


Abstract
Small interfering RNA (siRNA) are widely used to infer gene function. Here, insights in the equilibrium of siRNA-target hybridization are used for selection of efficient siRNA. The accessibilities of siRNA and target mRNA for hybridization, as measured by folding free energy change, are shown to be significantly correlated with efficacy. For this study, a partition function calculation that considers all possible secondary structures is used to predict target site accessibility; a significant improvement over calculations that consider only the predicted lowest free energy structure or a set of low free energy structures. The predicted thermodynamic features, in addition to siRNA sequence features, are used as input for a support vector machine that selects functional siRNA. The method works well for predicting efficient siRNA (efficacy >70%) in a large siRNA data set from Novartis. The positive predictive value (percentage of sites predicted to be efficient for silencing that are) is as high as 87.6%. The sensitivity and specificity are 22.7 and 96.5%, respectively. When tested on data from different sources, the positive predictive value increased 8.1% by adding equilibrium terms to 25 local sequence features. Prediction of hybridization affinity using partition functions is now available in the RNAstructure software package.
INTRODUCTION
It is now widely known that mRNA can be targeted and inhibited by short complementary oligonucleotides such as small interfering RNA (siRNA). The breakthrough study of this approach, called RNA interference (RNAi), was formally described in Caenorhabditis elegans as a response to double-stranded RNA (dsRNA) (1). It extensively changed our concept of gene regulation in animals, plants and fungi. In the RNAi pathway, dsRNA is processed by Dicer, a ribonuclease III-like enzyme, into 21–23 nucleotide long fragments, called siRNA. Then the antisense strand of siRNA is loaded onto RNA-induced silencing complex (RISC), which recognizes the target mRNA sequence via hybridization between the siRNA antisense strand and the complementary region of mRNA. Subsequently, cleavage or knock-down of the target mRNA is induced (2–4). For gene silencing, a 19 nucleotide duplex siRNA plus 3′ dinucleotide overhangs is commonly utilized (5).
Gene silencing with RNAi, however, does not work equally well for all siRNA complementary to different sites of mRNA. In response to this, a number of the rules to predict the silencing efficacy of a specific siRNA have been developed. These rules are commonly based on the features of the siRNA sequence: low G/C content, lack of self-structure, preference of A at position 3, preference of U at position 10, absence of G at position 13 and absence of G or C at position 19, etc. Although the mechanisms of most of these features are not understood, they are commonly utilized in methods for designing efficient siRNA (5–11). In one such study, a genome-wide siRNA library was designed with an artificial neural network using a large number of siRNA sequence features (12). But the conventional methods, which only focus on the sequence information of siRNA, cannot fully capture the mechanistic features of RNAi. Other factors, including protein binding, cellular localization and target mRNA secondary structure, may also influence the silencing efficacy of RNAi in vivo (13).
It has been demonstrated that the secondary structure of the target at the hybridization region is an important consideration for the effective hybridization of oligomers (14–17). Heale et al. (18) used this knowledge to predict functional siRNA according to predicted local structures, i.e. prediction of structure within 100 nucleotides in each direction from the binding site. In that study, only 55% of selected siRNA were efficient at silencing. With a larger data set, the linear correlation coefficient was found to be 0.149 between the local target stability and the silencing activity of siRNA (19). Recently, Ladunga (20) used 142 features to predict functional siRNAs. Among these features, mRNA accessibility was represented as p3 (the probability that each of four contiguous nucleotides is paired at the target's binding site) as predicted by sfold (21). In spite of the fact that the correlation of p3 and siRNA activity was found to be low (r = 0.0584), p3 was still highly weighted in the prediction by machine learning (20). An accurate prediction of mRNA accessibility is difficult mainly because the secondary structure cannot be well predicted for long RNA sequences (22,23); mRNA can be up to 8000 nucleotides in length. The number of possible secondary structures increases exponentially with the length increase of the sequence, and so it is difficult to predict the correct structure from so many possibilities.
Here, the effect of target mRNA structure on siRNA efficacy was investigated by use of a partition function calculation that considers all possible secondary structures of the mRNA to determine an ensemble folding free energy change. The correlation between the thermodynamic features of the target site and the efficacy of siRNA was significantly improved by optimization of the method for secondary structure prediction of mRNA. Free energies of binding of the siRNA antisense strand to the mRNA target were calculated using the OligoWalk algorithm (24), including terms that account for self-structure in both the siRNA and the target (Figure 1). These predicted thermodynamic features were utilized by a support vector machine (SVM) to predict functional siRNA.
