

Abstract
The development of gene-replacement therapy for inborn errors of metabolism has been hindered by the limited number of suitable large-animal models of these diseases and by inadequate methods of assessing the efficacy of treatment. Such methods should provide sensitive detection of expression in vivo and should be unaffected by concurrent pharmacologic and dietary regimens. We present the results of studies in a neonatal bovine model of citrullinemia, an inborn error of urea-cycle metabolism characterized by deficiency of argininosuccinate synthetase and consequent life-threatening hyperammonemia. Measurements of the flux of nitrogen from orally administered 15NH4 to [15N]urea were used to determine urea-cycle activity in vivo. In control animals, these isotopic measurements proved to be unaffected by pharmacologic treatments. Systemic administration of a first-generation E1-deleted adenoviral vector expressing human argininosuccinate synthetase resulted in transduction of hepatocytes and partial correction of the enzyme defect. The isotopic method showed significant restoration of urea synthesis. Moreover, the calves showed clinical improvement and normalization of plasma glutamine levels after treatment. The results show the clinical efficacy of treating a large-animal model of an inborn error of hepatocyte metabolism in conjunction with a method for sensitively measuring correction in vivo. These studies will be applicable to human trials of the treatment of this disorder and other related urea-cycle disorders.
Urea-cycle disorders are a primary cause of hyperammonemia in the pediatric population and are associated with very high neurologic morbidity and mortality (1). Moreover, they are representative of a large group of intrinsic inborn errors of hepatocyte metabolism. Current therapy consists of dietary protein restriction, the stimulation of alternative routes of nitrogen disposal with sodium phenylacetate and benzoate or with sodium phenylbutyrate, as well as arginine or citrulline supplementation to prevent arginine deficiency (2, 3). However, despite aggressive pharmacotherapy, patients are at risk for repeated episodes of hyperammonemia during catabolic crises, which are often precipitated by intercurrent illnesses. Lifetime morbidity and mortality is directly correlated with the duration and frequency of these episodes (4).
Gene-replacement therapy is a potential alternative to complement and ultimately replace pharmacotherapy of specific inborn errors of metabolism. The technique has promise as a chronic maintenance therapy and/or as an approach to metabolic rescue during acute crises. Because of the availability of high-titer preparations, their efficiency of transduction, and their relative specificity for the liver after intravascular delivery, adenoviral vectors are well suited for gene delivery in inborn errors that affect hepatic metabolism (5, 6). Several such conditions include ornithine transcarbamylase deficiency (7), factor IX deficiency (8, 9), phenylketonuria (10), and familial hypercholesterolemia (11, 12). However, clinical efficacy has yet to be achieved in humans.
Obstacles to the successful clinical application of adenoviral gene therapy include toxicity and the limited duration of expression, caused, in part, by the host immune response to viral proteins (13–16) and transgene expression (17–20). However, there is increasing evidence that both these problems may be circumvented by modification of the viral genome (21, 22), including complete deletion of all coding sequences in helper-dependent adenoviral vectors (23, 24). These vectors may lead to increased therapeutic index and prolonged transgene expression. Another obstacle to the institution of gene therapy for inborn errors of metabolism is the difficulty of measuring the efficacy and clinical benefit of treatment. This problem is particularly difficult to overcome, because conclusions about toxicity and efficacy based on measurements in small-animal models often do not translate to large-animal systems, including those of humans. The phenotypic expression of some transgenic mouse models for human diseases do not always compare with the human conditions. These issues, combined with the complications caused by necessary concurrent pharmacotherapy, make the interpretation of efficacy difficult in clinical trials.
We have addressed these obstacles by studying the efficacy of gene replacement in bovine citrullinemia (25). This large-animal model is very similar to the human urea-cycle disorder; citrullinemia manifests soon after birth and is characterized by poor feeding and altered behavior secondary to hyperammonemia. Affected animals are homozygous for a nonsense mutation in the bovine argininosuccinate synthetase (ASS) gene (26). They have plasma amino acid profiles similar to those seen in neonatal manifestation of the condition in humans, especially elevated concentrations of glutamine and citrulline. Without pharmacotherapy, the calves die in the first few days of life, having experienced a continuous neurologic decline after initial feeding. With sodium benzoate treatment (which directs nitrogen to hippuric acid) and arginine treatment (which provides substrate for ornithine and citrulline synthesis and also prevents arginine deficiency), the life span may be prolonged; however there is little improvement in the plasma amino acid profile, and the animals are still subject to frequent episodes of metabolic decompensation, as in the human condition. Because of their size (approximately 35 kg at birth) and the ease of tissue sampling, the calves are an excellent model for physiologic studies of the efficacy of therapies in citrullinemia (27).
