

Summary
Dmp1 (Dmtf1) is activated by oncogenic Ras-Raf-signaling and induces cell cycle arrest in an Arf, p53-dependent fashion. The survival of K-rasLA mice was shortened by ~15 weeks in both Dmp1+/− and Dmp1−/− backgrounds, the lung tumors of which showed significantly decreased frequency of p53 mutations compared to Dmp1+/+. Approximately 40 % of K-rasLA lung tumors from Dmp1+/+ mice lost one allele of the Dmp1 gene, suggesting the primary involvement of Dmp1 in K-ras-induced tumorigenesis. Loss of heterozygosity (LOH) of the hDMPI gene was detectable in ~35 % of human lung carcinomas, which was found in mutually exclusive fashion with LOH of INK4a/ARF or that of P53. Thus, DMP1 is a pivotal tumor suppressor for both human and murine lung cancers.
Keywords: Dmp1, K-ras, p19Arf, p16Ink4a, p53, lung cancer, loss of heterozygosity, deletion, promoter hypermethylation, haploid insufficiency
Significance
Dmp1 (Dmtf1) is activated by oncogenic Ras signaling and shows its tumor suppressor activity through the activation of the Arf-p53 pathway in mice. Here we show that Dmp1 deletion cooperates with oncogenic K-ras to form lung cancers in vivo and those tumors from Dmp1-knockout mice have significantly less frequent p53 mutations. Importantly, deletion of one allele of Dmp1 was found in 30~40 % of K-ras-induced murine lung tumors as well as in human non-small cell lung carcinomas, in which ARF and/or P53 remained intact. The present work provides evidence that hDMP1 is a physiological regulator of the ARF-P53 pathway in humans and is primarily involved in pulmonary carcinogenesis.
Introduction
Lung cancer is the leading cause of cancer deaths in the world, and accounts for more solid tumor deaths than any other carcinomas. More than 170,000 new cases are diagnosed each year in the United States alone, of whom ~160,000 will eventually die, representing 30 % of all cancer deaths (Jemal et al., 2006). Lung cancer can be categorized into two major histopathological groups: non-small-cell lung cancer (NSCLC) (Spira and Ettinger, 2004; Moran, 2006) and small-cell lung cancer (SCLC) (Schiller, 2001), the latter of which show neuroendocrine features. Approximately 80 % of lung cancers are NSCLC, and they are subcategorized into adenocarcinomas, squamous cell, adenosquamous, and large-cell carcinomas (Travis, 2002). SCLC and NSCLC show major differences in histopathologic characteristics that can be explained by the distinct patterns of genetic alterations found in both tumor classes (Zochbauer-Muller et al., 2002). For instance, the K-Ras gene is mutated in 20~30 % of NSCLC while its mutation is rare in SCLC; Rb inactivation is found in ~90 % of SCLC while p16INK4a is inactivated by deletion and/or promoter hypermethylation in ~50 % of NSCLC (for reviews, Fong et al., 2003; Meuwissen and Berns, 2005; Wistuba et al., 2001). Among dozens of the murine models of human lung cancer, the K-rasLA/+ (K-rasLA1/+, K-rasLA2/+) mouse model is one of the most sophisticated ones that mimic human NSCLC (Johnson et al., 2001). In this model, the K-ras gene is controlled by its own promoter and is activated only during spontaneous recombination events in the whole animal. Abnormality of the P53 gene is one of the most common events in human lung cancers (Toyooka et al., 2003), and accordingly, K-rasLA/+ lung carcinogenesis was strikingly accelerated in mice of both p53+/− and p53−/− backgrounds (Johnson et al., 2001).
The activity of p53 is positively regulated by p19Arf (p14ARF in humans) in response to oncogenic stress (Lowe and Sherr, 2003; Sherr 2001, 2006). p19Arf is an alternative reading frame gene product generated from the Ink4a/Arf locus which also encodes the cyclin-dependent kinase inhibitor p16Ink4a. p19Arf directly binds to Mdm2, thereby stabilizing and activating p53, whereas p16Ink4a binds to cyclin-dependent kinase 4 to inhibit Rb phosphorylation (Kim and Sharpless, 2006; Lowe and Sherr, 2003; Sherr, 2001). Since this single genetic locus encodes two independent tumor suppressor proteins that regulate the p53 and the Rb pathways, it is very frequently disrupted in human cancer (Ruas and Peters, 1998). Arf is induced by potentially harmful growth-promoting signals stemming from overexpression of various oncoproteins (Lowe and Sherr, 2003; Sherr, 2001). This Arf induction forces early-stage cancer cells to undergo p53-dependent and p53-independent cell cycle arrest or apoptosis, providing a powerful mode of tumor suppression. The Arf promoter monitors latent oncogenic signals in vivo (Zindy et al., 2003), and thus Arf-null mice are highly prone to spontaneous tumor development (Kamijo et al., 1999). There is a body of evidence showing the p53-independent functions of Arf (reviewed in Sherr, 2006). In human lung cancers, p14ARF is inactivated in 65 % of SCLC, while the gene is deleted in ~20 % of NSCLC. Promoter hypermethylation of ARF has been reported in ~10 % of NSCLC, but is less frequent than that of p16INK4a (~40 %) on the same locus (Meuwissen and Berns, 2005). The Arf transcription is negatively regulated by overexpression of nuclear proteins such as Bmi1, Twist, Tbx-2/3, and Pokemon, and overexpression of these proteins have been reported in human cancers (Brummelkamp et al., 2001; Maeda et al., 2005; Maestro et al., 1999; Jacobs et al., 1999, 2000; Yang et al., 2004).
Among known Arf activators, Dmp1 (cyclin D binding myb-like protein-1; also called Dmtf1: cyclin D binding myb-like transcription factor 1) is a unique tumor suppressor (Hirai and Sherr, 1996; Inoue and Sherr, 1998; for review, Inoue et al., 2007). Dmp1 was originally isolated in a yeast two-hybrid screen of a murine T-lymphocyte library with cyclin D2 as bait (Hirai and Sherr, 1996). Although Dmp1 is structurally related to Myb-family proteins, it binds to nonameric CCCG(G/T)ATG(T/C) DNA consensus sequences, a subset of which is also recognized by proteins of the Ets family. Importantly, Dmp1 directly binds to the Arf promoter to activate its expression, thereby inducing p53-dependent cell cycle arrest (Inoue et al., 1999). Dmp1-null mice are prone to spontaneous tumor development, which was accelerated when the animals were neonatally treated with ionizing radiation or dimethylbenzanthracene (Inoue et al., 2000, 2001). Lung adenomas/adenocarcinomas were the most common tumors found in Dmp1-knockout mice. The retention and expression of the wild-type Dmp1 allele in tumors arising in heterozygotes indicated that Dmp1 is haplo-insufficient for tumor suppression (Inoue et al., 2001; for reviews, Quon and Berns, 2001). The low frequency of Arf deletion and p53 mutation in tumors from Dmp1-knockout mice suggested that Dmp1 is a physiological regulator of the Arf-p53 pathway in vivo (Inoue et al., 2001). Information about the signaling cascades that regulate Dmp1 has been accumulating. The Dmp1 promoter is activated by the oncogenic Ras-Raf-MEK-ERK-Jun pathway, and the induction of Arf by Ras is Dmp1-dependent (Sreeramaneni et al., 2005). On the other hand, the Dmp1 promoter is repressed by overexpression of E2Fs and also by physiological mitogenic signaling. Thus, Dmp1 is a marker of cells that have exited from the cell cycle (Mallakin et al., 2006). Our recent study shows that the Dmp1 promoter is repressed by genotoxic stimuli that activate NF-κB (Taneja et al., 2007).
In striking contrast to the accumulating information on murine Dmp1, very little is known about the involvement of human DMP1 (hDMP1; hDMTF1) in cancer. The hDMP1 protein has very high structural homology with its murine counterpart (95 % identity with murine Dmp1 at the protein level). One allele of the genes at the hDMP1 locus was reportedly deleted in all the leukemic cells with chromosome 7q abnormalities regardless of the detailed karyotype at 7q, suggesting that one allelic loss of hDMP1 could contribute to 7q- malignancies that are refractory to conventional chemotherapy (Bodner et al., 1999). It has been reported that the hDMP1 locus encodes at least three splicing variants, hDMP1α β and γ (Tschan et al., 2003). The full-length hDMP1α gene corresponds to the murine Dmp1 gene that positively regulates the p19Arf-p53 pathway (Inoue et al., 1999; Tschan et al., 2003). By contrast, the hDMP1β and γ proteins do not bind to DNA and hDMP1β has dominant-negative effect on hDMP1α when it is overexpressed (Tschan et al., 2003).
