


Abstract
Mucosal human papillomaviruses (HPVs) are etiological agents of oral, anal and genital cancer. Properties of high- and low-risk HPV types cannot be reduced to discrete molecular traits. The E2 protein regulates viral replication and transcription through a finely tuned interaction with four sites at the upstream regulatory region of the genome. A computational study of the E2–DNA interaction in all 73 types within the alpha papillomavirus genus, including all known mucosal types, indicates that E2 proteins have similar DNA discrimination properties. Differences in E2–DNA interaction among HPV types lie mostly in the target DNA sequence, as opposed to the amino acid sequence of the conserved DNA-binding alpha helix of E2. Sequence logos of natural and in vitro selected sites show an asymmetric pattern of conservation arising from indirect readout, and reveal evolutionary pressure for a putative methylation site. Based on DNA sequences only, we could predict differences in binding energies with a standard deviation of 0.64 kcal/mol. These energies cluster into six discrete affinity hierarchies and uncovered a fifth E2-binding site in the genome of six HPV types. Finally, certain distances between sites, affinity hierarchies and their eventual changes upon methylation, are statistically associated with high-risk types.
INTRODUCTION
Human papillomaviruses (HPVs) are widespread pathogens that infect epithelia (1,2). There are over a hundred HPV types, of which roughly half can infect mucosal tissues and the other half produce common skin warts. All mucosal HPV types belong to the alpha papillomavirus genus, together with twelve cutaneous HPV types and two simian papillomaviruses (3,4). Mucosal HPV types are the etiological agents of cervical cancer, the second most common cancer in women with more than 200 000 deaths per year worldwide, and are also a causative agent of vaginal, anal, penile, and head and neck cancer (1,5). Mucosal HPV types differ widely in their oncogenic potential, with 19 types classified as ‘high-risk’ (types 16, 18, 26, 31, 33, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 68a, 73, 82, 82subtype) and 13 as ‘low-risk’ (types 6, 6a, 6b, 11, 40, 42, 43, 44, 54, 61, 70, 72 and 81) according to epidemiological evidence (2). Two prophylactic vaccines against types 6, 11, 16 and 18 have recently become available (6). However, they are unlikely to be introduced in the short term in developing countries, which account for 80% of the deaths due to cervical cancer (6). Moreover, it is not clear whether they protect against infection with all high-risk types and cannot cure the millions of people that are already infected. Therefore, there is still a need for understanding the oncogenicity of papillomaviruses in more detail.
HPVs are small viruses with an 8 kb double-stranded DNA genome that typically codes for only eight proteins (7). The E2 protein is a multifunctional polypeptide that plays a crucial role in HPV replication (8), regulation of transcription from the early promoter (7,8), and genome segregation (8). It is a multidomain protein formed by two globular domains linked by a flexible ‘hinge’ region (8). The C-terminal domain (E2C) functions as a dimerization (9,10) and DNA-binding domain (8,11) (Figure 1A). Several groups have studied the binding to DNA of E2 proteins from alpha HPV types 6, 11, 16, 18, 33 and 51 (8,12–29). All of these domains bind a pseudopalindromic target site with the consensus sequence aACCg(A/T)4cGGTt, where capital letters indicate strongly required bases, small letters weakly required bases and (A/T)4 a four-base long spacer often rich in A or T (8). The bases in the spacer do not make direct contact with the protein but contribute to the free energy of binding by indirect readout (8,12,16–19,30–36). The E2–DNA interaction is an important model system for the study of such effects in protein–DNA complexes (8,12,16–19,30–36). Strong binding to sequences not matching the consensus has also been described (27). E2 binds DNA as a homodimer (Figure 1A), with a helix of each monomer contacting two consecutive major grooves of its target site (8,19). The protein side chains contribute in an additive manner to the free energy of binding (22,23).
Figure 1.

Conserved features of the E2–DNA interaction. (A) Complex of the c-terminal domain of the HPV18 E2 protein with the idealized target DNA sequence CAACCGAATTCGGTTG. The two four-base half-sites in direct contact with the protein are shown in red, the four-base linker in silver and the two flanking bases in gold. The protein helices that contact the DNA directly in green. (B) Sequence logo (63,68) of the recognition helix for alpha papillomaviruses. Protein residues contributing more than 0.8 kcal/mol to the binding energy of HPV16 E2 (23) are indicated with asterisks. (C) Correlation between the free energies of binding of E2 proteins from HPV type 11 and 16 to four E2-BSs (open triangle) (16,19,20) and of E2 proteins from HPV types 18 and 16 to another set of four E2-BSs (filled square) (16,19,20). The correlation R-values are 0.87 (16/11 pair, dashed line) and 0.91 (16/18 pair, continuous line). The sequences of the DNA-binding helix of the three proteins are also shown, with the side chains contributing more than 0.8 kcal/mol to the binding energy of HPV16 E2 (23) in bold.
The E2 protein binds to four conserved sites (E2-BS) in the upstream regulatory region (URR) of the alpha papillomavirus genome (7), numbered according to their distance to the early promoter (Figure 2A). The function of each site in alpha HPV types 6, 11, 16, 18 and 31 has been studied by site mutation and deletion in replication and transcription assays. Binding of E2 to E2-BS1, E2-BS2 and E2-BS3 recruits the E1 protein to the origin of replication. E2-BS2 is the most important site for this function, followed by E2-BS1 and E2-BS3 (27,37–42). Binding of E2 to E2-BS4 induces transcription from the early promoter (43–45), leading to production of early viral proteins, including E2 itself and the oncogenic proteins E6 and E7. Unregulated transcription from the early promoter, usually due to disruption of the E2 gene upon integration of viral DNA into the host genome, leads to the accumulation of excessive amounts of E6 and E7 and is associated with cancer. Binding of E2 to E2-BS1, E2-BS2 and E2-BS3 represses transcription from the early promoter through displacement of Sp1 and TBP from their binding sites (Figure 2A), keeping the levels of E6 and E7 under control. E2-BS1 is the most important site for this function, followed by E2-BS2 and E2-BS3 (27,41,43–48).
Figure 2.

E2-binding sites in alpha papillomaviruses. (A) Schematic view of the upstream regulatory region of a prototypical alpha HPV genome. Shown are the flanking ORFs, L1 and E6, the start of the early promoter and its TATA box, the four binding sites for the E2 protein, the binding sites for the viral protein E1 and the host protein Sp1 and the silencer, enhancer and nuclear matrix attachment regions. (B) Sequence logos of the four E2-binding sites. Sites are shown in the 5′–3′ direction. (C) Histograms of the number of bases between E2-binding sites.