In attempting to evaluate phenotypic severity more accurately, we hypothesized that the measurement of the conversion of a [15N]-labeled urea-cycle precursor into urea would be an accurate measure of the rate of urea synthesis in vivo and would correlate with residual enzyme activity at the rate-limiting step in the pathway and with clinical severity; we also hypothesized that concurrent therapy would not affect the accuracy of this measurement. The usefulness of oral 15NH4Cl as an 15N precursor for urea synthesis has been shown in a murine model of ornithine transcarbamylase deficiency (28), and we have used [amide-15N]glutamine in continuous infusions in patients with urea-cycle disorder and control subjects to correlate phenotypic severity with urea synthetic rates (B.L., H.Y., F. Jahoor, W.O., A.L.B., and P.R., unpublished work). To explore the applicability and utility of bovine citrullinemia in gene-therapy studies, we determined the efficiency of hepatocyte transduction by adenoviral vectors after i.v. delivery and the effect of pharmacotherapy on nitrogen kinetics in unaffected age-matched calves. We also examined the clinical and metabolic efficacy of adenoviral mediated transduction of human ASS after i.v. delivery into neonatal citrullinemic calves.
MATERIALS AND METHODS
Citrullinemia Calves.
Calves were maintained in Australia. Embryos were recovered from heterozygous cows and transferred to recipients 7 days after insemination by a heterozygous bull. Calves weighing between 30 kg and 35 kg were born after a normal gestation and allowed to suck colostrum from their mothers. The calves were genotyped for the R86X mutation (26), and two homozygous wild-type (S6 and S7) and two homozygous mutant animals (S2 and S3) were isolated from their mothers and fed between 6 and 9 liters of milk per day. From 24 h after birth, the calves received daily oral supplements of 20 g of arginine and 20 g of sodium benzoate. Blood was collected daily for determination of plasma amino acids, ammonia, total bilirubin, and alanine aminotransferase, aspartate aminotransferase (AST), and alkaline phosphatase. The clinical condition of the calves was assessed every 6 h. The injection of the virus and blood sampling were accomplished via external jugular vein catheterization.
Adenoviral Vector Preparation and Administration.
AdΔE1CAGASS was prepared by cotransfection of the plasmid pBHGE3 and pΔE1CAGASS into 293 cells (a gift from F. Graham, McMaster University, Hamilton, Canada) by calcium phosphate coprecipitation by using a standard protocol (29). The 293 cells provide the E1a transcriptional factor in trans to initiate replication of the E1a-deficient adenovirus vector. pΔE1CAGASS contains the cytomegalovirus (CMV) enhancer/chicken β-actin promoter (30), human ASS cDNA (31), and rabbit β-globin poly-adenylation signal. Cytopathic effect was observed after 2 weeks, and the supernatant virus was purified twice on plaques on 293 cells. The virus was amplified and purified by CsCl centrifugation. Vector concentration was determined by both DNA content via OD at 260 nm and by plaque assay on 293 cells. Activity of the viral preparation was confirmed by infection of cultured ASS deficient XC fibroblasts at a multiplicity of infection of 100:1, followed by measurement of [14C]citrulline incorporation into arginine at 24 h after infection. After harvest, ASS enzymatic activity was also determined (32). AdΔE1CMVβgal has been described as a first-generation adenoviral vector deleted for E1 and wild-type for E3 expressing the β-galactosidase gene from a CMV promoter (19). All procedures for preparation of vectors were carried out in the Baylor College of Medicine Gene Vector Laboratory by using Good Laboratory Practice/Good Manufacturing Practice. Viral vectors were analyzed for sterility, endotoxin content, and mycoplasma. AdΔE1CMVβgal (1 × 1013 particles per kg) and AdΔE1CAGASS (1 × 1013 particles per kg) were diluted with PBS and then administered via i.v. injection into a control calf (S5) and two citrullinemic calves (S2 and S3), respectively. Before i.v. administration, calves were medicated with diphenhydramine and hydrocortisone to prevent acute non-IgE-mediated anaphylaxis, which we had observed in earlier experiments.