Figure 1.

Equilibrium considered in the OligoWalk algorithm for predicting the affinity of a structured oligonucleotide (siRNA) to a structured target (mRNA). Involved proteins are not shown and were neglected in the calculations. The free energy change of each equilibrium is Δ G° = − RT ln K, where, K is the equilibrium constant. K1, K2, K3 and K4 are related to 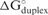 ,
, 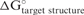 ,
,  and
and 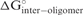 , respectively. K0 is also related to . Folding in the target (at the region of hybridization) and self-structure in the siRNA both compete with the formation of the siRNA-target complex needed for cleavage by RISC.
, respectively. K0 is also related to . Folding in the target (at the region of hybridization) and self-structure in the siRNA both compete with the formation of the siRNA-target complex needed for cleavage by RISC.
MATERIALS AND METHODS
Calculation of free energy costs of opening base pairs for hybridization
To predict the cost of opening base pairs in the mRNA for hybridization to siRNA, the structure is predicted once without constraints and then once with the constraint that the nucleotides in the hybridization site are forced single-stranded. This prediction assumes that siRNA binding results in the re-equilibration of the complete target secondary structure. The free energy cost,  , is then:
, is then:
where, 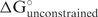 is the predicted folding free energy change for the native structure and is the predicted folding free energy change, where the nucleotides that will hybridize to the siRNA are forced single-stranded.
is the predicted folding free energy change for the native structure and is the predicted folding free energy change, where the nucleotides that will hybridize to the siRNA are forced single-stranded.
Different secondary structure prediction methods were investigated to calculate the free energy change terms. The lowest free energy structure prediction is a single predicted secondary structure. For suboptimal structure prediction, a heuristic method (25) is used to generate 1000 low free energy structures (with a window size of zero) and a weighted average free energy change is determined (24):
 |
where, the sum over s is over the set of predicted secondary structures, R is the gas constant and T is the absolute temperature (310.15 K).
A partition function (Q) calculation is a more rigorous method for examining secondary structure because information of the complete ensemble of possible secondary structures is included:
where, ΔG(s) is the free energy change of folding of structure s and the sum is over all possible secondary structures. Q can be calculated using a dynamic programming algorithm (26,27). The free energy cost to open the base pairs of the targeted RNA for oligonucleotide to hybridize is equal to the difference between the ensemble free energy change unconstrained and the ensemble free energy change with a constraint that nucleotides in complementary region are single stranded. The ensemble folding free energy change is:
Therefore,
The partition function code was optimized for calculating the constrained partition function using data from the unconstrained partition function. For example, many of the dynamic programming array positions in Qunconstrained are reused for Qconstrained because they are unchanged (27). Only the positions spanning the region of hybridization need to be recalculated in the arrays. This saved 70.6% computer time (8 h and 55 min down to 2 h and 37 min) for a complete scan of an mRNA of 730 nucleotides, running on a single core of a dual core AMD 270 processor.
Local structure prediction
Local and global (whole length of the targeted RNA) secondary structure prediction of the mRNA were compared. Local structure prediction only folds a certain total number of nucleotides centered at the binding region. When the target sequence was too close to the mRNA sequence end (5′ or 3′, end) for the folding region to be centered, the folding region was kept at the same length, but running to the end of the sequence, i.e. no longer centered on the siRNA binding site.
Available databases
Two databases provide experimental data for testing hypotheses. The first set is derived from a database of experiments performed by Huesken et al. (12) at Novartis and contains efficacy data for 2431 siRNA selected to random positions in 31 mRNA sequences. 2000 siRNAs induced >50% gene silencing, 1222 induced >70% and 369 induced >90%. The second set was assembled by Shabalina et al. (19) from a number of experiments reported in the literature and this database includes 653 siRNAs tested at different concentrations, targeting 52 distinct mRNAs. A total of 419 siRNAs induced >50% gene silencing, 293 induced >70% and 108 induced >90%. The databases have no targets in common.
Calculation of terminal siRNA base pairing stability
The 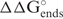 , the base pairing free energy difference between the 5′ end and 3′ end of the antisense strand in the duplex (6,7), was also calculated. Its best correlation with siRNA inhibition efficacy was found using a window size of two terminal base pairs (one nearest neighbor parameter), including the AU end penalty (28).
, the base pairing free energy difference between the 5′ end and 3′ end of the antisense strand in the duplex (6,7), was also calculated. Its best correlation with siRNA inhibition efficacy was found using a window size of two terminal base pairs (one nearest neighbor parameter), including the AU end penalty (28).