The E2–DNA interaction can be regulated by methylation of CG dinucleotides within the target site. This covalent modification is known to reduce the binding affinity of E2 for its binding sites (14) and the transcriptional activity of the protein (49). In vivo methylation can also modify the accessibility of papillomavirus DNA through chromatin remodeling (50). E2-BSs of types 16 and 18 are targeted by the host methylation machinery in a degree that changes with the differentiation state of the cell, the integrity of the viral genome and the progression of disease (49,51–55). It is not known whether methylation is a defense mechanism of the host, an integral part of the life cycle of the virus or a disease-related event (49,51–55).
Binding of E2 to its four target sites is hierarchical (7,17,20,44). The expression of E2 is finely regulated during the HPV life cycle (56,57), leading to changes in site occupancy that control transcription of early proteins and replication (7,17,20,44). Since cellular factors that compete with E2 for binding to the viral DNA have very low relative affinities (58) and binding of E2 to adjacent sites takes place with low cooperativity (58), the hierarchy of binding can in principle be described using only the affinity of E2 for the E2-BSs. The hierarchy of sites is not conserved between high- and low-risk HPV types and could be related to the development of cancer (17,20).
Many studies have compared proteins from prototypical high- and low-risk types looking for biochemical properties associated with the progression of disease. In the case of the E2 protein, these properties include changes in the nuclear localization signals (59), the mechanism of DNA binding (25,28), the hierarchy of sites (17,20), and tighter binding to DNA (60) and to p53 (61). To date, all the work on the interaction of E2 with its target DNA has focused on domains from a handful of HPV types. While it is important to elucidate how the most prevalent and oncogenic types function, a complete understanding of papillomaviruses and their role on disease should include all known types and integrate the full range of epidemiological and biochemical data (62). The study of the E2–DNA interaction in all HPV types would be a difficult experimental task. Here, we present a computational study on the E2–DNA interaction in all 73 known alpha papillomavirus types. We improved the description of the binding specificity of E2 using sequence logos (63) and revealed a strong selection for a methylation site within E2-binding sites. We showed that the affinity of E2 for its target DNA can be accurately predicted from an alignment of natural sites and used this result to infer the affinity hierarchy of sites for all types. Finally, we were able to identify molecular features of the interaction that are significantly over- and underrepresented in high-, low-risk and cutaneous HPV types.
MATERIALS AND METHODS
Database of E2-binding sites
All alfa papillomavirus genomes were obtained from the International Committee on Taxonomy of Viruses data Base (64) (taxonomy ID: 151340, see Table S1 for accession codes). Genomes with more than one entry were tested for redundancy with BLAST 2 Sequences (65). In most cases, the starting point for numbering of the genome falls inside the URR, which splits the upstream regulatory region into two stretches at the beginning and end of the given sequence. We aligned all genomes using ClustalW (66) and extracted the two partial URR sequences taking the E6 and L1 genes as reference. We then joined the two stretches to obtain a complete URR, which was degapped and realigned. Finally, the E2-binding sites were extracted and aligned manually. We used the alignment editors BioEdit v7.0.8 (Tom Hall, Ibis Biosciences) and Jalview v2.2.1 (67) for sequence manipulation.
Sequence logos
Sequence logos were generated with WebLogo (63,68) and the aligned DNA or protein sequences. The height of the stack of letters at a position i is calculated as:
| 1 |
where s is the number of symbols (4 for DNA and 20 for proteins) and f(b,i) are the fractions of each base or amino acid at position i. The third term is a small sample correction, where n is the number of sequences in the alignment. The maximum value of Rsequence is 2 for DNA and 4.32 for proteins, and the minimum is zero in both cases. The height of each letter within a stack is proportional to its abundance:
| 2 |
Two Sample Logo (69) was used for comparing the in vivo logo with the in vitro logo. The software takes as input a sample alignment and a background alignment and identifies positions that are enriched or depleted in a given base. We generated an alignment of in vitro selected sites using the reported base frequencies (27) and used it as a sequence background. We used the alignment in vivo sites as sample. We used the binomial test and a P-value cutoff of 0.05 to identify differences between the alignments.
Computational prediction of binding free energies
We have used the theory of Berg and von Hippel (70–72) and the alignment of natural E2-BSs to predict the binding energy of E2 to E2-BSs, relative to the binding energy of the consensus E2-BS. All calculations were carried out using in-house perl scripts and ProFit (Quantumsoft, Zurich). We used the direct sequences of E2-BS1 and E2-BS2 and the reverse complementary sequences of E2-BS3 and E2-BS4 in order to align all sites in the same orientation (see Results section). The theory assumes that the only selection pressure at natural binding sites is to have a binding energy above a threshold dictated by the amount of free protein in the cell and the required binding levels. Positions 4,5 of the E2-BS were excluded from our calculation because of the selection pressure for a methylation site at these bases (Figure 3A). Doing the calculation with the reported base frequencies from in vitro selection (27) does not change the results significantly (data not shown), confirming that selection for binding is the main evolutionary pressure at natural E2-BSs. A second assumption of the theory is that base pairs evolve independently. We used the Enologos software (73) to check that correlations between base-pair frequencies in natural E2-BSs are very weak or do not exist at all (data not shown).
Figure 3.

Influence of CG methylation in the evolution of E2-binding sites. (A) Top: Sequence logo for all four biological E2-binding sites. Middle: Sequence logo from in vitro binding selection experiments with HPV51 E2 (27). Bottom: Two sample logo, taking the biological logo as sample and the in vitro logo as background. Displayed bases are enriched in the biological sites compared with the sequences selected in vitro. (B) Presence of putative methylation sites (CG dinucleotides) in positions 4,5 and 10,11 for each of the four in vivo E2-binding sites.
The expected statistical noise in the correlation between experimental and calculated relative free energies of binding is around 1 kcal/mol (70–72). Thus, we chose experimental datasets that span at least 2 kcal/mol in order to be able to observe a correlation. Each experimental dataset was measured under different solvent conditions (16,18–20,27). Since such changes influence the sequence discrimination capacity of the E2 protein (12), we made a separate correlation for each dataset.
We also used the Berg–von Hippel theory to look for unreported E2-BSs in the URR of alpha papillomaviruses. We calculated the relative binding energy of all possible sites of 14 bases using the base frequencies from the alignment of natural E2-BSs. In some cases, the base present in the putative binding site was absent from the corresponding position in the alignment of natural sites. Although the contribution of such a base to the binding energy cannot be calculated in a straightforward manner, it can be assumed to be highly detrimental to binding. Thus, we postulated that sites with bases not present in natural sites were not E2-BSs. We considered a sequence to be an E2-BSs if its predicted relative binding energy was lower than 4 kcal/mol, that is, a KD up to 830-fold worse than the consensus sequence. This setup detects 95% of the known E2-BSs.