Nitrogen-Flux Stable-Isotope Protocol.
Isotope studies on citrullinemic and control calves were performed over 2-day intervals (Fig. 1A). Calves were fed milk every 6 h. Feedings were supplemented with 400 mg of 15NH4Cl (Cambridge Isotope Laboratories, Cambridge, MA) after the “zero-hour” time point. Before feeding, blood was obtained by venipuncture for analysis of plasma amino acids and ammonia. In addition, 4 ml of whole blood treated with the anticoagulant EDTA was snap-frozen and stored at −70°C (Fig. 1A) for measurement of [15N]glutamine and [15N]urea. Citrullinemic calves were studied before vector treatment and then over days 2 and 3, days 6 and 7, and days 17 and 18 after treatment. One of the calves (S3) had gastroenteritis on days 6 and 7 after treatment, and that particular isotope study was not performed. Calf S3 was treated with both sodium benzoate and arginine until day 4 after vector administration, at which time the sodium benzoate was withdrawn. Treatment with sodium benzoate was discontinued for calf S2 7 days after vector administration. Because there is evidence that even liver transplantation does not correct the arginine requirement in citrullinemic patients (33), both calves were continued on arginine supplementation for the duration of the study.
Figure 1.

Schematics of stable-isotope studies. (A) This 2-day stable-isotope study (represented by ovals in B) consists of 15NH4Cl feedings every 6 h, preceded by blood sampling. Metabolic parameters were measured every 6 h and are listed under the time line. (B) Time line (in days) of citrullinemic-calf studies (S2 and S3) and control-calf studies (S6 and S7). The durations of arginine and/or sodium benzoate treatments are shown above the time lines. Ovals represent 2-day stable-isotope studies. For citrullinemic calves (S2 and S3), the days are listed relative to the day of virus infusion (day 0). For control calves (S6 and S7), the days are listed relative to the start of pharmacotherapy with benzoate and arginine (day 0).
Flux studies were first performed in control milk-fed calves S6 and S7 without medication. Both sodium benzoate and arginine were then added to the diets (at day 0), and the flux studies were repeated over days 2 and 3 after initiation of pharmacotherapy. Although treatment with arginine was continued throughout the study, treatment with sodium benzoate was discontinued on day 4, and, finally, both calves were studied a third time on arginine supplementation alone over days 6 and 7 (Fig. 1B).
Metabolite, Biochemical, and Histological Analyses.
Plasma samples taken during the intermittent oral administration of 15NH4Cl were mixed with an equal volume of water and centrifuged through a 10-kDa cutoff filter at 16,000 × g for 4 h. The sample then was separated into two aliquots. The n-propyl-ester heptafluorobutyramide derivative of glutamine was prepared according to a published method (34). Urea was analyzed as the 2-pyrimidinol tert-butylmethylsilyl derivative. Briefly, dried ultrafiltrate was mixed with 550 μl of malonaldehyde bis-methyl acetal and incubated at room temperature for 2 h. After drying in a nitrogen stream, the residue was mixed with 300 μl of tert-butyldimethylsilane and incubated for 24 h at room temperature. Mass spectrometry for both derivatives was performed on a Hewlett Packard 9890A quadrupole gas chromatograph/mass spectrometer. The urea derivative was analyzed by electron-impact ionization, and the glutamine derivative was analyzed by methane-negative chemical ionization. We monitored ions with a mass-to-charge ratio of 153 and 154 for urea and 346 and 347 for glutamine. The results were expressed as tracer/tracee ratios (mol of M + 1:100 mol of M + 0) after correction for baseline enrichment of the M + 1 isotopomer.
