

Abstract
Arterial smooth muscle cell large-conductance Ca2+-activated potassium (KCa) channels have been implicated in modulating hypoxic dilation of systemic arteries, although this is controversial. KCa channel activity in arterial smooth muscle cells is controlled by localized intracellular Ca2+ transients, termed Ca2+ sparks, but hypoxic regulation of Ca2+ sparks and KCa channel activation by Ca2+ sparks has not been investigated. We report here that in voltage-clamped (−40 mV) cerebral artery smooth muscle cells, a reduction in dissolved O2 partial pressure from 150 to 15 mmHg reversibly decreased Ca2+ spark-induced transient KCa current frequency and amplitude to 61% and 76% of control, respectively. In contrast, hypoxia did not alter Ca2+ spark frequency, amplitude, global intracellular Ca2+ concentration, or sarcoplasmic reticulum Ca2+ load. Hypoxia reduced transient KCa current frequency by decreasing the percentage of Ca2+ sparks that activated a transient KCa current from 89% to 63%. Hypoxia reduced transient KCa current amplitude by attenuating the amplitude relationship between Ca2+ sparks that remained coupled and the evoked transient KCa currents. Consistent with these data, in inside-out patches at −40 mV hypoxia reduced KCa channel apparent Ca2+ sensitivity and increased the Kd for Ca2+ from ∼17 to 32 μM, but did not alter single-channel amplitude. In summary, data indicate that hypoxia reduces KCa channel apparent Ca2+ sensitivity via a mechanism that is independent of cytosolic signaling messengers, and this leads to uncoupling of KCa channels from Ca2+ sparks. Transient KCa current inhibition due to uncoupling would oppose hypoxic cerebrovascular dilation.
Keywords: transient, calcium-activated, potassium, current
CHANGES IN OXYGEN (O2) partial pressure (PO2) modulate tissue perfusion and regional blood flow, but reactivity varies depending on the anatomic origin of the vasculature (9, 23, 25, 30). In general, hypoxia constricts small distal pulmonary arteries and arterioles but dilates small systemic arteries and arterioles, including those in the brain (2, 25, 31). Hypoxic pulmonary vasoconstriction acts to divert blood supply to O2-rich regions of the lung. In contrast, in the systemic vasculature hypoxic vasodilation functions to match O2 supply to metabolic demand. Although O2 regulation of vascular reactivity serves diverse physiological roles, mechanisms that mediate these responses are poorly understood.
Multiple signaling messengers have been proposed to mediate hypoxia-induced systemic artery dilation, including endothelium-dependent nitric oxide, cyclooxygenase products, adenosine 3′,5′-cyclic monophosphate (cAMP), and guanosine 3′,5′-cyclic monophosphate (cGMP) (12, 13, 25). Activation of potassium channels, including ATP-sensitive and large-conductance calcium (Ca2+)-activated potassium (KCa) channels, is also proposed to contribute to hypoxic vasodilation (3, 4, 14, 16, 18, 35). However, there are conflicting reports of arterial smooth muscle cell KCa channel regulation by hypoxia, with studies reporting activation, inhibition, or no modulation (3, 16, 25, 35, 41, 45). Thus the role of KCa channels in hypoxic vasodilation is unclear.
In arterial smooth muscle cells, KCa channels are activated by localized intracellular Ca2+ transients, termed Ca2+ sparks, which occur because of the opening of ryanodine-sensitive Ca2+ release (RyR) channels in the sarcoplasmic reticulum (SR) membrane (21, 36). Ca2+ sparks generate the micromolar subsarcolemmal intracellular Ca2+ concentration ([Ca2+]i) elevation necessary for KCa channel activation and therefore are critical modulators of KCa channel activity (39, 51). Because of their rapid and localized temporal and spatial properties, Ca2+ sparks do not contribute directly to [Ca2+]i (21). Ca2+ spark-induced KCa channel activation causes membrane hyperpolarization, leading to a decrease in voltage-dependent Ca2+ influx, a reduction in global [Ca2+]i, and vasodilation. Conversely, Ca2+ spark inhibition or a reduction in the coupling of Ca2+ sparks to KCa channels results in vasoconstriction (21, 36).
O2 regulation of KCa channels in arterial smooth muscle cells has primarily been studied by measuring single-channel activity or whole cell currents (9, 16, 20, 38). Thus hypoxic regulation of Ca2+ sparks and KCa channels that are under Ca2+ spark control is unclear. Since Ca2+ sparks are a principal regulator of KCa channel activity in cerebral artery smooth muscle cells, the present study was undertaken to study hypoxic regulation of Ca2+ sparks, KCa channels, and the coupling relationship between Ca2+ sparks and KCa channels. Our data indicate that in cerebral artery smooth muscle cells hypoxia reduces the apparent micromolar Ca2+ sensitivity of KCa channels, which leads to Ca2+ spark to KCa channel uncoupling and a decrease in the frequency and amplitude of transient KCa currents. In contrast, in voltage-clamped cells, hypoxia does not alter Ca2+ spark frequency or amplitude or global [Ca2+]i. These data suggest that, in response to hypoxia, uncoupling of KCa channels from Ca2+ sparks would oppose the cerebral artery dilation.
MATERIALS AND METHODS
Arterial smooth muscle cell isolation
Individual smooth muscle cells were enzymatically dissociated as previously described (7, 49, 50). Briefly, Sprague-Dawley rats (∼200−250 g) of either sex were anesthetized by an intraperitoneally injected overdose of pentobarbital sodium (150 mg/kg body wt). Animal protocols used were reviewed and approved by the Animal Care and Use Committee of the University of Tennessee. The brain was then removed and placed into ice-cold (4°C) HEPES-buffered physiological salt solution containing (in mM) 134 NaCl, 6 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose (pH 7.4 with NaOH). Posterior cerebral, middle cerebral, and cerebellar arteries (100−200 μm in diameter) were removed and cleaned of connective tissue. Individual smooth muscle cells were dissociated from arteries with a HEPES-buffered isolation solution containing (in mM) 55 NaCl, 80 sodium glutamate, 5.6 KCl, 2 MgCl2, 10 HEPES, and 10 glucose (pH 7.3 with NaOH), which was supplemented with papain (0.7 mg/ml) and collagenase (1.0 mg/ml), as described previously (19). Smooth muscle cells were maintained in ice-cold (4°C) HEPES-buffered isolation solution and used for experiments between 1 and 8 h after isolation.