Clustering of predicted binding energies
We have used the k-means algorithm (74) to cluster alpha papillomavirus types according to the predicted binding energies of natural E2-BSs. Virus types are defined as i points in a 4D space using the four predicted binding energies. The algorithm uses as input the number of clusters, j. First, it defines a centroid in the 4D space for each cluster. Next, clusters are defined by associating each point to the nearest centroid. Then, j new centroids are calculated as the centers of mass of the clusters. The association of each point and calculation of new centroids are repeated until the centroids do not move. This algorithm minimizes the sum of the square distances J between the i data points xi and the j centroids cj:
| 3 |
This procedure will converge to a minimum that depends significantly on the initial randomly selected cluster centers. In order to reach the global minimum, we run the procedure 1000 times until convergence and kept the solution with the lowest J. We run the algorithm using the software R (The R project for statistical computing, http://www.r-project.org/).
Association studies
We tested the association between virus type genotypes and phenotypes using the hypergeometric test (75). We have a total population of 72 virus types, of which a number x ≤ 72 has a certain epidemiological property. We draw from this population y ≤ 72 types having a molecular property, and z ≤ (x, y) of them have the epidemiological property of interest. We used the hypergeometric function to calculate the probability of having z successes at drawing without replacement of y objects from a total population of 72, given that the success population is x. In the case of a positive association, the P-value is defined as the sum of the probabilities of having z or more successes. Conversely, for a negative association, the P-value is defined as the sum of the probabilities of having z or less successes. A positive or negative association is reported if the P-value is smaller than the chosen 0.05 cutoff. All calculations were done using the hypergeometric function implemented in MS Excel.
RESULTS
Low protein variability in the E2–DNA interaction
We have examined the conservation of the E2–DNA interaction across alpha HPV types. Figure 1A shows the complex of the E2 protein from HPV18 with its target DNA (19). HPV6 and HPV16 E2 proteins form similar 1:1 complexes with modest changes in the quaternary structure of the protein (8,22,28). The structures of the isolated E2 proteins from HPV16 and HPV31 are also conserved (8,24,32,76). The E2 recognition helix is the main determinant of DNA binding (Figure 1A), (23). The sequence logo (63) in Figure 1B shows the conservation of residues in the recognition helix in alpha HPV types, in a conservation scale that goes from 0 to 4.3 bits for proteins (see Methods section). This region of the protein is highly conserved, as indicated by the tall letter stacks that approach the maximum conservation value (63). The eight E2 residues that contribute more than 0.8 kcal/mol to the binding energy of HPV16 E2 (23) are indicated with an asterisk in Figure 1B. Four of them (N294, K297, C298 and R302) are invariable in all 73 alpha papillomaviruses, and two (R300 and Y301) are more than 94% conserved.
The absolute binding energy of E2 to DNA is conserved across alpha HPV types. Remarkably, the dissociation constants of E2 proteins from the types 6, 11, 16 and 18 from the ACCGAAAACGGT site, measured by different laboratories and with different solvent conditions and flanking nucleotides, vary only from 1.7 to 17 nanomolar (12.0 to 10.6 kcal/mol in free energy of binding) (17–20). We have also compared the sequence discrimination ability of E2 proteins from HPV types 11, 16 and 18 (Figure 1C), that is, if the relative binding energies of different E2-BSs are the same for different E2 proteins. We have correlated the binding energies of E2 proteins from HPV11 and HPV16 to a set of E2-BSs (triangles and dashed line) (16,19,20) and of E2 proteins from HPV18 and HPV16 to a different set of E2-BSs (squares and continuous line) (16,19,20) (Figure 1C). The correlation R-values for the 16/11 and 16/18 datasets are 0.87 and 0.91, clearly showing that the three domains discriminate between different DNA sites in a similar manner. The 11, 16 and 18 types are phylogenetically distant, belonging to different alpha papillomavirus species (4). The overall sequence identities for the DNA-binding domains of the E2 proteins are 54% for the 16/11 pair and 61% for the 16/18 pair. Regarding the side chains that contribute more than 0.8 kcal/mol to the binding energy of HPV16 E2 (23), six of them are conserved in the three proteins and the other two vary (Figure 1C). Overall, these results strongly suggest that the sequence discrimination ability of E2 proteins is determined to a great extent by the side chains of residues N294, K297, C298, R302, R300 and Y301, which are highly conserved in alpha papillomaviruses (Figure 1B).
We conclude that, to a first approximation, the differences in the E2–DNA interaction across alpha HPV types are due to a great extent to changes in the DNA part of the complex and not to the E2 protein. From now on, we will focus our analysis in the variability of the E2-binding sites in the HPV genome. Since the function of each E2-binding site is also conserved across types 6, 11, 16, 18 and 31 (27,37–48), we hypothesize that phenotypical differences between types due to the E2–DNA interaction will be due to differences between their E2-binding sites.
Conservation and asymmetry of the four E2-binding sites
In this study, we have considered the E2-binding sites of 73 alpha papillomavirus types: HPV 2, 2a, 2isoC2, 3, 6, 6a, 6b, 7, 10, 11, 13, 13b, 16, 18, 26, 27, 27b, 28, 29, 30, 31, 32, 33, 34, 35, 35H, 39, 40, 42, 43, 44, 45, 51, 52, 53, 54, 55, 56, 57, 57b, 58, 59, 61, 62cand, 66, 67, 68a, 69, 70, 71, 72, 73, 74subtype, 77, 81, 82, 82subtype, 83, 84, 85cand, 86cand, 87cand, 89cand, 90cand, 91, 94, 94korean, 97, 97iso624, 102 and 106, PCPV1 and RHPV1 (see Supplementary Table S1). We have extracted all sites from the genomes and aligned them (see Methods section). All four sites were present in all genomes analyzed, with the exception of E2-BS2 in type 94korean. We have chosen to display the variability in E2-binding sites using sequence logos for clarity (63,68). Logos represent an alignment of DNA sites as a row of letter stacks. The height of a stack is proportional to the information content at that position of the alignment, which can be taken as a measure of conservation. For DNA, it takes values between 0 and 2 bits (see Methods section). The heights of the letters within a stack are proportional to the abundance of each base. On the other hand, consensus sequences are limited to only one base per position and thus display less information than sequence logos (63).