Histologic analyses were performed on 10% formalin-fixed, paraffin-embedded tissues stained with hematoxylin and eosin. Tissues analyzed include brain (cerebral cortex), muscle, heart, lungs, liver, spleen, kidney, small and large intestine, and gonad. Tissues were also processed by cryofixation and stained with 5-bromo-4-chloro-3-indolyl β-d-galactoside for β-galactosidase activity. Efficacy of transduction was estimated by counting the number of positively staining cells per total cells per high-power field (at ×400 magnification). Serum chemistries were analyzed in the Baylor College of Medicine Clinical Pathology Laboratory of the Center for Comparative Medicine. Plasma amino acid analyses were performed by using standard methods on a Beckman–Spinco 6300 amino acid analyzer.
RESULTS
In Vivo Transduction of Bovine Hepatocytes.
The efficiency of hepatocyte transduction and toxicity was determined after i.v. delivery of a first-generation adenoviral vector expressing β-galactosidase (AdE1ΔCMVβGal). AdE1ΔCMVβGal (1 × 1013 particles per kg) was administered into the external carotid vein of a 1-week-old wild-type newborn calf. The animals were killed three days after injection, and 5-bromo-4-chloro-3-indolyl β-d-galactoside staining of tissues identified the expression of β-galactosidase in hepatocytes (Fig. 2). Approximately 20% of hepatocytes were transduced. The pattern of staining suggested greater transduction of central venous rather than periportal hepatocytes. No other tissues stained for β-galactosidase. No elevations in alanine aminotransferase (16 ± 2 international units/liter; n = 4), AST (48 ± 19 international units/liter; n = 4), or alkaline phosphatase (441 ± 273 international units/liter; n = 4) or clinical signs of illness were noted. Hematoxylin and eosin stain of liver and tissue sections indicated the absence of inflammatory cell infiltrates (data not shown).
Figure 2.

The efficiency of adenoviral transduction after i.v. delivery. Staining for β-galactosidase expression in the right lobe of the liver (A), the left lobe of the liver (B), the spleen (C), and the lungs (D) shows only transduction of hepatocytes (×100).
Effects of Pharmacotherapy on Nitrogen-Flux Measurements in Normal Calves.
Whole-blood [15N]urea enrichment during the intermittent oral administration of 15NH4Cl was measured over consecutive 2-day periods in two control calves (S6 and S7). The labeling of the plasma glutamine (2.55 ± 0.43 mol% excess) and urea (2.32 ± 0.43 mol% excess) pools reached similar isotopic enrichments. Treatment of the calves with pharmacological doses of sodium benzoate/arginine or arginine alone had only a small effect on 15N-labeling of urea (Fig. 3). These data indicate that in normal calves, steady-state measurements of [15N]urea enrichment derived from oral doses of 15NH4Cl precursor are relatively insensitive to the pharmacologic doses of sodium benzoate and arginine.
Figure 3.

Blood [15N]urea enrichment in control calves and the effects of arginine (Arg) and/or sodium benzoate (Benz) therapy (Rx). (A) Absolute [15N]urea enrichment (mol% excess) shown as time course of each 2-day stable-isotope study. Measurements were taken at 6-h intervals before feedings with milk and 15NH4Cl. Studies without medications (♦), with arginine and sodium benzoate (■), and with only arginine (▴) show that steady-state levels are achieved after three to five doses of 15NH4Cl. (B) Average [15N]urea enrichments over each 2-day isotope study from S6 and S7 on various therapeutic regimens. Determinations were made from blood samples obtained after the third dose of 15NH4Cl at steady state. Error bars represent SD.
In Vivo Correction of Bovine Citrullinemia.
