Afzal V, Feeney L, McDowell M, Sadinski W, Volpe JPG, Busch D, Yu Y, Nagasawa H, Little JB. Increased UV sensitivity and chromosomal instability related to p53 function in the xeroderma pigmentosum variant. Cancer Research. 1999;59:1102–1108. [PubMed] [Google Scholar]
- 24.Cleaver JE, Bartholomew J, Char S, Crowley E, Feeney L, Limoli CL. Polymerase η and p53 jointly regulate cell survival, apoptosis and Mre11 recombination during S phase checkpoint arrest after UV irradiation. DNA repair. 2002;3:1–17. doi: 10.1016/s1568-7864(01)00004-0. [DOI] [PubMed] [Google Scholar]
- 25.Limoli CL, Giedzinski E, Morgan WF, Cleaver JE. Polymerase η deficiency in the XP variant uncovers an overlap between the S phase checkpoint and double strand break repair. Proc Nat Acad Sci USA. 2000;97:7939–7946. doi: 10.1073/pnas.130182897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Limoli CL, Giedzinski E, Bonner WM, Cleaver JE. UV-induced replication arrest in the xeroderma pigmentosum variant leads to double strand breaks, γ-H2Ax formation, and Mre11 relocalization. Proc Natl Acad Sci USA. 2002;99:233–238. doi: 10.1073/pnas.231611798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Limoli CL, Laposa R, Cleaver JE. DNA Replication arrest in XP Variant cells after UV exposure is diverted into an Mre11-dependent recombination pathway by the kinase inhibitor Wortmannin. Mutat Res. 2002;510:121–129. doi: 10.1016/s0027-5107(02)00257-9. [DOI] [PubMed] [Google Scholar]
- 28.Thakur M, Wernick M, Collins C, Limoli C, Crowley E, Cleaver JE. DNA polymerase η undergoes alternative splicing, protects against UV sensitivity and apoptosis, and suppresses Mre11-dependent recombination. Genes, Chromosomes and Cancer. 2001;32:222–235. doi: 10.1002/gcc.1186. [DOI] [PubMed] [Google Scholar]
- 29.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 30.Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, Beeche M, Bodnar AG, Wahl GM, Tlsty TD, Chiu CP. Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat Genet. 1999;21:111–114. doi: 10.1038/5056. [DOI] [PubMed] [Google Scholar]
- 31.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 32.Vaziri H, Squire JA, Pandita TK, Bradley G, Kuba RM, Zhang H, Gulyas S, Hill RP, Nolan GP, Benchimol S. Analysis of genomic integrity and p53-dependent G1 checkpoint in telomerase-induced extended-life-span human fibroblasts. Mol Cell Biol. 1999;19:2373–2379. doi: 10.1128/mcb.19.3.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toouli CD, Huschtscha LI, Neumann AA, Noble JR, Colgin LM, Hukku B, Reddel RR. Comparison of human mammary epithelial cells immortalized by simian virus 40 T-Antigen or by the telomerase catalytic subunit. Oncogene. 2002;21:128–139. doi: 10.1038/sj.onc.1205014. [DOI] [PubMed] [Google Scholar]
- 34.Thompson LH, Humphrey RM. Proliferation kinetics of mouse L-959 cells irradiated with ultraviolet light: a time-lapse photographic study. Radiation Research. 1970;41:183–201. [PubMed] [Google Scholar]
- 35.Hessel A, Siegel RJ, Mitchell DL, Cleaver JE. Xeroderma pigmentosum variant with multisystem involvement. Archives of Dermatology. 1992;128:1233–1237. [PubMed] [Google Scholar]
- 36.Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double strand breaks. Proc Natl Acad Sci USA. 2006;103:9891–9896. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- 38.Lee J-H, Pauli TT. ATM activation by DNA double-stand breaks through the Mre11-Rad05-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 39.Lowndes NF, Toh GW-L. DNA repair: the importance of phosphorylating histone H2AX. Curr Biol. 2005;15:R99–R102. doi: 10.1016/j.cub.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 40.Snijders AM, Nowak N, Segraves R, Blackwood S, Brown N, Conroy J, Hamilton G, Hindle AK, Huey B, Kimura K, Law S, Myambo K, Palmer J, Ylstra B, Yue JP, Gray JW, Jain AN, Pinkel D, Albertson DG. Assembly of microarrays for genome-wide measurement of DNA copy number. Nat Genet. 2001:263–264. doi: 10.1038/ng754. [DOI] [PubMed] [Google Scholar]
- 41.Cordeiro-Stone M, Zaritskaya LS, Price LK, Kaufmann WK. Replication fork bypass of a pyrimidine dimer blocking leading strand DNA synthesis. Journal of Biological Chemistry. 1997;272:13945–13954. doi: 10.1074/jbc.272.21.13945. [DOI] [PubMed] [Google Scholar]