Training and validation sets with SVM
The LIBSVM (29) implementation of SVM was used for binary classification with a radial basis function kernel. The input values were scaled and the model was optimized by LIBSVM's optimization program. The silencing efficacies of 50% or 70% were used as the classification boundary in separate tests. Classification was done with –b 1 parameter to output probabilities. Cutoff of probabilities were varied for construction of ROC curves and curves of positive predictive values as a function of sensitivity. For predictions in which the Novartis database was split into training and testing databases, the training set was randomly generated six times and the results were averaged.
Statistical analysis
The linear correlation coefficients (r) were calculated between siRNA silencing efficacy (from experimental results) and different features, among different databases. In the database of Shabalina et al. (19), the silencing efficacy is represented as ln (activity), where activity is the percentage amount of the targeted mRNA expression after RNA interference treatment as compared to the control. The inhibition efficacies reported in the Novartis database (12) are transformed to activity as well (activity = 1− inhibition efficacy). The activity is reset to 0.001 if it is reported to be less than or equal to 0 and reset to 0.999 if it is reported to be larger than or equal to 1. A two tailed t-test was used to test the significance of the linear correlation (calculated with Statistics-Basic-0.42 perl module downloaded from http://www.cpan.org). Every coefficient is shown along with a t-test P-value (Tables 1 and 3). Correlations were considered significant for P-values of <0.05, which means the siRNA efficacy is very unlikely to be randomly distributed by its position on the targeted mRNA sequence.
Table 1.
Thermodynamic features predicted by OligoWalk algorithm
| Free energy type | Correlation between ln(Activity) and different free energy changesa | |
|---|---|---|
| r | t-test P-valueb | |
|
−0.2298 | 1.78 × 10−15 |
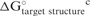 c c
|
−0.1949 (−0.1799)d | 2.66 × 10−15 (3.33 × 10−15)d |
|
−0.1882 (−0.1873)d | 3.11 × 10−15 (2.89 × 10−15)d |
|
−0.1812 (−0.1790)d | 3.11 × 10−15 (3.11 × 10−15)d |
|
e
|
−0.3507 | 8.88 × 10−16 |
aThe correlations were calculated within Novartis data set (12) plus the data sets collected by Shabalina et al. (19). Activity is the percentage amount of the targeted mRNA after RNA interference compared to the control. Here, r is the correlation coefficient. Negative correlations indicate that decreasing each folding free energy change (increased stability) results in increased ln (activity) (decreased silencing efficiency).
bA P-value (probability) <0.05 is statistically significant.
cThe values were calculated from partition function method with folding size of 800 nucleotides centered on the binding site.
dThe values in parenthesis are calculated with the optimal structure prediction method.
eThe best correlation was found by considering 2bp at the end, including the AU end penalty (28).
Table 3.
Prediction performance for efficient siRNA (inhibition efficacy >70%)
| Parameters for SVM | PPV (%) | Sensitivity (%) | Specificity (%) |
|---|---|---|---|
| All 28 features | 78.6 | 22.9 | 95.1 |
| Not considering siRNA's self-structure free energy changes | 77.0 | 19.8 | 95.5 |
| Not considering mRNA's self-structure free energy change | 73.5 | 21.2 | 94.0 |
| Not considering either siRNA or mRNA self-structure free energy changes | 70.5 | 19.1 | 93.7 |
The SVM was trained with Novartis data set (12) and tested on the data sets from different sources, which are collected by Shabalina et al. (19). Positive predictive value (PPV), the percent of selected siRNA sequences that are efficient at silencing, is the main criterion to show the best prediction performance because it measures how well a set of efficient siRNA sequences can be selected.
RESULTS
Prediction of mRNA binding accessibility
Both experiments (17) and computational predictions (18) demonstrate that siRNA efficacy is affected by the secondary structure accessibility at the siRNA binding site. If the nucleotides at the binding site are base paired in the native structure, the binding affinity of the siRNA is lowered by the cost of displacing the existing pairs. At equilibrium, the conformation and stability of the local binding site may also be influenced by the conformations of parts of the target mRNA that are distant in sequence. Therefore, the characteristics of global structure need to be considered in the calculation of the binding-site accessibility. Furthermore, the secondary structure prediction of the whole mRNA sequence is difficult to predict, because mRNA is longer than most structured non-coding RNAs and because the coding region of an mRNA might not be selected for a single structure. As the length of an RNA increases, there are many more possible secondary structures and the number of secondary structures with free energies within RT of the lowest free energy structure also increases exponentially (30). Using the free energy change nearest neighbor model (22,28,31), this is observed as a decrease in the structure prediction accuracy of the lowest free energy structure for longer sequences. In this study, both the need for global secondary structure prediction and for predicting ensembles of structures were specifically examined.