Patch-clamp electrophysiology
Potassium currents were measured with the perforated-patch or the excised inside-out patch-clamp configuration (Axopatch 200B, Clampex 8.2). For perforated-patch experiments, HEPES-buffered physiological salt solution was used as the bath solution. The pipette solution contained (in mM) 110 KAsp, 30 KCl, 10 NaCl, 1 MgCl2, 10 HEPES, and 0.05 EGTA (pH 7.2, KOH). For inside-out patch recordings, the bath solution contained (in mM) 130 KCl, 10 HEPES, 1 MgCl2, 5 EGTA, and 1.6 HEDTA (pH 7.2 with KOH), with free Ca2+ concentrations of 1, 3, 10, 30, 100, or 300 μM. The pipette solution for inside-out patch experiments contained (in mM) 130 KCl, 10 HEPES, 1 MgCl2, 5 EGTA, and 1.6 HEDTA, with 10 μM free Ca2+ (pH 7.4 with KOH). Free Ca2+ concentrations were measured with a Ca2+-sensitive (Corning no. 476041) and a reference (Corning no. 476370) electrode. Hypoxic solutions were obtained by purging bath solution with 100% N2 in a gas-impermeant container for at least 1 h before use. Experimental chambers were continuously perfused with normoxic or hypoxic solution at a rate of 5−10 ml/min. Dissolved PO2 was monitored in experimental chambers with an O2-sensitive electrode (Extech Instruments). Changing the perfusion solution from normoxic to hypoxic reduced the dissolved PO2 in the chamber from ∼150 to 15 mmHg. K+ currents were filtered at 1 kHz and digitized at 4 kHz. KCa current analysis was performed off-line with custom analysis software or Clampfit 9.2. A transient KCa current was defined as the simultaneous opening of three KCa channels, as previously defined (7, 27). Single-KCa channel amplitude was measured in normoxia and hypoxia with histograms.
Confocal Ca2+ imaging
Cells were incubated in HEPES-buffered isolation solution and fluo-4 AM (10 μM) for 25 min at room temperature, followed by a 30-min wash. Imaging was performed with HEPES-buffered physiological salt solution in the experimental chamber. Fluo-4 fluorescence was imaged with a Noran Oz laser scanning confocal microscope with a ×60 water-immersion objective (numerical aperture = 1.2) by illuminating with 488-nm light and collecting emitted light >500 nm. Images (256 × 240 pixels, 56.3 × 52.8 μm) were recorded every 8.3 ms (i.e., at 120 images/s). Simultaneous current and fluorescence measurements were synchronized with a light-emitting diode placed above the recording chamber that was triggered during acquisition. Each cell was imaged for at least 10 s under each condition. Ca2+ sparks and global Ca2+ concentration were analyzed off-line with custom software written with IDL 5.3 that was a kind gift from Dr. M. T. Nelson (University of Vermont, Burlington, VT). Ca2+ sparks were detected by dividing off an area 1.54 μm (7 pixels) × 1.54 μm (7 pixels) (i.e., 2.37 μm2) in each image (F) by a baseline (F0) that was determined by averaging 10 images without Ca2+ spark activity. The entire area of each image was analyzed to detect Ca2+ sparks. A Ca2+ spark was defined as a local increase in F/F0 > 1.2. Global Ca2+ fluorescence was calculated from the same images used for Ca2+ spark analysis and was the mean pixel value of 100 different images acquired during a 10-s period (7, 8, 49, 50).
Fura-2 imaging
Isolated smooth muscle cells were incubated in HEPES-buffered isolation solution containing fura-2 AM (5 μM) and 0.05% Pluronic F-127 for 20 min, followed by a 15-min wash. Experiments were performed with HEPES-buffered physiological salt solution in the chamber. Fura-2 was alternately excited at 340 or 380 nm with a PC-driven hyperswitch (Ionoptix, Milton, MA). Background-corrected ratios were collected every 1 s at 510 nm with a Dage MTI integrating CCD camera (Ionoptix). SR Ca2+ load ([Ca2+]SR) was estimated by measuring the amplitude of caffeine (10 mM)-induced [Ca2+]i transients (7, 8, 49, 50).
Reagents
Unless otherwise specified, all reagents were purchased from Sigma-Aldrich (St. Louis, MO). Fluo-4 AM, fura-2 AM, and Pluronic F-127 were purchased from Molecular Probes (Eugene, OR) and papain from Worthington Biochemical (Lakewood, NJ).
Statistical analysis
Values are expressed as means ± SE. Student's t-test and Student-Newman-Keuls test were used for comparing paired or unpaired data and multiple data sets, respectively. Simultaneous Ca2+ spark and transient KCa current amplitude data were fit with a linear regression function and the slope ± SE of each fit was compared with a Student's t-test. The relationship between KCa channel open probability (Po) and free Ca2+ concentration was fit with a Hill equation, y = Vmax * xn/ (xn + kn), where Vmax is the maximal Po of KCa; n is the Hill coefficient (nH), and k is the dissociation constant (Kd). Vmax, nH, and Kd were compared between normoxia and hypoxia with a Student's t-test. P < 0.05 was considered significant.
RESULTS
Hypoxia inhibits transient KCa currents in cerebral artery smooth muscle cells
Transient KCa currents were measured in isolated smooth muscle cells using the perforated-patch clamp configuration. At −40 mV, a reduction in dissolved PO2 from ∼150 (normoxia) to 15 (hypoxia) mmHg reversibly reduced the frequency and amplitude of transient KCa currents (Fig. 1). Specifically, hypoxia decreased mean transient KCa current frequency from 0.79 ± 0.16 to 0.48 ± 0.11 Hz, or to ∼61% of normoxia (n = 10 cells, P < 0.05). Hypoxia reduced mean transient KCa current amplitude from 33.0 ± 6.3 to 25.1 ± 5.7 pA, or to ∼76% of normoxia (n = 10 cells, P < 0.05).
