


Abstract
Understanding regulatory mechanisms of protein synthesis in eukaryotes is essential for the accurate annotation of genome sequences. Kozak reported that the nucleotide sequence GCCGCC(A/G)CCAUGG (AUG is the initiation codon) was frequently observed in vertebrate genes and that this ‘consensus’ sequence enhanced translation initiation. However, later studies using invertebrate, fungal and plant genes reported different ‘consensus’ sequences. In this study, we conducted extensive comparative analyses of nucleotide sequences around the initiation codon by using genomic data from 47 eukaryote species including animals, fungi, plants and protists. The analyses revealed that preferred nucleotide sequences are quite diverse among different species, but differences between patterns of nucleotide bias roughly reflect the evolutionary relationships of the species. We also found strong biases of A/G at position −3, A/C at position −2 and C at position +5 that were commonly observed in all species examined. Genes with higher expression levels showed stronger signals, suggesting that these nucleotides are responsible for the regulation of translation initiation. The diversity of preferred nucleotide sequences around the initiation codon might be explained by differences in relative contributions from two distinct patterns, GCCGCCAUG and AAAAAAAUG, which implies the presence of multiple molecular mechanisms for controlling translation initiation.
INTRODUCTION
The control of translation initiation is one of the most fundamental processes in the regulation of gene expression. In 1978, Kozak (1,2) proposed the scanning model for translation initiation in eukaryotes. According to this model, the 40S ribosomal subunit with several initiation factors binds the 7-methyl guanosine cap at the 5′ end of an mRNA and moves along the mRNA until it encounters an AUG codon. It was also proposed that when the AUG codon is in the context of GCCGCC(A/G)CCAUGG (A/G represents A or G and AUG represents the translation initiation codon), which is called the ‘Kozak consensus sequence’, the efficiency of translation initiation is enhanced. However, the detailed molecular mechanism of translation initiation in eukaryotes is still unclear. Moreover, although the sequence is described as a ‘consensus’ sequence, the extent of conservation is quite low. It was reported that only 0.2% of vertebrate genes contain precisely the sequence GCCGCC(A/G)CCAUGG (3). We therefore avoid using the word ‘consensus’ in this context, and instead refer to the sequence as ‘preferred’ sequence.
Kozak compiled 211 genes (4) and 699 genes (5) primarily from vertebrates and obtained the above sequence. This sequence was initially thought to be essential for all eukaryotes (4). Later, however, it was revealed that a preferred nucleotide sequence around the initiation codon varies considerably among different species. The preferred sequences are GCGGC(A/C)(A/G)(A/C)CAUGGCG for Monocots (1127 genes), AAAAAAA(A/C)AAUGGCU for Dicots (derived from 3643 genes) (6), ACAACCAAAAUGGC for Drosophila melanogaster (192 genes), UAAAT(A/C)AACAUG(A/G)C for other invertebrates (155 genes), and AAAAAAAAAAUGTC for Saccharomyces cerevisiae (461 genes) (3). Kozak also reported that replacement of A/G at position −3 (three bases before the initiation codon) and G at position +4 (one base after the initiation codon) strongly impaired translation initiation in mammals (7,8). However, in S. cerevisiae nucleotide substitutions at position −3 did not substantially affect the rate of translation initiation (9,10), although there is a nucleotide bias towards A at this position (3). It therefore appears that the molecular mechanisms for recognizing the initiation codon vary among species.
There have been two limitations to previous studies aimed at identifying preferred sequences around the initiation codon. First, the number of species and genes examined was limited. In this study, we used whole-genome expression data and gene sequences from diverse eukaryote species. The second issue has been that the GC contents in genomes are known to differ from species to species. The preference for A before the initiation codon in Dicots and S. cerevisiae can be partially explained by the AT-richness of their genomes. To compare nucleotide sequences responsible for translation initiation among various species, differences in the usage of nucleotides in each genome must be considered. We previously invented a method of graphically representing nucleotide appearance biases at each position in a gene on the basis of the deviation from the expected values that are calculated for a given genome sequence (11,12). Application of this method to bacterial genomic data led to the successful identification of the Shine-Dalgarno (SD) sequence, a well-characterized signal for translation initiation in prokaryotes (11). We have also reported that the nucleotides appearing at the second codon (the codon next to the initiation codon) are highly biased in eukaryote genes and that a preferred second codon is characteristic of each species (e.g. GCG for mammals and plants) (12).
To obtain additional insight into the molecular mechanisms of translation initiation in eukaryotes, we extensively examined the nucleotide sequences around the initiation codon by using the method introduced above. We conducted comparative analyses of the biases in nucleotides located in positions proximal to the initiation codon among 47 eukaryote species including animals, fungi, plants and protists. We thereby were able to identify both universal and species-specific features, and these features possibly reflect the evolution of the mechanism of translation initiation.
MATERIALS AND METHODS
Data
We used cDNA or genome sequence data from 47 eukaryote species including 22 metazoans, eight plants, nine fungi and eight protists. Species names and the database used are shown in Table 1. We used only protein-coding genes that start from the AUG codon and end with a stop codon. As for human genes, we used genes in categories I–IV provided by the H-Invitational Database (13). When information about alternative splicing variants was available, only one representative sequence with the longest coding sequence (CDS) was used. Otherwise, all of the protein-coding genes were used [for UniGene database (14)]. The amount of expressed mRNAs in humans and S. cerevisiae, obtained by serial analysis of gene expression (SAGE), were downloaded from H-ANGEL (http://jbirc.jbic.or.jp/hinv/h-angel/) (15) and Holstege's web site (http://www.wi.mit.edu/young/expression.html) (16), respectively.
Table 1.