Although Dmp1 plays critical roles as a mediator of ras signaling to Arf induction in cultured cells, the role of Dmp1 in ras signaling has not been demonstrated in vivo. This study was conducted to demonstrate the collaborative effects of Dmp1 inactivation and K-ras activation in pulmonary carcinogenesis. We found that one allele of Dmp1 was deleted in a significant percentage of lung tumors from Dmp1+/+; K-rasLA mice, showing the primary role of Dmp1 in murine lung cancer. We also analyzed 51 human NSCLC samples for hDMP1, INK4a/ARF, and P53 and show that LOH of the hDMP1 gene is frequently found in NSCLC, especially those which retain an intact INK4a/ARF and/or P53 locus.
Results
K-rasLA induced tumorigenesis is significantly accelerated in both Dmp1−/− and Dmp1+/− mice
In order to investigate the cooperation of Dmp1-inactivation and K-ras activation in vivo, we backcrossed Dmp1-knockout mice for six generations with a C57BL/6 male, and then crossed the Dmp1+/− mice with K-rasLA2/+ or K-rasLA1/+ mice (Johnson et al., 2001). The average survival of wild-type K-rasLA mice was 10–15 weeks longer in our C57BL/6 strain than in the original report due to the difference of the genetic background. K-rasLA2/+ induced tumorigenesis was greatly accelerated in both Dmp1−/− and Dmp1+/− mice, with no differences between groups of Dmp1−/− and Dmp1+/− (Figure 1A; mean survival, 21 weeks in Dmp1−/− and Dmp1+/− vs. 35 weeks in Dmp1+/+, P< 0.005 in both cases). K-rasLA1/+ induced tumor formation was also accelerated in both Dmp1−/− and Dmp1+/− backgrounds (mean survival, 36 weeks in Dmp1−/− (P< 0.001), 41 weeks in Dmp1+/− (P= 0.001), and 55 weeks for Dmp1+/+) (Figure 1B). Although the average survival was slightly shorter in Dmp1−/−; K-rasLA1/+ mice than in Dmp1+/−; K-rasLA1/+ mice, there was no statistically significant differences between these two cohorts (P= 0.13). All of the K-rasLA mice generated lung tumors (adenomas and adenocarcinomas) regardless of the Dmp1 genotype, with minor differences in the incidence of thymic lymphomas (~30 %) and tail papillomas (20~30 %) (Figure S1A). Some of the Dmp1+/−, Dmp1−/−; K-rasLA/+ mice developed other types of tumors than lung carcinomas, thymus lymphomas, or papillomas. They included a case of cholangiocarcinoma, osteosarcoma, neurofibrosarcoma, and ovarian tumor (Figure S1; a picture of cholangiocarcinoma is shown in Figure 2E). Lung tumors from Dmp1+/−; K-rasLA1/+ and Dmp1+/−; K-rasLA2/+ mice retained the wild-type Dmp1 allele, when examined by genomic DNA PCR (Figure 1C). The levels of Dmp1 mRNA expression was 2–5 times higher in 8 of 11 lung tumors from Dmp1+/+; K-rasLA mice than in normal lungs, suggesting the activation of the endogenous Dmp1 promoter by oncogenic K-ras (Figure 1D, middle left panel, gray bars). However, the Dmp1 mRNA levels were at comparable levels in 3 of 11 Dmp1+/+; K-rasLA lung tumors, suggesting the disruption of the ras-Dmp1 signaling pathway (Figure 1D, middle left panel, white bars). Likewise, the level of Dmp1 mRNA was 2–4 times higher in 6 of 11 Dmp1+/−; K-rasLA lung tumors than in Dmp1+/− lungs while they were at the same or lower levels in 5 of 11 cases (Figure 1D, middle right panel). Nucleotide sequencing of Dmp1 RT-PCR products from five lung tumors from different mice identified no mutation of Dmp1 in the DNA-binding domain (data not shown). Immunohistochemical staining of Dmp1+/− lung carcinomas with Dmp1-specific antibody (RAX; Mallakin et al., 2006) showed that Dmp1 protein is expressed in lung tumor cells from a Dmp1+/−; K-rasLA mice (Figure 1E, F, and G). These data indicate haploid insufficiency of Dmp1 in suppressing K-ras-induced tumor formation.
Figure 1. Tumor-free survival in cohorts of (A) Dmp1+/+, Dmpl+/−, and Dmp1−/−; K-rasLA2/+ mice and (B) Dmp1+/+, Dmp1+/−, and Dmp1−/−; K-rasLA1/+ mice.

A: Statistically significant difference in survival was found between Dmp1+/+ vs. Dmp1+/−; K-rasLA2/+ (P< 0.005) and Dmp1+/+ vs. Dmp1−/−; K-rasLA2/+ mice (P< 0.005). There were no significant differences in survival between Dmp1+/− vs. Dmp1−/−; K-rasLA2/+ (P= 0.38).
B: Significant difference of survival was found between Dmp1+/+ vs. Dmp1−/−; K-rasLA1/+ (P< 0.001) and Dmp1+/+ vs. Dmp1+/−; K-rasLA1/+ mice (P= 0.001), but not between Dmp1+/− vs. Dmp1−/−; K-rasLA1/+ (P= 0.13).
C: Retention of the Dmp1 wild-type allele in lung tumors from Dmp1+/−; K-rasLA/+ mice as examined by genomic DNA PCR.
D: Expression of the Dmp1 mRNA in lung tumors from Dmp1+/+, Dmp1+/−, and Dmp1−/−; K-rasLA/+ mice. Real-time PCR was conducted to quantitate the Dmp1 mRNA expression in K-rasLA/+ lung tumors. Black bars show the average level of Dmp1 expression in the lung of each genotype. Gray bars are the samples that showed higher level of Dmp1 expression in K-rasLA lung tumors than in their normal lung controls. The means Dmp1+/− SD for three experiments are shown.
E: Well-differentiated lung adenocarcinoma found in a Dmp1+/−; K-rasLA1/+ mouse. H&E stain.
F: Detection of the Dmp1 protein in the nuclei of Dmp1+/−; K-rasLA1/+ mouse tumor cells with the Dmp1-specific antibody, RAX.
G: Negative staining of the lung tumor section of Dmp1−/−; K-rasLA1/+ mouse with RAX antibody. Scale bar in E, F, and G is 100 µm.
Figure 2. Pathological examination of tumors found in K-rasLA/+ mice.

A: Multiple lung adenomas and adenocarcinomas in a Dmp1+/+; K-rasLA1/+ mouse (42-week-old).
B: Advanced lung adenocarcinoma in a Dmp1+/−; K-rasLA1/+ mouse (39-week-old).
C: Disseminated lung adenocarcinoma in a Dmp1−/−; K-rasLA1/+ mouse (38-week-old).
D: Leg metastasis of lung adenocarcinoma in a Dmp1+/−; K-rasLA1/+ mouse (40-week-old).
E: Cholangiocarcinoma of the liver in a Dmp1+/−; K-rasLA1/+ mouse (40-week-old).
F: Lung nodule number and size (mean +/− SEM) in K-rasLA mice. Ranges were compared with unpaired Student’s t-tests.
G: Well-differentiated adenocarcinoma found in a Dmp1+/+; K-rasLA2/+ mouse.
H: Poorly differentiated adenocarcinoma in a Dmp1+/−; K-rasLA2/+ mouse. The tumor cells are very pleomorphic and are invading into blood vessels.
I: Intrabronchial invasion of a Dmp1−/−; K-rasLA2/+ lung carcinoma.
J: Liver metastasis of lung adenocarcinoma in a Dmp1+/−; K-rasLA2/+ mouse. Scale bar in G, H, I, and J is 100 µm.