Fig. 1.

Hypoxia inhibits transient Ca2+-activated potassium (KCa) currents in voltage-clamped cerebral artery smooth muscle cells. A: original trace illustrating reversible transient KCa current inhibition by a reduction in dissolved O2 from ∼150 (normoxia) to ∼15 (hypoxia) mmHg. Expanded segments are shown below the full trace to illustrate individual transient KCa currents in normoxia and hypoxia. B and C: hypoxia reduces mean transient KCa current frequency (B) and amplitude (C) (n = 10 for each). *P < 0.05 compared with normoxia.
Hypoxia attenuates the coupling ratio and amplitude relationship between Ca2+ sparks and transient KCa currents
To investigate mechanisms mediating transient KCa current inhibition by hypoxia, simultaneous measurements of Ca2+ sparks and transient KCa currents were obtained by performing confocal Ca2+ imaging in combination with patch-clamp electrophysiology. At −40 mV, in normoxia, 89.1 ± 3.5% of Ca2+ sparks activated a transient KCa current (n = 155 sparks, 16 cells). In the same cells, hypoxia reduced Ca2+ spark coupling to 62.6 ± 9.1% (n = 123 sparks). Hypoxia also reduced the slope of the amplitude relationship between coupled Ca2+ sparks and evoked transient KCa currents from 66 ± 4 to 45 ± 5 pA/F/F0 (P < 0.05, Fig. 2B). In contrast, hypoxia did not alter mean Ca2+ spark frequency (normoxia 0.74 ± 0.13 Hz, hypoxia 0.76 ± 0.17 Hz; n = 16 cells, P > 0.05) or amplitude (F/F0, 1.70 ± 0.05 vs. 1.72 ± 0.05; n = 155 and 123 sparks, respectively, P > 0.05) (Fig. 2, C and D). In hypoxia, global fluo-4 fluorescence was 1.03 ± 0.04 that in normoxia, indicating that global Ca2+ did not change (n = 16 cells, P > 0.05).
Fig. 2.

Hypoxic regulation of Ca2+ sparks and the effective coupling of Ca2+ sparks to KCa channels. A: original simultaneous recordings of transient KCa currents (top) and Ca2+ sparks that occurred at 2 locations (bottom) in the same cell voltage-clamped at −40 mV in normoxia (left) and hypoxia (right). * and #, coupled and uncoupled Ca2+ sparks, respectively. B: amplitude correlation of coupled Ca2+ sparks and evoked transient KCa currents in normoxia (black squares; n = 134) and hypoxia (gray circles; n = 86) with linear regression and 95% confidence bands fit to each data set (slope ± SE: normoxia 66 ± 4, hypoxia 45 ± 5). Hypoxia decreased the effective coupling of Ca2+ sparks to KCa channels (P = 0.0037). C and D: hypoxia did not change mean Ca2+ spark frequency (C) or amplitude (D) (n = 155 and 123 sparks in normoxia and hypoxia, respectively; n = 16 cells).
Consistent with the observation that hypoxia did not alter Ca2+ spark frequency, hypoxia also did not change [Ca2+]SR, as determined by measuring caffeine (10 mM)-induced [Ca2+]i transients in isolated smooth muscle cells (Fig. 3). Collectively, these data suggest that in cerebral artery smooth muscle cells hypoxia reduces the effective coupling of Ca2+ sparks to KCa channels but does not alter Ca2+ spark properties or global [Ca2+]i.
Fig. 3.
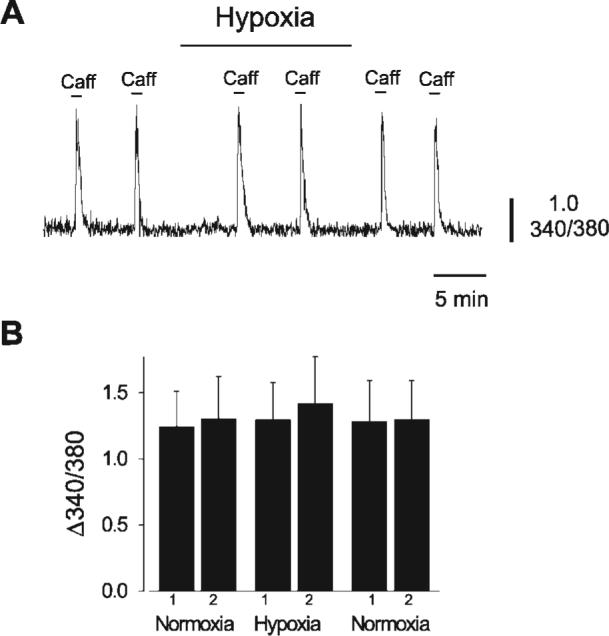
Hypoxia does not alter cerebral artery smooth muscle cell sarcoplasmic reticulum (SR) Ca2+ load. A: caffeine (10 mM)-induced intracellular Ca2+ concentration ([Ca2+]i) transients were similar in normoxia and hypoxia in an isolated cerebral artery smooth muscle cell. 340/380, 340- to 380-nm ratio. B: mean change in fura-2 ratio in normoxia and hypoxia (n = 6 cells).
Hypoxia decreases the apparent Ca2+ sensitivity of cerebral artery smooth muscle cell KCa channels
To determine mechanisms mediating hypoxia-induced Ca2+ spark uncoupling and attenuation of the amplitude relationship between Ca2+ sparks and KCa channels, KCa channel activity was measured in cerebral artery smooth muscle cells. To block transient KCa currents, Ca2+ sparks were inhibited with thapsigargin (100 nM), which depletes [Ca2+]SR.KCa channel activity was measured with the perforated-patch clamp configuration with myocytes voltage-clamped at 0 mV to improve the signal-to-noise ratio. In the same smooth muscle cells, hypoxia reduced mean KCa channel activity (NPo) from 0.18 ± 0.03 to 0.08 ± 0.03, or to ∼44% of that in normoxia (n = 7 cells, P < 0.05; Fig. 4). In contrast, hypoxia did not alter KCa channel amplitude (normoxia 5.2 ± 0.3 pA, hypoxia 5.2 ± 0.4 pA; n = 7 cells, P > 0.05).