The OligoWalk algorithm (24) can be used to predict the equilibrium binding stability of siRNA to an RNA target. It explicitly considers self-structures for both the siRNA and the target that compete with the hybridization for the equilibrium shown in Figure 1. Binding stability is quantified by equilibrium free energy changes using the free energy change nearest neighbor model at 37°C (22,28). For this work, the OligoWalk algorithm was enhanced to consider not only one predicted lowest free energy secondary structure, but the complete ensemble of structures for an RNA sequence with a partition function. Partition function calculations of RNA secondary structure compensate for incomplete knowledge of the folding rules by emphasizing the importance of predicted base pairs that are well-determined (27).
The binding-site accessibility is quantified as the free energy change to be overcome to open the base pairs of the targeted mRNA for siRNA hybridization (). It is defined as the difference in free energy of the mRNA in the native state and the mRNA with the hybridization site single-stranded, i.e. accessible to siRNA binding. Secondary structure prediction is used to predict the structures of both the native state and the open state. This means that the secondary structure of the target message is assumed to remain at equilibrium and therefore the secondary structure of the target changes in response to having the target-site nucleotides paired to the antisense siRNA.
To demonstrate the benefit of considering all possible structures, was predicted using three different methods. In one method, only the optimal structure (lowest free energy structure) was considered for each structure prediction. In the second method, 1000 suboptimal structures (within 10% of the optimal structure's free energy change) were sampled heuristically (22). In the third method, all possible structures were considered using a partition function.
As explained earlier, global structure prediction is expected to be more accurate than local structure prediction. Local structure prediction, however, saves significant computer time because the secondary structure prediction algorithms scale O(N3), where N is the length of sequence folded. Therefore, for example, using a region of 800 nucleotides reduces the calculation time by a factor of eight compared to predicting the global secondary structure of a 1600 nucleotide mRNA. To test whether local secondary structure prediction is adequate, different total lengths of flanking sequence from 100 to 2000 nucleotides were tested for correlation to experimental data and compared to global folding.
Figure 2 plots correlation of to the natural log of experimentally determined activity, defined as the fraction of mRNA levels after siRNA treatment as compared to a negative control. As expected, predicted free energy changes for longer folding sizes achieved a better correlation to siRNA efficiency data. Furthermore, for any length of structure prediction, the partition function provides better correlation than either optimal or suboptimal structure prediction. The correlation for suboptimal structure prediction was even worse than optimal structure prediction because the suboptimal structures generated heuristically cannot be used to accurately calculate the equilibrium constant. The improvement in correlation slows as a function of length of folding region once the size reaches 800 nucleotides. This is partially because few base pairs occur in RNA between nucleotides that are separated farther in sequence than 800 nucleotides. For example, in rRNA, where the secondary structure is known (32), over 75% of base pairs occur between nucleotides separated by fewer than 100 nucleotides and 99% base pairs occur between nucleotides separated by fewer than 800 nucleotides. The correlation also does not significantly improve for folding regions longer than 800 nucleotides because the accuracy of secondary structure prediction is also known to decrease for sequences longer than 800 nucleotides (22,23). Apparently, an 800 nucleotide folding size is a good compromise between calculation cost and correlation to siRNA efficacy. Additionally, the calculated from global folding (data not shown) was not better than the 800 nucleotide folding size for the selection of efficient siRNA with the method described subsequently.
Figure 2.

The correlation between the ln(activity) and the free energy cost of opening local structure of mRNA (). Three prediction methods are used, optimal structure prediction (lowest free energy structure), suboptimal structure prediction (a set of heuristically generated low free energy structures) and the partition function calculation. Activity is the fraction of the targeted mRNA expression after RNA interference treatment as compared to the control. Different sizes of local structure centered on the binding region were folded. 4000 nucleotides of flanking sequence are folded if the sequence is larger than 4000 nucleotides in global folding. The y-axis, r, is the correlation coefficient. The correlations were calculated within Novartis data set (12) plus all other data sets collected by Shabalina et al. (19).