Neonatal calves were genotyped by PCR amplification on the day of birth, and citrullinemic animals were fed arginine/benzoate. At 1 week of age, a 2-day baseline nitrogen-flux study was performed with 15NH4Cl administered with feedings every 6 h. Even though blood glutamine was highly labeled (2.31 ± 0.39 mol% excess), [15N]urea enrichment was undetectable in the initial study of both citrullinemic calves (S2 and S3), consistent with the absence of urea-cycle-specific urea synthesis (Fig. 4A). A day after completion of this first study, AdΔE1CAGASS (1 × 1013 particles per kg) was administered by i.v. infusion. Over days 2 and 3 and days 16 and 17 after treatment, the 2-day nitrogen-flux studies were repeated. Treatment with the vector did not alter the isotopic enrichment of circulating glutamine (2.18 ± 0.24 mol% excess), but urea was now enriched with 15N to values that ranged from 0.8 to 1.5 mol% excess, a finding consistent with adenoviral transduction of hepatocytes and de novo ASS activity (Fig. 4A). On the basis of [15N]urea labeling, urea synthesis was maintained for at least 18 days after vector administration. Moreover, both calves (S2 and S3) fed normally during this period, consistent with improved clinical condition. Plasma citrulline levels remained elevated (for S2, 2,037 ± 362 μM; for S3, 2,228 ± 175 μM; for normal calves, 46 ± 37 μM, n = 5). However, this continued elevation was not unexpected, because net citrulline clearance occurs because of renal ASS activity and selective restoration of this activity in the liver would not affect the urea cycle in the kidney. Importantly, plasma glutamine levels decreased to normal levels after administration of the vector (Fig. 4B). This decrease presumably reflects the de novo urea-cycle flux, which relieved the accumulation of precursor in the total-body nitrogen pool. Arginine levels remained constant within the normal range (129 ± 75 μM, n = 5), whereas plasma ammonia fluctuated (for S2, 106 ± 84 μM; for S3: 216 ± 36 μM). AST levels remained normal except for two elevations during the course of the study. S2 and S3 had elevations of serum AST to 510 international units/liter on day 7 and 585 international units/liter on day 14, respectively. Both of these values normalized over the next 2 days. However, serum alanine aminotransferase and alkaline phosphatase levels remained normal throughout the study. Histological analysis of hematoxylin and eosin stains did not show evidence of inflammatory infiltrate.
Figure 4.
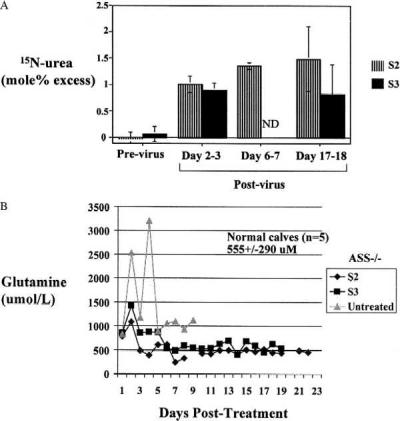
Efficacy of treatment of citrullinemic calves with AdΔE1CAGASS. (A) Average [15N]urea enrichment in S2 and S3 before vector treatment and at time points after vector treatments (error bars represent SD). The posttreatment (days 6 and 7) study on calf S3 was not done (ND), because the calf experienced gastroenteritis. (B) Time course of plasma glutamine levels in an untreated citrullinemic calf (▴) and in treated S2 (♦) and S3 (■) citrullinemic calves (ASS −/−). Control normal calves have average plasma glutamine levels as shown.
DISCUSSION
Urea-cycle disorders account for significant neurologic morbidity and mortality in pediatrics and provide a paradigm for defects of hepatocyte intermediary metabolism. Development of gene-replacement therapy for this condition and other conditions has been hindered by a shortage of animal models of target human diseases and by poor assays for metabolic correction. We show the application of in vivo gene-therapeutic correction of citrullinemia in a neonatal large-animal model that resembles both the pathologic condition and the therapeutic approach to the human condition. Systemic delivery of 1 × 1013 particles per kg of a first-generation adenoviral vector expressing β-galactosidase resulted in specific transduction of 20% of hepatocytes. This percentage is 2–5 times less than the efficiency seen with the same preparation in mice (ref. 19 and unpublished data). Although this difference is likely to be species-specific, it shows that hepatocyte-directed expression can be achieved with i.v. delivery but also that achieving a high percentage of transduction in larger species may be more difficult. Interestingly, transduction was preferential to central venous hepatocytes. The converse would be expected, given that, after systemic i.v. injection, the virus is delivered initially to liver parenchyma at periportal hepatocytes via the portal and hepatic arterial circulation. In the mouse, at least, this apparent difference in distribution is lost at higher doses, at which almost all hepatocytes are transduced (unpublished data). The nature of this effect at lower levels of transduction is unclear.