The 47 eukaryote species used for analysis
| Species | Common name | Databasea |
|---|---|---|
| Animals, Vertebrates | ||
| Homo sapiensb | Human | H-Invitational Database 3.0 (13) |
| Pan troglodytesb | Chimpanzee | Ensembl (CHIMP1A) (35) |
| Macaca fascicularisb | Crab-eating macaque | UniGene (14) |
| Macaca mulattab | Rhesus monkey | Ensembl (MMUL_0_1) |
| Mus musculusb | Mouse | FANTOM3 (36) |
| Rattus norvegicusb | Rat | Mammalian Gene Collection (37) |
| Oryctolagus cuniculusb | Rabbit | UniGene |
| Canis familiarisb | Dog | Ensembl (BROADD1) |
| Bos taurusb | Cattle | Mammalian Gene Collection |
| Sus scrofab | Pig | UniGene |
| Gallus gallusb | Chicken | Ensemble (WASHUC1) |
| Xenopus laevisb | African clawed frog | Xenopus Gene Collection (38) |
| Xenopus tropicalisb | Western clawed frog | Xenopus Gene Collection |
| Danio reriob | Zebrafish | Zebrafish Gene Collection (39) |
| Animals, Invertebrates | ||
| Ciona intestinalisb | Sea squirt | UniGene |
| Drosophila melanogasterb | Fruit fly | Ensemble (BDGP4) |
| Anopheles gambiaeb | African malaria mosquito | Ensemble (AgamP3) |
| Apis melliferab | Honeybee | Ensemble (AMEL2.0) |
| Bombyx morib | Domestic silkworm | UniGene |
| Tribolium castaneum | Red flour beetle | RefSeq (14) |
| Caenorhabditis elegansb | Ensemble (CEL150) | |
| Schistosoma japonicumb | UniGene | |
| Plants, Monocots | ||
| Oryza sativab | Rice | KOME (released on 24 December 2004) (40) |
| Hordeum vulgareb | Barley | UniGene |
| Triticum aestivumb | Bread wheat | UniGene |
| Zea maysb | Indian corn | UniGene |
| Plants, Dicots | ||
| Arabidopsis thalianab | Thale cress | TAIR (released on 28 February 2004) (41) |
| Glycine maxb | Soybean | UniGene |
| Lycopersicon esculentumb | Tomato | UniGene |
| Solanum tuberosumb | Potato | UniGene |
| Fungi | ||
| Saccharomyces cerevisiae | Budding yeast | MIPS (18) |
| Debaryomyces hansenii | RefSeq | |
| Eremothecium gossypii | RefSeq | |
| Kluyveromyces lactis | RefSeq | |
| Yarrowia lipolytica | RefSeq | |
| Candida glabrata | RefSeq | |
| Schizosaccharomyces pombe | Fission yeast | GeneDB (Version 2.1) (19) |
| Aspergillus fumigatus | RefSeq | |
| Cryptococcus neoformans | RefSeq | |
| Protists | ||
| Theileria parva | RefSeq | |
| Theileria annulata | RefSeq | |
| Cryptosporidium parvum | RefSeq | |
| Plasmodium falciparum | GeneDB (released on 26 January 2006) | |
| Leishmania major | RefSeq | |
| Trypanosoma brucei | RefSeq | |
| Dictyostelium discoideum | Slime mold | dictyBase (released on 3, May, 2006) (42) |
| Cyanidioschyzon merolae | Cyanidioschyzon merolae Genome Project (43) |
aThese data were downloaded from the following websites. H-Invitational Database 3.0, http://www.jbirc.jbic.or.jp/hinv/ahg-db/; Ensembl, http://www.ensembl.org/; UniGene, http://www.ncbi.nlm.nih.gov/sites/entrez?db=unigene; FANTOM3, http://fantom.gsc.riken.go.jp/; Mammalian Gene Collection, http://mgc.nci.nih.gov/; Xenopus Gene Collection, http://xgc.nci.nih.gov/; Zebrafish Gene Collection, http://zgc.nci.nih.gov/; RefSeq, http://www.ncbi.nlm.nih.gov/RefSeq/; KOME, http://cdna01.dna.affrc.go.jp/cDNA/; TAIR, http://www.arabidopsis.org/; MIPS, http://mips.gsf.de/; GeneDB, http://www.sanger.ac.uk/; dictyBase, http://dictybase.org/; Cyanidioschyzon merolae Genome Project, http://merolae.biol.s.u-tokyo.ac.jp/.
bSpecies for which cDNA data were used.
