

Abstract
Intravascular pressure-induced vasoconstriction (the “myogenic response”) is intrinsic to smooth muscle cells, but mechanisms that underlie this response are unresolved. Here we investigated the physiological function of arterial smooth muscle cell caveolae in mediating the myogenic response. Since caveolin-1 (cav-1) ablation abolishes caveolae formation in arterial smooth muscle cells, myogenic mechanisms were compared in cerebral arteries from control (cav-1+/+) and cav-1-deficient (cav-1−/−) mice. At low intravascular pressure (10 mmHg), wall membrane potential, intracellular calcium concentration ([Ca2+]i), and myogenic tone were similar in cav-1+/+ and cav-1−/− arteries. In contrast, pressure elevations to between 30 and 70 mmHg induced a smaller depolarization, [Ca2+]i elevation, and myogenic response in cav-1−/− arteries. Depolarization induced by 60 mM K+ also produced an attenuated [Ca2+]i elevation and constriction in cav-1−/− arteries, whereas extracellular Ca2+ removal and diltiazem, an L-type Ca2+ channel blocker, similarly dilated cav-1+/+ and cav-1−/− arteries. Nω-nitro-l-arginine, an nitric oxide synthase inhibitor, did not restore myogenic tone in cav-1−/− arteries. Iberiotoxin, a selective Ca2+-activated K+ (KCa) channel blocker, induced a similar depolarization and constriction in pressurized cav-1+/+ and cav-1−/− arteries. Since pressurized cav-1−/− arteries are more hyperpolarized and this effect would reduce KCa current, these data suggest that cav-1 ablation leads to functional KCa channel activation, an effect that should contribute to the attenuated myogenic constriction. In summary, data indicate that cav-1 ablation reduces pressure-induced depolarization and depolarization-induced Ca2+ influx, and these effects combine to produce a diminished arterial wall [Ca2+]i elevation and constriction.
Keywords: calcium-activated potassium channels, membrane depolarization, arterial wall calcium
In small, resistance-sized arteries, an increase in intravascular pressure induces vasoconstriction (3, 10). This reaction, termed the “myogenic response,” occurs independent of endothelium, neurogenic, or hormonal factors, and thus, is intrinsic to smooth muscle cells (10). The myogenic response plays a crucial role in circulatory blood flow autoregulation and in providing baseline tone that can be altered by vasoconstrictors or vasodilators (10).
An intravascular pressure elevation induces an arterial wall membrane potential depolarization that activates smooth muscle cell voltage-dependent calcium (Ca2+) channels, leading to an elevation in Ca2+ influx and an increase in global intracellular Ca2+ concentration ([Ca2+]i) (10, 29). The [Ca2+]i elevation activates Ca2+/calmodulin-dependent myosin light chain kinase, resulting in phosphorylation of myosin light chain, myosin cross bridging with actin, and the sustained constriction (10, 29). In small arteries at high intravascular pressures, Rho kinase (ROK) and protein kinase C activation increase contractile apparatus Ca2+ sensitivity and contribute to myogenic constriction (10, 17, 18). Intravascular pressure also activates negative-feedback mechanisms that limit myogenic constriction, including those mediated by large conductance Ca2+-activated K+ (KCa) and voltage-dependent K+ (Kv) channels (4, 28). Thus the level of constriction induced by intravascular pressure is the equilibrium between positive- and negative-feedback mechanisms that can be activated through multiple signaling pathways. To integrate signaling pathways that regulate the myogenic response, smooth muscle cells are likely to structurally and spatially organize functional components. One cellular structure known to localize signaling proteins in the plasma membrane is a caveolae.
Caveolae are flask-shaped, 50- to 100-nm sized plasma membrane invaginations that are enriched in cholesterol and sphingolipids (for review, see Ref. 38). Caveolae are functionally important membrane microdomains because many cellular signaling molecules localize within these structures (38). Structural protein markers required for caveolae formation are the caveolins (38). Three isoforms of caveolin (cav-1, -2, and -3) have been identified (for reviews, see Refs. 33 and 38). Cav-1 and -2 are expressed in many different cell types, including smooth muscle cells, whereas cav-3 is expressed primarily in cardiac and skeletal muscle cells (33, 38).
Evidence indicates that caveolae regulate vascular contractility. Disruption of caveolae by cav-1 ablation or with dextrin, a cholesterol-depleting agent, inhibited agonist-induced arterial constriction (12, 13, 27, 41). Nitric oxide (NO)-dependent relaxation was augmented in cav-1-deficient (cav-1−/−) aortic rings, suggesting NO generation is upregulated in this tissue (12, 37). Similarly, phenylephrine-induced vasoconstriction was attenuated in murine cav-1−/− aortic rings, and this effect was reversed by NG-nitro-l-arginine methyl ester (l-NAME), a NO synthase (NOS) inhibitor (12, 37). Cerebral artery smooth muscle cells of cav-1−/− mice also exhibited higher Ca2+ spark and transient KCa current frequency, deficient coupling between voltage-dependent Ca2+ channels, and RyR channels that produce Ca2+ sparks, and an elevation in sarcolemmal KCa and Kv current density (7). These studies indicate that cav-1 ablation and caveolae abolishment alters vascular contractility, implicating these membrane microdomains as functionally important in the regulation of arterial tone. However, the physiological function of caveolae in mediating the myogenic response is unclear.
The goal of the present study was to investigate the functional involvement of caveolae in the cerebral artery myogenic response by using a murine model in which cav-1 is genetically ablated and caveolae are abolished (7). We tested the hypothesis that cav-1 ablation attenuates pressure-induced vasoconstriction. Data indicate that in the absence of cav-1, pressure induces a diminished membrane depolarization and depolarization-induced Ca2+ influx, resulting in a smaller arterial wall [Ca2+]i elevation and an attenuated vasoconstriction. Given that cav-1 abolishment elevates NO generation in endothelial cells and KCa channel activity in arterial smooth muscle cells (7, 30), we investigated the contribution of each of these potential modifications to the attenuated myogenic response. Data indicate that NOS inhibition does not restore myogenic tone in cav-1−/− arteries and suggest that KCa channel activation contributes to attenuated pressure-induced depolarization and constriction.
MATERIALS AND METHODS
Animals
Animal protocols used were reviewed and approved by the Animal Care and Use Committee of the University of Tennessee Health Science Center. Cav-1 knockout (cav-1−/−, strain: Cav1tm 1 Mls; genetic background: 129/Sv, C57BL/6J and SJL) or control (cav-1+/+, strain: C57BL/6J) mice (7, 37) of 4–9 wk age were purchased from Jackson Laboratories (Bar Harbor, ME) and used for these studies.
Tissue preparation
Mice were euthanized with pentobarbital sodium (130 mg/kg ip). The brain was removed and placed into oxygenated ice-cold (4°C) physiological saline solution (PSS) of the following composition (in mM): 112 NaCl, 4.8 KCl, 24 NaHCO3, 1.8 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, and 10 glucose, which was gassed with 21% O2-5% CO2-74% N2 to pH 7.4. Posterior cerebral, cerebellar, and middle cerebral arteries (∼150 μm in diameter) were dissected from the brain and cleaned of adventitial connective tissue.