In the citrullinemic calves, we show at least a temporarily rescue with a single treatment of an adenoviral vector expressing the human ASS cDNA. The calves improved clinically and had decreased serum-glutamine profiles, consistent with de novo urea synthesis. It was particularly important to note that they continued to survive in spite of withdrawal of sodium benzoate pharmacotherapy. Sodium benzoate is critical in the treatment of this condition, because it conjugates glycine to form hippurate. Subsequently, hippurate is excreted in the urine, thereby providing an alternative route of disposal for the circulating nitrogen pool. Arginine therapy was continued, because correction of hepatic urea-cycle activity likely does not restore net arginine production. Similarly, net citrulline clearance occurs via the kidney urea cycle. In humans with citrullinemia who have had liver transplants, citrulline levels remain elevated after transplantation, because correction of the hepatic enzyme deficiency does not affect net citrulline clearance and arginine synthesis by the kidneys, which are still enzyme-deficient (35). In this study, because i.v. injection of adenovirus selectively transduces hepatocytes, ASS activity is restored only in the liver and not in the kidney, hence the continued elevation in citrulline and the requirement for arginine.
The treated calves experienced two transient episodes of elevated serum AST. The etiology of this short-term inflammation is unclear, but its course is unlike the later and more chronic host immune-related toxicity that has been observed in toxicity studies in mice and nonhuman primates (36). Ultimately, as was expected with the use of first-generation adenoviral vectors, the therapeutic correction was transient with deterioration of the clinical status 3 weeks after treatment. These vectors, which are deficient in the E1a transactivation factor, are replication-deficient, but continue to express late viral structural proteins that initiate immune clearance of the transduced cells, dose-related inflammation, and a subsequent loss of enzymatic correction. The duration of expression may be increased with the use of the latest helper-dependent adenoviral vector systems that eliminate the immunogenicity and toxicity associated with viral gene expression. These vectors are devoid of viral coding sequences and do not express viral proteins after transduction.
Importantly, we demonstrate the use of a stable-isotope nitrogen precursor/product protocol for correlating [15N]urea enrichment with in vivo urea-cycle activity and clinical improvement after viral gene therapy. At steady state, the flux from 15NH4Cl to [15N]urea was unaffected by administration of sodium benzoate and/or arginine, with the [15N]urea enrichment remaining between 2–2.5 mol% excess. The lack of effect observed with concurrent drug treatment is especially important, because, as in the bovine model, human studies would be performed with necessary concurrent pharmacotherapy that may normalize other measures of efficacy, such as plasma amino acids and ammonia. Transduction of approximately 20% of hepatocytes with an adenoviral vector expressing human ASS from an ubiquitously active promoter resulted in 43% and 50% of the [15N]urea enrichment measured in control calves. This level of transduction was sufficient to achieve clinical improvement, albeit on a temporary basis. These studies show that bovine citrullinemia is a useful model for evaluating in vivo gene therapy for a classic inborn error of hepatocyte metabolism. Moreover, measurement of the flux of 15NH4Cl to [15N]urea in these calves has enabled us to correlate de novo expression of human ASS with urea-cycle activity and clinical efficacy in spite of potentially confounding variables such as concurrent pharmacotherapy. Together, these data lend insight into a large-animal model of a urea-cycle defect and form the basis of designing future human clinical trials.
Acknowledgments
The authors thank A. Warman and J. Heidorn for technical assistance. This work was supported in part by National Institutes of Health Grants DK02407 (to B.L.) and HL51754 (to A.L.B.), by the U.S. Department of Agriculture/Agriculture Research Service under Cooperative Agreement 58-6258-6100, and by the Baylor College of Medicine Mental Retardation Research Center and Child Health Research Center.