Fig. 4.
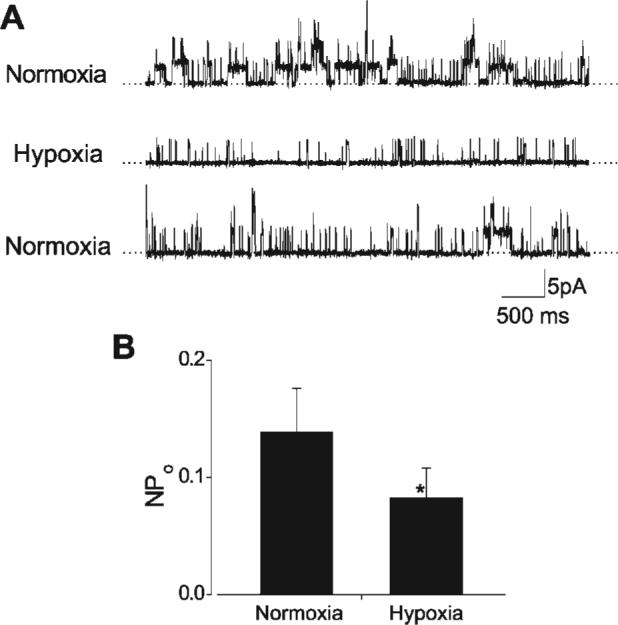
Hypoxia inhibits KCa channels in rat cerebral artery smooth muscle cells. A: original traces obtained from the same smooth muscle cell illustrating reversible KCa channel inhibition by hypoxia at 0 mV. Transient KCa currents were blocked by pretreatment with thapsigargin (100 nM), to deplete SR Ca2+. B: mean data illustrating hypoxic inhibition of KCa channel activity (NPo, n = 7 cells). *P < 0.05 compared with normoxia.
We then tested the hypothesis that hypoxia inhibits transient KCa currents by decreasing KCa channel apparent Ca2+ sensitivity. We also sought to determine whether KCa channel inhibition by hypoxia requires cytosolic signaling pathways. To test these hypotheses, cerebral artery smooth muscle cell KCa channel open probability (Po) was measured over a range of intracellular free Ca2+ concentrations from 1 to 300 μM in excised inside-out membrane patches voltage-clamped at −40 mV. Hypoxia increased the mean Kd for Ca2+ of KCa channels from 17.3 ± 1.3 to 31.9 ± 0.81 μM without altering nH or the maximum Po (Fig. 5A and B). Relative KCa channel inhibition by hypoxia was also Ca2+ dependent. With 3, 10, and 30 μM free Ca2+, which were the Ca2+ concentrations where inhibition was greatest, hypoxia decreased KCa channel Po to 29%, 38%, and 65% of normoxia, respectively (Fig. 5A).
Fig. 5.
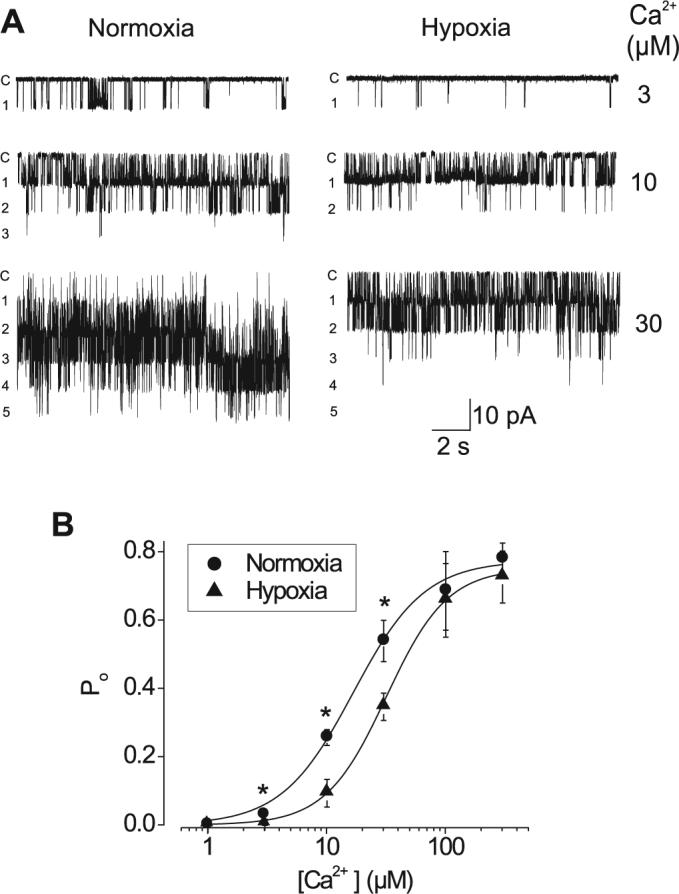
Hypoxia decreases the apparent Ca2+ sensitivity of KCa channels in excised membrane patches from rat cerebral artery smooth muscle cells. A: original recordings obtained from the same membrane patch voltage-clamped at −40 mV, illustrating KCa channel inhibition by hypoxia at 3, 10, and 30 μM Ca2+. c, Closed level, with number of open levels also shown. B: average single-KCa channel open probability (Po) vs. free [Ca2+] in normoxia and hypoxia. Data were fit with a Hill equation. In normoxia, the mean apparent dissociation constant (Kd) for Ca2+ at −40 mV was 17.3 ± 1.29 μM, with a Hill coefficient of 1.52 ± 0.14 and a maximum Po of 0.80 ± 0.02 (n = 3−6 cells). In the same patches, hypoxia increased the mean Kd for Ca2+ of KCa channels to 31.9 ± 0.81 μM but did not alter the Hill coefficient (1.68 ± 0.06) or the maximum Po (0.75 ± 0.01).