Biological features of Dmp1+/− and Dmp1−/− lung tumors
All K-rasLA mice developed multi-focal lung tumors when they showed signs of distress and were sacrificed, regardless of the Dmp1 genotype (Figure 2A–C; Figure S1). Lung carcinomas were found in more than half of Dmp1+/− or Dmp1−/−; K-rasLA mice and were significantly larger than those found in Dmp1+/+ mice of the same age (Figure 2A–C; all the mice were ~40 weeks old). It was common to find nearly entire replacement of the lung by tumors in Dmp1+/− or Dmp1−/− lung tumors (Figure 2B, C). The number of lung tumor nodules significantly increased in both Dmp1+/− and Dmp1−/− mice when the mice became sick and sacrificed (P = 0.008 and P = 0.016, respectively; Figure 2F). Also a trend toward increased nodule size was seen in both Dmp1+/− and Dmp1−/− mice; however, the difference was not statistically significant due to relatively large variation of tumor size (P = 0.093 and P = 0.077, respectively). Two of 36 Dmp1+/−; K-rasLA2/+ mice and 2 of 38 Dmp1+/−; K-rasLA1/+ developed macroscopically recognized distant metastases (three liver/intra-abdominal metastases and one leg metastasis) of their lung tumors (Figure 2D, leg metastasis; Figure 2J, liver metastasis), while none of 50 Dmp1+/+; K-rasLA/+ mice showed macroscopic metastatic lesions. The leg or abdominal tumors were negative for markers of carcinoid tumors (chromogranin, synaptophysin), but were positive for a marker for the lung surfactant protein C (Figure S2), indicating that they were metastases of the lung adenocarcinomas. When examined under the light microscope, tumors from Dmp1+/+; K-rasLA mice were adenomas or well-differentiated adenocarcinomas that mimic human papillary adenocarcinomas (~20 % when the largest lung tumor nodules were studied) (Figure 2G). Approximately 50 % of the largest lung tumors from Dmp1+/− or Dmp1−/− mice were well, moderately, or poorly differentiated adenocarcinomas, many of which showed signs of intravascular (Figure 2H) or intrabronchial invasion (Figure 2I). There was a trend of increased frequency of intravascular or intrabronchial invasion in Dmp1+/− (8/28, 28.6 %) or Dmp1−/− (6/16, 37.5 %); K-rasLA lung tumors than in Dmp1+/+; K-rasLA lung tumors (4/22, 18.2 %) when they became sick and sacrificed. However, the difference was not statistically different (P = 0.244 for Dmp1+/− vs. Dmp1+/+ and P = 0.102 for Dmp1−/− vs. Dmp1+/+, respectively) possibly because Dmp1+/+; K-rasLA mice lived 14–19 weeks longer than Dmp1+/− or Dmp1−/−; K-rasLA mice (Figure 1A, B), and by that time many of Dmp1+/+; K-rasLA lung tumors have corrupted the Dmp1-p53 pathway (see the following molecular analyses).
p53 mutation is rare in lung tumors from Dmp1+/− or Dmp1−/−; K-rasLA/+ mice
Previous work had suggested that Dmp1 is a physiological regulator of the Arf-Mdm2-p53 tumor surveillance pathway in Eµ-Myc lympholeukemias (Inoue et al., 2001). Thus we studied the frequency of p53 mutations, Mdm2 overexpression, and Ink4a/Arf deletions in Dmp1 wild-type K-rasLA lung carcinomas. p53 was highly (#356, #59, and #844) or moderately (#208) overexpressed in 4 of 11 Dmp1+/+ lung tumors (36 %) randomly chosen for analysis (Figure 3A). These patterns of p53 protein expression are typical of mutant forms of p53, which neither transcriptionally activate Mdm2 to trigger p53 destruction (Haupt et al., 1997; Kubbutat et al. 1997) nor repress Arf transcription (Stott et al. 1998). Cloning and sequencing of the cDNA for p53 demonstrated the existence of mutations within the DNA-binding domain in all the four lung cancer samples that overexpressed p53 (data not shown). Three Dmp1+/+; K-rasLA/+ lung tumors expressed significant levels of p19Arf (#356, #59, #844), suggesting the disruption of the p53-Arf feedback loop in these tumors. On the other hand, none of the Dmp1+/− or Dmp1−/−; K-rasLA/+ lung tumors showed high levels of expression of p53, suggesting that the vast majority of the p53 protein expressed in Dmp1-knockout tumors was wild-type (Figure 3A, top panels). p19Arf was not upregulated in any of the Dmp1+/−, Dmp1−/−; K-rasLA/+ lung tumors, consistent with the wild-type p53 expression in these tumors (Figure 3A, third panels). Mdm2 was not overexpressed in any of the K-rasLA lung tumors compared with the murine cell line that overexpresses Mdm2 (Figure 3A, second panels). None of the p53, Arf, or Ink4a genes showed biallelic deletion in any of the lung tumors examined, regardless of their Dmp1 status as studied by semi-quantitative PCR analyses (representative data for Arf Exon 1β are shown in Figure 3B; for p16Ink4a and p53, data not shown). However, Arf was hemizygously deleted in ~25 % of K-rasLA lung tumors irrespective of the Dmp1 genotype as studied by real-time PCR (Figure S3A). The Ink4a/Arf modulators, Bmi1, Twist, Tbx2/3, and Pokemon have been reported to downregulate p19Arf levels and contribute to tumor formation. However, none of these proteins was overexpressed in K-rasLA lung tumors in comparison to 3T3 cells as examined by Western blotting with specific antibodies (Figure S3B).
Figure 3.

Analysis of the Arf-Mdm2-p53 pathway in K-rasLA/+ lung tumors.
A: Western blotting of lung tumors for p53, Mdm2, p19Arf, and p16Ink4a. Tumor cells were resected from the center of well-circumscribed lung carcinomas under the light microscope and proteins were extracted. The control cell lysates were obtained from spontaneously immortalized p53-mutant MEFs for p53, p19Arf, p16Ink4a, and Actin; dm3T3 cells for Mdm2. The p53 mutations were detected in 4 of 11 Dmp1+/+; K-rasLA/+ mice (36 %) while they were not found in the Dmp1+/− and Dmp1−/−; K-rasLA/+ lung tumors.
B: Detection of the p19Arf Exon1β genomic DNA by semi-quantitative-PCR. The Arf gene was not homozygously deleted in K-rasLA lung tumors regardless of the Dmp1 genotype.
C: Quantification of the Dmp1 genomic DNA by real-time PCR. The black bars show the relative copy number of the Dmp1 genomic DNA in Dmp1+/+, Dmp1+/−, and Dmp1−/− mice tails, with β-actin as an internal control (mean +/− SEM). The Dmp1 locus is deleted in 5 of 12 randomly chosen lung tumors from Dmp1+/+; K-rasLA/+ mice. The Dmp1 gene was hemizygously deleted in 5 of 8 cases of K-rasLA/+ lung tumors with wild-type p53 (#465, #381, #154, #395, and #205).
D: Quantification of the Dmp1 genomic DNA in lung tumors from p53+/− or p53−/−; K-rasLA mice (mean +/− SEM). The Dmp1 gene was not deleted in any one of these 14 lung tumors from p53-knockout mice.
Since neither homozygous Arf deletion nor Arf repressor overexpression was found in lung tumors from Dmp1+/+; K-rasLA/+ mice, we hypothesized that the Dmp1 gene might be deleted in some of these lung tumors. Quantitative real-time PCR showed that 5 of 12 lung tumors from Dmp1 wild-type mice showed single allelic deletion of the Dmp1 gene (#465, #381, #154, #395, and #205) (Figure 3C, white bars). Interestingly, 5 of 8 (63 %) p53 wild-type lung tumors showed deletion of the Dmp1 gene, while none of the p53-mutant lung tumors showed hemizygous deletion of Dmp1 (#356, #59, #208, and #844) (Figure 3C; P = 0.038, χ² = 4.29). The Dmp1 locus was selectively deleted in K-rasLA lung tumors in most cases since the gram3 gene located ~430 kb upstream of the Dmp1 locus was deleted in only one of 12 K-rasLA lung tumors and the abcb1 gene located ~430 kb downstream from the Dmp1 locus was never deleted in any of the 12 lung tumors (Figure S4). The Dmp1 gene was not deleted in any of the lung tumor DNAs isolated from p53+/−; K-rasLA or p53−/−; K-rasLA mice, showing mutually exclusive inactivation of Dmp1 and p53 in K-rasLA lung tumors (Figure 3D). Collectively, our data indicate that when lung carcinomas arise from wild-type K-rasLA mice, the cells undergo either p53 mutation or Dmp1 deletion to inactivate the Arf-p53 pathway.