Western blot analysis
Cerebral arteries collected from four mice were homogenized in Laemmli sample buffer containing 2.5% SDS, 10% glycerol, 0.01% bromphenol blue, and 5% β-mercaptoethanol in 100 mM Tris·HCl (pH 6.8). Homogenates were then centrifuged at 10,000 g for 5 min at 4°C. A 5-μl aliquot of supernatant was placed on a nitrocellulose membrane, and following amido black staining, protein concentration was measured by spectrophotometric quantification at 630 nm. Proteins (20 μg/lane) were separated by electrophoresis on 7.5% SDS-polyacrylamide gels and transferred onto polyvinyldifluoride membranes using a Mini Trans Blot Cell (Bio-Rad, Hercules, CA). Membranes were first washed in Tris-buffered solution (composition in g/l: 1.31 Tris and 9 NaCl; pH 7.4) supplemented with 0.1% Tween 20 and then blocked for 1 h in Tris-buffered solution that was supplemented with 5% nonfat dry milk. Membranes were incubated overnight at 4°C with rabbit polyclonal anti-KCa channel α-subunit antibody (Alomone, Jerusalem, Israel) or mouse monoclonal anti-cav-1 antibody (BD Transduction), both used at a dilution of 1:500. Blots were washed three times with 0.1% Tween 20 and then incubated for 1 h with respective horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies (Pierce Biotechnology). To determine the specificity of the KCa channel α-subunit antibody, a control blocking peptide was preadsorbed with the primary antibody in a peptide-to-antibody ratio of 3:1 (wt/wt). Protein expression was normalized by reprobing membranes with monoclonal antibodies raised against a highly conserved region of actin (1:5,000, Chemicon International), followed by incubation with horseradish peroxidase-conjugated anti-mouse IgG (1:10,000, Pierce Biotechnology I). Membranes were developed for 1 min using enhanced chemiluminescence (Amersham, Arlington Heights, IL). Band intensity was quantified by digital densitometry using Quantity One software version 4.4.1 (Bio-Rad).
Pressurized artery diameter measurements
An arterial segment 1–2 mm in length was cannulated at each end in a temperature-controlled perfusion chamber (Living Systems Instrumentation; Burlington, VT). The chamber was continuously perfused with PSS equilibrated with a mixture of 21% O2-5% CO2-74% N2 and maintained at 37°C. Arteries were observed with a charge-coupled device (CCD) camera attached to an inverted microscope (Nikon TE 200). Arterial diameter was measured by using the automatic edge-detection function of IonWizard software (Ionoptix; Milton, MA) and digitized at 1 Hz using a personal computer. Steady-state changes in intravascular pressure were achieved by elevating and lowering an attached reservoir and monitored using a pressure transducer. No intraluminal flow was present during the experiments. Tested compounds were applied via chamber perfusion. K+ (60 mM)-containing PSS was prepared by replacement of equimolar Na+ with K+. Posterior cerebral, cerebellar, and middle cerebral arteries developed similar levels of myogenic tone, and thus data with these arteries were pooled to generate cav-1+/+ and cav-1−/− data sets.
Measurement of membrane potential
Before membrane potential recordings were obtained, arteries were maintained at either 10 or 50 mmHg for 2 h to ensure steady-state myogenic tone had occurred. Membrane potential was measured by inserting glass microelectrodes filled with 3 M KCl (50–90 mΩ) into the adventitial side of pressurized arteries. Membrane potential was recorded using a WPI FD223 amplifier and digitized using pClamp 9.2 software (Axon Instruments) and a personal computer. Criteria for successful intracellular impalements were 1) a sharp negative change in potential upon insertion; 2) stable voltage for at least 1 min after entry; 3) a sharp positive voltage deflection upon exit from the recorded cell; and 4) a <10% change in tip resistance after the impalement.
Simultaneous arterial wall [Ca2+]i and diameter measurements
Before cannulation, arterial segments were incubated in PSS containing fura-2 AM (5 μM) and pluronic F-127 (0.05%) for 45 min at room temperature. After being washed with PSS, arteries were cannulated and allowed to de-esterify fura-2 AM for an additional 20 min before experimentation. Fura-2 was alternately excited at 340 or 380 nm using a PC-driven hyperswitch (Ionoptix). Arteries were also illuminated with 665-nm incident light to allow arterial wall imaging using a CCD camera (Ionoptix). A 585LP dichroic mirror was used to split emitted light to the photomultiplier tube and the CCD camera. Arterial diameter and fura-2 fluorescence were measured simultaneously. Background-corrected fura-2 ratios were collected every 1 s at 510 nm using the photomultiplier tube, and arterial wall diameter measurements were recorded every 0.2 s. Arterial wall [Ca2+]i was calculated using the following equation (20):
where R is the 340/380 nm ratio, Rmin and Rmax are the minimum and maximum ratios determined in Ca2+-free and saturating Ca2+ solutions, respectively, β is the ratio of Ca2+ free to Ca2+ replete of emissions at 380 nm excitation, and Kd is the dissociation constant for fura-2 [224 nM, (20)]. Rmin, Rmax, and β were determined by increasing Ca2+ permeability with ionomycin (10 μM) and perfusing arteries with 10 mM Ca2+ or Ca2+-free PSS (no added Ca2+ with 10 mM EGTA). Rmin, Rmax, and β were 0.58 ± 0.02, 1.31 ± 0.09, and 1.84 ± 0.09 for cav-1+/+ arteries (n = 6), and 0.59 ± 0.04, 1.22 ± 0.07, and 1.72 ± 0.05 for cav-1−/− arteries (n = 5), respectively.
Statistics
Graphpad Instat software (Graphpad Prism, San Diego, CA) was used for statistical analysis. Results are expressed as means ± SE. Statistical significance was calculated using Student's t-tests for paired or unpaired data, or one-way analysis of variance followed by Student-Newman-Keuls test for multiple comparisons. P < 0.05 was considered significant. The magnitude of myogenic tone was calculated using the following equation:
Chemicals
Unless otherwise stated, all chemicals were purchased from Sigma Chemical (St. Louis, MO). Fura-2 and pluronic F-127 were purchased from Molecular Probes (Eugene, OR).
RESULTS
Pressure-induced constriction is attenuated in cav-1−/− arteries
Western blot analysis was performed to measure cav-1 expression in cerebral arteries of cav-1+/+ and cav-1−/− mice. Data indicate that cav-1 is expressed in cav-1+/+ mouse cerebral arteries but is absent in cav-1−/− mouse arteries (Fig. 1A). These data are consistent with studies demonstrating that caveolae are present in cerebral artery smooth muscle cells of cav-1+/+ mice but are absent in the cells of cav-1−/− mice (7).
Fig. 1.