The logos for the four E2-binding sites are displayed in Figure 2B. E2-BS1, E2-BS2 and E2-BS4 are much more conserved than E2-BS3. The logos of all sites comply with the established consensus sequence aACCg(A/T)4cGGTt. Both bases in the two palindromic half-sites making direct contacts with the protein (direct readout) (23) and in the four-base linker [indirect readout (16)] are significantly conserved. To date, it is believed that all bases in the linker bear similar importance. In spite of this, there is a clear conservation gradient from the most conserved position 6 to the least conserved position 9 in E2-BS1, E2-BS2 and from 9 to 6 in E2-BS4 (Figure 2B). E2-BSs are generally asymmetric, with only 11 (4%) being full palindromes. Due to this lack of symmetry, the direct and inverse orientations of a site are not equivalent (70, 77–83). E2-BS1 and E2-BS2 have a consensus linker sequence AAA(A/T) in the 5′–3′ direction, while E2-BS3 and E2-BS4 meet that consensus only when the reverse complementary sequences are considered (Figure 2B). Interestingly, the E2-BSs seem to be oriented in different directions in the HPV genome. The results in Figure 2B indicate that E2-binding sites differ in their levels of conservation and their orientation, in agreement with their different physiological roles.
Distances between E2-binding sites
The relative positions of the E2-binding sites in the alpha HPV genomes could influence the cooperativity of protein–DNA binding (58). We examined the conservation of the relative positions of the E2-binding sites in the alpha HPV genomes (Figure 2C). The number of bases between E2-BS1 and E2-BS2 is very conserved, ranging only from 0 to 3 and being in most cases 1 or 2. The distance between sites 2 and 3 is more variable, being in most cases between 60 and 70 bases. Intriguingly, the distance is 95 bases for 6 types, all of them cutaneous. Finally, approximately 275 to 400 bases separate E2-binding sites 3 and 4, without a preferred value. We did not observe any correlation between the different distances (data not shown). In conclusion, distances between E2-BSs 1, 2 and 3 are evolutionarily restricted in alpha papillomaviruses.
Evolutionary pressure for a CpG methylation site within the E2-binding site
The main evolutionary pressure at many DNA-binding sites is to maintain the binding energy above a certain threshold (70–72). We have investigated whether this is the case for the alpha papillomavirus E2-binding sites by comparing the naturally occurring sites with a set of sites selected in vitro with affinity as the only constraint (27). The top sequence logo in Figure 3A corresponds to all naturally occurring E2-binding sites, using the reverse complement sequences of E2-BS3 and E2-BS4 in the alignment (see above). The middle logo in Figure 3A corresponds to a set of sites resulting from in vitro amplification and selection for affinity to HPV51 E2 (27). The consensus in vitro site AACACAAATCGGTT binds strongly to both HPV51 and HPV16 E2 domains (27), suggesting that the results of the experiment can be extrapolated to other alpha E2 proteins. There is a qualitative agreement between in vivo and in vitro selection in most positions of the site. However, the most frequent bases differ at positions 4 and 5. We can better visualize this difference using a two sample logo (Figure 3A, bottom) (69). This logo displays the bases that are overrepresented in the in vivo selected sites relative to the in vitro selected sites. The main difference between the two sets of sites is the presence of a CG dinucleotide at positions 4,5 in the naturally occurring sites. Thus, there seems to be evolutionary pressure for conservation of a methylation site at positions 4,5 in the E2-binding site.
Since each E2-BS has different effects on papillomavirus replication, genome maintenance and transcription, we looked for putative methylation sites at positions 4,5 of all four E2-BSs. The CG dinucleotide is present in nearly all E2-BS1, E2-BS2 and E2-BS4 and a majority of E2-BS3 (Figure 3B). A second CG dinucleotide is present at positions 10,11 of the E2-binding site in both the in vivo and in vitro logos (Figure 3A). This indicates that the second CG site is present in vivo due to affinity constraints, although simultaneous evolution for a methylation site cannot be excluded. The putative methylation site at positions 10,11 is as conserved as the positions 4 and 5 in E2-BS4 and E2-BS3 and less conserved in E2-BS1 and E2-BS2 (Figure 3B). Overall, our results suggest that the four E2-BSs would differ in their ability to be regulated by methylation.
Computational prediction of the affinity of E2 proteins for E2-binding sites
We have shown that binding affinity is the main evolutionary pressure at 12 of the 14 positions of the E2 target DNA, while at the two remaining positions the main evolutionary pressure is for the presence of a methylation site and not for binding (Figure 3A). We have chosen to study the evolution of these twelve positions in terms of binding affinity and in a global manner, rather than analyzing each position separately with sequence logos. In this way, the results can be related to the affinity hierarchy of the four binding sites and its role in regulating viral replication and transcription.
We have quantified the evolutionary pressure for binding at individual sites using the statistical mechanical theory from Berg and von Hippel (70–72) (see Methods section). Briefly, the theory assumes that (i) the DNA target sites of a protein have been selected by evolution to maintain binding affinity above a certain threshold and (ii) the bases of the binding site make independent, additive contributions to the free energy of binding. Given these two postulates, there is a simple relationship between the frequency of occurrence of two bases (i and j) at a given position in the repertoire of natural sites and the difference in their contributions to the free energy of binding:
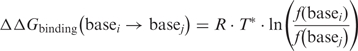 |
4 |
Where R is the universal gas constant [1.987 cal/(molK)]. T* is a pseudotemperature term related to the tolerance of the protein–DNA interaction to mutations and unrelated to physical temperature (70–72). The exact value of T* determines the slope of the correlations in Figure 4 but not their R-value. In our calculations, T* was arbitrarily set to 298 K (70–72). The difference in affinity between two sites can be calculated by summing the differences in affinity over all positions of the site:
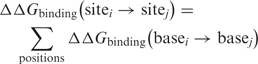 |
5 |
This theory has been successfully applied to the quantitative prediction of relative binding constants for several protein–DNA interactions (70–72).
Figure 4.

Correlation between observed and predicted free energies of binding for E2–DNA complexes for E2 proteins. (A) HPV type 16, data from Ref. (18). (B) HPV type 16, data from Ref. (16). (C) HPV type 11, data from Ref. (20). (D) HPV type 18, data from Ref. (19). (E) HPV type 51, data from Ref. (27). Units are kcal/mol in all cases. The binding energy of the consensus target sequence was arbitrarily set to zero. All other sequences have positive predicted values of ▵▵Gbinding, indicative of a reduced predicted binding affinity. The total number of points is 38, the standard deviation between observed and predicted values is 0.64 kcal/mol or 2.9-fold in KD.