Evaluation of nucleotide frequency bias
To examine biases in nucleotide appearance around the initiation codon, all genes from each species were aligned at the initiation codons without any alignment gaps. The number of each nucleotide [A, U (T), G and C] was counted at each position in the alignment. The observed numbers of nucleotides were compared with the expected numbers using the likelihood-ratio statistic or the G-statistic, which is used for a test for goodness-of-fit (17). The expectations were calculated for each species in four separate categories, namely, the 5′ untranslated regions (UTRs) and the first, second and third positions in a codon in CDSs, because nucleotide frequencies are different among these categories. The G-value at position i was calculated by the formula:
| 1 |
where 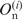 is the observed number of nucleotide n (A, U, G and C) at position i, and
is the observed number of nucleotide n (A, U, G and C) at position i, and 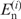 is the expected number of nucleotide n in the category to which position i belongs (5′ UTRs or the first, second or third positions in a codon). As regards the genomic data [RefSeq, MIPS and GeneDB (14,18,19)], 100 base-pair (bp) regions upstream from the initiation codon were regarded as the 5′ UTRs and data from these regions were used for the computation of the expectations. It is known that the distribution of the G-statistic is approximated by the χ2-distribution with f − 1 degrees of freedom when the sample size is large, where f is the number of different classes (f = 4). Each term in Formula 1 represents the contribution of each nucleotide to the bias. When
is the expected number of nucleotide n in the category to which position i belongs (5′ UTRs or the first, second or third positions in a codon). As regards the genomic data [RefSeq, MIPS and GeneDB (14,18,19)], 100 base-pair (bp) regions upstream from the initiation codon were regarded as the 5′ UTRs and data from these regions were used for the computation of the expectations. It is known that the distribution of the G-statistic is approximated by the χ2-distribution with f − 1 degrees of freedom when the sample size is large, where f is the number of different classes (f = 4). Each term in Formula 1 represents the contribution of each nucleotide to the bias. When 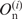 is larger and smaller than
is larger and smaller than 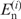 , the values of
, the values of 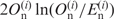 become positive and negative, respectively. For this reason, we regarded each term in Formula 1 as a measure of the bias for each nucleotide at a given position. G-values are proportional to the number of genes (N) when the fractions of observed and expected numbers of nucleotides are the same. To compare nucleotide biases among different species with different numbers of genes, we defined a value that is not affected by the number of genes,
become positive and negative, respectively. For this reason, we regarded each term in Formula 1 as a measure of the bias for each nucleotide at a given position. G-values are proportional to the number of genes (N) when the fractions of observed and expected numbers of nucleotides are the same. To compare nucleotide biases among different species with different numbers of genes, we defined a value that is not affected by the number of genes, 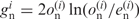 , where
, where 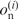 and
and 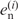 are the fractions of the observed and expected numbers of nucleotide n at position i. When
are the fractions of the observed and expected numbers of nucleotide n at position i. When 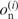 is zero,
is zero, 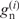 is defined to be zero. The G-value divided by N is equal to the sum of
is defined to be zero. The G-value divided by N is equal to the sum of 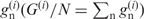 .
.
Cluster analysis of the patterns in nucleotide biases
We quantified similarities between the patterns in nucleotide bias around initiation codons by using the Pearson's correlation coefficient. The correlation coefficient rXY between species X and Y was calculated from the gn values from positions −9 to −1 in the 5′ UTRs, and from positions +4 to +6 in the CDSs (the second codon) as follows:
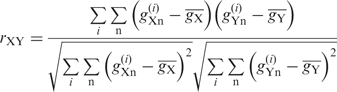 |
where 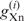 and
and 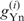 represent gn values of nucleotide n (A, U, G or C) at position i in species X and Y, respectively, and
represent gn values of nucleotide n (A, U, G or C) at position i in species X and Y, respectively, and 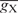 and
and 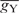 represent the average of gn values among all positions (from –9 to −1 and from +4 to +6) and nucleotides in species X and Y, respectively. We calculated r-values for all combinations among the 47 species examined and defined the similarity score D as 1 − r. Using the similarity scores, the cluster analysis was conducted by the group average method (Figure 3), the centroid method and the Ward method (Figure S1). Note that D is free from the absolute values of gn. When the number of genes used is small, the gn values tend to become large, apparently because highly expressed genes are more likely to be contained in a small gene set than are genes expressed at low levels. Therefore, although the gn values are affected by the number of genes, the values of D are expected to be robust against the difference in the number of genes for each species.
represent the average of gn values among all positions (from –9 to −1 and from +4 to +6) and nucleotides in species X and Y, respectively. We calculated r-values for all combinations among the 47 species examined and defined the similarity score D as 1 − r. Using the similarity scores, the cluster analysis was conducted by the group average method (Figure 3), the centroid method and the Ward method (Figure S1). Note that D is free from the absolute values of gn. When the number of genes used is small, the gn values tend to become large, apparently because highly expressed genes are more likely to be contained in a small gene set than are genes expressed at low levels. Therefore, although the gn values are affected by the number of genes, the values of D are expected to be robust against the difference in the number of genes for each species.
Figure 3.

Cluster dendrogram showing similarities between patterns of nucleotide bias. The distance D was calculated from the gn values from positions −9 to +6 (see Materials and Methods section). The group average method was used for the construction of the cluster dendrogram. An asterisk (*) indicates a cluster that is conserved among the dendrograms constructed by three different clustering methods (Figure S1).
Evaluation of hexanucleotide biases
We evaluated the deviation of the observed number of a particular combination of nucleotides from the expected number by using the Z-value (Tables 3 and 4 and Table S1 that is available as Supplementary Data). The Z-value was calculated by Z = (O − E)/[E (1 − E/N)]1/2, where N is the number of genes, and O and E are observed and expected numbers of a particular combination of nucleotides, respectively. The expected number was calculated by assuming that a nucleotide at each position appears independently (see the legend of Table 3). We calculated the Z-values for all possible combinations of six nucleotides (46 = 4096 combinations) in the region upstream of the initiation codon (from positions −6 to −1) and ranked them according to the Z-value. To see whether a sequence that is a mixture of GCCGCCAUG and AAAAAAAUG is suppressed in genes or not, we examined hexanucleotide sequences generated by combining three nucleotides from GCCGCCAUG and three nucleotides from AAAAAAAUG (e.g. GAAACCAUG or ACAGACAUG). There are 20 (6C3) such combinations (Table 4). From them, we excluded AAAGCCAUG and GCCAAAAUG, because GCCAUG and AAAAUG were observed much more frequently than the expectations (Table S1). We regarded the remaining 18 sequences as ‘mixed sequences’. We conducted the Wilcoxon rank sum test to see whether the ranks of the 18 mixed sequences are significantly low among the 4096 sequences or not.
Table 3.