Loss of heterozygosity (LOH) and promoter hypermethylation analyses of hDMP1 in human non-small cell lung cancer (NSCLC)
The hDMP1 gene is located on human chromosome 7q21, a region which is often deleted in therapy-induced acute leukemias and myelodysplastic syndromes (Bodner et al., 1999). Whether the human DMP1 gene (hDMP1) is inactivated in human carcinomas has never been investigated. We therefore extracted genomic DNA from 51 non-small cell lung cancer samples (33 adenocarcinomas, 16 squamous cell carcinomas, and 2 adenosquamous carcinomas) and studied LOH for hDMP1 with two different sets of primers that amplify the dinucleotide repeats located at the 5’ (#92465, 34 kb from hDMP1) and 3’ ends of the hDMP1 gene (#198004, 28 kb from hDMP1; Figure 4A). For INK4a/ARF LOH analysis, we designed primers that amplify the repetitive sequences within 500 bps from human ARF Exon 1β (#33647) and those that amplify the sequences between Exon 1β and Exonα (#27251;Figure 4B). There are at least 4 high-affinity hDMP1 binding sites in this region (Figure 4B, inverted red triangles). For P53, two sets of primers were selected that amplify the dinucleotide repeats located 26 kb 5’ (#158111) and 11 kb 3’ (#89737) of the P53 gene (Figure 4C). Two major area peaks were quantified from paired samples (normal tissue and lung cancer) and the qLOH values were calculated (see equation described in the Experimental Procedures). When the qLOH values were more than 2.0 or less than 0.5 with either one of the two sets of LOH primers, the tumor sample was found to have LOH for the locus (Figure 4 and Figure 5). Typical cases of LOH positive samples are shown in Figure 4D and E for hDMP1 (with #92465 and #198004 primer sets), Figure 4F and G for INK4a/ARF (with #33647 and #27251), and Figure 4H and I for P53 (with #158111 and #89737). We also conducted hDMP1, p14ARF, and p16INK4a promoter hypermethylation assays (Figure 5 and Figure S5). The sequences of primers for LOH and promoter hypermethylation assays are shown in Table S1.
Figure 4. Loss of heterozygosity (LOH) analysis of the hDMP1, INK4a/ARF, and P53 loci in human non-small cell lung carcinomas.

A: Genomic locus of the hDMP1 gene. The two different primer sets were designed to amplify the dinucleotide repeat sequences located on the 5’ and 3’ end of the hDMP1 gene. The non-coding exons were colored silver and the coding exons were colored gold.
B: Genomic structure of the human INK4a/ARF locus. The two sets of PCR primers were designed to detect the dinucleotide repeats within 500 bps of Exon 1β (#33647) and those between Exon 1β and Exon 1α (#27251). The inverted triangles shown in red indicate the location of high-affinity hDMP1-binding sites.
C: Genomic structure of the human P53 gene and the location of the PCR primers used for LOH analyses.
D–I: Representative patterns of LOH for hDMP1, INK4a/ARF, and P53 in human non-small cell lung carcinoma. Genomic DNA was extracted from lung carcinomas and their normal counterparts and PCR was conducted with 6-FAM-labeled primers that amplify the dinucleotide repeats within (or close to) each locus. The area peaks of the PCR products were quantitated by ABI 3700 DNA analyzer. The qLOH values were determined through the following equation: qLOH = Area Peak 1/Area Peak 2 (normal tissue) divided by Area Peak 1’/Area Peak 2’ (tumor tissue). The arrows indicate the peak that was lost in tumor cells. The sample was considered to have LOH when the value was >2.0 or <0.5.
D: LOH analysis of NSCLC with 5’ hDMP1 primer set, #92465.
E: LOH analysis of NSCLC with 3’ hDMP1 primer set, #198004.
F: LOH analysis with INK4a/ARF 5’ probe, #33647.
G: LOH analysis with INK4a/ARF 3’ probe, #27251.
H: LOH analysis with P53 5’ #158111 primers.
I: LOH analysis with P53 3’ #89737 primers.
Figure 5. Summary of the results for qLOH values and promoter hypermethylation in 51 cases of human non-small cell lung cancer.

The positive results for LOH (qLOH >2.0 or <0.5) were shown in bold red characters. When one of the two markers (5’ or 3’) showed qLOH value >2.0 or <0.5, the sample was found to be positive for LOH for the tumor suppressor locus. Cases of mutually exclusive inactivation of hDMP1 and INK4a/ARF or hDMP1 and P53 are shown “yes” in bold blue characters. Abbreviations: LC adeno, adenocarcinoma of the lung; LC squam, squamous cell carcinoma of the lung; LC adenosq, adenosquamous carcinoma of the lung; hDMP1 Met, human DMP1 promoter hypermethylation; ARF Met, p14ARF promoter hypermethylation; INK4a Met, p16INK4a promoter hypermethylation. Exclusive of DMP1 LOH, LOH of INK4a/ARF (or P53) not overlapping with that of hDMP1 in the same sample. Del, homozygous deletion; single, LOH was not evaluated due to a single peak result; partial, weak promoter methylation. N.D., not determined. N.D.*, not determined due to positive signals from histologically normal tissue (Holst et al., 2003).
With the 5’ set of hDMP1 primers (#92465), 14 of 42 cases (33.3 %) were positive for LOH; with the 3’ set of hDMP1 primers (#198004), 13 of 36 cases were positive (36.1 %). Seven of 29 cases (24.1 %) were positive for LOH with both of these hDMP1 primer sets (Figure 5). We then conducted detailed mapping of the genomic locus on human chromosome 7q21 deleted in human NSCLC with 5 other sets of LOH primers (Figure 6A). We also conducted quantification of the hDMP1 genomic DNA at exons 8 and 20 by real-time PCR to supplement the LOH study (Figure 6A, B). Interestingly, the genomic region deleted in NSCLC was confined to the hDMP1/MGC4175 locus (i.e., from #69164 to #259145) in 15 of 19 hDMP1 LOH(+) cases (Figure 6B, #2000-3 was not included). Thus, the hDMP1/MGC4175 locus was specifically targeted in NSCLC samples although there were a small number of cases that showed broader deletion including the hDMP1/MGC4175 locus.
Figure 6. Detailed mapping of the chromosomal 7q21 region deleted in human NSCLC.
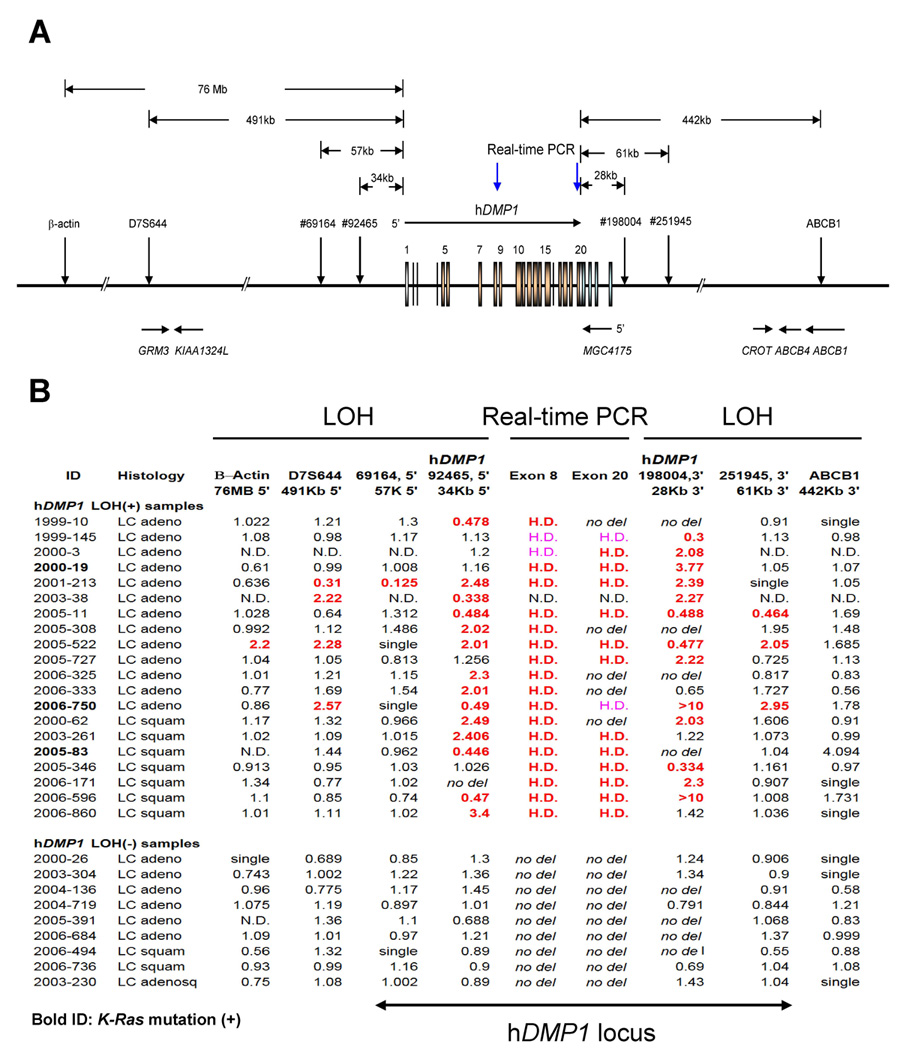
A: Genomic structure of the human hDMP1 locus and the primers used for the LOH analyses. GRM3: glutamate receptor 3; KIAA1324L: KIAA1324-like; MGC4175: Mammalian Gene Collection 4175; CROT: carnitine O-octanoyltransferase; ABCB4: ATP-binding cassette, sub-family B (MDR/TAP), member 4; ABCB1: ATP-binding cassette, sub-family B (MDR/TAP), member 1.