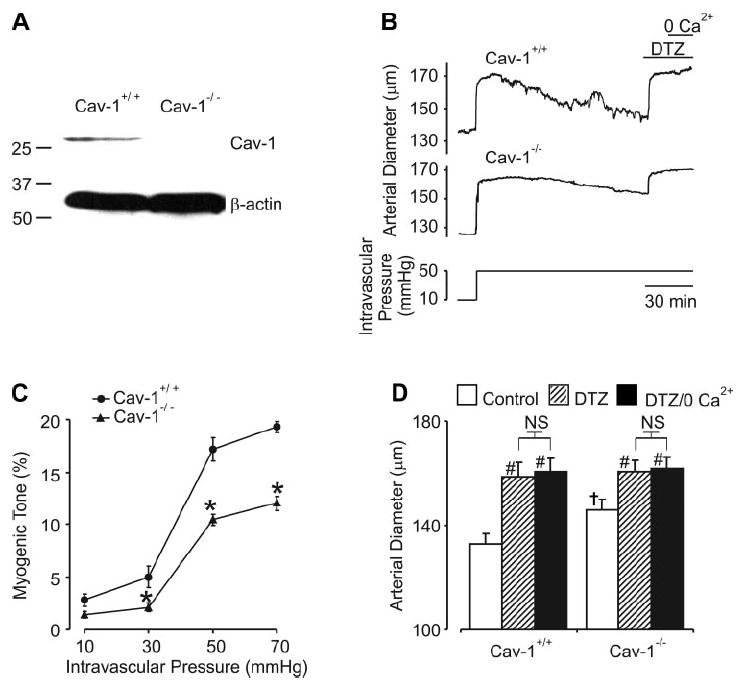
Myogenic constriction is attenuated in caveolin-1 (cav-1)−/− arteries. A: Western immunoblot illustrates the presence of cav-1 in cerebral arteries of control mice and the absence of cav-1 in cerebral arteries of cav-1−/− mice (data are representative of 4 experiments). In contrast, β-actin was detected in both cav-1+/+ and cav-1−/− arteries. B: representative traces illustrate that an elevation in intravascular pressure from 10 to 50 mmHg induces an attenuated myogenic constriction in a cav-1−/− artery, when compared with the response in a cav-1+/+ artery. C: relationship between intravascular pressure and myogenic tone in cav-1+/+ (n = 6) and cav-1−/− (n = 6) arteries. *P < 0.05 when compared with cav-1+/+ arteries at the same pressure. B and D: diltiazem (DTZ) significantly increased the diameter of both cav-1+/+ (n = 6) and cav-1−/− (n = 6) arteries. In the continued presence of DTZ, removal of extracellular Ca2+ did not cause any further increase in diameter. #P < 0.05 when compared with the respective control; †P < 0.05 when compared with cav-1+/+ at the same pressure. NS, nonsignificant difference.
Pressure-induced constriction was measured at steady-state intravascular pressures between 10 and 70 mmHg in cav-1+/+ and cav-1−/− cerebral arteries. At 10 mmHg, myogenic tone was similar in cav-1+/+ and cav-1−/− arteries (Fig. 1, B and C). However, at pressures between 30 and 70 mmHg, cav-1−/− arteries developed less myogenic tone (Fig. 1, B and C). For example, at 50 mmHg, mean steady-state tone in cav-1+/+ arteries was 17 ± 1% and 10 ± 0.5% in cav-1−/− arteries. Attenuated myogenic tone in cav-1−/− arteries was not due to differences in vessel size because passive diameter, as determined in a Ca2+-free solution, was similar to cav-1+/+ arteries (50 mmHg, cav-1+/+, 162 ± 4; cav-1−/−,175 ± 8 μm; P > 0.05).
To investigate mechanisms mediating myogenic constriction, diameter regulation by diltiazem, an L-type Ca2+ channel blocker, was studied. At 50 mmHg, diltiazem (50 μM) increased the mean diameter of both cav-1+/+ and cav-1−/− arteries (Fig. 1, B and D). In the continued presence of diltiazem, removal of extracellular Ca2+ did not cause any further dilation of either cav-1+/+ or cav-1−/− arteries (Fig. 1, B and D). In summary, these data indicate that pressure induces a smaller constriction in cav-1−/− arteries than in cav-1+/+ arteries. Data also indicate that pressure-induced constriction in both cav-1+/+ and cav-1−/− arteries occurs due to voltage-dependent Ca2+ channel activation.
Pressure induces a smaller depolarization in cav-1−/− arteries
Membrane potential was measured in cav-1+/+ and cav-1−/− arteries pressurized to either 10 or 50 mmHg. At 10 mmHg, mean membrane potential was similar in cav-1+/+ (approximately −59 mV) and cav-1−/− (approximately −62 mV) arteries (Fig. 2, A and B). In contrast, a pressure elevation to 50 mmHg depolarized cav-1+/+ arteries to approximately −31 mV, whereas cav-1−/− arteries depolarized to only approximately −44 mV (Fig. 2, A and B). These data indicate that pressure-induced membrane depolarization is attenuated in cav-1−/− arteries.
Fig. 2.
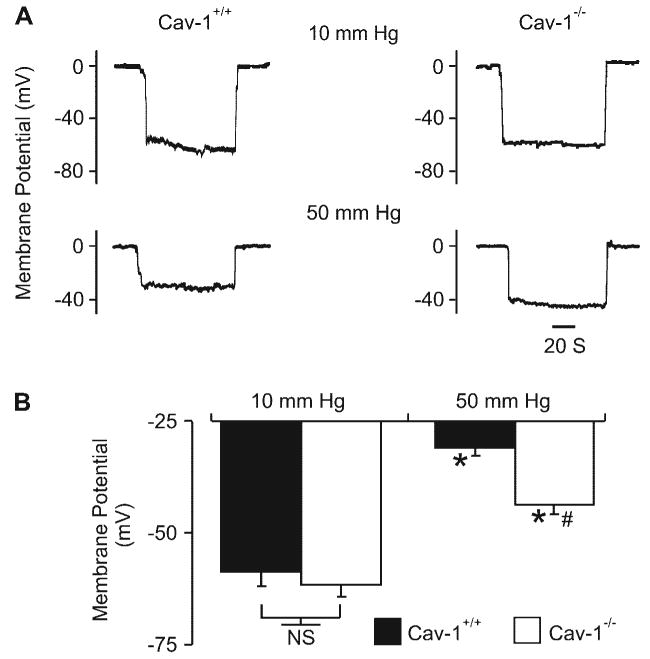
Pressure induces a smaller depolarization in cav-1−/− arteries. A: original membrane potential recordings in pressurized (10 and 50 mmHg) cav-1+/+ and cav-1−/− arteries. B: regulation of arterial wall membrane potential by intravascular pressure in cav-1+/+ (10 mmHg, n = 6, 50 mmHg, n = 14) and cav-1−/− (10 mmHg, n = 7, 50 mmHg, n = 14) arteries. *P < 0.05 when compared with membrane potential at 10 mmHg; #P < 0.05 when compared with membrane potential of cav-1+/+ arteries at 50 mmHg.
Pressure induces a smaller arterial wall [Ca2+]i elevation in cav-1−/− arteries
We sought to test the hypothesis that in cav-1−/− arteries, an attenuated pressure-induced membrane depolarization results in a diminished [Ca2+]i elevation and, thus, a reduced myogenic constriction. Arterial wall [Ca2+]i and diameter were simultaneously measured in pressurized cav-1+/+ and cav-1−/− arteries. As shown in Fig. 3, at 10 mmHg mean arterial wall, [Ca2+]i was similar in cav-1+/+ (∼84 nM) and cav-1−/− (∼80 nM) arteries. Increasing intravascular pressure from 10 to 70 mmHg caused an immediate increase in arterial wall [Ca2+]i in both cav-1+/+ and cav-1−/− arteries. However, mean arterial wall [Ca2+]i was higher in cav-1+/+ (∼296 nM) than in cav-1−/− (∼206 nM) arteries. Extracellular Ca2+ removal similarly reduced both [Ca2+]i and myogenic constriction in pressurized cav-1+/+ and cav-1−/− arteries (Fig. 3C). These data indicate that pressure induces a diminished arterial wall [Ca2+]i elevation and constriction in cav-1−/− arteries.