Observed and expected numbers of genes containing a preferred sequence
| H. sapiens | C. elegans | O. sativa | A. thaliana | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pattern | O | E | Z | O | E | Z | O | E | Z | O | E | Z |
| GCCGCCAUG | 79 | 16.7 | 15.3* | 4 | 0.4 | 5.5* | 262 | 44.6 | 32.6* | 20 | 1.5 | 14.9* |
| AAAAAAAUG | 13 | 2.7 | 6.3* | 62 | 36.3 | 4.3* | 33 | 2.8 | 17.9* | 235 | 73.2 | 19.0* |
O and E represent the observed and expected numbers of genes containing a given sequence, respectively. E was calculated under the assumption that each nucleotide at each position appears independently. For example, E for AAAAAAAUG in humans was calculated as 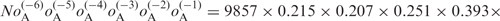
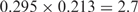 , where N is the number of human genes with 5′ UTRs that are six or more bases long, and oA(i) is the observed fraction of A at position i. The deviation of O from E was evaluated by the Z-value (see Materials and Methods section). An asterisk indicates P < 10−4.
, where N is the number of human genes with 5′ UTRs that are six or more bases long, and oA(i) is the observed fraction of A at position i. The deviation of O from E was evaluated by the Z-value (see Materials and Methods section). An asterisk indicates P < 10−4.
Table 4.
Observed and expected numbers of genes that contain a preferred sequence or a mixed sequence
| Pattern | O | E | Z | Rank |
|---|---|---|---|---|
| AAAAAAAUG | 1725 | 337.3 | 75.6 | 1 |
| GCCGCCAUG | 841 | 113.5 | 68.3 | 2 |
| ACAGCAAUG | 190 | 142.1 | 4.0 | 523 |
| AACACCAUG | 242 | 215.6 | 1.8 | 1008 |
| GAAGCAAUG | 158 | 139.6 | 1.6 | 1093 |
| GCAACAAUG | 274 | 261.9 | 0.7 | 1398 |
| ACAACCAUG | 225 | 240.4 | −1.0 | 2302 |
| ACAGACAUG | 146 | 162.9 | −1.3 | 2473 |
| GCACAAAUG | 54 | 69.1 | −1.8 | 2718 |
| GCAAACAUG | 261 | 300.2 | −2.3 | 2946 |
| GACAACAUG | 226 | 269.4 | −2.6 | 3113 |
| GACACAAUG | 185 | 234.9 | −3.3 | 3345 |
| GAAACCAUG | 186 | 236.3 | −3.3 | 3349 |
| ACCACAAUG | 185 | 239.0 | −3.5 | 3438 |
| GAAGACAUG | 115 | 160.1 | −3.6 | 3465 |
| ACCAACAUG | 204 | 274.0 | −4.2 | 3655 |
| AACGCAAUG | 72 | 127.4 | −4.9 | 3788 |
| AACGACAUG | 67 | 146.1 | −6.5 | 3972 |
| GACGAAAUG | 68 | 159.2 | −7.2 | 4027 |
| ACCGAAAUG | 50 | 162.0 | −8.8 | 4075 |
These numbers were obtained from 219 496 genes in all of the 47 species examined.
RESULTS
We evaluated biases in nucleotide appearance at each position around the initiation codon by using the G-statistic (see Materials and Methods section). Figure 1 shows the results obtained for 10 012 human genes. As shown in the upper diagram of this figure, the fractions of nucleotides A, T, G and C vary considerably in a position-dependent manner. The largest deviation of nucleotide frequencies from the expected values was observed at position −3, which is indicated by the largest G-value at this position (middle diagram). At this position, the values of gn are positive for A and G (lower diagram), which indicates that A and G appear more frequently than the expectations (see Materials and Methods section). In fact, the fractions of A and G at position −3 are 39.3% and 34.6%, respectively, which are much larger than those in the entire 5′ UTRs of 10 012 human genes (23.7% and 26.6%, respectively). The results depicted in the lower diagram suggest that the preferred sequence in humans is GCCGCC(A/G)(C/A)CAUGGCG, which is nearly the same as the sequence reported by Kozak (4,5). Note that the bias of GCG at the second codon is also quite strong, as we previously reported (12).
Figure 1.

Biases in nucleotide appearance for 10 012 human genes. Top, fractions of nucleotides appearing at each position in 24 base-pair (bp) regions in 5′ UTRs and CDSs. A, U, G and C are shown in green, magenta, yellow and blue, respectively. Middle, G-values divided by the number of genes (N = 10 012), showing the deviation from the expected values. Bottom, the values of gn for n = A, U, G or C at each position. The color scheme is the same as that used in the diagram at the top. Colored bars above and below the horizontal line indicate positive and negative gn values, respectively, and these bars were drawn without overlapping. In the middle and bottom diagrams, the values for the initiation codon (AUG) are omitted. Note that the biases shown in this figure are statistically highly significant (P << 10−10 from positions −9 to +6) because of the large sample size.
We applied the method described here to investigate the genes of 47 eukaryote species for which full-length cDNA or whole-genome data are available (Figure 2). These species included a wide variety of eukaryotes such as the soil-dwelling social amoeba Dictyostelium discoideum and the unicellular red alga Cyanidioschyzon merolae (Table 1). We used cDNA data when they are available, because gene annotation based on expression data is expected to be more accurate than that predicted from genome sequences. For most of the animal and plant species examined, cDNA data were used (Table 1). In Figure 2, only the region from positions −9 to +6 is shown, in which the G-values are relatively large. This figure reveals that the preferred nucleotide sequences around the initiation codon, as well as the extent of deviation from the expectations, vary among species. However, several features were commonly observed among species. For example, A is preferred at position −3 in all species examined. To compare the patterns of bias in nucleotide frequencies among different species, we quantified the similarity of gn values in the region from positions −9 to +6 between two species (see Materials and Methods section). By using a similarity score (D), we conducted the cluster analysis (Figure 3). The results showed that vertebrates, Monocots and Dicots each formed a cluster, thus indicating that the patterns of nucleotide bias are similar within each of these groups of organisms. Although the cluster dendrogram changed depending on the method of cluster analysis used, the clustering of vertebrates, Monocots and Dicots was robust (Figure S1). Fungi, invertebrates (containing urochordates, arthropods, nematodes and platyhelminithes) and each taxonomic group of protists also tended to form a cluster. These observations suggest that the patterns of nucleotide bias around the initiation codon roughly reflect the evolutionary relationships of eukaryote species.