We have tested whether the Berg–von Hippel theory holds for the interaction between E2 and its target DNA. We extracted five sets of free energies of binding between E2 proteins from HPV types 11, 16, 18 and 51 and four to fifteen target sites from the literature (16,18–20,27). We have calculated the binding energy of all sites relative to the binding energy of the consensus sequence using the alignment of all natural E2-BSs (see Methods section). The binding energy of the consensus sequence was arbitrarily set to zero. All other sequences have positive predicted values of ΔΔGbinding, indicative of a reduced predicted relative binding affinity. Positions 4,5 did not evolve according to affinity constraints (see above) and were excluded from the calculation. The correlation between experimental and calculated relative binding energies for five experimental datasets and four different E2 proteins is shown in Figure 4. The correlation R-values range from 0.74 to 0.96. The average standard deviation between observed and predicted values is 0.64 kcal/mol for 38 points, or 2.9-fold in KD. The performance of our sequence-based method is close to the standard deviation of 0.57 kcal/mol (16 points) obtained for the HPV16 E2 protein using structure-based modeling (16). The remarkable agreement between experiment and theory implies that E2-binding sites in the papillomavirus genome evolve as postulated in the theory from Berg and von Hippel. Furthermore, the success in predicting relative binding energies for four different E2 proteins using a model built from the binding sites of all alpha papillomaviruses supports our approximation that E2 proteins have similar DNA discrimination properties. We therefore propose that the theory can be used to predict the relative binding affinity of E2 proteins for any variation of an E2-binding site.
Six prototypical affinity hierarchies in alpha papillomaviruses
Similarly, we have used the Berg–von Hippel theory to calculate the affinity of E2 for all binding sites in alpha papillomaviruses relative to the affinity for the consensus sequence (see Methods section). E2-BS4 is the site with the tighter binding, only 0.61 ± 0.53 kcal/mol (average ± standard deviation) worse than consensus. It is followed by E2-BS2 (0.91 ± 0.69 kcal/mol), E2-BS1 (1.56 ± 1.47 kcal/mol) and E2-BS3 (1.94 ± 1.60 kcal/mol). This hierarchy is in qualitative agreement with the conservation levels in the logo for each site (Figure 2B).
A manual inspection of all affinity hierarchies revealed the existence of several groups of very similar types. We have clustered all virus types into hierarchy groups using the k-means algorithm (see Methods section). In this procedure, each virus type is represented by its four predicted relative binding free energies. The types are grouped by minimizing the sum of all differences between the predicted energies and the group averages. Clustering into less than six groups led to types with different hierarchies being grouped together, indicating that five or less groups were not enough to describe the data adequately (data not shown). Clustering into more than six groups led to two or more very similar groups, indicating that types in these groups belong to the same group (data not shown). The differences between the predicted relative binding free energies of the clusters are well above the 0.64 kcal/mol standard deviation of the prediction (Figure 5), supporting the clustering procedure. The distribution of the predicted changes in the free energy of binding upon substitution of a single base of the E2-BS show a continuous and broad distribution (Supplementary Figure 1). Thus, our model has the potential to generate a continuum of affinity hierarchies. We conclude that the discrete affinity hierarchies predicted for the known E2 sites is not an artifact of our model but a feature of alpha papillomavirus biology.
Figure 5.

Six classes of relative binding affinity hierarchies for the E2–DNA interaction. For each group of types, we represent the average relative predicted affinity and standard deviation for each site (thick black line, points) and the values for each type (high-risk types in red, low-risk types in green, cutaneous types in blue and other types in grey). The types were grouped using the k-means algorithm (see Methods section). The binding energy of the consensus target sequence was arbitrarily set to zero. All other sequences have positive predicted values of ΔΔGbinding, indicative of a reduced predicted binding affinity. (A) High-risk types 16, 35, 52, 53, 56, 66 and 73; low-risk types 6b, 40, 42, 43 and 44; cutaneous types 2, 2a, 2isoC2, 27 and 27b; and types 13, 13b, 30, 34, 35H, 55, 57, 57b, 67, 71, 74subtype, 90cand, 91, 106, PCPV1 and RHPV1. (B) High-risk types 18, 26, 33, 39, 45, 51, 58, 59, 68a, 82 and 82subtype; low-risk types 6, 6a, 11 and 54; cutaneous type 7; and types 32, 85cand, 97 and 97iso624. (C) Cutaneous types 3, 10, 28, 29 and 94; and types 84, 86cand, 87cand and 89cand. (D) Low-risk types 61, 72 and 81; and type 62cand. (E) High-risk type 31; low-risk type 70; and type 69. (F) Cutaneous type 77; and types 83 and 102.
The outcome of clustering into six groups is shown in Figure 5. Thirty-three of the 72 types belong to the first group, in which E2-BS1, E2-BS2 and E2-BS4 have an affinity close to that of the consensus sequence and binding to E2-BS3 is clearly weaker (Figure 5A; high-risk types 16, 35, 52, 53, 56, 66 and 73; low-risk types 6b, 40, 42, 43 and 44; cutaneous types 2, 2a, 2isoC2, 27 and 27b; and types 13, 13b, 30, 34, 35H, 55, 57, 57b, 67, 71, 74subtype, 90cand, 91, 106, PCPV1 and RHPV1). For the second largest group, all E2-BSs have a predicted relative binding energy very close to that of the consensus sequence (20 types, Figure 5B; high-risk types 18, 26, 33, 39, 45, 51, 58, 59, 68a, 82 and 82subtype; low-risk types 6, 6a, 11 and 54; cutaneous type 7; and types 32, 85cand, 97 and 97iso624). In the third largest group, E2-BS2 and E2-BS4 have a good predicted relative binding energy and E2-BS1 and E2-BS3 have less affinity for E2 (9 types, Figure 5C; cutaneous types 3, 10, 28, 29 and 94; and types 84, 86cand, 87cand and 89cand). Four types belong to a fourth group, in which E2-BS2 and E2-BS4 are predicted to be good binders, E2-BS1 to bind E2 weakly and E2-BS3 to have only marginal affinity for the protein (Figure 5D; low-risk types 61, 72 and 81; and type 62cand). In the three types in the fifth group, all sites but E2-BS2 have a good predicted relative binding affinity (Figure 5E; high-risk type 31; low-risk type 70; and type 69). Finally, the three types in the sixth group, E2-BS2 and E2-BS4 are predicted to be good binders, E2-BS3 to bind E2 weakly and E2-BS1 to have only marginal affinity for the protein (Figure 5F; cutaneous type 77; and types 83 and 102).