Correlation between silencing efficiency and RNA thermodynamic features
To consider the effect of self-structure in the siRNA antisense strand, which can decrease the equilibrium affinity to the mRNA target, free energy changes were predicted for both unimolecular and bimolecular siRNA folding,  and
and  , respectively (Figure 1). These terms were also calculated with a partition function and this is fast because the siRNA sequence considered is the 19 bases that will hybridize to the target.
, respectively (Figure 1). These terms were also calculated with a partition function and this is fast because the siRNA sequence considered is the 19 bases that will hybridize to the target.
Table 1 shows the correlation between the predicted thermodynamic stabilities from the equilibrium terms shown in Figure 1 and siRNA efficacy using the two different siRNA efficacy data sets (12,19). Table 1 also includes the well-known orientation effect () in which the 3′ end of the antisense strand should be more stable than the 5′ end of the duplex with the sense strand (6,7). Each of the equilibrium stability terms as calculated by OligoWalk is statistically significant as tested by a t-test.
Predicting efficient siRNA with a SVM
In the selection of efficient siRNA sequences, each of the thermodynamic features predicted by OligoWalk need to be weighted because they have different extent of influence on siRNA silencing. Therefore, a classification SVM, implemented with LIBSVM (29), was trained to utilize the free energy changes to predict efficient siRNA. An SVM is a machine learning method capable of making classifications, including providing a confidence on the classification. Twenty-three other sequence features were also added as input parameters (Table 2) to the SVM. These features were chosen from the most correlated sequence features found by Ladunga using the Novartis database (20). 2182 siRNAs were used in the training set and the generated models were tested on the remaining 249 siRNAs. Using different confidence thresholds for classification by the SVM, the prediction method can optimize either sensitivity or specificity of siRNA selection. To show the tradeoffs, receiver operator characteristic (ROC) curves and curves of positive predictive value (PPV) as a function of sensitivity are shown in Figure 3a and b, respectively. The plots were generated using two different experimental silencing efficacies (50% and 70%) as classification boundaries for the experimental data. PPV is defined as the percent of siRNAs predicted to be efficient that are experimentally shown to be efficient at silencing. Sensitivity is the percent of efficient siRNA predicted to be efficient. Specificity is the percent of inefficient siRNAs that are correctly predicted to be inefficient. In the design of a method for selecting siRNA, PPV is more important than sensitivity because it is more important to reduce the number of siRNA sequences that need to be tested to find one that is efficient in silencing. It is less important to find all efficient siRNA for a long mRNA sequence because there is almost always a large pool of possible siRNA that can efficiently silence a given mRNA. For efficient siRNA prediction (inhibition efficacy larger than 70%), the best PPV, sensitivity and specificity are 87.6%, 22.7% and 96.5%, respectively, with the Novartis data set (12).
Table 2.
Correlations between ln (activity)a of siRNA and different features
| Individual feature | Position | r | t-test P-value |
|---|---|---|---|
|
b
|
mRNA | −0.1971 | 1.11 × 10−15 |
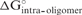 |
all | −0.1895 | 1.55 × 10−15 |
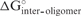 |
all | −0.1974 | 2.89 × 10−15 |
|
all | −0.2501 | 1.78 × 10−15 |
|
1 versus 19 | −0.3507 | 6.66 × 10−16 |
| ΔG° | 1 | −0.3427 | 4.44 × 10−16 |
| ΔH° | 1 | −0.3215 | 1.11 × 10−15 |
| U | 1 | −0.2625 | 1.33 × 10−15 |
| G | 1 | 0.2385 | 2.22 × 10−15 |
| ΔH° | all | −0.2473 | 1.78 × 10−15 |
| U | all | −0.1962 | 2.22 × 10−15 |
| UU | 1 | −0.193 | 1.78 × 10−15 |
| G | all | 0.1838 | 3.11 × 10−15 |
| GG | 1 | 0.1434 | 1.20 × 10−12 |
| GC | 1 | 0.1301 | 1.21 × 10−10 |
| GG | all | 0.1605 | 4.88 × 10−15 |
| ΔG° | 2 | −0.1659 | 4.22 × 10−15 |
| UA | all | −0.1267 | 3.61 × 10−10 |
| U | 2 | −0.1332 | 4.26 × 10−11 |
| C | 1 | 0.1434 | 1.21 × 10−12 |
| CC | all | 0.1447 | 7.58 × 10−13 |
| ΔG° | 18 | 0.1024 | 4.22 × 10−07 |
| CC | 1 | 0.1116 | 3.46 × 10−08 |
| GC | all | 0.1403 | 3.63 × 10−12 |
| CG | 1 | 0.1018 | 4.86 × 10−07 |
| ΔG° | 13 | −0.1092 | 6.81 × 10−08 |
| UU | all | −0.1414 | 2.49 × 10−12 |
| A | 19 | 0.0804 | 7.29 × 10−05 |
The siRNA (19 base pairs) sequence features are chosen from the most correlated features found by Ladunga (20) in Novartis data set (12). They are compared with the thermodynamic features predicted by the OligoWalk algorithm. The correlations are calculated within Novartis data set.