Fig. 3.
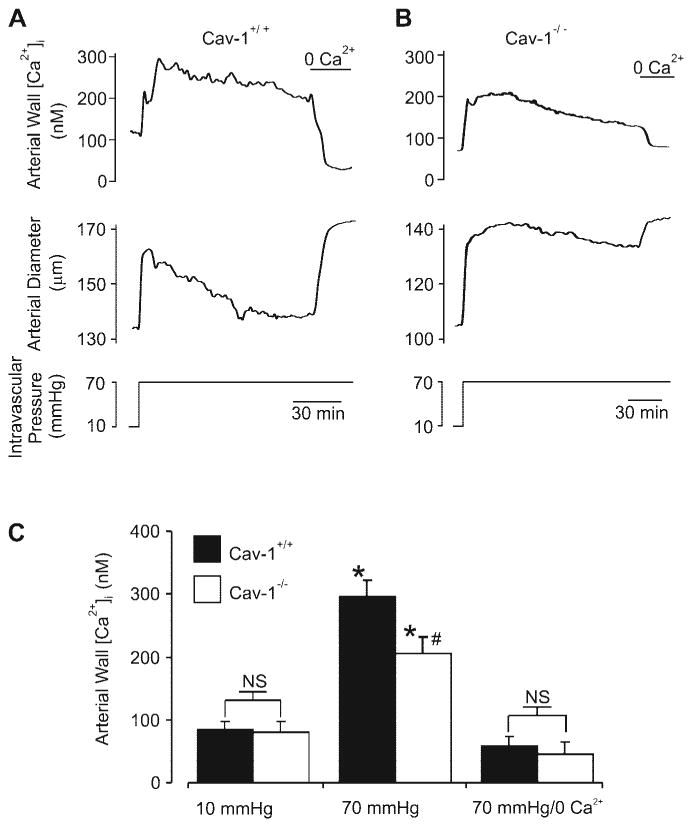
Pressure induces a smaller arterial wall intracellular Ca2+ concentration ([Ca2+]i) elevation in cav-1−/− arteries. A and B: simultaneous measurement of arterial wall [Ca2+]i and diameter in cav-1+/+ and cav-1−/− arteries. C: regulation by intravascular pressure of mean arterial wall [Ca2+]i in cav-1+/+ (n = 7) and cav-1−/− (n = 5) cerebral arteries. *P < 0.05 when compared with arterial wall [Ca2+]i at 10 mmHg; #P < 0.05 when compared with arterial wall [Ca2+]i in cav-1+/+ arteries at 70 mmHg.
Membrane depolarization induces an attenuated arterial wall [Ca2+]i elevation and constriction in cav-1−/− arteries
To investigate mechanisms mediating attenuated pressure-induced [Ca2+]i elevations and constrictions in cav-1−/− arteries, depolarization was induced using a 60 mM K+-containing PSS solution. In cav-1+/+ arteries pressurized to 70 mmHg, 60 mM K+ elevated mean wall [Ca2+]i to ∼434 nM and constricted to ∼65% of passive diameter. In contrast, in cav-1−/− arteries, 60 mM K+ elevated mean arterial wall [Ca2+]i to only ∼242 nM and constricted to only ∼80% of passive diameter (Fig. 4, A and B). These data suggest that cav-1 ablation attenuates depolarization-induced arterial wall [Ca2+]i elevations and constrictions.
Fig. 4.
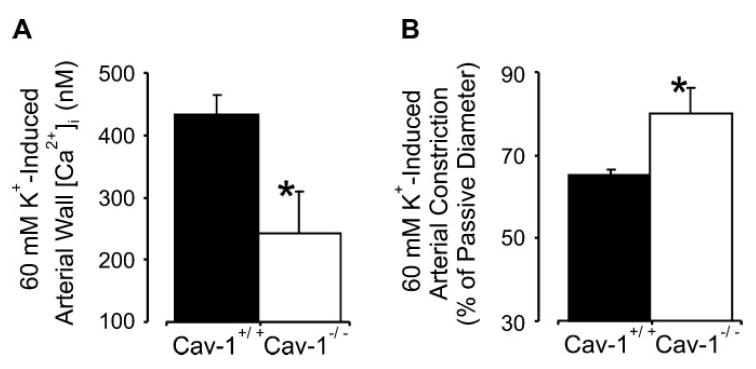
Membrane depolarization induces an attenuated [Ca2+]i elevation and constriction in cav-1−/− arteries. Mean arterial wall [Ca2+]i (A) and constriction (B) induced by 60 mM K+ in pressurized (70 mmHg) cav-1+/+ (n = 8) and cav-1−/− (n = 6) cerebral arteries. *P < 0.05 when compared with cav-1+/+ arteries.
NOS inhibition does not restore myogenic tone in cav-1−/− arteries
Cav-1 is an endothelial NOS (eNOS) inhibitor (30). Cav-1 ablation leads to eNOS activation and an increase in endothelial cell NO production (30). Because eNOS-derived NO is a vasodilator (40), we sought to investigate whether NOS activation contributes to the attenuated myogenic response in cav-1−/− cerebral arteries. Bradykinin (10 μM), an endothelium-dependent vasodilator (39), induced similar magnitude vasodilations in cav-1+/+ and cav-1−/− arteries, indicating the presence of functional endothelium (Fig. 5A; P > 0.05). However, although Nω-nitro-l-arginine (l-NNA, 1 mM), an NOS inhibitor, constricted both cav-1+/+ and cav-1−/− arteries, l-NNA did not restore the myogenic tone difference (Fig. 5B; P < 0.05). These data indicate that NOS activation and NO generation do not contribute significantly to the attenuated myogenic response in cav-1−/− arteries.
Fig. 5.
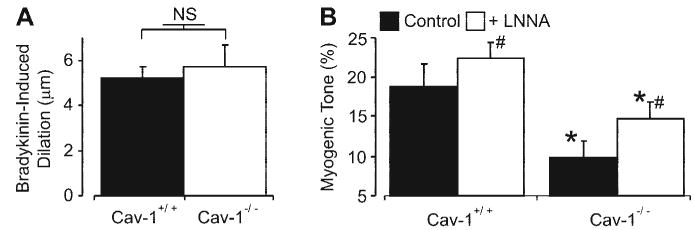
Bradykinin-induced dilations are similar in cav-1+/+ and cav-1−/− arteries, and Nω-nitro-l-arginine (l-NNA) does not restore myogenic tone in cav-1−/− arteries. A: mean bradykinin (10 μM)-induced dilations in cav-1+/+ (n = 6) and cav-1−/− (n = 6) arteries. B: mean myogenic tone at 50 mmHg in cav-1+/+ (n = 5) and cav-1−/− (n = 5) cerebral arteries in the absence and presence of l-NNA (1 mM). #P < 0.05 when compared with myogenic tone in the absence of l-NNA; *P < 0.05 when compared with cav-1+/+ arteries under the same experimental condition.