Our results agree with the current knowledge on the role of each site. In all hierarchies, the affinity of E2-BS4 is as least as good as for other sites (Figure 5), ensuring that the early genes of the virus are transcribed (43–45). E2-BS1, E2-BS2 or both have also a good affinity in all hierarchies (Figure 5), guaranteeing viral replication and the repression of oncogene transcription (27,37–48). E2-BS3, that has a secondary role in both transcription and replication, has a lower predicted affinity than the other three sites in the most abundant hierarchy (Figure 5A).
A fifth E2-binding site in the alpha papillomavirus upstream regulatory region
It is generally accepted that alpha papillomaviruses have four E2-BSs in the upstream regulatory region of their genome, while beta papillomaviruses may have five (84) and bovine papillomavirus type 1 has eleven (85). On the other hand, E2-BS2 is absent from type 94korean (see above). This prompted us to look for unreported E2-BSs in the URR of alpha papillomaviruses. We have used the Berg–von Hippel algorithm to predict the binding of all possible 14-base sites in the URRs of all alpha papillomaviruses (see Methods section). Known E2-BSs of 95% have predicted binding energies up to 4 kcal/mol worse than the binding energy of the consensus sequence. If we use this number as a cutoff, we predict six novel E2-BSs, in types 30, 44, 54, 61, 90 and 102 (Table 1), with predicted binding energies ranging from 0.29 to 3.40 kcal/mol. The novel E2-BS for HPV type 44 is known to bind the E2 proteins from types 6, 16 and 18 with affinities ranging from 1.4 to 18 nM (17–19), with a binding energy 0.2 kcal/mol worse than the consensus sequence (19). This agreement with experimental data suggests that E2 binds the new predicted sites with significant affinity. The position of the six sites is fairly conserved: five of them are <30 bases 3′ of E2-BS4, and the other is <100 bases 5′ of it. We tentatively name these new sites as E2-BS5.
Table 1.
Newly identified E2-binding sites
| HPV type | Sequencea | Distance to site 4b | ΔΔGbinding (Predicted)c (kcal/mol) |
|---|---|---|---|
| 30 | AACCAAAAAGGGTG | 93 | 3.11 |
| 44 | AACCGAAAACGGTT | −15 | 0.29 |
| 54 | AACCGAAACCGTTT | Overlapping site 4 | 2.48 |
| 61 | GACCGAAACCGGTC | −19 | 1.52 |
| 90 | GACCGAAACCGGGA | −2 | 3.40 |
| 102 | GACCGAAACCGGTC | −25 | 1.52 |
aIn the orientation with the best predicted energy. The sites from types 30, 44, 61, 90 and 102 are in the 3′–5′ direction, the site from type 54 is in the 5′–3′ direction.
bDistance is negative if site 5 is closer to the early promoter than site 4 and positive otherwise.
cRelative to the consensus sequence.
Possible association between molecular properties of the E2–DNA interaction and epidemiology of alpha papillomaviruses
The alpha papillomavirus genus includes all mucosal types as well as 12 cutaneous types (2, 2a, 2isoc2, 3, 7, 10, 27, 27b, 28, 29, 77 and 94). Mucosal alpha papillomavirus types are commonly classified as high-risk (types 16, 18, 26, 31, 33, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 68a, 73, 82, 82subtype) or low-risk (types 6, 6a, 6b, 11, 40, 42, 43, 44, 54, 61, 70, 72 and 81) according to the ratio of the odds of cervical cancer in patients infected with a certain type and in HPV-negative patients (2). We have tested whether the epidemiological behavior of mucosal high-risk, mucosal low-risk and cutaneous types is phenomenologically associated with the molecular properties of the E2–DNA interaction using the hypergeometric test (see Methods section). For example, the distance between E2-BS1 and E2-BS2 is 2 in 43 of 72 types, and in 10 of the 19 high-risk types. We use the hypergeometric probability distribution to calculate the probability of picking 10 or more high-risk types by choosing 43 types at random (75). If this probability is lower than the chosen significance level (0.05), we can conclude that the phenotype ‘high-risk’ is associated with a distance of two bases between E2-BS1 and E2-BS2.
The results for the association between phenotypes and molecular properties are shown in Table 2. High-risk types are associated with the affinity hierarchy of E2-BSs in Figure 5B, low-risk types with the affinity hierarchy in Figure 5D and cutaneous types with the affinity hierarchy in Figure 5C. Since a majority E2-BSs has two putative methylation sites at positions 4,5 and 10,11 (Figure 3), we have looked for the association of epidemiological behavior with the absence of methylation sites (‘Methylation defects’). High-risk types are positively associated with a missing methylation site in positions 10,11 of E2-BS2 and positions 4,5 of E2-BS3 and negatively associated with a missing methylation site in positions 10,11 of E2-BS1 and E2-BS3. Cutaneous types are positively associated with a missing methylation site in positions 10,11 of E2-BS1 and E2-BS3 and negatively associated with a missing methylation site in positions 4,5 of E2-BS3. With respect to the number of bases between sites, high-risk types are positively associated with the presence of two bases between E2-BS1 and E2-BS2 and negatively associated with the presence of a single base. The opposite is true for cutaneous types. This group of types is also positively associated with a distance of 95 bases between E2-BS2 and E2-BS3 (Table 2 and Figure 2). We did not observe a statistically significant association at the 0.05 level between epidemiological behavior and the presence of a E2-BS5 or the distance between sites 3 and 4 (data not shown). Altogether, we have been able to associate high-risk types with seven different molecular properties, low-risk types for only one molecular property and cutaneous types with seven molecular properties.
Table 2.