- 42.Cordeiro-stone M, Makhov AM, Zaritskaya LS, Griffith JD. Analysis of DNA replication forks encountering a pyrimidine dimer in the template to the leading strand. Journal of Molecular Biology. 1999;289:1207–1218. doi: 10.1006/jmbi.1999.2847. [DOI] [PubMed] [Google Scholar]
- 43.Cordeiro-Stone M, Frank A, Bryant M, Oguejiofor I, Hatch SB, McDaniel LD, Kaufmann WK. DNA damage responses protect xeroderma pigmentosum variant from UVC-induced clastogenesis. Carcinogenesis. 2002;23:959–965. doi: 10.1093/carcin/23.6.959. [DOI] [PubMed] [Google Scholar]
- 44.Cleaver JE, Laposa RR, Limoli CL. DNA replication in the face of (In)surmountable odds. Cell Cycle. 2003;2:310–315. [PubMed] [Google Scholar]
- 45.Lehmann AR, Kirk-Bell S, Arlett CA, Paterson MC, Lohman PHM, de Weerd-Kastelein EA, Bootsma D. Xeroderma pigmentosum cells with normal levels of excision repair have a defect on DNA synthesis after UV-irradiation. Proc. Natl. Acad. Sci. USA. 1975;72:219–235. doi: 10.1073/pnas.72.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lehmann AR. The relationship between pyrimidine dimers and replicating DNA in ultraviolet light-irradiated human fibroblasts. Nucleic Acids Research. 1979;7:1901–1912. doi: 10.1093/nar/7.7.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 48.Dutta A, Ruppert JM, Aster JC, Winchester E. Inhibition of DNA replication factor RPA by p53. Nature. 1993;365:79–82. doi: 10.1038/365079a0. [DOI] [PubMed] [Google Scholar]
- 49.Squires S, Coates JA, Goldberg M, Toji LH, Jackson SP, Clarke DJ, Johnson RT. p53 prevents the accumulation of double-strand DNA breaks at stalled-replication forks induced by UV in human cells. Cell Cycle. 2004;3:1543–1557. doi: 10.4161/cc.3.12.1272. [DOI] [PubMed] [Google Scholar]
- 50.Cleaver JE. Caffeine toxicity is inversely related to DNA repair in simian virus-40 transformed xeroderma pigmentosum cells irradiated with ultraviolet light. Teratogenesis, Carcinogenesis and Mutagenesis. 1989;9:147–155. doi: 10.1002/tcm.1770090303. [DOI] [PubMed] [Google Scholar]
- 51.Kumari A, Schultz N, Helleday T. p53 protects from replication-associated DNA double-strand breaks in mammalian cells. Oncogene. 2004;23:2324–2329. doi: 10.1038/sj.onc.1207379. [DOI] [PubMed] [Google Scholar]
- 52.Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates J.R.r., Hays L, Morgan WM, Petrini JH. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–486. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 53.Maser RS, Monsen KJ, Nelms BE, Petrini JH. hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol Cell Biol. 1997;17:6087–6096. doi: 10.1128/mcb.17.10.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nelms BE, Maser RS, MacKay JF, Lagally MG, Petrini JH. In situ visualization of DNA double-strand break repair in human fibroblasts. Science. 1998;280:590–592. doi: 10.1126/science.280.5363.590. [DOI] [PubMed] [Google Scholar]
- 55.Bunz F, Fauth C, Speicher MR, Dutriaux A, Sedivy JM, Kinzler KW, Vogelstein B, Lengauer C. Targeted inactivation of p53 in human cells does not result in aneuploidy. Cancer res. 2002;62:1129–1133. [PubMed] [Google Scholar]
- 56.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 57.Bastian BC, Olshen A, LeBoit PE, Pinkel D. Classification of melanocytic tumors by DNA copy number changes. Am J Pathol. 2003;163:1765–1770. doi: 10.1016/S0002-9440(10)63536-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 59.Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Research. 1999;59:4375–4382. [PubMed] [Google Scholar]
- 60.Sarkaria JN, Tibbetts RS, Busby EC, Kennedy AP, Hill DE, Abraham RT. Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res. 1998;58:4375–4382. [PubMed] [Google Scholar]
- 61.Cleaver JE, Thomas GH. Rapid diagnosis of sensitivity to ultraviolet light in fibroblasts from dermatological disorders with particular reference to xeroderma pigmentosum. J Invest Dermatology. 1988;90:467–471. doi: 10.1111/1523-1747.ep12460917. [DOI] [PubMed] [Google Scholar]
- 62.Jain AN, Tokuyasu TA, Snijders AM, Segraves R, Albertson DG, Pinkel D. Fully automatic quantification of microarray image data. Genome Research. 2002;12:325–332. doi: 10.1101/gr.210902. [DOI] [PMC free article] [PubMed] [Google Scholar]