B: Summary of the qLOH and real-time PCR values with 9 different markers (7 LOH and 2 real-time PCR) in the 20 hDMP1 LOH(+) samples and 9 hDMP1 LOH(−) samples. The positive results for LOH (qLOH >2.0 or <0.5) are shown in bold red. In real-time PCR, the samples were found to have hemizygous deletion of hDMP1 when the genomic DNA level was 0.25–0.65 (H.D. in bold red, DNA in the normal lung = 1.00). The borderline cases (0.66–0.75) are shown in pink. Samples that showed point mutation for K-Ras at codon 12 or 13 are shown in bold ID. H.D., hemizygous deletion; no del, no deletion as studied by genomic DNA real-time PCR; N.D., not done; single, single peak in LOH analysis. Note that there are only two genes at the hDMP1 locus between markers #69164 and #251945 (hDMP1 and MGC4175) and this locus is selectively deleted in 15 of 19 NSCLC samples.
Next we conducted hDMP1 promoter methylation assay by using enzymatically methylated human genomic DNA as a positive control. We found only one case of hDMP1 promoter hypermethylation (#2000-3; Figure 5 and Figure S5). We then isolated RNAs from lung cancer samples from seven different patients in whom duplicate tumor samples were available and conducted sequencing of the hDMP1 cDNA. All of the seven samples expressed the hDMP1 mRNA, but none of them, including those which showed LOH for hDMP1 (#1999-10, #2005-308, and #2005-522), showed mutations for hDMP1 (data not shown). We then investigated if lung cancer cells had splicing alterations for hDMP1 by real-time TaqMan PCR in 11 samples that had been randomly chosen. However, we could not detect any lung cancer-specific overexpression of the hDMP1β isoform that has dominant-negative effect on hDMP1α (Figure S6; Tschan et al., 2003). Thus, hemizygous gene deletion was considered to be the major mechanism of hDMP1 inactivation in NSCLC.
Mutually exclusive deletion of hDMP1 and INK4a/ARF, P53 in human NSCLC
The 51 pairs of NSCLC samples pairs were also studied for LOH of INK4a/ARF and P53. With INK4a/ARF primers, 12 of 40 cases (30 %) were positive for LOH or showed biallelic deletion with the #33647 primers, and 16 of 45 samples (35.6 %) were positive for LOH or showed biallelic deletion with #27251 primers (Figure 4 and Figure 5). Ten of 35 cases (28.6 %) showed LOH or homozygous deletion with both sets of the INK4a/ARF probe. Promoter hypermethylation was found in 3 cases (3/46, 6.5 %) for p14ARF and 25 cases (25/47, 53.2 %) for p16INK4a, compatible with previous reports from other groups (Meuwissen and Berns, 2005) (Figure 5 and Figure S5). Ten cases of the p16INK4a promoter hypermethylation were observed simultaneously with LOH of the locus (Figure 5). Three tumor samples showed homozygous deletion of Exon 1β for p14ARF (#2003-86, #2003-442, and #2003-246). Together, the results suggest that these two genes behaved as classical tumor suppressors in the NSCLC samples. Interestingly, 32 out of 34 cases showed mutually exclusive inactivation of the hDMP1 and the INK4a/ARF loci (94.1 %; 95 % confidence interval, 86.2 % to 100 %; P = 0.0035, χ² = 8.52 based on mutually exclusive hypothesis) compatible with the cluster of hDMP1-binding sites throughout the INK4a/ARF locus (Figure 4B). LOH of P53 was found in 30.4 % (14/46) with 5’ primers, 46.2 % (18/39) with 3’ primers, and 24.3 % (9/37) with both primers. Sequencing analysis of P53 cDNAs from six lung cancer samples demonstrated the presence of point mutations of P53 in LOH(+) samples (#2005-308, #2005-391, #1995-95, and #2005-242), but not in LOH(−) samples (#2005-522, #2005-346), indicating that both alleles of P53 were inactivated in the lung cancer cells that showed LOH for P53 (data not shown). Again, LOH of hDMP1 and that of P53 tend not to overlap each other (Figure 5, 30/35 = 85.7 % exclusive; 95 % confidence interval, 74.1 % to 97.3 %; P = 0.027, χ² = 4.88 based on mutually exclusive hypothesis). On the other hand, inactivation of the INK4a/ARF locus and that of the P53 locus was found to actually occur more frequently together rather than mutually exclusively (Figure 5; 14/27, 51.9 % exclusive; 95 % confidence interval, 33.0 % to 70.7 %; P = 0.0045, χ² = 8.08 against mutually exclusive hypothesis). We then genotyped NSCLC samples for K-Ras mutation. Point mutations involving codons 12 or 13 of K-Ras were found in 7 of 48 samples analyzed (14.6 %), and 3 of the 7 K-Ras mutations were found in hDMP1 LOH(+) samples (#2000-19, #2006-750, and #2005-83; shown in bold ID in Figure 6). Taken together, LOH of the hDMP1 gene was found in ~35 % of human NSCLC, especially those that retain a wild-type INK4a/ARF and/or P53 locus and ~15 % of hDMP1 LOH occurred simultaneously with K-Ras mutation.
Detection of the hDMP1 protein in human lung cancer samples and growth inhibition of lung cancer cell lines by activated Dmp1:ER
To investigate the consequences of hemizygous hDMP1 deletion in human NSCLC cells, formalin-fixed, paraffin embedded lung cancer sections were stained with Dmp1-specific antibody, RAX (Mallakin et al., 2006; Figure S7). Strong signals were detectable in the nuclei of P53-mutant NSCLC cell line H727, which was dramatically downregulated by infecting the cells with hDMP1-specific shRNA retroviruses (Figure S7A–C). Then we randomly chose 9 hDMP1 LOH(+) and 8 hDMP1 LOH(−) lung cancers and stained them with the antibody to Dmp1. Positive nuclear staining (grade 3++ to 2+) was obtained in 8 of 8 hDMP1 LOH(−) lung cancer samples while the staining was very weak (grade 1+/−) or negative (grade 0) in 7 of 9 hDMP1 LOH(+) lung cancers (Figure S7H). In one case (#2006-171) the hDMP1 signal was negative, suggesting the complete inactivation of the hDMP1 protein in lung cancer cells. Thus, our immunohistochemistry results are quite consistent with those of hDMP1 LOH analyses (P< 0.001).
We then studied if Dmp1 overexpression inhibits the growth of human NSCLC cell lines with different genetic backgrounds of ARF and P53. In H460 cells with wild-type ARF and P53, activation of Dmp1:ER with 4-hydroxytamoxifen (4-HT) (Inoue et al., 1999) efficiently inhibited the growth (Figure 7A, pink line). On the other hand, lung cancer cell lines that lack INK4a/ARF (A549) or P53 (H1299, H358) proliferated exponentially even with activation of Dmp1:ER although their growth was slightly slower than control cells (Figure 7B–D; pink lines, Dmp1:ER virus-infected cells with 4-HT; blue lines, mock-infected cells with 4-HT). Western analysis showed accumulation of p14ARF, p53, and its target hDM2 and p21Cip1 in H460 cells where Dmp1:ER was activated with 4-HT (Figure 7E, left panel). In H1299 cells, only p14ARF increased with stimulation of Dmp1:ER (Figure 7E, right panel). Interestingly, real-time PCR analysis showed that the hDMP1 gene was hemizygously deleted in H460 cells, but not in other cell lines (data not shown). Together, the results indicate that overexpression of activated Dmp1:ER efficiently inhibits the growth of the human lung cancer cell line that has wild-type ARF and P53.
Figure 7. Proliferation assay of human non-small cell lung cancer cell lines by overexpressing Dmp1:ER.