aActivity is the fraction of the targeted mRNA after RNA interference compared to the control.
bThe values were calculated from partition function method with folding size as 800 nucleotides centered on the binding site.
Figure 3.

ROC curve and PPV of SVM prediction (a) ROC curves and (b) PPV as a function of sensitivity: all 28 features (listed in Table 2) are used to train the SVM. siRNA with different silencing efficacies (>50% and >70%) within Novartis data set (12) are predicted (see Methods section). (c) ROC curves and (d) PPV as a function of sensitivity: the SVM is trained on the whole Novartis data set and tested on the database collected by Shabalina et al. (19). Plots are shown for selecting efficient siRNA (silencing efficacies >70%) both with and without self-structure folding free energy terms. There are 28 features in total (Table 2) when including local sequences terms and folding free energy changes. Thermodynamic features are those predicted by OligoWalk (Table 1).
In order to test the robustness of the method, the SVM was trained on the whole Novartis data set and tested on the database collected by Shabalina et al. (19). Different sets of features were used to train the SVM, resulting in different performances (Figure 3c and d). Both ROC curves and PPV as a function of sensitivity were plotted and the plot of PPV captured some details that the ROC curves did not represent clearly. The best prediction results from the combination of sequence preferences and thermodynamic features (Table 2). The SVM performance with the Shabalina et al. database is not as good as the performance with the Novartis data set. It is not surprising because the Shabalina et al. database is more diverse in the way that experiments were performed. Furthermore, the sequence parameters were derived from the Novartis data set and, in spite of the cross-validation procedure used, there is still chance that the data set was over-trained. Finally, an SVM was also trained on the database from Shabalina et al. and tested on Novartis data set. The performance of this SVM was between the performance of the former two training and testing methods (data not shown).
Because the RNA (siRNA and mRNA) self-structure energies (, and ) and duplex free energy () are correlated with one another, different combinations of these thermodynamic features were used with the classification SVM to see their effect on siRNA selection accuracy. Figure 3d shows that removing the self-structure free energy terms (, or ) from the set of input parameters lowers the PPV at any sensitivity. The accuracy of the best prediction results for selecting efficient siRNA (inhibition efficacy larger than 70%) is listed in Table 3. As siRNA sequence features correlate with siRNA self-structure free energies (,), SVM prediction with all other 26 parameters still performs reasonably. But the self-structure of mRNA () cannot be predicted by local siRNA sequence information, therefore only considering local features lowers the PPV by 5.1%. The PPV increased as much as 8.1% by using all three self-structure free energies with the other 25 local sequence parameters (Table 2). The predicted free energy changes associated with RNA structure as described earlier are among the most correlated features of functional siRNA (Table 2).
DISCUSSION
In this work, siRNA sequences were successfully selected using a SVM trained with equilibrium binding thermodynamics and siRNA sequence features. The equilibrium predictions explicitly account for the duplex stability, the self-structure in the target message and self-structure in the antisense siRNA strand. The equilibrium features provide improved siRNA selection as judged by the positive predictive value and sensitivity of the selection method.
Significant correlations were observed between siRNA efficacy and different thermodynamic parameters, although RNA secondary structure prediction itself is not perfect. The strongest correlation between target structure stability and efficacy was found when a complete ensemble of structures of RNA sequences was predicted with a rigorous partition function calculation, which has not been previously utilized. The correlation of target accessibility and siRNA efficacy was shown to be adequately predicted using 800 nucleotides of total sequence, centered on the binding region. The partition function calculation for predicting accessibility for binding is a new methodology that could also be applied widely, such as with microRNA target prediction, antisense oligonucleotide design and microarray analysis and design.