DISCUSSION
The major novel findings of our study investigating O2 regulation of cerebral artery smooth muscle cell KCa channels that are under Ca2+ spark control indicate that hypoxia: 1) reversibly reduces transient KCa current frequency and amplitude, 2) does not alter Ca2+ spark frequency or amplitude or change global [Ca2+]i in voltage-clamped cells, 3) attenuates both the coupling ratio and effective coupling of Ca2+ sparks to KCa channels, and 4) reduces the apparent Ca2+ sensitivity of KCa channels through a mechanism that is independent of cytosolic signaling pathways. These data indicate that a reduction in PO2 causes a decrease in KCa channel apparent Ca2+ sensitivity, leading to KCa channel uncoupling from Ca2+ sparks and a decrease in transient KCa current frequency and amplitude. These data suggest that hypoxia-induced KCa channel uncoupling would oppose the resulting vasodilation.
Previous studies have demonstrated that hypoxia inhibits KCa channels in rabbit and lamb pulmonary artery smooth muscle cells, rat cerebral artery smooth muscle cells, and rat carotid body chemoreceptor cells (1, 9, 20, 30, 31, 38, 42, 47). Similarly, recombinant human and rat KCa channels expressed in immortalized cell lines were inhibited by hypoxia (24, 26, 32, 48). In contrast, hypoxia activated cat cerebral artery smooth muscle cell and piglet pial artery KCa channels, leading to vasodilation (3, 4, 16). Our data indicate that in rat cerebral artery smooth muscle cells, hypoxia inhibits transient KCa currents in intact cells and KCa channels in both intact cells and excised membrane patches. Conceivably, KCa channel responses to O2 may be species specific, but further studies will be required to investigate this hypothesis.
Hypoxia may have blocked transient KCa currents by inhibiting Ca2+ sparks. Data obtained with confocal imaging indicated that hypoxia did not alter Ca2+ spark frequency or amplitude. An elevation in global [Ca2+]i activates Ca2+ sparks, whereas a reduction in global [Ca2+]i inhibits Ca2+ sparks (21). Hypoxic regulation of global [Ca2+]i was measured to determine whether mechanisms that regulate Ca2+ sparks were altered by a reduction in PO2. However, hypoxia did not change global [Ca2+]i. Since a [Ca2+]SR reduction inhibits Ca2+ sparks, and blocking SR Ca2+ release elevates [Ca2+]SR, we also measured hypoxic regulation of [Ca2+]SR (6, 28). Hypoxia did not alter [Ca2+]SR, providing further support for our finding that hypoxia did not regulate Ca2+ sparks. Rather, hypoxia decreased transient KCa current frequency by reducing the percentage of Ca2+ sparks that activated a transient KCa current. Hypoxia also reduced the amplitude relationship between Ca2+ sparks and KCa channels that remained coupled, resulting in a decrease in transient KCa current amplitude. Therefore, hypoxia reduced KCa channel activity in cerebral artery smooth muscle cells by reducing both the coupling percentage and the effective coupling of Ca2+ sparks to KCa channels. In smooth muscle cells, Ca2+ sparks activate transient KCa currents by elevating subsarcolemmal [Ca2+]i to within the micromolar concentration range (39, 51). Here, hypoxia reduced KCa channel coupling in intact smooth muscle cells where KCa channels were exposed to subsarcolemmal micromolar Ca2+ concentrations generated by Ca2+ sparks. Hypoxia also reduced KCa channel apparent Ca2+ sensitivity in excised membrane patches that were exposed to micromolar Ca2+ concentrations. These data demonstrate that hypoxia reduces KCa channel apparent Ca2+ sensitivity via a mechanism that is independent of cytosolic signaling pathways, and this a primary mechanism leading to Ca2+ spark uncoupling.
Mechanisms by which acute changes in PO2 regulate KCa channel activity are unclear. Heme oxygenase-2 (HO-2) is physically coupled to KCa channels and is proposed to act as an oxygen sensor in carotid body glomus cells (23, 48). However, genetic ablation of HO-2 did not change O2 sensitivity of carotid body glomus cells or chromaffin cells (37). Oxygen sensitivity of KCa channels may also depend on the presence of a cysteine-rich, stress-regulated exon (STREX) present within the channel COOH terminus (32). However, KCa channels expressed in rat cerebral artery smooth muscle cells were recently cloned by our group and do not contain a STREX motif (20). Thus O2 sensing in cerebral artery smooth muscle cell KCa channels occurs through a STREX-independent mechanism. NADPH oxidase and AMP-activated protein kinase (AMPK) may also act as O2 sensors in pulmonary artery smooth muscle and carotid body type I cells (10, 22, 23). In the present study, the membrane-delimited effect of hypoxia on KCa channels that occurs in excised patches is unlikely to involve soluble signaling messengers or enzymes that require cofactors other than the ions present in the bath solution, arguing against a role for NADPH. In addition, phosphorylation is unlikely to be necessary since ATP was not present, suggesting that AMPK is not involved. Chronic hypoxia also reduces cerebral artery KCa channel β1-subunit expression (35), an effect that would reduce KCa channel apparent Ca2+ sensitivity (5). However, in our experiments, hypoxic inhibition of transient KCa currents and KCa channels was immediate and unlikely to occur through a reduction in protein expression. Mitochondria are another potential O2 sensor in vascular smooth muscle cells (33, 46). Hypoxia depolarizes mitochondria in renal artery smooth muscle cells but hyperpolarizes mitochondria in pulmonary artery smooth muscle cells (33). A small mitochondrial depolarization, such as that induced by diazoxide, an ATP-sensitive potassium (KATP) channel opener, or a nanomolar concentration of CCCP, a protonophore, activates Ca2+ sparks and transient KCa currents in cerebral artery smooth muscle cells (49). In contrast, a large mitochondrial depolarization induced by micromolar CCCP or rotenone, an electron transport chain complex I blocker, inhibits Ca2+ sparks and transient KCa currents (8, 49). Here, the primary effect of hypoxia was mediated by an effect on KCa channels, since hypoxia did not change Ca2+ spark frequency or amplitude. These data suggest that if hypoxia alters mitochondrial potential in cerebral artery smooth muscle cells, the net effects on Ca2+ sparks and KCa channels are very small and secondary to a direct membrane-delimited effect of the PO2 reduction on KCa channels. While the KCa channel O2 sensor is unclear, data indicate that the interaction between O2 and KCa channels can occur in the absence of cytosolic signaling pathways and suggest that the KCa channel itself or a closely associated regulatory molecule is the O2 sensor.