A: H460; hDMP1+/−, ARF+, p16del, P53+, Rb+
B: H1299; hDMP1+, ARF+, p16Met, P53del, Rb+
C: A549; hDMP1+, ARFdel, p16del, P53+, Rb+
D: H358; hDMP1+, ARF+, p16Met, P53del, Rb+
Pink lines show the growth curves of Dmp1:ER virus-infected cells treated with 2µM 4-HT, blue lines show those of mock-infected cells with 4-HT (mean +/− SEM). Activation of Dmp1:ER by 4-HT inhibited the growth of H460 cells with wild-type ARF and P53, but had little effects on other lung cancer cell lines that showed deletion of ARF or P53.
E: Western blotting analyses of H460 and H1299 cells expressing activated Dmp1:ER or empty vector with specific antibodies to Dmp1, p14ARF, p53, hDM2, and p21Cip1. The numbers show hours after addition of 4-HT.
Discussion
It has now become clear that the DMP1 transcription factor is primarily involved in both human and murine lung cancer. The hDMP1 gene is located on human chromosome 7q21, a locus often deleted in human malignancies (Bieche et al., 1992; Kerr et al., 1996; Oriola et al., 2001). One previous report showed that the hDMP1 locus was lost in 9 out of 9 leukemic cells with chromosome 7q abnormalities as studied by FISH, regardless of the detailed karyotype of leukemic cells (Bodner et al., 1999). However, it was difficult to point to the roles of hDMP1 in human 7q- leukemias since the probe contained sequences of several other genes as well. In this study, we designed unique primers that specifically amplify the repetitive sequences located within 35 kb of the hDMP1 gene. Thus, the experimental results dependably reflected the events occurring within the locus. With these probes, we could detect the LOH of the hDMP1 locus in ~35 % of NSCLC samples, the frequency of which was even close to that of the INK4a/ARF or P53 locus of the same samples (30~45 %). Our detailed analysis showed that the deletion of chromosome 7q21 was limited to the hDMP1/MGC4175 locus in more than 75% of the samples in human NSCLC. Likewise, in K-rasLA lung tumors, the deletion was limited to the Dmp1/MGC4175 locus in more than 90 % of the cases (Figure S4). It was not possible to distinguish the deletion of hDMP1 and that of MGC4175 by LOH analyses since these two genes are too closely located. However, it is very unlikely that MGC4175 deletion contributes to pulmonary carcinogenesis since it encodes a mitochondrial protein that is associated with taxoland doxorubicin-resistant malignant phenotypes in human cancer cell lines (Duan et al., 2004). Most importantly, our current K-rasLA; Dmp1-knockout mice model clearly points to the role of Dmp1-deletion in pulmonary carcinogenesis since MGC4175 remains intact in the Dmp1-targeting vector (Inoue et al., 2000). Thus, we speculate that hDMP1 is the critical gene for suppression of pulmonary carcinogenesis located on human chromosome 7q21.
Human DMP1 promoter hypermethylation was found only in one of the 23 lung cancer samples we examined, and none of them showed bi-allelic deletion of the hDMP1 gene. The second hDMP1 allele was expressed and not mutated in the tumor cells that showed LOH, although there was one exceptional case where lung tumor cells showed both LOH and promoter methylation for hDMP1. Likewise, splicing alterations that result in the overexpression of the dominant-negative hDMP1β isoform was not found in the 11 samples we have randomly analyzed (Figure S6). Generally, promoter hypermethylation or point mutation of the second allele is a feature of classical tumor suppressor genes and is very rare in haplo-insufficient tumor suppressors, such as p27Kip1 (Chim et al., 2005; Kibel et al., 2001; for review, Herman and Baylin, 2003). Thus, our data are consistent with our hypothesis that hDMP1 may be haplo-insufficient for tumor suppression in human lung cancer. On the other hand, 3 of 40 NSCLC cases showed bi-allelic deletion of ARF Exon 1β and 10 of 25 cases of p16INK4a promoter methylation occurred simultaneously with LOH of the INK4a/ARF locus (Figure 5), compatible with the concept of INK4a/ARF being classical tumor suppressors.
Our data indicate that LOH of the hDMP1 gene and that of the INK4a/ARF locus occurred in mutually exclusive fashion in >90 % of the cases (P< 0.01), although there were two exceptional cases that showed inactivation of both the hDMP1 and the INK4a/ARF loci. One possible explanation is that the deletion or methylation of the INK4a/ARF locus will result in the absence or modification of the hDMP1-binding sites: thus, tumors that have inactivated INK4a/ARF do not have to delete the hDMP1 gene at the same time. Likewise, LOH of hDMP1 also occurred significantly less frequently in lung tumors that showed LOH for P53 (P< 0.05). Interestingly, one of the four human non-small cell lung cancer cell lines (H460) showed hemizygous deletion of hDMP1, where both ARF and P53 are wild-type (Somasundaram et al., 1999) and K-Ras is mutant. Activated Dmp1:ER efficiently induced p14ARF and inhibited the growth of H460 cells while other lung cancer cell lines with deletion of ARF (A549) or P53 (H1299, H358) were resistant to Dmp1 overexpression. Consistent with the human data, the Dmp1 gene was deleted only in K-rasLA lung tumors with wild-type p53, and but not in any of the lung tumors from p53+/− or p53−/−; K-rasLA mice, suggesting the mutually exclusive inactivation of Dmp1 and p53 in K-rasLA lung tumors. Collectively, the DMP1 gene is frequently deleted in lung tumors where the INK4a/ARF locus and/or the P53 locus remains wild-type since hDMP1, ARF, and P53 are in the same signaling pathway.
Although homozygous deletion of Arf was not observed in any K-rasLA lung tumors, hemizygous deletion of Arf was found in ~25 % of lung tumors regardless of the genotype of Dmp1. Haploid insufficiency of p19Arf has been reported in murine solid tumors only when p16Ink4a was homozygously inactivated (Krimpenfort et al., 2001). However, inactivation of p16Ink4a by gene deletion or promoter hypermethylation is rare in K-rasLA lung tumors (Johnson et al., 2001; J. Sage, personal communication). Therefore, it is unlikely that the hemizygous Arf deletion contributed to lung tumorigenesis. If Arf does not contribute to K-rasLA lung tumors, Dmp1 should be regulating the p53 activity by a yet unknown mechanism. On the other hand, p14ARF is apparently involved in human NSCLC either by 1) biallelic deletion (#2003-86, #2003-442, and #2003-246)(Figure 5) or by 2) hemizygous deletion with simultaneous biallelic inactivation of the p16INK4a locus (#2000-96, #2003-422, #2004-719, #2004-983, #1999-95, and #2006-172).
K-rasLA lung tumors are different from Eµ-Myc lymphomas in that bi-allelic Arf deletion or Mdm2 overexpression was not found in any tumors regardless of the genotype of Dmp1 (Inoue et al., 2001). None of the Ink4a/Arf modulators, such as Bmi1, Twist, Tbx2/3, and Pokemon were overexpressed in K-rasLA lung tumors, ruling out the possibility of the involvement of these Ink4a/Arf modulators for K-ras-induced tumor formation. p53 mutation was less frequent in lung tumors from Dmpl+/−, Dmp1−/−; K-rasLA mice, thus Dmp1 deletions and p53 mutations might have similar effects. In fact, we have found that tumors that showed deletion of Dmp1 tend to show the phenotype of adenocarcinomas (5/7, 71 %; the number includes two other cases of Dmp1 deletion that were not shown in Figure 3). All the lung tumors that showed mutation of p53 were adenocarcinomas (4/4, 100 %). On the other hand, lung tumors that did not show Dmp1 or p53 alterations were mostly adenomas, and there was only one case of adenocarcinoma in this group (1/5, 20 %). This phenotypic difference was statistically significant (P = 0.036 for Dmp1 deletion, P = 0.016 for p53 mutation). Thus, deletions of Dmp1 or mutations of p53 are frequently associated with malignant phenotypes of K-rasLA lung tumors.
Dmp1 showed typical haploid insufficiency in suppression of K-rasLA lung cancers, especially in K-rasLA2/+ tumors. Although many other tumor suppressor genes have now been reported to show haploid-insufficiency to some extent, this level of strong haploid insufficiency was observed only in p27Kip1 and Dmp1 (reviewed in Payne and Kemp, 2006; Quon and Berns, 2001). In the case of p27Kip1, it was explained that complete loss of p27Kip1 results in decreased assembly of cyclin D1 and Cdk4 especially in mammary epithelial cells, thus MMTV-neu-induced breast cancer was not accelerated in a p27Kip1−/− background (Muraoka et al., 2002). Although the exact molecular mechanisms of haploid insufficiency of Dmp1 remain to be determined, apparently one copy loss of Dmp1 is enough to inactivate the p53 pathway in K-ras as well as in Eµ-Myc-induced tumor formation (Figure 3A in this study; Inoue et al., 2001).