Figure 2.

Nucleotide biases around the initiation codon in 47 eukaryote species. Each diagram shows gn values from positions −9 to +6 in each species. The name of the species and the number of genes used are also given. The color scheme is the same as that used in Figure 1. The initiation codon (AUG) is not shown.
Figure 4 shows the preferred sequences for each taxonomic group of eukaryotes. These sequences were obtained by taking the average of the patterns of nucleotide bias for all species belonging to each group. The sequences obtained here are similar to those previously reported. For example, for Monocots the preferred sequence obtained is G(A/C)(G/A)GC(A/C/G)(G/A)(C/A)(G/C)AUGGCG, which is similar to that reported in Joshi et al. (6) (see Introduction section). The following biases in nucleotide appearance were commonly observed among all taxonomic groups examined: −6G (G at position −6), −3A/G, −2A/C and +5C. Of these biases, −3A is the most remarkable. Moreover, a general tendency toward the under-representation of U around the initiation codon was observed. The biases in protists are relatively weak, reflecting highly variable patterns of nucleotide bias in these species (Figure 2).
Figure 4.

Nucleotide biases around the initiation codon for each taxonomic group of eukaryotes. The gn value at each position for each nucleotide was calculated from the average of on values and that of en values among all species belonging to a given taxonomic group. The number of species used is shown for each group. sp., species.
Figure 4 also suggests that A-rich biases are present in the region from positions −4 to −1 in almost all species examined. These biases are clearly observed even in species with very low GC content. For example, the fraction of A in the D. discoideum genome is 38.8% (the GC content is 22.4%) (20), while the fraction of A at position −3 is as high as 85.9% (Figure S2). Moreover, we identified signals that had not been reported to date. Monocots showed a signal of GC(C/G)GC(C/G)AUG as mentioned above, but a similar pattern was also observed in Dicots. Furthermore, a relatively weak but clear signal of GCCGCCAUG was detected from invertebrates and fungi, which is similar to the sequence for vertebrates. It therefore appears that the preferred sequences in eukaryotes can be regarded as a summation of the repetition of GCC and that of A (see Discussion section).
To determine whether the biases described above are responsible for the efficiency of translation initiation, we examined the correlation of the strengths of the biases with gene expression levels. It is reasonable to assume that the translation rates would be high for highly expressed genes and low for genes expressed at low levels. In fact, we have conducted a genome-wide microarray analysis and shown that the efficiency of translation initiation is correlated with the expression level of mRNAs for S. cerevisiae genes (Akiyama et al., unpublished data). Therefore, the signals for efficient translation initiation are assumed to be more conspicuous for highly expressed genes. Figure 5A indicates the results for 1000 genes with high expression levels and those for 1000 genes with low expression levels in humans and S. cerevisiae. Table 2 gives the fractions of −3A and +5G for genes expressed at high and low levels, and those for the entire set of genes in the two species. These results clearly show that the biases identified by using an entire set of genes became stronger when highly expressed genes were used. These results suggest that some of the preferred sequences identified in this study are responsible for the efficiency of translation initiation.
Figure 5.

Several features of nucleotide bias around the initiation codon. (A) Nucleotide bias around the initiation codon for genes expressed at high and low levels in humans (left) and S. cerevisiae (right). The diagram for each species shown at the left is the same as that in Figure 2. The middle and right diagrams for each species were calculated by using the top 1000 genes with higher expression levels and the bottom 1000 genes with lower expression levels, respectively, in each species. (B) Three-base periodicity observed in humans (left) and Oryza sativa (right). Arrows indicate every three bases. (C) U- and A-rich biases observed at positions around −40 and around −15, respectively, which are indicated by arrows. The gn values are shown from positions −100 to −1 for Xenopus laevis, Danio rerio, D. melanogaster and Anopheles gambiae.
Table 2.
Fractions (%) of -3A and +5G for genes expressed at high and low levels
| H. sapiens | S. cerevisiae | |||||
|---|---|---|---|---|---|---|
| All | Top 1000 | Bottom 1000 | All | Top 1000 | Bottom 1000 | |
| −3A | 39.3 | 46.3 | 36.8 | 58.2 | 72.2 | 46.0 |
| +5C | 35.8 | 41.5 | 34.4 | 38.0 | 50.6 | 29.2 |
We also identified a clear pattern of three-base periodicity from several vertebrate and Monocot species (Figure 5B). Interestingly, a similar pattern of a GCC or GCG repeat was observed in both 5′ UTRs and CDS regions, and the biases were more prominent in the regions near the initiation codon. One might suspect that the three-base periodicity in the 5′ UTRs is due to an artifact, i.e. that the CDSs are wrongly annotated as UTRs because of an inaccurate assignment of initiation codons. To determine whether this observation is due to an artifact or not, we conducted the same analysis using only the genes containing an in-frame stop codon in the 5′ UTR, but which do not contain any in-frame AUG codons between the initiation codon and the closest upstream stop codon from the initiation codon (Figure S3A). For such genes, there are no possibilities that wrongly annotated 5′ UTRs used in the analyses contain CDSs. The results clearly show that the periodic pattern described above is also observed in such genes, suggesting that this pattern is not an artifact, but rather a signal for the initiation of translation (Figure S3B).
As shown in Figure 5C, U- and A-rich biases were commonly observed around positions −40 and −15, respectively, in amphibians, fishes and insects. In D. melanogaster, for example, the average fraction of U in the region from positions −45 to −35 is 28.2%, and the average fraction of A from positions −20 to −10 is 35.6%. These values are considerably larger than the averages in the entire 5′ UTRs (21.4% for U and 31.5% for A). However, these biases are not clear in the other species.