There is still room for improving the prediction of target accessibility. For example, the co-axial stacking between the hybrid helix and the helix of the target RNA were not included in these calculations (33). The kinetic control of binding also affects the efficacy of siRNA, which is considered as a local disruption free energy change in the Shao et al. (34) local model. This cannot be predicted by partition function calculation because the partition function predicts the RNA ensemble behavior at equilibrium. Furthermore, many tertiary interactions and protein binding on mRNA are yet unpredictable. The sequence identity of the 3′ overhangs of siRNA can also be considered for the design of siRNA. Although the two overhangs appear to have little or no effect on interference activity (8), they were also suggested to be involved in the interaction with proteins like the Paz domain of EIF2C2 (35).
A negative correlation coefficient was found between siRNA efficacy and the free energy change of the oligonucleotide-target duplex () (r = −0.2501, see details in Table 2). This result indicates that the siRNA would be less efficient if the direct binding between siRNA and mRNA were stronger. In other words, in general, low G/C content would be preferred by functional siRNA as has been noted previously (36). The same feature was shown for microRNAs (19). One simple explanation is that the free energy cost to unwind the siRNA (or microRNA) is more important than the strength of siRNA-target duplex formation. Alternatively, the RISC complex must be able to dissociate readily from targets after cleavage for multiple turnover and this may be improved by weaker binding by the antisense siRNA strand to the target or another single sense siRNA strand in the solution. The siRNA bimolecular stability was also found to have positive correlation with siRNA efficency, therefore, the propensity of siRNA to dimerize is disfavored. There is no clear mechanistic explanation for this effect, but it may be that RISC bound with antisense siRNA strand could be inhibited if an antisense strand hybridizes to a second antisense strand.
Efficient selection of siRNA is now incorporated in the RNAstructure software package for Microsoft Windows. This package is available for download at http://rna.urmc.rochester.edu.
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health grant R01GM076485 to D.H.M. D.H.M. is an Alfred P. Sloan Foundation Research Fellow. Funding to pay the Open Access publication charges for this article was provided by the National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 2.Tijsterman M, Plasterk RH. Dicers at RISC; the mechanism of RNAi. Cell. 2004;117:1–3. doi: 10.1016/s0092-8674(04)00293-4. [DOI] [PubMed] [Google Scholar]
- 3.Hannon GJ. RNA interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 4.Murchison EP, Hannon GJ. miRNAs on the move: miRNA biogenesis and the RNAi machinery. Curr. Opin. Cell Biol. 2004;16:223–229. doi: 10.1016/j.ceb.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 5.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat. Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- 6.Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 7.Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 8.Amarzguioui M, Prydz H. An algorithm for selection of functional siRNA sequences. Biochem. Biophys. Res. Commun. 2004;316:1050–1058. doi: 10.1016/j.bbrc.2004.02.157. [DOI] [PubMed] [Google Scholar]
- 9.Harborth J, Elbashir SM, Vandenburgh K, Manninga H, Scaringe SA, Weber K, Tuschl T. Sequence, chemical, and structural variation of small interfering RNAs and short hairpin RNAs and the effect on mammalian gene silencing. Antisense Nucleic A. 2003;13:83–105. doi: 10.1089/108729003321629638. [DOI] [PubMed] [Google Scholar]
- 10.Ui-Tei K, Naito Y, Takahashi F, Haraguchi T, Ohki-Hamazaki H, Juni A, Ueda R, Saigo K. Guidelines for the selection of highly effective siRNA sequences for mammalian and chick RNA interference. Nucleic Acids Res. 2004;32:936–948. doi: 10.1093/nar/gkh247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan B, Latek R, Hossbach M, Tuschl T, Lewitter F. siRNA selection server: an automated siRNA oligonucleotide prediction server. Nucleic Acids Res. 2004;32:W130–134. doi: 10.1093/nar/gkh366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huesken D, Lange J, Mickanin C, Weiler J, Asselbergs F, Warner J, Meloon B, Engel S, Rosenberg A, et al. Design of a genome-wide siRNA library using an artificial neural network. Nat. Biotechnol. 2005;23:995–1001. doi: 10.1038/nbt1118. [DOI] [PubMed] [Google Scholar]
- 13.Miyagishi M, Taira K. siRNA becomes smart and intelligent. Nat. Biotechnol. 2005;23:946–947. doi: 10.1038/nbt0805-946. [DOI] [PubMed] [Google Scholar]