In conclusion, we have demonstrated that the Dmp1 (hDMP1) gene is hemizygously deleted in a significant percentage of murine and human non-small cell lung carcinomas, especially those which retained the intact Arf-p53 pathway. Thus, DMP1 is principally involved in pulmonary carcinogenesis. Dmp1 showed haploid insufficiency in K-rasLA murine lung tumors, and our data with lung cancer patients’ samples are compatible with haploid insufficiency of hDMP1 in NSCLC. Since hDMP1 LOH (+) lung cancer cells retain one allele of the hDMP1 locus, this gene might be a promising target for future drug development.
Experimental Procedures
Creation of Dmp1+/−, Dmp1−/−; K-rasLA/+ mice
Dmp1-heterozygous females were backcrossed to the same C57BL/6 male for more than six generations to obtain Dmp1+/− mice that were >98 % C57BL/6 background overall. The mouse Dmp1 gene is localized on chromosome 5. All the seven markers on mouse chromosome 5 had been replaced with the C57BL/6 markers at G6. One male K-rasLA1/+ or K-rasLA2/+ mouse was crossed with two Dmp1+/− females to obtain Dmp1+/−; K-rasLA1/+ or K-rasLA2/+ mice. Then these Dmp1+/−; K-rasLA/+ transgenic mice were further crossed with Dmp1+/− mice to obtain more than 25 mice with each genetic background. Mice were observed daily, and were euthanized when they showed any signs of tumor formation. Mice were maintained in accordance with the Guide for the Care and Use of Laboratory Animals.
Statistical analyses
Statistical differences of survival in Dmp1+/+, Dmp1+/−, and Dmp1−/−; K-rasLA1/+ or K-rasLA2/+ mice were analyzed by XLSTAT-Life software (Addinsoft, New York, NY). Mann-Whitney test (two-sided) were used to generate the P values (significance level, α = 0.05). For each comparison of LOH, we performed a 1 degree of freedom test to determine whether the LOH of hDMP1, INK4a/ARF, and P53 in NSCLC samples were more likely to occur mutually exclusively or together. To do this, three separate chi-square tests were performed - one for each pair of data (i.e., hDMP1 and INK4a/ARF, hDMP1 and P53, and INK4a/ARF and P53). In these analyses, we examined the expected cell count versus the observed cell count in order to determine whether there was evidence of mutual exclusivity or not (evidence of mutual exclusivity is supported when the off-diagonal elements of the 2×2 table have higher observed counts than expected, whereas mutual exclusivity is not supported if the main diagonal cells of the 2×2 table have higher observed counts than expected). In addition to performing chi-square tests, we estimate 95 % confidence intervals for binomial proportions (using a normal approximation). These intervals were calculated using data where at least one of the two markers showed LOH (shown in bold red in Figure 5) and we estimated the probability that co-occurrence does not happen given that at least one marker has occurred. Unpaired Student’s t-tests were conducted for the analyses of number and size of K-rasLA lung tumors as well as to demonstrate the difference of intensity of hDMP1 signals in immunohistochemistry.
Human lung cancer samples
Fifty-one pairs of frozen human lung cancer tissues (33 cases of adenocarcinoma, 16 cases of squamous cell carcinoma, and 2 cases of adenosquamous carcinoma) and their normal counterparts were obtained from the Tissue Procurement Core Facility at the Wake Forest University Comprehensive Cancer Center. The samples had already been resected from patients with informed consent and had been stored in liquid nitrogen. The samples do not contain any subject identifiers. The human protocol had been approved by the Institutional Review Board.
Loss of heterozygosity (LOH) and sequencing analyses of human lung cancer specimen
Two sets of markers for microsatellite analysis of hDMP1 (#92465, #198004), two sets of markers for INK4a/ARF (#33647, #27251), and two sets of markers for P53 (#158111, #89737) were selected by using the software at http://www.gramene.org (Ware et al., 2002) (Figure 4A–C). These markers were chosen because they are polymorphic between individuals in more than 60 % of the cases. For detailed mapping of the region deleted in human NSCLC, 4 other sets of primers were designed by our selves (B-Actin, #69164, #251945, and ABCB1) (Figure 6A, Table S1). D7S644 sequences were obtained from the NCBI. The forward primer of each pair was labeled on the 5' end with the fluorescent dye FAM (Operon Technologies, Huntsville, AL) and PCR amplification was performed with DNA isolated from the tumor and normal tissues. PCR products were visualized on a 1.2 % agarose gel. Genotypes were identified by peak analysis of the fluorescent signal detected on an ABI 3700 DNA analyzer (Applied Biosystems). The qLOH values were determined through the following equation. qLOH = Area Peak 1/Area Peak 2 (normal tissue) divided by Area Peak 1’/Area Peak 2’ (tumor tissue). LOH was assessed if the qLOH value was found to be >2.0 or <0.5 (So et al., 2004). Each sample was found to be positive for LOH of the locus when the qLOH values were >2.0 or <0.5 in one of the two sets of primers. When one of the two sets of LOH primers showed a single peak, LOH was determined by the other set. Whenever duplicate samples were available, total RNA was extracted from lung cancer tissues by using RNAlater®-ICE (Ambion, Applied Biosystems) and TRIzol® (Invitrogen, Carlsbad, CA), and RT-PCR was conducted with PfuUltra™ Hotstart DNA polymerase (Stratagene, La Jolla, CA).
Other experimental procedures are described in Supplemental Data.
Supplementary Material
Acknowledgements
We are very grateful to T. Jacks, D. Tuveson, N. Young, K. Mercer, and A. Deconinck for K-rasLA mice and paraffin blocks for p53+/−, p53−/−; K-rasLA mice; J. Sage for unpublished data; G. Sui and K. Klein and for critical reading of the manuscript. We thank C. Sherr and M. Roussel for Dmp1-knockout mice and plasmids. We also thank J. Garvin for murine pathology and J. Clark, S. Lagedrost, and S. Barton for technical assistance. This work was supported by NIH/NCI 5R01CA106314 (K. Inoue). D. Frazier is supported by NIH Institutional Research Training Grant 5T32CA079448 (F. Torti).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bieche I, Champeme MH, Matifas F, Hacene K, Callahan R, Lidereau R. Loss of heterozygosity on chromosome 7q and aggressive primary breast cancer. Lancet. 1992;339:139–143. doi: 10.1016/0140-6736(92)90208-k. [DOI] [PubMed] [Google Scholar]
- Bodner SM, Naeve CW, Rakestraw KM, Jones BG, Valentine VA, Valentine MB, Luthardt FW, Willman CL, Raimondi SC, Downing JR, et al. Cloning and chromosomal localization of the gene encoding human cyclin D-binding Myb-like protein (hDMP1) Gene. 1999;229:223–228. doi: 10.1016/s0378-1119(98)00591-5. [DOI] [PubMed] [Google Scholar]
- Chim CS, Wong AS, Kwong YL. Epigenetic inactivation of the CIP/KIP cell-cycle control pathway in acute leukemias. Am. J. Hematol. 2005;80:282–287. doi: 10.1002/ajh.20503. [DOI] [PubMed] [Google Scholar]
- Duan Z, Brakora KA, Seiden MV. MM-TRAG (MGC4175), a novel intracellular mitochondrial protein, is associated with the taxol- and doxorubicin-resistant phenotype in human cancer cell lines. Gene. 2004;340:53–59. doi: 10.1016/j.gene.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Fong KM, Sekido Y, Gazdar AF, Minna JD. Lung cancer. 9: Molecular biology of lung cancer: Clinical implications. Thorax. 2003;58:892–900. doi: 10.1136/thorax.58.10.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. New Engl. J. Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- Hirai H, Sherr CJ. Interaction of D-type cyclins with a novel myb-like transcription factor. DMP1. Mol. Cell. Biol. 1996;16:6457–6467. doi: 10.1128/mcb.16.11.6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Sherr CJ. Gene expression and cell cycle arrest mediated by transcription factor DMP1 is antagonized by D-type cyclins through a cyclin-dependent-kinase-independent mechanism. Mol. Cell. Biol. 1998;18:1590–1600. doi: 10.1128/mcb.18.3.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Sherr CJ, Shapiro LH. Regulation of the CD13/aminopeptidase N gene by DMP1, a transcription factor antagonized by D-type cyclins. J. Biol. Chem. 1998;273:29188–29194. doi: 10.1074/jbc.273.44.29188. [DOI] [PubMed] [Google Scholar]