DISCUSSION
In this study, we revealed that the signals −3A/G, −2A/C and +5C are common among various eukaryote species (Figure 4). Several studies have shown that −3A/G plays the most crucial role in enhancing translation initiation (7,21–24). In accord with these studies, our results indicated that the signal of −3A/G is the most remarkable in almost all eukaryote species examined, and this signal is even stronger in highly expressed genes (Figure 5A). Recently, Pisarev et al. (24) demonstrated that −3G in an mRNA interacts with a eukaryotic initiation factor eIF2α by using a rabbit cell system, although the amino acids in an eIF2α that are involved in the interaction are still unknown. Based on this observation, a purine base at position −3 was proposed to interact with an eIF2α as a key element in translation initiation. Since the amino acid sequences of this protein are highly conserved among various eukaryotes, the interaction between the nucleotide at position −3 and an eIF2α may be a common mechanism for translation initiation.
Although Kozak (25) reported that recognition of the initiation codon was not augmented by a nucleotide at position +5, other researchers suggested that +5A/C in mammals or +5C in plants affect the efficiency of translation initiation (26,27). Our analyses using various eukaryotes (Figures 2 and 4) and highly expressed genes (Figure 5A and Table 2) suggest the importance of +5C for translation initiation. However, since the nucleotide at position +5 determines the chemical properties of the second amino acid, it is also possible that this nucleotide is under the functional constraint of the amino acid sequences (12). As regards the −2A/C signal, to our knowledge, there has been no experimental data suggestive of its role in the initiation of translation.
Although +4G has been described as important for translation initiation in vertebrates and plants, the effect of +4G is relatively minor compared with that of −3A/G (21,24,25). Our study showed that the nucleotide appearing at position +4 is highly biased, but a preferred nucleotide is not common among all eukaryotes. In vertebrates and plants, G is preferred at this position, whereas in invertebrates, fungi and protists, T is generally preferred. The biases of +4G in humans and +4U in S. cerevisiae were even more conspicuous when highly expressed genes were examined, suggesting the possibility that position +4 is involved in enhancing translation initiation; however, the nucleotide at the position required for effective translation initiation appear to be diversified among eukaryotes.
In the original scanning model, it was postulated that translation is initiated from the first AUG codon in an mRNA (8). However, it has since been revealed that the actual mechanism of translation initiation is much more complicated than previously thought. It is known that AUG trinucleotides referred to as upstream AUGs (uAUGs) are frequently observed in 5′ UTRs, and that short open reading frames designated as upstream ORFs (uORFs) are often also present (28). It has been reported that ∼55% and ∼25% of mammalian genes have one or more uAUGs and uORFs, respectively (29). These uAUGs and uORFs are apparently involved in the down-regulation of translation (30). Moreover, even if the first AUG codon is located within a context of a ‘Kozak consensus sequence’, translation is not necessarily initiated from it (31,32). Dresios et al. (33) suggested that a short element in a eukaryotic mRNA directly base pairs with an 18S rRNA to enhance translation initiation, which is similar to the interaction of the SD sequence with a 16S rRNA in a prokaryotic mRNA. It should be noted that the original scanning model cannot account for these observations.
Our results are consistent with the previous assertion that preferred sequences around the initiation codon vary among different eukaryote species (Figure 2) (5,6). However, Figure 4 suggests that the sequences could generally be decomposed into two distinct patterns, the repetition of GCC and that of A. To examine this possibility in more detail, we compared the observed and expected numbers of genes containing the sequences GCCGCCAUG and AAAAAAAUG in several species (Table 3). The expected number was calculated based on the assumption that an observed nucleotide at each position will appear in a manner independent of a nucleotide at another position. The results clearly showed that the observed numbers are significantly larger than the expected numbers for these sequences. It is therefore suggested that the sequence GCCGCCAUG or AAAAAAAUG, and not a particular nucleotide at each position, may play a role as a whole in translation initiation. We further examined the existence of genes containing a hexanucleotide sequence that is a mixture of these two sequences (e.g. GAAACCAUG or ACAGACAUG). We then found that such mixed sequences are significantly suppressed in genes (P < 0.01), while AAAAAAAUG and GCCGCCAUG are the most and the second most over-represented patterns, respectively, among all hexanucleotide sequences (Table 4). These observations support the idea that there are two distinct patterns of signals for translation initiation.
If we assume the presence of the two distinct patterns of signals, then the variation in preferred sequences among different species could be accounted for by differences in the relative contribution from each pattern. In vertebrates or Monocots, the signal of GCC repeats is relatively strong, whereas in invertebrates or Dicots, the signal of repetition of A is more conspicuous. What, then, determines the relative contribution of each pattern to the preferred sequence in a given species? One factor might be the GC content in the genome. Figure 6A shows the GC content in 5′ UTRs and that in the whole genome sequences in 25 species with data for more than 3000 genes. This figure suggests that these species can be classified into two groups, i.e. GC-rich and AT-rich. As shown in Figure 6B, a species belonging to the GC-rich group shows a clear signal of GCC repeats, while an AT-rich species frequently exhibits very strong signals of A. These distinct signals might be recognized by different molecular mechanisms. Kozak (34) herself pointed out that the ‘Kozak consensus sequence’ is repetitious and that the unit of recognition may be a three-base motif. The three-base periodicity observed in this study might help a ribosome locate the correct reading frame. However, the mechanisms for recognizing the abovementioned signals remain unknown at this stage of research. Additional experimental studies will be required to gain a more precise understanding of the molecular mechanisms of translation initiation in eukaryotes.
Figure 6.