Association of molecular and epidemiological properties in alpha papillomaviruses
| Molecular property | High risk (19) | Low risk (13) | Cutaneous (12) | |||||
|---|---|---|---|---|---|---|---|---|
| n | P-value | n | P-value | n | P-value | |||
| Affinity hierarchy | 7 | >0.05 | 5 | >0.05 | 5 | >0.05 | ||
| 11 | 1.2 × 10−3 (+) | 4 | >0.05 | 1 | >0.05 | |||
| 0 | >0.05 | 0 | >0.05 | 5 | 4.9 × 10−3 (+) | |||
| 0 | >0.05 | 3 | 1.7 × 10−2 (+) | 0 | >0.05 | |||
| 1 | >0.05 | 1 | >0.05 | 0 | >0.05 | |||
| 0 | >0.05 | 0 | >0.05 | 1 | >0.05 | |||
| Methylation defect | Site 1 | Position 4 (1) | 1 | >0.05 | 0 | >0.05 | 0 | >0.05 |
| Position 10 (22) | 0 | 3.6 × 10−3 (−) | 3 | >0.05 | 7 | 2.9 × 10−2 (+) | ||
| Site 2 | Position 4 (0) | 0 | >0.05 | 0 | >0.05 | 0 | >0.05 | |
| Position 10 (8) | 7 | 2.3 × 10−4 (+) | 0 | >0.05 | 0 | >0.05 | ||
| Site 3 | Position 4 (23) | 14 | 1.4 × 10−5 (+) | 2 | >0.05 | 0 | 6.0 × 10−3 (−) | |
| Position 10 (26) | 1 | 6.4 × 10−4 (−) | 2 | >0.05 | 12 | 6.3 × 10−7 (+) | ||
| Site 4 | Position 4 (0) | 0 | >0.05 | 0 | >0.05 | 0 | >0.05 | |
| Position 10 (1) | 0 | >0.05 | 0 | >0.05 | 1 | >0.05 | ||
| Distance between sites | d12 = 0 (4) | 1 | >0.05 | 1 | >0.05 | 0 | >0.05 | |
| d12 = 1 (43) | 5 | 7.2 × 10−4 (−) | 10 | >0.05 | 12 | 1.0 × 10−3 (+) | ||
| d12 = 2 (21) | 10 | 1.1 × 10−2 (+) | 2 | >0.05 | 0 | 1.0 × 10−2 (−) | ||
| d12 = 3 (4) | 3 | >0.05 | 0 | >0.05 | 0 | >0.05 | ||
| d23 = 95 (6) | 0 | >0.05 | 0 | >0.05 | 6 | 5.9 × 10−6 (+) | ||
Epidemiologial properties are shown as columns and molecular properties as rows, with the number of types between brackets. For a given combination of properties, we indicate the observed number of types and the probability that the observation occurs by chance. Plus and minus signs indicate which combinations of molecular and epidemiological properties occur together more or less often than at random, respectively.
DISCUSSION
The modulated interaction of the C-terminal domain of the alpha papillomavirus E2 protein with its four target sites is crucial for the regulation of viral replication and transcription from the early promoter. We have shown that the sequence of E2-BSs varies significantly, leading to differences in predicted relative binding affinity (Figure 5), orientation and asymmetry (Figure 2) and putative methylation sites (Figure 3) across types. In this section, we will discuss the variability in connection to the evolution and biology of alpha papillomaviruses.
E2 and its target DNA form a complex with a highly dynamic, water-mediated interface (8,23) in which the energetic contributions of protein residues are additive (23). In agreement with this, we were able to describe the evolution of the DNA site using a model that postulates additive contributions of the DNA bases to binding (Figure 4) (70–72). We propose that the flexible protein–DNA interface allows the DNA to evolve much faster than the protein and into four well-differentiated binding sites (Figures 1 and 2). The four bases within the E2-BS spacer responsible for indirect readout are significantly conserved (Figures 2 and 3), suggesting that their energetic contribution to binding plays a role in papillomavirus evolution. The conservation gradient within the spacer (Figures 2 and 3) contradicts the general belief that the four bases make approximately equal contributions to binding (8,12,16–19,30–36) and shows that indirect readout can give rise to complex patterns of conservation.
Due to the asymmetric conservation gradient within the E2-BS spacer, most naturally occurring E2-BSs are not perfect palindromes. In principle, this is in contrast with the homodimeric nature of the E2 protein. However, natural DNA-binding sites of homodimeric transcription factors are sometimes asymmetric (70,77–83,86) and can have higher affinity than symmetric sites (81,83). Asymmetric target sites may bend in an asymmetric manner upon protein binding and/or lead to asymmetric changes in the structure of the protein (77–80), as reviewed in Ref. (87). This, in turn, can regulate the DNA structure in the vicinity of the target site and/or the binding of other proteins (70,77,78,86). The asymmetric E2-BSs are oriented in a conserved manner that varies from site to site (Figure 2). The replication function of an engineered E2-BS2 was shown to depend on its orientation (27). We propose that the orientation of naturally occurring E2-BSs induces asymmetric DNA bending and/or changes in the structure of E2 and plays a functional role by regulating the binding of other proteins or domains to E2 and/or to nearby DNA target sites, such as the HPV helicase E1 (37,38,40).
E2-BS1 and E2-BS2 are nearly side-by-side in the papillomavirus genome (Figure 2). These two sites are flanked by a TATA box and an Sp1-binding site at very conserved distances (Dellarole,M., unpublished data). All four sites are involved in the regulation of transcription of early viral proteins (27,41,43–48), suggesting that they act as a functional unit in alpha papillomaviruses. Cooperative effects have indeed been observed for binding of HPV16 E2 to E2-BS1 and E2-BS2 (58) and may also be present in the binding of Sp1 and TBP. Insertion of a base correlates with high risk in a virus type (Table 2), suggesting that the exact distances within this functional unit have functional significance. The distance between E2-BS2 and E2-BS3 is also restricted in 66 out of 72 types (Figure 2), suggesting that they form another functional unit involved in replication, together with the binding site for the E1 protein (27,37–42). The variability of the position of E2-BS4 may be linked to the diversity of binding sites for cellular proteins in the enhancer region (62).
CG dinucleotides are underrepresented in the HPV genome (55), suggesting that the virus has eliminated disadvantageous methylation sites. However, there is evolutionary pressure in favor of at least one methylation site within the E2-BS (Figure 3A). This pressure varies among E2-BSs (Figure 3B), suggesting that it is related to the function of each site. Interestingly, the alpha papillomavirus URR has binding sites for human proteins involved in local DNA demethylation close to an E2-BS (58,88–92), such as the glucocorticoid receptor, Sp1 and NF-κB. Furthermore, the HPV E2 protein can act as a cofactor of the glucocorticoid receptor (93). We deduce that papillomaviruses have integrated methylation of E2-BSs by the host cell in their life cycle, turning a potential mechanism of defense into an additional layer of regulation. This regulation may take place through changes in the affinity hierarchy of E2-BSs upon methylation (14). Remarkably, these changes may be different in high-risk and cutaneous types due do missing methylation sites (Figure 3 and Table 2).