In human and porcine coronary and rabbit cerebral artery smooth muscle cells, hypoxia inhibited L-type Ca2+ channels and reduced [Ca2+]i (43). In contrast, in pulmonary artery smooth muscle cells hypoxia stimulated SR Ca2+ release and increased cytosolic [Ca2+]i (11, 17, 31, 34, 44). In fetal sheep pulmonary artery smooth muscle cells, a PO2 elevation stimulated transient KCa currents (40), whereas in rabbit pulmonary artery smooth muscle cells hypoxia irreversibly blocked transient outward currents (45). While opposing responses to O2 have been reported in pulmonary artery smooth muscle cells, in the present study hypoxia did not alter either cytosolic [Ca2+]i in voltage-clamped cerebral artery smooth muscle cells or [Ca2+]SR in isolated myocytes.
Hypoxia leads to cerebral artery dilation, a response that functions to match blood flow to metabolic requirements (15, 25). Although the signaling mechanisms mediating hypoxic vasodilation may depend on the precise reduction in PO2, hypoxia induces systemic artery hyperpolarization, which would reduce smooth muscle cell voltage-dependent Ca2+ channel activity, leading to a reduction in global [Ca2+]i and vasodilation (13, 29, 30, 43). Data here indicate that a hypoxic reduction in KCa channel activity due to Ca2+ spark uncoupling would attenuate, rather than contribute to, the hypoxic hyperpolarization and vasodilation. In contrast to effects in systemic arteries, hypoxia depolarizes and constricts pulmonary arteries (31). In pulmonary artery smooth muscle cells, hypoxia-induced KCa channel inhibition would contribute to the membrane depolarization, which would activate voltage-dependent Ca2+ channels, leading to vasoconstriction (31, 34). It remains to be determined whether hypoxia also modulates Ca2+ sparks and the coupling relationship between Ca2+ sparks and KCa channels in pulmonary artery smooth muscle cells, and whether such changes are mediated by the mechanism we describe here.
In summary, data indicate that hypoxia reduces KCa channel apparent Ca2+ sensitivity through a membrane-delimited mechanism, leading to a decrease in the effective coupling of Ca2+ sparks to KCa channels and a reduction in transient KCa current frequency and amplitude. These data indicate that KCa channel inhibition caused by uncoupling from Ca2+ sparks would oppose the hypoxic vasodilation.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants HL-67061 and HL-77678 to J. H. Jaggar. A. Adebiyi is a recipient of a postdoctoral fellowship from the Southeast Affiliate of the American Heart Association.
REFERENCES
- 1.Aaronson PI, Robertson TP, Knock GA, Becker S, Lewis TH, Snetkov V, Ward JP. Hypoxic pulmonary vasoconstriction: mechanisms and controversies. J Physiol. 2006;570:53–58. doi: 10.1113/jphysiol.2005.098855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Archer SL, Huang JM, Reeve HL, Hampl V, Tolarova S, Michelakis E, Weir EK. Differential distribution of electrophysiologically distinct myocytes in conduit and resistance arteries determines their response to nitric oxide and hypoxia. Circ Res. 1996;78:431–442. doi: 10.1161/01.res.78.3.431. [DOI] [PubMed] [Google Scholar]
- 3.Armstead WM. Contribution of KCa channel activation to hypoxic cerebrovasodilation does not involve NO. Brain Res. 1998;799:44–48. doi: 10.1016/s0006-8993(98)00462-4. [DOI] [PubMed] [Google Scholar]
- 4.Armstead WM. Relationship among NO, the KATP channel, and opioids in hypoxic pial artery dilation. Am J Physiol Heart Circ Physiol. 1998;275:H988–H994. doi: 10.1152/ajpheart.1998.275.3.H988. [DOI] [PubMed] [Google Scholar]
- 5.Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the β1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–876. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- 6.Cheranov SY, Jaggar JH. Sarcoplasmic reticulum calcium load regulates rat arterial smooth muscle calcium sparks and transient KCa currents. J Physiol. 2002;544:71–84. doi: 10.1113/jphysiol.2002.025197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheranov SY, Jaggar JH. Mitochondrial modulation of Ca2+ sparks and transient KCa currents in smooth muscle cells of rat cerebral arteries. J Physiol. 2004;556:755–771. doi: 10.1113/jphysiol.2003.059568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheranov SY, Jaggar JH. TNF-α dilates cerebral arteries via NADPH oxidase-dependent Ca2+ spark activation. Am J Physiol Cell Physiol. 2006;290:C964–C971. doi: 10.1152/ajpcell.00499.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cornfield DN, Reeve HL, Tolarova S, Weir EK, Archer S. Oxygen causes fetal pulmonary vasodilation through activation of a calcium-dependent potassium channel. Proc Natl Acad Sci USA. 1996;93:8089–8094. doi: 10.1073/pnas.93.15.8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans AM. AMP-activated protein kinase and the regulation of Ca2+ signalling in O2-sensing cells. J Physiol. 2006;574:113–123. doi: 10.1113/jphysiol.2006.108381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franco-Obregon A, Lopez-Barneo J. Differential oxygen sensitivity of calcium channels in rabbit smooth muscle cells of conduit and resistance pulmonary arteries. J Physiol. 1996;491:511–518. doi: 10.1113/jphysiol.1996.sp021235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fredricks KT, Liu Y, Rusch NJ, Lombard JH. Role of endothelium and arterial K+ channels in mediating hypoxic dilation of middle cerebral arteries. Am J Physiol Heart Circ Physiol. 1994;267:H580–H586. doi: 10.1152/ajpheart.1994.267.2.H580. [DOI] [PubMed] [Google Scholar]