Correlation between GC contents and nucleotide bias around the initiation codon. (A) Horizontal and vertical axes represent the GC contents in the whole genome and in the 5′ UTRs, respectively. All species with data for >3000 genes were used. These species can be classified into two groups, i.e. GC-rich (yellow circle) and AT-rich (green circle). We used the genomic GC contents in X. laevis (35) and Schistosoma japonicum (14) for those in Xenopus tropicalis and Schistosoma mansoni, respectively, because the genome sequences of X. laevis and S. japonicum are not available. The name of each species is represented by the initial letter of the generic name and the first three letters of the specific name (Table 1). (B) Bias in nucleotide appearance around the initiation codon for 13 GC-rich species and for 12 AT-rich species. These diagrams were created in the same manner as those in Figure 4.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We would like to thank Tadashi Imanishi, Motohiko Tanino, Kaoru Mogushi, Takeshi Fukuhara, Emilio Campos, Takeshi Hase, Yutaka Fukuoka, Tadashi Masuda, Soichi Ogishima, and Fengrong Ren for their helpful comments and discussion. Funding for this work was provided by the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Genome Information Integration Project of the Ministry of Economy, Trade and Industry of Japan, and the Japan Biological Informatics Consortium (17710162 to Y.N.). Funding to pay the Open Access publication charges for this article was provided by Tokyo Medical and Dental University.
Conflict of interest statement. None declared.
REFERENCES
- 1.Kozak M. How do eucaryotic ribosomes select initiation regions in messenger RNA? Cell. 1978;15:1109–1123. doi: 10.1016/0092-8674(78)90039-9. [DOI] [PubMed] [Google Scholar]
- 2.Kozak M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 2002;299:1–34. doi: 10.1016/S0378-1119(02)01056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cavener D, Ray S. Eukaryotic start and stop translation sites. Nucleic Acids Res. 1991;19:3185–3192. doi: 10.1093/nar/19.12.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kozak M. Compilation and analysis of sequences upstream from the translational start site in eukaryotic mRNAs. Nucleic Acids Res. 1984;12:857–872. doi: 10.1093/nar/12.2.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kozak M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987;15:8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joshi C, Zhou H, Huang X, Chiang V. Context sequences of translation initiation codon in plants. Plant Mol. Biol. 1997;35:993–1001. doi: 10.1023/a:1005816823636. [DOI] [PubMed] [Google Scholar]
- 7.Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- 8.Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234:187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- 9.Cigan A, Donahue T. Sequence and structural features associated with translational initiator regions in yeast – a review. Gene. 1987;59:1–18. doi: 10.1016/0378-1119(87)90261-7. [DOI] [PubMed] [Google Scholar]
- 10.Yun D, Laz T, Clements J, Sherman F. mRNA sequences influencing translation and the selection of AUG initiator codons in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 1996;19:1225–1239. doi: 10.1111/j.1365-2958.1996.tb02468.x. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe H, Gojobori T, Miura K. Bacterial features in the genome of Methanococcus jannaschii in terms of gene composition and biased base composition in ORFs and their surrounding regions. Gene. 1997;205:7–18. doi: 10.1016/s0378-1119(97)00517-9. [DOI] [PubMed] [Google Scholar]
- 12.Niimura Y, Terabe M, Gojobori T, Miura K. Comparative analysis of the base biases at the gene terminal portions in seven eukaryote genomes. Nucleic Acids Res. 2003;31:5195–5201. doi: 10.1093/nar/gkg701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imanishi T, Itoh T, Suzuki Y, O’Donovan C, Fukuchi S, Koyanagi K, Barrero R, Tamura T, Yamaguchi-Kabata Y, et al. Integrative annotation of 21,037 human genes validated by full-length cDNA clones. PLoS Biol. 2004;2:e162. doi: 10.1371/journal.pbio.0020162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wheeler D, Barrett T, Benson D, Bryant S, Canese K, Chetvernin V, Church D, DiCuccio M, Edgar R, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2006;34:D173–D180. doi: 10.1093/nar/gkj158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanino M, Debily M, Tamura T, Hishiki T, Ogasawara O, Murakawa K, Kawamoto S, Itoh K, Watanabe S, et al. The Human Anatomic Gene Expression Library (H-ANGEL), the H-Inv integrative display of human gene expression across disparate technologies and platforms. Nucleic Acids Res. 2005;33:D567–D572. doi: 10.1093/nar/gki104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holstege F, Jennings E, Wyrick J, Lee T, Hengartner C, Green M, Golub T, Lander E, Young R. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- 17.Sokal RR, Rohlf FJ. Biometry. 3rd. New York: W.H.Freeman and Co.; 1993. pp. 689–697. [Google Scholar]
- 18.Mewes H, Frishman D, Mayer K, Münsterkötter M, Noubibou O, Pagel P, Rattei T, Oesterheld M, Ruepp A, et al. MIPS: analysis and annotation of proteins from whole genomes in 2005. Nucleic Acids Res. 2006;34:D169–D172. doi: 10.1093/nar/gkj148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hertz-Fowler C, Peacock C, Wood V, Aslett M, Kerhornou A, Mooney P, Tivey A, Berriman M, Hall N, et al. GeneDB: a resource for prokaryotic and eukaryotic organisms. Nucleic Acids Res. 2004;32:D339–D343. doi: 10.1093/nar/gkh007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eichinger L, Pachebat J, Glöckner G, Rajandream M, Sucgang R, Berriman M, Song J, Olsen R, Szafranski K, et al. The genome of the social amoeba Dictyostelium discoideum. Nature. 2005;435:43–57. doi: 10.1038/nature03481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lukaszewicz M, Feuermann1 M, Jérouville B, Stas A, Boutry M. In vivo evaluation of the context sequence of the translation initiation codon in plants. Plant Sci. 2000;154:89–98. doi: 10.1016/s0168-9452(00)00195-3. [DOI] [PubMed] [Google Scholar]