ABBREVIATIONS
- ASS
argininosuccinate synthetase
- AST
aspartate aminotransferase
- CMV
cytomegalovirus
References
- 1.Brusilow S W, Horwich A L. In: The Molecular and Metabolic Basis of Inherited Disease. Scriver C R, Beaudet A L, Sly W S, Valle D, editors. New York: McGraw–Hill; 1995. pp. 1187–1232. [Google Scholar]
- 2.Batshaw M L, Brusilow S, Waber L, Blom W, Brubakk A M, Burton B K, Cann H M, Kerr D, Mamunes P, Matalon R, et al. N Engl J Med. 1982;306:1387–1392. doi: 10.1056/NEJM198206103062303. [DOI] [PubMed] [Google Scholar]
- 3.Brusilow S W, Danney M, Waber L J, Batshaw M, Burton B, Levitsky L, Roth K, McKeethren C, Ward J. N Engl J Med. 1984;310:1630–1634. doi: 10.1056/NEJM198406213102503. [DOI] [PubMed] [Google Scholar]
- 4.Msall M, Batshaw M L, Suss R, Brusilow S W, Mellits E D. N Engl J Med. 1984;310:1500–1505. doi: 10.1056/NEJM198406073102304. [DOI] [PubMed] [Google Scholar]
- 5.Bramson J L, Graham F L, Gauldie J. Curr Opin Biotechnol. 1995;6:590–595. doi: 10.1016/0958-1669(95)80097-2. [DOI] [PubMed] [Google Scholar]
- 6.Graham F L, Prevec L. In: Vaccines: New Approaches to Immunological Problems. Ellis R W, editor. Boston: Butterworth–Heinemann; 1992. pp. 363–390. [Google Scholar]
- 7.Ye X, Robinson M B, Batshaw M L, Furth E E, Smith I, Wilson J M. J Biol Chem. 1996;271:3639–3646. doi: 10.1074/jbc.271.7.3639. [DOI] [PubMed] [Google Scholar]
- 8.Smith T A, Mehaffey M G, Kayda D B, Saunders J M, Yei S, Trapnell B C, McClelland A, Kaleko M. Nat Genet. 1993;5:397–402. doi: 10.1038/ng1293-397. [DOI] [PubMed] [Google Scholar]
- 9.Kay M A, Landen C N, Rothenberg S R, Taylor L A, Leland F, Wiehle S, Fang B, Bellinger D, Finegold M, Thompson A R, et al. Proc Natl Acad Sci USA. 1994;91:2353–2357. doi: 10.1073/pnas.91.6.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eisensmith R C, Woo S L. J Inherited Metab Dis. 1996;19:412–423. doi: 10.1007/BF01799102. [DOI] [PubMed] [Google Scholar]
- 11.Kobayashi K, Oka K, Forte T, Ishida B, Teng B, Ishimura-Oka K, Nakamuta M, Chan L. J Biol Chem. 1996;271:6852–6860. doi: 10.1074/jbc.271.12.6852. [DOI] [PubMed] [Google Scholar]
- 12.Kozarsky K F, McKinley D R, Austin L L, Raper S E, Stratford-Perricaudet L D, Wilson J M. J Biol Chem. 1994;269:13695–13702. [PubMed] [Google Scholar]
- 13.Yang Y, Jooss K U, Su Q, Ertl H C, Wilson J M. Gene Ther. 1996;3:137–144. [PubMed] [Google Scholar]
- 14.Yang Y, Su Q, Wilson J M. J Virol. 1996;70:7209–7212. doi: 10.1128/jvi.70.10.7209-7212.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y, Wilson J M. J Immunol. 1995;155:2564–2570. [PubMed] [Google Scholar]
- 16.Yang Y, Nunes F A, Berencsi K, Furth E E, Gonczol E, Wilson J M. Proc Natl Acad Sci USA. 1994;91:4407–4411. doi: 10.1073/pnas.91.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Ginkel F W, Liu C, Simecka J W, Dong J Y, Greenway T, Frizzell R A, Kiyono H, McGhee J R, Pascual D W. Hum Gene Ther. 1995;6:895–903. doi: 10.1089/hum.1995.6.7-895. [DOI] [PubMed] [Google Scholar]