- Inoue K, Roussel MF, Sherr CJ. Induction of ARF tumor suppressor gene expression and cell cycle arrest by transcription factor DMP1. Proc. Natl. Acad. Sci. USA. 1999;96:3993–3998. doi: 10.1073/pnas.96.7.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Wen R, Rehg JE, Adachi M, Cleveland JL, Roussel MF, Sherr CJ. Disruption of the ARF transcriptional activator DMP1 facilitates cell immortalization, Ras transformation, and tumorigenesis. Genes Dev. 2000;14:1797–1809. [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Zindy F, Randle DH, Rehg JE, Sherr CJ. Dmp1 is haplo-insufficient for tumor suppression and modifies the frequencies of Arf and p53 mutations in Myc-induced lymphomas. Genes Dev. 2001;15:2934–2939. doi: 10.1101/gad.929901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Mallakin A, Frazier DP. Dmp1 and tumor suppression. Oncogene. 2007;26:4329–4335. doi: 10.1038/sj.onc.1210226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, Keblusek P, Robanus-Maandag E, Kristel P, Lingbeek M, Nederlof PM, van Welsem T, van de Vijver MJ, Koh EY, Daley GQ, et al. Senescence bypass screen identifies TBX2, which represses Cdkn2a (p19(ARF)) and is amplified in a subset of human breast cancers. Nat Genet. 2000;26:291–299. doi: 10.1038/81583. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J. Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59:2217–2222. [PubMed] [Google Scholar]
- Kerr J, Leary JA, Hurst T, Shih YC, Antalis TM, Friedlander M, Crawford E, Khoo SK, Ward B, Chenevix-Trench G. Allelic loss on chromosome 7q in ovarian adenocarcinomas: two critical regions and a rearrangement of the PLANH1 locus. Oncogene. 1996;13:1815–1818. [PubMed] [Google Scholar]
- Kibel AS, Christopher M, Faith DA, Bova GS, Goodfellow PJ, Isaacs WB. Methylation and mutational analysis of p27(kip1) in prostate carcinoma. Prostate. 2001;48:248–253. doi: 10.1002/pros.1104. [DOI] [PubMed] [Google Scholar]
- Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature. 2001;413:83–86. doi: 10.1038/35092584. [DOI] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Lowe S, Sherr CJ. Tumor suppression by Ink4a-Arf: Progress and puzzles. Curr. Opin. Genet. Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- Maeda T, Hobbs RM, Merghoub T, Guernah I, Zelent A, Cordon-Cardo C, Teruya-Feldstein J, Pandolfi PP. Role of the proto-oncogene Pokemon in cellular transformation and ARF repression. Nature. 2005;433:278–285. doi: 10.1038/nature03203. [DOI] [PubMed] [Google Scholar]
- Maestro R, DeiTos AP, Hamamori Y, Krasnokutsky S, Sartorelli V, Kedes L, Doglioni C, Beach DH, Hannon GJ. Twist is a potential oncogene that inhibits apoptosis. Genes Dev. 1999;13:2207–2217. doi: 10.1101/gad.13.17.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallakin A, Taneja P, Matise LA, Willingham MC, Inoue K. Expression of Dmp1 in specific differentiated, nonproliferating cells and its repression by E2Fs. Oncogene. 2006;25:7703–7713. doi: 10.1038/sj.onc.1209750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen R, Berns A. Mouse models for human lung cancer. Genes Dev. 2005;19:643–664. doi: 10.1101/gad.1284505. [DOI] [PubMed] [Google Scholar]
- Moran CA. Pulmonary adenocarcinoma: The expanding spectrum of histologic variants. Arch. Pathol. Lab. Med. 2006;130:958–962. doi: 10.5858/2006-130-958-PATESO. [DOI] [PubMed] [Google Scholar]
- Muraoka RS, Lenferink AE, Law B, Hamilton E, Brantley DM, Roebuck LR, Arteaga CL. ErbB2/Neu-induced, cyclin D1-dependent transformation is accelerated in p27-haploinsufficient mammary epithelial cells but impaired in p27-null cells. Mol. Cell. Biol. 2002;22:2204–2219. doi: 10.1128/MCB.22.7.2204-2219.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oriola J, Halperin I, Mallofre C, Muntane J, Angel M, Rivera-Fillat F. Screening of selected genomic areas potentially involved in thyroid neoplasms. Eur. J. Cancer. 2001;37:2470–2474. doi: 10.1016/s0959-8049(01)00302-1. [DOI] [PubMed] [Google Scholar]
- Payne SR, Kemp CJ. Tumor suppressor genetics. Carcinogenesis. 2006;26:2031–2045. doi: 10.1093/carcin/bgi223. [DOI] [PubMed] [Google Scholar]
- Quon KC, Berns A. Haplo-insufficiency? Let me count the ways. Genes Dev. 2001;15:2917–2921. doi: 10.1101/gad.949001. [DOI] [PubMed] [Google Scholar]
- Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim. Biophys. Acta Rev. Cancer. 1998;1378:F115–F177. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- Schiller JH. Current standards of care in small-cell and non-small-cell lung cancer. Oncology. 2001;61 Suppl 1:3–13. doi: 10.1159/000055386. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. The INK4a/ARF network in tumor suppression. Nat. Rev. Mol. Cell. Biol. 2001;2:731–737. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat. Rev. Cancer. 2006;6:663–673. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- So CK, Nie Y, Song Y, Yang GY, Chen S, Wei C, Wang LD, Doggett NA, Yang CS. Loss of heterozygosity and internal tandem duplication mutations of the CBP gene are frequent events in human esophageal squamous cell carcinoma. Clin. Cancer Res. 2004;10:19–27. doi: 10.1158/1078-0432.ccr-03-0160. [DOI] [PubMed] [Google Scholar]
- Somasundaram K, MacLachlan TK, Burns TF, Sgagias M, Cowan KH, Weber BL, el-Deiry WS. BRCA1 signals ARF-dependent stabilization and coactivation of p53. Oncogene. 1999;18:6605–6614. doi: 10.1038/sj.onc.1203284. [DOI] [PubMed] [Google Scholar]
- Spira A, Ettinger DS. Multidisciplinary management of lung cancer. N. Engl. J. Med. 2004;350:379–392. doi: 10.1056/NEJMra035536. [DOI] [PubMed] [Google Scholar]
- Sreeramaneni R, Chaudhry A, McMahon M, Sherr CJ, Inoue K. Ras-Raf-Arf signaling critically depends on Dmp1 transcription factor. Mol. Cell. Biol. 2005;25:220–232. doi: 10.1128/MCB.25.1.220-232.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, Palmero I, Ryan K, Hara E, Vousden KH, et al. The alternative product from the human CDKN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998;17:5001–5014. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja P, Mallakin A, Matise LA, Frazier DP, Choudhary M, Inoue K. Repression of Dmp1 and Arf transcription by anthracyclins: critical roles of the NF-kappaB subunit p65. Oncogene. 2007 Jun 4; doi: 10.1038/sj.onc.1210568. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyooka S, Tsuda T, Gazdar AF. The TP53 gene, tobacco exposure, and lung cancer. Hum. Mutat. 2003;21:229–239. doi: 10.1002/humu.10177. [DOI] [PubMed] [Google Scholar]
- Travis WD. Pathology of lung cancer. Clin. Chest Med. 2002;23:65–81. doi: 10.1016/s0272-5231(03)00061-3. [DOI] [PubMed] [Google Scholar]
- Tschan MP, Fischer KM, Fung VS, Pirnia F, Borner MM, Fey MF, Tobler A, Torbett BE. Alternative splicing of the human cyclin D-binding Myb-like protein (hDMP1) yields a truncated protein isoform that alters macrophage differentiation patterns. J. Biol. Chem. 2003;278:42750–42760. doi: 10.1074/jbc.M307067200. [DOI] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Wistuba I, Gazdar AF, Minna JD. Molecular genetics of small cell lung carcinoma. Semin. Oncol. 2001;28:3–13. [PubMed] [Google Scholar]
- Zindy F, Williams RT, Baudino TA, Rehg JE, Skapek SX, Cleveland JL, Roussel MF, Sherr CJ. Arf tumor suppressor promoter monitors latent oncogenic signals in vivo. Proc. Natl. Acad. Sci. USA. 2003;100:15930–15935. doi: 10.1073/pnas.2536808100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zochbauer-Muller S, Gazdar AF, Minna JD. Molecular pathogenesis of lung cancer. Annu. Rev. Physiol. 2002;64:681–708. doi: 10.1146/annurev.physiol.64.081501.155828. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.