Iberiotoxin similarly depolarizes and constricts cav-1+/+ and cav-1−/− arteries
Cav-1 ablation elevates Ca2+ spark and transient KCa current frequency and sarcolemmal KCa current density in cerebral artery smooth muscle cells (7). Since KCa channel activation opposes myogenic constriction (22, 31), we investigated whether a KCa current elevation contributes to the attenuated myogenic constriction in cav-1−/− arteries. Membrane potential and diameter regulation by iberiotoxin, a selective KCa channel blocker, was studied. Iberiotoxin (100 nM) similarly depolarized pressurized (50 mmHg) cav-1+/+ and cav-1−/− arteries by ∼5.7 and 5.1 mV, respectively (Fig. 6A, P > 0.05). Iberiotoxin also similarly constricted cav-1+/+ and cav-1−/− arteries by ∼4 and 4.2 μm, respectively (Fig. 6B, P > 0.05). Given that the hyperpolarized membrane potential of cav-1−/− arteries should reduce KCa channel activity, these data suggest that KCa channel activity is elevated in cav-1−/− arteries and contributes to the attenuated pressure-induced vasoconstriction (4).
Fig. 6.
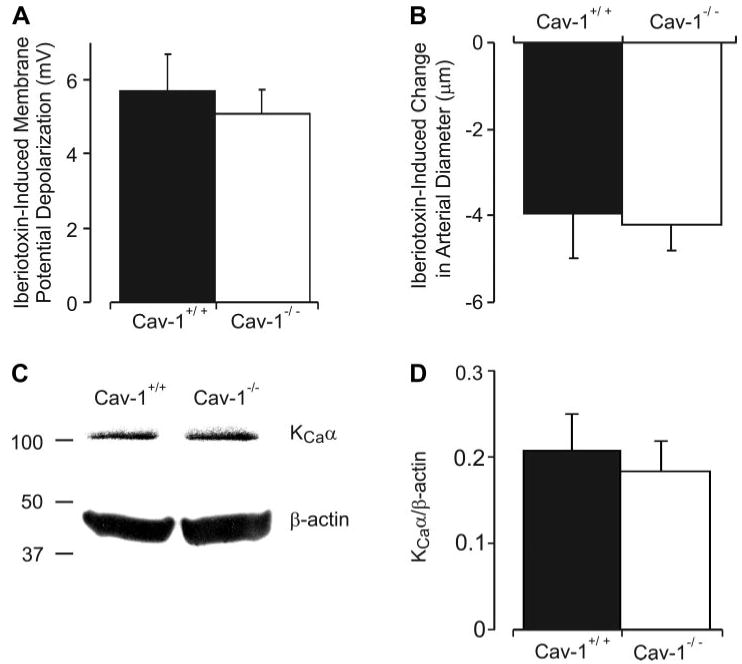
Iberiotoxin induces a similar depolarization and constriction in cav-1+/+ and cav-1−/− arteries, but total arterial KCa channel protein is similar. A: membrane depolarization induced by iberiotoxin (100 nM) is similar in pressurized (50 mmHg) cav-1+/+ (n = 6) and cav-1−/− (n = 6) arteries. B: iberiotoxin (100 nM) induces a similar constriction in cav-1+/+ (n = 6) and cav-1−/− (n = 6) arteries pressurized to 50 mmHg. C: representative Western immunoblots for KCa channel α-subunit and β-actin protein in cav-1+/+ and cav-1−/− cerebral artery lysates. D: mean data illustrating digital densitometric analysis of bands obtained by Western immuno-blotting for KCa channel α-subunit normalized to β-actin (n = 4 separate experiments, with each experiment performed using arteries pooled from 4 mice, giving a total of 16 cav-1+/+ and 16 cav-1−/− mice used to obtain data).
Cav-1 ablation does not alter total KCa channel protein in cerebral arteries
Sarcolemmal KCa channel density is higher in cerebral artery smooth muscle cells of cav-1−/− mice than that in cav-1+/+ mice (7). To investigate whether the KCa current density elevation occurs due to an increase in KCa channel expression, quantitative Western blot analysis of cerebral artery lysates was performed. Total KCa channel protein, when normalized to β-actin, was similar in cav-1+/+ and cav-1−/− arteries (Fig. 6, C and D; P > 0.05). These data indicate that cav-1 ablation does not alter total KCa channel protein in cerebral arteries.
DISCUSSION
Novel findings are presented in this study demonstrating that cav-1 ablation attenuates myogenic constriction over a wide range of intravascular pressures in cerebral arteries. We show that the suppressed myogenic constriction in cav-1−/− arteries occurs due to a reduced pressure-induced depolarization and depolarization-induced [Ca2+]i elevation, both of which attenuate the subsequent arterial wall [Ca2+]i elevation. We also show that NOS inhibition does not restore myogenic constriction in cav-1−/− arteries. Similarly, although KCa current inhibition did not restore myogenic tone, data indicate that this negative feedback pathway is upregulated. These findings indicate that cav-1 expression is necessary for pressure to induce a full myogenic vasoconstriction and suggest that KCa channel activation contributes to the attenuated myogenic response.
Cav-1 expression is necessary for caveolae formation in cerebral artery smooth muscle cells (7). Previous studies have demonstrated that cav-1 ablation and chemical abolishment of caveolae via cholesterol depletion modifies agonist-induced vascular contractions (12, 13, 27, 37, 41). Cav-1 also regulates flow-induced mechanotransduction in carotid arteries (47). However, regulation of the myogenic response by cav-1 has not been studied. In the present study, pressure induced a smaller depolarization in cav-1−/− arteries than in cav-1+/+ arteries. Conceivably, one or more components of the mechanism that senses changes in intravascular pressure, the pathways that transduce the signal, and the ion channels that are activated may undergo delocalization or dysregulation in response to cav-1 ablation, resulting in an attenuated pressure-induced depolarization. For example, cav-1 regulates signaling mediated by integrins, a family of transmembrane glycoproteins that act as arterial pressure sensors (11, 44). Phospholipase C, an enzyme that may be localized to caveolae in cardiac myocytes and immortalized cells, has also been proposed to contribute to the cerebral artery myogenic response (16, 26, 34). Studies have also implicated transient receptor potential (45), chloride (32), and degenerin/epithelial Na+ (14) channels in myogenic constriction. Transient receptor potential and chloride channels may also be associated with caveolae (1, 42). Thus one or more cav-1- or caveolae-dependent pathways that contribute to pressure-induced constriction may be dysfunctional in the absence of cav-1.