- 13.Frisbee JC, Maier KG, Falck JR, Roman RJ, Lombard JH. Integration of hypoxic dilation signaling pathways for skeletal muscle resistance arteries. Am J Physiol Regul Integr Comp Physiol. 2002;283:R309–R319. doi: 10.1152/ajpregu.00741.2001. [DOI] [PubMed] [Google Scholar]
- 14.Gauthier KM. Hypoxia-induced vascular smooth muscle relaxation: increased ATP-sensitive K+ efflux or decreased voltage-sensitive Ca2+ influx? Am J Physiol Heart Circ Physiol. 2006;291:H24–H25. doi: 10.1152/ajpheart.00260.2006. [DOI] [PubMed] [Google Scholar]
- 15.Gauthier-Rein KM, Bizub DM, Lombard JH, Rusch NJ. Hypoxia-induced hyperpolarization is not associated with vasodilation of bovine coronary resistance arteries. Am J Physiol Heart Circ Physiol. 1997;272:H1462–H1469. doi: 10.1152/ajpheart.1997.272.3.H1462. [DOI] [PubMed] [Google Scholar]
- 16.Gebremedhin D, Bonnet P, Greene AS, England SK, Rusch NJ, Lombard JH, Harder DR. Hypoxia increases the activity of Ca2+-sensitive K+ channels in cat cerebral arterial muscle cell membranes. Pflügers Arch. 1994;428:621–630. doi: 10.1007/BF00374586. [DOI] [PubMed] [Google Scholar]
- 17.Gelband CH, Gelband H. Ca2+ release from intracellular stores is an initial step in hypoxic pulmonary vasoconstriction of rat pulmonary artery resistance vessels. Circulation. 1997;96:3647–3654. doi: 10.1161/01.cir.96.10.3647. [DOI] [PubMed] [Google Scholar]
- 18.Haddad GG, Jiang C. O2-sensing mechanisms in excitable cells: role of plasma membrane K+ channels. Annu Rev Physiol. 1997;59:23–42. doi: 10.1146/annurev.physiol.59.1.23. [DOI] [PubMed] [Google Scholar]
- 19.Jaggar JH. Intravascular pressure regulates local and global Ca2+ signaling in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol. 2001;281:C439–C448. doi: 10.1152/ajpcell.2001.281.2.C439. [DOI] [PubMed] [Google Scholar]
- 20.Jaggar JH, Li A, Parfenova H, Liu J, Umstot ES, Dopico AM, Leffler CW. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ Res. 2005;97:805–812. doi: 10.1161/01.RES.0000186180.47148.7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol Cell Physiol. 2000;278:C235–C256. doi: 10.1152/ajpcell.2000.278.2.C235. [DOI] [PubMed] [Google Scholar]
- 22.Jones RD, Hancock JT, Morice AH. NADPH oxidase: a universal oxygen sensor? Free Radic Biol Med. 2000;29:416–424. doi: 10.1016/s0891-5849(00)00320-8. [DOI] [PubMed] [Google Scholar]
- 23.Kemp PJ. Detecting acute changes in oxygen: will the real sensor please stand up? Exp Physiol. 2006;91:829–834. doi: 10.1113/expphysiol.2006.034587. [DOI] [PubMed] [Google Scholar]
- 24.Kemp PJ, Peers C, Lewis A. Oxygen sensing by human recombinant large conductance, calcium-activated potassium channels. Regulation by acute hypoxia. Adv Exp Med Biol. 2003;536:209–215. doi: 10.1007/978-1-4419-9280-2_27. [DOI] [PubMed] [Google Scholar]
- 25.Leffler CW, Smith JS, Edrington JL, Zuckerman SL, Parfenova H. Mechanisms of hypoxia-induced cerebrovascular dilation in the newborn pig. Am J Physiol Heart Circ Physiol. 1997;272:H1323–H1332. doi: 10.1152/ajpheart.1997.272.3.H1323. [DOI] [PubMed] [Google Scholar]
- 26.Lewis A, Peers C, Ashford ML, Kemp PJ. Hypoxia inhibits human recombinant large conductance, Ca2+-activated K+ (maxi-K) channels by a mechanism which is membrane delimited and Ca2+ sensitive. J Physiol. 2002;540:771–780. doi: 10.1113/jphysiol.2001.013888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li A, Adebiyi A, Leffler CW, Jaggar JH. KCa channel insensitivity to Ca2+ sparks underlies fractional uncoupling in newborn cerebral artery smooth muscle cells. Am J Physiol Heart Circ Physiol. 2006;291:H1118–H1125. doi: 10.1152/ajpheart.01308.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu P, Xi Q, Ahmed A, Jaggar JH, Dopico AM. Essential role for smooth muscle BK channels in alcohol-induced cerebrovascular constriction. Proc Natl Acad Sci USA. 2004;101:18217–18222. doi: 10.1073/pnas.0406096102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lombard JH, Liu Y, Fredricks KT, Bizub DM, Roman RJ, Rusch NJ. Electrical and mechanical responses of rat middle cerebral arteries to reduced PO2 and prostacyclin. Am J Physiol Heart Circ Physiol. 1999;276:H509–H516. doi: 10.1152/ajpheart.1999.276.2.H509. [DOI] [PubMed] [Google Scholar]
- 30.Lopez-Barneo J, del Toro R, Levitsky KL, Chiara MD, Ortega-Saenz P. Regulation of oxygen sensing by ion channels. J Appl Physiol. 2004;96:1187–1195. doi: 10.1152/japplphysiol.00929.2003. [DOI] [PubMed] [Google Scholar]
- 31.Mauban JR, Remillard CV, Yuan JX. Hypoxic pulmonary vasoconstriction: role of ion channels. J Appl Physiol. 2005;98:415–420. doi: 10.1152/japplphysiol.00732.2004. [DOI] [PubMed] [Google Scholar]