- 22.Kochetov A. AUG codons at the beginning of protein coding sequences are frequent in eukaryotic mRNAs with a suboptimal start codon context. Bioinformatics. 2005;21:837–840. doi: 10.1093/bioinformatics/bti136. [DOI] [PubMed] [Google Scholar]
- 23.Pesole G, Gissi C, Grillo G, Licciulli F, Liuni S, Saccone C. Analysis of oligonucleotide AUG start codon context in eukariotic mRNAs. Gene. 2000;261:85–91. doi: 10.1016/s0378-1119(00)00471-6. [DOI] [PubMed] [Google Scholar]
- 24.Pisarev A, Kolupaeva V, Pisareva V, Merrick W, Hellen C, Pestova T. Specific functional interactions of nucleotides at key -3 and +4 positions flanking the initiation codon with components of the mammalian 48S translation initiation complex. Genes Dev. 2006;20:624–636. doi: 10.1101/gad.1397906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kozak M. Recognition of AUG and alternative initiator codons is augmented by G in position +4 but is not generally affected by the nucleotides in positions +5 and +6. EMBO J. 1997;16:2482–2492. doi: 10.1093/emboj/16.9.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grünert S, Jackson R. The immediate downstream codon strongly influences the efficiency of utilization of eukaryotic translation initiation codons. EMBO J. 1994;13:3618–3630. doi: 10.1002/j.1460-2075.1994.tb06669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sawant S, Kiran K, Singh P, Tuli R. Sequence architecture downstream of the initiator codon enhances gene expression and protein stability in plants. Plant Physiol. 2001;126:1630–1636. doi: 10.1104/pp.126.4.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris D, Geballe A. Upstream open reading frames as regulators of mRNA translation. Mol. Cell. Biol. 2000;20:8635–8642. doi: 10.1128/mcb.20.23.8635-8642.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crowe M, Wang X, Rothnagel J. Evidence for conservation and selection of upstream open reading frames suggests probable encoding of bioactive peptides. BMC Genomics. 2006;7:16. doi: 10.1186/1471-2164-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meijer H, Thomas A. Control of eukaryotic protein synthesis by upstream open reading frames in the 5′-untranslated region of an mRNA. Biochem. J. 2002;367:1–11. doi: 10.1042/BJ20011706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rogers GW, Edelman GM, Mauro VP. Differential utilization of upstream AUGs in the beta-secretase mRNA suggests that a shunting mechanism regulates translation. Proc. Natl Acad. Sci. USA. 2004;101:2794–2799. doi: 10.1073/pnas.0308576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lammich S, Schöbel S, Zimmer A, Lichtenthaler S, Haass C. Expression of the Alzheimer protease BACE1 is suppressed via its 5′-untranslated region. EMBO Rep. 2004;5:620–625. doi: 10.1038/sj.embor.7400166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dresios J, Chappell SA, Zhou W, Mauro VP. An mRNA-rRNA base-pairing mechanism for translation initiation in eukaryotes. Nat. Struct. Mol. Biol. 2006;13:30–34. doi: 10.1038/nsmb1031. [DOI] [PubMed] [Google Scholar]
- 34.Kozak M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J. Mol. Biol. 1987;196:947–950. doi: 10.1016/0022-2836(87)90418-9. [DOI] [PubMed] [Google Scholar]
- 35.Birney E, Andrews D, Caccamo M, Chen Y, Clarke L, Coates G, Cox T, Cunningham F, Curwen V, et al. Ensembl 2006. Nucleic Acids Res. 2006;34:D556–D561. doi: 10.1093/nar/gkj133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carninci P, Kasukawa T, Katayama S, Gough J, Frith M, Maeda N, Oyama R, Ravasi T, Lenhard B, et al. The transcriptional landscape of the mammalian genome. Science. 309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 37.Gerhard D, Wagner L, Feingold E, Shenmen C, Grouse L, Schuler G, Klein S, Old S, Rasooly R, et al. The status, quality, and expansion of the NIH full-length cDNA project: the Mammalian Gene Collection (MGC) Genome Res. 2004;14:2121–2127. doi: 10.1101/gr.2596504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morin R, Chang E, Petrescu A, Liao N, Griffith M, Chow W, Kirkpatrick R, Butterfield Y, Young A, et al. Sequencing and analysis of 10,967 full-length cDNA clones from Xenopus laevis and Xenopus tropicalis reveals post-tetraploidization transcriptome remodeling. Genome Res. 2006;16:796–803. doi: 10.1101/gr.4871006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rasooly R, Henken D, Freeman N, Tompkins L, Badman D, Briggs J, Hewitt AT The National Institutes of Health Trans-NIH Zebrafish Coordinating Committee. Genetic and genomic tools for zebrafish research: the NIH zebrafish initiative. Dev. Dyn. 2003;228:490–496. doi: 10.1002/dvdy.10366. [DOI] [PubMed] [Google Scholar]
- 40.Rhee S, Beavis W, Berardini T, Chen G, Dixon D, Doyle A, Garcia-Hernandez M, Huala E, Lander G, et al. The Arabidopsis Information Resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res. 2003;31:224–228. doi: 10.1093/nar/gkg076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kikuchi S, Satoh K, Nagata T, Kawagashira N, Doi K, Kishimoto N, Yazaki J, Ishikawa M, Yamada H, et al. Collection, mapping, and annotation of over 28,000 cDNA clones from japonica rice. Science. 2003;301:376–379. doi: 10.1126/science.1081288. [DOI] [PubMed] [Google Scholar]
- 42.Chisholm R, Gaudet P, Just E, Pilcher K, Fey P, Merchant S, Kibbe W. dictyBase, the model organism database for Dictyostelium discoideum. Nucleic Acids Res. 2006;34:D423–D427. doi: 10.1093/nar/gkj090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsuzaki M, Misumi O, Shin-i T, Maruyama S, Takahara M, Miyagishima S, Mori T, Nishida K, Yagisawa F, et al. Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D. Nature. 2004;428:653–657. doi: 10.1038/nature02398. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.