Cav-1 abolishment may also upregulate negative-feedback pathways that limit pressure-induced depolarization, leading to a diminished vasoconstriction. Indeed, in cav-1−/− cerebral artery smooth muscle cells, Ca2+ spark and transient KCa current frequency are higher and KCa and KV current density are elevated (7). Notably, Ca2+ spark and transient KCa current activation in cav-1−/− arterial smooth muscle cells does not occur due to NOS activation (7). Data presented here show that cav-1−/− arteries have a more negative membrane potential, which should reduce KCa channel activity and K+ driving force (i.e., single channel conductance) and, therefore, decrease the hyperpolarizing influence of KCa channels on membrane potential (4, 22, 25). However, iberiotoxin similarly depolarized and constricted cav-1+/+ and cav-1−/− arteries. Thus, even though the membrane potential of pressurized cav-1−/− arteries is more negative, the hyperpolarizing influence of KCa channels on membrane potential is similar to that in cav-1+/+ arteries. These data suggest that KCa channel activity is higher in cav-1−/− than in cav-1+/+ arteries.
In cerebral artery smooth muscle cells, Ca2+ sparks control KCa channel activity (24, 31). Modifications in KCa channel Ca2+ sensitivity or density alter the functional effectiveness of Ca2+ spark coupling (5, 23). However, in cav-1+/+ and cav-1−/− arterial smooth muscle cells, KCa channel apparent Ca2+ sensitivity is similar (7). Similarly, the KCa channel density elevation in cav-1−/− cells does not elevate the amplitude of Ca2+ spark-induced transient KCa currents but instead maintains KCa channel to Ca2+ spark coupling similar to that in cav-1+/+ cells (7). Thus alterations in KCa channel Ca2+ sensitivity and density cannot explain the increase in KCa channel activity in cav-1−/− cells. Rather, the KCa current increase in cav-1−/− arteries appears to occur due to an elevation in smooth muscle cell Ca2+ spark frequency (7). In cav-1−/− cells, Ca2+ spark frequency is independent of voltage-dependent Ca2+ channel activity and thus would be membrane potential independent, in contrast to the membrane potential dependence of transient KCa current frequency in cav-1+/+ cells (7, 22, 25). Thus the more negative membrane potential of cav-1−/− arteries would produce a slight reduction in transient KCa current amplitude when compared with cav-1+/+ cells but should not alter transient KCa current frequency, which is twofold higher than in cav-1+/+ cells (7, 22). Thus the higher smooth muscle cell Ca2+ spark frequency in cav-1−/− arteries appears to underlie the upregulated KCa current. However, although KCa channel activity appears to be higher in cav-1−/− arteries, iberiotoxin did not rectify the membrane potential or myogenic tone difference when compared with cav-1+/+ arteries. Thus additional mechanisms also contribute to the attenuated pressure-induced depolarization and constriction in cav-1−/− cerebral arteries.
Although sarcolemmal KCa channel density was elevated in cav-1−/− arterial smooth muscle cells, total KCa channel protein was similar in cav-1+/+ and cav-1−/− arteries as measured using Western blot analysis (7). Several factors may explain these findings, including that increased plasma membrane insertion of KCa channels in smooth muscle cells of cav-1−/− arteries may lead to a consequent reduction in protein stored in intracellular compartments such as the golgi apparatus, leading to no change in total KCa channel protein. In addition, Western blot analysis of whole arteries, which would measure KCa channel protein expressed in all cells of the wall, may not be able to detect the ∼ 1.5-fold increase in sarcolemmal KCa channel protein in cav-1−/− arterial smooth muscle cells (7). Finally, cav-1 ablation may reduce KCa channel expression in arterial wall cell types other than smooth muscle cells, e.g., perivascular neurons, leading to no net change in total wall KCa channel protein. Further studies will be required to investigate why cav-1 ablation elevates plasma membrane KCa channel density in arterial smooth muscle cells but does not alter total arterial wall KCa channel protein.
In cerebral arteries, pressure-induced depolarization activates smooth muscle cell voltage-dependent Ca2+ channels (29). In mesenteric arteries and skeletal muscle arterioles, intravascular pressure also stimulates Ca2+ influx via store-operated Ca2+ channels or capacitative Ca2+ entry, respectively (36, 48). Diltiazem profoundly reduced arterial wall [Ca2+]i and fully dilated cav-1+/+ and cav-1−/− arteries, and subsequent removal of extracellular Ca2+ did not cause any further dilation. These data indicate that voltage-dependent Ca2+ channels are the principal Ca2+ entry pathway that stimulates constriction in both cav-1+/+ and cav-1−/− arteries. However, intravascular pressure and a 60 mM K+-induced membrane depolarization both induced a smaller [Ca2+]i elevation in cav-1−/− arteries. Thus cav-1 ablation attenuates pressure-induced [Ca2+]i elevations through two mechanisms, by 1) suppressing the level of membrane depolarization, leading to a smaller increase in voltage-dependent Ca2+ channel open probability; and 2) lowering the gain of the depolarization-induced arterial wall [Ca2+]i elevation. KCl-induced contraction is also attenuated in the cav-1−/− bladder (46). In contrast, caveolae disruption with dextrin did not alter KCl-induced contraction in the rat tail artery, and a synthetic cav-1 scaffolding domain peptide did not alter KCl-induced aortic contraction (13, 27). These differing effects on KCl-induced contractions may relate to the experimental methods used to alter cav-1 expression, cav-1 function, or caveolae formation, the smooth muscle cell type studied, or the experimental techniques used to measure contractility.
Mechanisms by which cav-1 ablation attenuates depolarization-induced [Ca2+]i elevations may be multifactorial. Steady-state arterial wall [Ca2+]i represents an equilibrium between Ca2+ influx and extrusion, and either or both of these mechanisms may be modified in cav-1−/− arterial smooth muscle cells, leading to a net [Ca2+]i reduction. Voltage-dependent Ca2+ channels may be localized to caveolae in airway smooth muscle cells (9). Caveolae may also store and transport Ca2+ during cellular signaling (2). Voltage-dependent Ca2+ current density and current-voltage relationships are similar in cav-1+/+ and cav-1−/− cerebral artery smooth muscle cells (7). However, cav-1−/− cells have a smaller surface area than cav-1+/+ cells, even though cellular dimensions are similar (7). Thus the smaller total number of voltage-dependent Ca2+ channels present in the plasma membrane of cav-1−/− arterial smooth muscle cells may explain attenuated depolarization-induced [Ca2+]i elevations in cav-1−/− arterial smooth muscle cells.
Signaling components that mediate Ca2+ sensitization may also be modified in cav-1−/− smooth muscle cells, leading to an attenuated Ca2+-induced constriction. ROK translocation to the caveolae of rat femoral artery smooth muscle cells may mediate the Ca2+-sensitization mechanism (43). Similarly, a synthetic cav-1 scaffolding domain peptide inhibited PKC-dependent contractions and kinase activity in ferret aortic smooth muscle (27). Thus, in addition to attenuating pressure- and depolarization-induced [Ca2+]i elevations, cav-1 ablation may also reduce Ca2+ sensitization of constriction.