- 32.McCartney CE, McClafferty H, Huibant JM, Rowan EG, Shipston MJ, Rowe IC. A cysteine-rich motif confers hypoxia sensitivity to mammalian large conductance voltage- and Ca-activated K (BK) channel alpha-subunits. Proc Natl Acad Sci USA. 2005;102:17870–17876. doi: 10.1073/pnas.0505270102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michelakis ED, Hampl V, Nsair A, Wu X, Harry G, Haromy A, Gurtu R, Archer SL. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ Res. 2002;90:1307–1315. doi: 10.1161/01.res.0000024689.07590.c2. [DOI] [PubMed] [Google Scholar]
- 34.Morio Y, McMurtry IF. Ca2+ release from ryanodine-sensitive store contributes to mechanism of hypoxic vasoconstriction in rat lungs. J Appl Physiol. 2002;92:527–534. doi: 10.1152/jappl.2002.92.2.527. [DOI] [PubMed] [Google Scholar]
- 35.Navarro-Antolin J, Levitsky KL, Calderon E, Ordonez A, Lopez-Barneo J. Decreased expression of maxi-K+ channel β1-subunit and altered vasoregulation in hypoxia. Circulation. 2005;112:1309–1315. doi: 10.1161/CIRCULATIONAHA.104.529404. [DOI] [PubMed] [Google Scholar]
- 36.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 37.Ortega-Saenz P, Pascual A, Gomez-Diaz R, Lopez-Barneo J. Acute oxygen sensing in heme oxygenase-2 null mice. J Gen Physiol. 2006;128:405–411. doi: 10.1085/jgp.200609591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park MK, Lee SH, Lee SJ, Ho WK, Earm YE. Different modulation of Ca-activated K channels by the intracellular redox potential in pulmonary and ear arterial smooth muscle cells of the rabbit. Pflügers Arch. 1995;430:308–314. doi: 10.1007/BF00373904. [DOI] [PubMed] [Google Scholar]
- 39.Perez GJ, Bonev AD, Nelson MT. Micromolar Ca2+ from sparks activates Ca2+-sensitive K+ channels in rat cerebral artery smooth muscle. Am J Physiol Cell Physiol. 2001;281:C1769–C1775. doi: 10.1152/ajpcell.2001.281.6.C1769. [DOI] [PubMed] [Google Scholar]
- 40.Porter VA, Rhodes MT, Reeve HL, Cornfield DN. Oxygen-induced fetal pulmonary vasodilation is mediated by intracellular calcium activation of KCa channels. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1379–L1385. doi: 10.1152/ajplung.2001.281.6.L1379. [DOI] [PubMed] [Google Scholar]
- 41.Resnik E, Herron J, Fu R, Ivy DD, Cornfield DN. Oxygen tension modulates the expression of pulmonary vascular BKCa channel α- and β-subunits. Am J Physiol Lung Cell Mol Physiol. 2006;290:L761–L768. doi: 10.1152/ajplung.00283.2005. [DOI] [PubMed] [Google Scholar]
- 42.Riesco-Fagundo AM, Perez-Garcia MT, Gonzalez C, Lopez-Lopez JR. O2 modulates large-conductance Ca2+-dependent K+ channels of rat chemoreceptor cells by a membrane-restricted and CO-sensitive mechanism. Circ Res. 2001;89:430–436. doi: 10.1161/hh1701.095632. [DOI] [PubMed] [Google Scholar]
- 43.Smani T, Hernandez A, Urena J, Castellano AG, Franco-Obregon A, Ordonez A, Lopez-Barneo J. Reduction of Ca2+ channel activity by hypoxia in human and porcine coronary myocytes. Cardiovasc Res. 2002;53:97–104. doi: 10.1016/s0008-6363(01)00422-9. [DOI] [PubMed] [Google Scholar]
- 44.Vadula MS, Kleinman JG, Madden JA. Effect of hypoxia and norepinephrine on cytoplasmic free Ca2+ in pulmonary and cerebral arterial myocytes. Am J Physiol Lung Cell Mol Physiol. 1993;265:L591–L597. doi: 10.1152/ajplung.1993.265.6.L591. [DOI] [PubMed] [Google Scholar]
- 45.Vandier C, Delpech M, Bonnet P. Spontaneous transient outward currents and delayed rectifier K+ current: effects of hypoxia. Am J Physiol Lung Cell Mol Physiol. 1998;275:L145–L154. doi: 10.1152/ajplung.1998.275.1.L145. [DOI] [PubMed] [Google Scholar]
- 46.Waypa GB, Schumacker PT. Hypoxic pulmonary vasoconstriction: redox events in oxygen sensing. J Appl Physiol. 2005;98:404–414. doi: 10.1152/japplphysiol.00722.2004. [DOI] [PubMed] [Google Scholar]
- 47.Weir EK, Lopez-Barneo J, Buckler KJ, Archer SL. Acute oxygen-sensing mechanisms. N Engl J Med. 2005;353:2042–2055. doi: 10.1056/NEJMra050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Williams SE, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, Peers C, Kemp PJ. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science. 2004;306:2093–2097. doi: 10.1126/science.1105010. [DOI] [PubMed] [Google Scholar]
- 49.Xi Q, Cheranov SY, Jaggar JH. Mitochondria-derived reactive oxygen species dilate cerebral arteries by activating Ca2+ sparks. Circ Res. 2005;97:354–362. doi: 10.1161/01.RES.0000177669.29525.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xi Q, Tcheranova D, Parfenova H, Horowitz B, Leffler CW, Jaggar JH. Carbon monoxide activates KCa channels in newborn arteriole smooth muscle cells by increasing apparent Ca2+ sensitivity of α-subunits. Am J Physiol Heart Circ Physiol. 2004;286:H610–H618. doi: 10.1152/ajpheart.00782.2003. [DOI] [PubMed] [Google Scholar]
- 51.Zhuge R, Fogarty KE, Tuft RA, Walsh JV., Jr Spontaneous transient outward currents arise from microdomains where BK channels are exposed to a mean Ca2+ concentration on the order of 10 μM during a Ca2+ spark. J Gen Physiol. 2002;120:15–27. doi: 10.1085/jgp.20028571. [DOI] [PMC free article] [PubMed] [Google Scholar]