- 14.Vickers TA, Wyatt JR, Freier SM. Effects of RNA secondary structure on cellular antisense activity. Nucleic Acids Res. 2000;28:1340–1347. doi: 10.1093/nar/28.6.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bohula EA, Salisbury AJ, Sohail M, Playford MP, Riedemann J, Southern EM, Macaulay VM. The efficacy of small interfering RNAs targeted to the type 1 insulin-like growth factor receptor (IGF1R) is influenced by secondary structure in the IGF1R transcript. J. Biol. Chem. 2003;278:15991–15997. doi: 10.1074/jbc.M300714200. [DOI] [PubMed] [Google Scholar]
- 16.Far RK, Sczakiel G. The activity of siRNA in mammalian cells is related to structural target accessibility: a comparison with antisense oligonucleotides. Nucleic Acids Res. 2003;31:4417–4424. doi: 10.1093/nar/gkg649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schubert S, Grunweller A, Erdmann VA, Kurreck J. Local RNA target structure influences siRNA efficacy: systematic analysis of intentionally designed binding regions. J. Mol. Biol. 2005;348:883–893. doi: 10.1016/j.jmb.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 18.Heale BS, Soifer HS, Bowers C, Rossi JJ. siRNA target site secondary structure predictions using local stable substructures. Nucleic Acids Res. 2005;33:e30. doi: 10.1093/nar/gni026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shabalina SA, Spiridonov AN, Ogurtsov AY. Computational models with thermodynamic and composition features improve siRNA design. BMC Bioinformatics. 2006;7:65. doi: 10.1186/1471-2105-7-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ladunga I. More complete gene silencing by fewer siRNAs: transparent optimized design and biophysical signature. Nucleic Acids Res. 2007;35:433–440. doi: 10.1093/nar/gkl1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ding Y, Lawrence CE. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res. 2003;31:7280–7301. doi: 10.1093/nar/gkg938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl Acad. Sci. USA. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doshi KJ, Cannone JJ, Cobaugh CW, Gutell RR. Evaluation of the suitability of free-energy minimization using nearest-neighbor energy parameters for RNA secondary structure prediction. BMC Bioinformatics. 2004;5:105. doi: 10.1186/1471-2105-5-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mathews DH, Burkard ME, Freier SM, Wyatt JR, Turner DH. Predicting oligonucleotide affinity to nucleic acid targets. RNA. 1999;5:1458–1469. doi: 10.1017/s1355838299991148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zuker M. The use of dynamic programming algorithms in RNA secondary structure prediction. In: Waterman MS, editor. Mathematical Methods for DNA Sequences. Boca Raton: CRC Press; 1989. [Google Scholar]
- 26.McCaskill JS. The equilibrium partition function and base pair probabilities for RNA secondary structure. Biopolymers. 1990;29:1105–1119. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- 27.Mathews DH. Using an RNA secondary structure partition function to determine confidence in base pairs predicted by free energy minimization. RNA. 2004;10:1178–1190. doi: 10.1261/rna.7650904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia T, SantaLucia J., Jr, Burkard ME, Kierzek R, Schroeder SJ, Jiao X, Cox C, Turner DH. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick pairs. Biochemistry. 1998;37:14719–14735. doi: 10.1021/bi9809425. [DOI] [PubMed] [Google Scholar]
- 29.Chang C, Lin C. LIBSVM: a library for support vector machines. 2001. http://www.csie.ntu.edu.tw/~cjlin/libsvm/.
- 30.Wuchty S, Fontana W, Hofacker IL, Schuster P. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers. 1999;49:145–165. doi: 10.1002/(SICI)1097-0282(199902)49:2<145::AID-BIP4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 31.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters provides improved prediction of RNA Secondary Structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 32.Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'Souza LM, Du Y, Feng B, Lin N, Madabusi LV, et al. The comparative RNA web (CRW) site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3 doi: 10.1186/1471-2105-3-2. article 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mir KU, Southern EM. Determining the influence of structure on hybridization using oligonucleotide arrays. Nat. Biotechnol. 1999;17:788–792. doi: 10.1038/11732. [DOI] [PubMed] [Google Scholar]
- 34.Shao Y, Chan CY, Maliyekkel A, Lawrence CE, Roninson IB, Ding Y. Effect of target secondary structure on RNAi efficiency. RNA. 2007;13:1631–1640. doi: 10.1261/rna.546207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma JB, Ye K, Patel DJ. Structural basis for overhang-specific small interfering RNA recognition by the PAZ domain. Nature. 2004;429:318–322. doi: 10.1038/nature02519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holen T, Amarzguioui M, Wiiger MT, Babaie E, Prydz H. Positional effects of short interfering RNAs targeting the human coagulation trigger tissue factor. Nucleic Acids Res. 2002;30:1757–1766. doi: 10.1093/nar/30.8.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]