Association with the cav-1 scaffolding domain inhibits eNOS and reduces eNOS-dependent NO production in cultured bovine aortic endothelial cells (6). Consistent with these observations, NO-dependent relaxation was enhanced in cav-1−/− aortic rings (12, 37). Here NOS inhibition failed to restore myogenic tone in cav-1−/− arteries. Therefore, in contrast to inhibition of agonist-induced force generation (12, 37), NOS activation and an increase in wall NO do not appear to significantly contribute to the attenuated myogenic constriction in cav-1−/− cerebral arteries. Bradykinin, an endothelium-dependent vasodilator, dilated pressurized cav-1+/+ and cav-1−/− arteries, indicating the presence of functional endothelium (39). Indeed, bradykinin induced similar magnitude dilations in cav-1+/+ and cav-1−/− pressurized arteries, which is unexpected given the different resting conditions in these tissues. Bradykinin induces vasodilation through multiple mechanisms, including the generation of NO, endothelium-derived hyperpolarizing factor, prostacyclin, and reactive oxygen species (15). Thus further experiments will be required to study the involvement of cav-1 in endothelium-mediated vasodilation in pressurized cerebral arteries.
Caveolins regulate physiological functions and have been implicated in the pathogenesis of several diseases, including muscular dystrophy, Alzheimer's disease, and breast cancer (21). With the use of scanning electron microscopy, abnormal caveolae aggregates were found in the myocardium of spontaneously hypertensive rats (19). Cav-1−/− mice exhibited ventricular and myocyte hypertrophy, pulmonary hypertension, diminished left ventricular systolic function, and a reduced tolerance to exercise (8, 12, 35, 37, 49). An increase in endothelial cav-1 in diseased blood vessels has also been described (50). Our study supports accumulating evidence that caveolins regulate cardiovascular physiology, and an alteration in caveolin expression may contribute to vascular pathologies.
In summary, the present findings support a functional role for cav-1 in the cerebral artery myogenic response. In cerebral arteries, cav-1 ablation reduces both the pressure-induced membrane depolarization and depolarization-induced [Ca2+]i elevation, resulting in an attenuated arterial wall [Ca2+]i increase and a diminished myogenic constriction. Data also suggest that KCa channel activation should contribute to the attenuated myogenic response in cav-1−/− arteries.
Acknowledgments
GRANTS This study was supported by National Heart, Lung, and Blood Institute Grants HL-077678 and HL-67061 to J. H. Jaggar. A. Adebiyi is a recipient of a Postdoctoral Fellowship from the Southeast Affiliate of the American Heart Association.
References
- 1.Ambudkar IS, Brazer SC, Liu X, Lockwich T, Singh B. Plasma membrane localization of TRPC channels: role of caveolar lipid rafts. Novartis Found Symp. 2004;258:63–70. [PubMed] [Google Scholar]
- 2.Anderson RG. Caveolae: where incoming and outgoing messengers meet. Proc Natl Acad Sci USA. 1993;90:10909–10913. doi: 10.1073/pnas.90.23.10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bayliss WM. On the local reactions of the arterial wall to changes in internal pressure. J Physiol. 1902;28:220–231. doi: 10.1113/jphysiol.1902.sp000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–535. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- 5.Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the β1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–876. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- 6.Bucci M, Gratton JP, Rudic RD, Acevedo L, Roviezzo F, Cirino G, Sessa WC. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat Med. 2000;6:1362–1367. doi: 10.1038/82176. [DOI] [PubMed] [Google Scholar]
- 7.Cheng X, Jaggar JH. Genetic ablation of caveolin-1 modifies Ca2+ spark coupling in murine arterial smooth muscle cells. Am J Physiol Heart Circ Physiol. 2006;290:H2309–H2319. doi: 10.1152/ajpheart.01226.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen AW, Park DS, Woodman SE, Williams TM, Chandra M, Shirani J, Pereira de SA, Kitsis RN, Russell RG, Weiss LM, Tang B, Jelicks LA, Factor SM, Shtutin V, Tanowitz HB, Lisanti MP. Caveolin-1 null mice develop cardiac hypertrophy with hyperactivation of p42/44 MAP kinase in cardiac fibroblasts. Am J Physiol Cell Physiol. 2003;284:C457–C474. doi: 10.1152/ajpcell.00380.2002. [DOI] [PubMed] [Google Scholar]
- 9.Darby PJ, Kwan CY, Daniel EE. Caveolae from canine airway smooth muscle contain the necessary components for a role in Ca2+ handling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1226–L1235. doi: 10.1152/ajplung.2000.279.6.L1226. [DOI] [PubMed] [Google Scholar]
- 10.Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- 11.Davis MJ, Wu X, Nurkiewicz TR, Kawasaki J, Davis GE, Hill MA, Meininger GA. Integrins and mechanotransduction of the vascular myogenic response. Am J Physiol Heart Circ Physiol. 2001;280:H1427–H1433. doi: 10.1152/ajpheart.2001.280.4.H1427. [DOI] [PubMed] [Google Scholar]
- 12.Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- 13.Dreja K, Voldstedlund M, Vinten J, Tranum-Jensen J, Hellstrand P, Sward K. Cholesterol depletion disrupts caveolae and differentially impairs agonist-induced arterial contraction. Arterioscler Thromb Vasc Biol. 2002;22:1267–1272. doi: 10.1161/01.atv.0000023438.32585.a1. [DOI] [PubMed] [Google Scholar]
- 14.Drummond HA, Gebremedhin D, Harder DR. Degenerin/epithelial Na+ channel proteins: components of a vascular mechanosensor. Hypertension. 2004;44:643–648. doi: 10.1161/01.HYP.0000144465.56360.ad. [DOI] [PubMed] [Google Scholar]
- 15.Faraci FM, Heistad DD. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol Rev. 1998;78:53–97. doi: 10.1152/physrev.1998.78.1.53. [DOI] [PubMed] [Google Scholar]
- 16.Fujita T, Toya Y, Iwatsubo K, Onda T, Kimura K, Umemura S, Ishikawa Y. Accumulation of molecules involved in alpha1-adrenergic signal within caveolae: caveolin expression and the development of cardiac hypertrophy. Cardiovasc Res. 2001;51:709–716. doi: 10.1016/s0008-6363(01)00348-0. [DOI] [PubMed] [Google Scholar]
- 17.Gokina NI, Knot HJ, Nelson MT, Osol G. Increased Ca2+ sensitivity as a key mechanism of PKC-induced constriction in pressurized cerebral arteries. Am J Physiol Heart Circ Physiol. 1999;277:H1178–H1188. doi: 10.1152/ajpheart.1999.277.3.H1178. [DOI] [PubMed] [Google Scholar]
- 18.Gokina NI, Park KM, Elroy-Yaggy K, Osol G. Effects of Rho kinase inhibition on cerebral artery myogenic tone and reactivity. J Appl Physiol. 2005;98:1940–1948. doi: 10.1152/japplphysiol.01104.2004. [DOI] [PubMed] [Google Scholar]
- 19.Goto Y, Yoshikane H, Honda M, Morioka S, Yamori Y, Moriyama K. Three-dimensional observation on sarcoplasmic reticulum and caveolae in myocardium of spontaneously hypertensive rats. J Submicrosc Cytol Pathol. 1990;22:535–542. [PubMed] [Google Scholar]