- 18.Tripathy S K, Black H B, Goldwasser E, Leiden J M. Nat Med. 1996;2:545–550. doi: 10.1038/nm0596-545. [DOI] [PubMed] [Google Scholar]
- 19.Morral N, O’Neal W, Zhou H, Langston C, Beaudet A. Hum Gene Ther. 1997;8:1275–1286. doi: 10.1089/hum.1997.8.10-1275. [DOI] [PubMed] [Google Scholar]
- 20.Dai Y, Schwarz E M, Gu D, Zhang W, Sarvetnick N. Proc Natl Acad Sci USA. 1995;92:1401–1405. doi: 10.1073/pnas.92.5.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou H, O’Neal W, Morral N, Beaudet A L. J Virol. 1996;70:7030–7038. doi: 10.1128/jvi.70.10.7030-7038.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao G P, Yang Y, Wilson J M. J Virol. 1996;70:8934–8943. doi: 10.1128/jvi.70.12.8934-8943.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schiedner G, Morral N, Parks R J, Wu Y, Koopmans S C, Langston C, Graham F L, Beaudet A L, Kochanek S. Nat Genet. 1998;18:180–183. doi: 10.1038/ng0298-180. [DOI] [PubMed] [Google Scholar]
- 24.Fisher K J, Choi H, Burda J, Chen S, Wilson J M. Virology. 1996;217:11–22. doi: 10.1006/viro.1996.0088. [DOI] [PubMed] [Google Scholar]
- 25.Harper P A W, Healy P J, Dennis J A, O’Brien J J, Rayward D H. Aust Vet J. 1986;63:378–379. doi: 10.1111/j.1751-0813.1986.tb02907.x. [DOI] [PubMed] [Google Scholar]
- 26.Dennis J A, Healy P J, Beaudet A L, O’Brien W E. Proc Natl Acad Sci USA. 1989;86:7947–7951. doi: 10.1073/pnas.86.20.7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patejunas G, Lee B, Dennis J A, Healy P, Reeds P J, Yu J, Frazer M, Mull B, Warman A W, Beaudet A L, et al. J Inherited Metab Dis. 1998;21:138–150. doi: 10.1023/a:1005322010854. [DOI] [PubMed] [Google Scholar]
- 28.Batshaw M L, Yudkoff M, McLaughlin B A, Gorry E, Anegawa N J, Smith I A S, Hyman S L, Robinson M B. Gene Ther. 1995;2:743–749. [PubMed] [Google Scholar]
- 29.Graham F L, Prevec L. In: Methods in Molecular Biology. Murray E J, editor. Clifton, NJ: Humana; 1991. pp. 109–128. [DOI] [PubMed] [Google Scholar]
- 30.Niwa H, Yamamura K, Miyazaki J. Gene. 1991;108:193–200. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi K, Saheki T, Imamura Y, Noda T, Inoue I, Matuo S, Hagihara S, Nomiyama H, Jinno Y, Shimada K. Am J Hum Genet. 1986;38:667–680. [PMC free article] [PubMed] [Google Scholar]
- 32.Northrup H, Beaudet A L, O’Brien W E. Prenat Diagn. 1990;10:771–779. doi: 10.1002/pd.1970101203. [DOI] [PubMed] [Google Scholar]
- 33.Rabier D, Narcy C, Bardet J, Parvy P, Saudubray J M, Kamoun P. J Inherited Metab Dis. 1991;14:277–280. doi: 10.1007/BF01811681. [DOI] [PubMed] [Google Scholar]
- 34.Reeds P J, Burrin D G, Stoll B, Jahoor F, Henry J, Frazer M E. Am J Physiol. 1997;273:E408–E415. doi: 10.1152/ajpendo.1997.273.2.E408. [DOI] [PubMed] [Google Scholar]
- 35.Jan D, Poggi F, Jouvet P, Rabier D, Laurent J, Beringer A, Hubert P, Saudubray J M, Revillon Y. Transplant Proc. 1994;26:188. [PubMed] [Google Scholar]
- 36.O’Neal W K, Zhou H, Morral N, Aguilar-Cordova E, Pestaner J, Langston C, Mull B, Wang Y, Beaudet A L, Lee B. Hum Gene Ther. 1998;9:1587–1598. doi: 10.1089/hum.1998.9.11-1587. [DOI] [PubMed] [Google Scholar]