- 20.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 21.Hnasko R, Lisanti MP. The biology of caveolae: lessons from caveolin knockout mice and implications for human disease. Mol Interv. 2003;3:445–464. doi: 10.1124/mi.3.8.445. [DOI] [PubMed] [Google Scholar]
- 22.Jaggar JH. Intravascular pressure regulates local and global Ca2+ signaling in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol. 2001;281:C439–C448. doi: 10.1152/ajpcell.2001.281.2.C439. [DOI] [PubMed] [Google Scholar]
- 23.Jaggar JH, Leffler CW, Cheranov SY, Tcheranova D, Shuyu E, Cheng X. Carbon monoxide dilates cerebral arterioles by enhancing the coupling of Ca2+ sparks to Ca2+-activated K+ channels. Circ Res. 2002;91:610–617. doi: 10.1161/01.res.0000036900.76780.95. [DOI] [PubMed] [Google Scholar]
- 24.Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol Cell Physiol. 2000;278:C235–C256. doi: 10.1152/ajpcell.2000.278.2.C235. [DOI] [PubMed] [Google Scholar]
- 25.Jaggar JH, Stevenson AS, Nelson MT. Voltage dependence of Ca2+ sparks in intact cerebral arteries. Am J Physiol Cell Physiol. 1998;274:C1755–C1761. doi: 10.1152/ajpcell.1998.274.6.C1755. [DOI] [PubMed] [Google Scholar]
- 26.Jang IH, Kim JH, Lee BD, Bae SS, Park MH, Suh PG, Ryu SH. Localization of phospholipase C-gamma1 signaling in caveolae: importance in EGF-induced phosphoinositide hydrolysis but not in tyrosine phosphorylation. FEBS Lett. 2001;491:4–8. doi: 10.1016/s0014-5793(01)02165-2. [DOI] [PubMed] [Google Scholar]
- 27.Je HD, Gallant C, Leavis PC, Morgan KG. Caveolin-1 regulates contractility in differentiated vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2004;286:H91–H98. doi: 10.1152/ajpheart.00472.2003. [DOI] [PubMed] [Google Scholar]
- 28.Knot HJ, Nelson MT. Regulation of membrane potential and diameter by voltage-dependent K+ channels in rabbit myogenic cerebral arteries. Am J Physiol Heart Circ Physiol. 1995;269:H348–H355. doi: 10.1152/ajpheart.1995.269.1.H348. [DOI] [PubMed] [Google Scholar]
- 29.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508:199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minshall RD, Sessa WC, Stan RV, Anderson RG, Malik AB. Caveolin regulation of endothelial function. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1179–L1183. doi: 10.1152/ajplung.00242.2003. [DOI] [PubMed] [Google Scholar]
- 31.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 32.Nelson MT, Conway MA, Knot HJ, Brayden JE. Chloride channel blockers inhibit myogenic tone in rat cerebral arteries. J Physiol. 1997;502:259–264. doi: 10.1111/j.1469-7793.1997.259bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J Biol Chem. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- 34.Osol G, Laher I, Kelley M. Myogenic tone is coupled to phospholipase C and G protein activation in small cerebral arteries. Am J Physiol Heart Circ Physiol. 1993;265:H415–H420. doi: 10.1152/ajpheart.1993.265.1.H415. [DOI] [PubMed] [Google Scholar]
- 35.Park DS, Woodman SE, Schubert W, Cohen AW, Frank PG, Chandra M, Shirani J, Razani B, Tang B, Jelicks LA, Factor SM, Weiss LM, Tanowitz HB, Lisanti MP. Caveolin-1/3 double-knockout mice are viable, but lack both muscle and non-muscle caveolae, and develop a severe cardiomyopathic phenotype. Am J Pathol. 2002;160:2207–2217. doi: 10.1016/S0002-9440(10)61168-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Potocnik SJ, Hill MA. Pharmacological evidence for capacitative Ca2+ entry in cannulated and pressurized skeletal muscle arterioles. Br J Pharmacol. 2001;134:247–256. doi: 10.1038/sj.bjp.0704270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di VD, Hou H, Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- 38.Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002;54:431–467. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 39.Rosenblum WI, Nelson GH, Povlishock JT. Laser-induced endothelial damage inhibits endothelium-dependent relaxation in the cerebral microcirculation of the mouse. Circ Res. 1987;60:169–176. doi: 10.1161/01.res.60.2.169. [DOI] [PubMed] [Google Scholar]
- 40.Schulz R, Triggle CR. Role of NO in vascular smooth muscle and cardiac muscle function. Trends Pharmacol Sci. 1994;15:255–259. doi: 10.1016/0165-6147(94)90321-2. [DOI] [PubMed] [Google Scholar]
- 41.Shaw L, Sweeney MA, O'Neill SC, Jones CJ, Austin C, Taggart MJ. Caveolae and sarcoplasmic reticular coupling in smooth muscle cells of pressurised arteries: the relevance for Ca2+ oscillations and tone. Cardiovasc Res. 2006;69:825–835. doi: 10.1016/j.cardiores.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 42.Suginta W, Karoulias N, Aitken A, Ashley RH. Chloride intracellular channel protein CLIC4 (p64H1) binds directly to brain dynamin I in a complex containing actin, tubulin and 14-3-3 isoforms. Biochem J. 2001;359:55–64. doi: 10.1042/0264-6021:3590055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Urban NH, Berg KM, Ratz PH. K+ depolarization induces RhoA kinase translocation to caveolae and Ca2+ sensitization of arterial muscle. Am J Physiol Cell Physiol. 2003;285:C1377–C1385. doi: 10.1152/ajpcell.00501.2002. [DOI] [PubMed] [Google Scholar]
- 44.Wei Y, Yang X, Liu Q, Wilkins JA, Chapman HA. A role for caveolin and the urokinase receptor in integrin-mediated adhesion and signaling. J Cell Biol. 1999;144:1285–1294. doi: 10.1083/jcb.144.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Welsh DG, Morielli AD, Nelson MT, Brayden JE. Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ Res. 2002;90:248–250. doi: 10.1161/hh0302.105662. [DOI] [PubMed] [Google Scholar]
- 46.Woodman SE, Cheung MW, Tarr M, North AC, Schubert W, Lagaud G, Marks CB, Russell RG, Hassan GS, Factor SM, Christ GJ, Lisanti MP. Urogenital alterations in aged male caveolin-1 knockout mice. J Urol. 2004;171:950–957. doi: 10.1097/01.ju.0000105102.72295.b8. [DOI] [PubMed] [Google Scholar]
- 47.Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, Lin MI, Drab M, Kurzchalia TV, Stan RV, Sessa WC. Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest. 2006;116:1284–1291. doi: 10.1172/JCI27100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J, Wier WG, Blaustein MP. Mg2+ blocks myogenic tone but not K+-induced constriction: role for SOCs in small arteries. Am J Physiol Heart Circ Physiol. 2002;283:H2692–H2705. doi: 10.1152/ajpheart.00260.2002. [DOI] [PubMed] [Google Scholar]
- 49.Zhao YY, Liu Y, Stan RV, Fan L, Gu Y, Dalton N, Chu PH, Peterson K, Ross J, Jr, Chien KR. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci USA. 2002;99:11375–11380. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zulli A, Buxton BF, Black MJ, Ming Z, Cameron A, Hare DL. The immunoquantification of caveolin-1 and eNOS in human and rabbit diseased blood vessels. J Histochem Cytochem. 2006;54:151–159. doi: 10.1369/jhc.5A6677.2005. [DOI] [PubMed] [Google Scholar]