The DNA-binding sites for the E2 protein in the 72 alpha papillomavirus types may in principle have a continuum of many different affinity hierarchies. Nevertheless, the predicted free energies of binding can be clustered in only six prototypical relative affinity hierarchies, well differentiated from each other (Figure 5). The three most common hierarchies include 86% of the types. This unexpected simplicity implies that only a small number of well-defined affinity hierarchies render a functional virus. This poses two challenges to our understanding of alpha papillomaviruses. First, how the balanced regulatory roles of the four E2-BSs in transcription of the early proteins and replication along the life cycle of the virus determine the observed affinity hierarchies. Second, the mechanism by which these hierarchies are related to the development of cancer.
The alpha papillomavirus genus includes not only all genital HPV types, but also 12 cutaneous types (4,94). About one half of the types in the cutaneous beta papillomavirus genus have a fifth binding site for E2 in the vicinity of E2-BS4 (77) (Dellarole,M., unpublished data). This is reminiscent of the novel E2-BS5 we have identified in six alpha papillomavirus types (Table 1). However, these types are phylogenetically unrelated (4,94) and none of them causes cutaneous warts. A closer examination of the E2-BSs in beta papillomaviruses reveals that their function (95), relative positions (96) and consensus sequences (DellaroleM., unpublished data) are significantly different to both genital and cutaneous alfa papillomaviruses. We speculate that cutaneous alpha papillomaviruses may have developed their own strategy to persistently infect non-mucosal epithelia. Both the binding of cellular transcription factors (62) and the characteristics of the E2-BSs associated with cutaneous alpha papillomavirus types (Table 2: affinity hierarchy in Figure 5C; methylation defect at position 10 of E2-BS1 and E2-BS3; lack of methylation defect at position 4 of E2-BS3; distance of one base and not of two bases between E2-BS1 and E2-BS2; distance of 95 bases between E2-BS2 and E2-BS3) may be part of this strategy.
Genital papillomavirus types are usually labeled as ‘high-risk’ or ‘low-risk’ according to the odds ratio of developing cervical cancer (2). However, the odds ratios for the different types do not cluster at a high value for high-risk types and a value of one for low-risk types. If the uncertainty in the odds ratio is taken into account, they cover a continuum of values that go from 1 to ∼400 (1,2). This suggests that the risk phenotype associated with a type comes from the sum of a large number of small genotypic contributions (‘grains of sand’ model) and not from a small number of highly decisive genetic features (‘few switches’ model). In conformity with the ‘grains of sand’ model, we have found that high-risk types are weakly associated with seven different molecular properties of the E2-BSs (Table 2: affinity hierarchy in Figure 5B; methylation defects at position 10 of E2-BS2 and position 4 of E2-BS3; lack of methylation defects at position 10 of E2-BS1 and E2-BS3; distance of two bases and not of one base between E2-BS1 and E2-BS2). Conversely, low-risk types are weakly associated with only one molecular property (Table 2: affinity hierarchy in Figure 5D) and thus do not differ much from the alpha papillomavirus background in terms of the E2–DNA interaction. We hypothesize that genital papillomaviruses are low-risk types by default and that E2-BS genotypes are only able to increase the risk and not to decrease it. In agreement with this, coinfection with a low-risk type does not decrease the odds ratio of a high-risk type (1,2).
The HPV E2 protein regulates the transcription of the HPV E6 and E7 oncogenes through its interaction with DNA. In agreement with this, we were able to associate some properties of its DNA target sites to high-risk types. Our results confirm previous hypothesis about the importance of the hierarchy of affinities of the E2-BSs (17,20) and binding site methylation (49,51–55) and extrapolate them from a handful of types to the full alpha papillomavirus genus. Additionally, we can now add the distance between E2-BS1 and E2-BS2 to the list of genotypes linked to the development of disease (25,28,59–61). It will be of interest to analyze the interplay of these properties of E2 with the molecular properties of the E6 and E7 oncogenes known to be associated with cancer. The results from this work may also help us understand the epidemiological behavior of molecular variants of HPV (97).
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online.
ACKNOWLEDGEMENTS
We would like to thank Pedro Beltrao, Emanuelle Raineri and Jesús Tejero for technical advice. This work was funded by Wellcome Trust Grant [GR077355AYA]; Agencia Nacional de Promoción Científica y Tecnológica PICT [2000 01-08959]; doctoral fellowship from Consejo Nacional de Investigaciones Científicas y Técnicas to M.D.; MUTIS postdoctoral fellowship from the Agencia Española de Cooperación Internacional to I.E.S.; G.P.G. is a Career Investigator from Consejo Nacional de Investigaciones Científicas y Técnicas. Funding to pay the Open Access publication charges for this article was provided by XXX.
Conflict of interest statement. None declared.
REFERENCES
- 1.Bosch FX, Sanjosé S, Castellsagué X, Moreno V, Muñoz N. In: Papillomavirus Research: From Natural History to Vaccines and Beyond. Saveria Campo M, editor. Chapter 3. Wymondham: Caisreir Academic Press; 2006. pp. 19–40. [Google Scholar]
- 2.Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, Shah KV, Snijders PJ, Meijer CJ. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003;348:518–527. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- 3.de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H. Classification of papillomaviruses. Virology. 2004;324:17–27. doi: 10.1016/j.virol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 4.Bravo IG, Alonso A. Phylogeny and evolution of papillomaviruses based on the E1 and E2 proteins. Virus Genes. 2007;34:249–262. doi: 10.1007/s11262-006-0017-4. [DOI] [PubMed] [Google Scholar]
- 5.D'Souza G, Kreimer AR, Viscidi R, Pawlita M, Fakhry C, Koch WM, Westra WH, Gillison ML. Case-control study of human papillomavirus and oropharyngeal cancer. N. Engl. J. Med. 2007;356:1944–1956. doi: 10.1056/NEJMoa065497. [DOI] [PubMed] [Google Scholar]
- 6.Cohen J. Public health. High hopes and dilemmas for a cervical cancer vaccine. Science. 2005;308:618–621. doi: 10.1126/science.308.5722.618. [DOI] [PubMed] [Google Scholar]
- 7.Kalantari M, Bernard H-U. In: Papillomavirus Research: From Natural History to Vaccines and Beyond. Saveria Campo M, editor. Chapter 4. Wymondham: Caisreir Academic Press; 2006. pp. 41–52. [Google Scholar]
- 8.Hegde RS. The papillomavirus E2 proteins: structure, function, and biology. Annu. Rev. Biophys. Biomol. Struct. 2002;31:343–360. doi: 10.1146/annurev.biophys.31.100901.142129. [DOI] [PubMed] [Google Scholar]
- 9.McBride AA, Byrne JC, Howley PM. E2 polypeptides encoded by bovine papillomavirus type 1 form dimers through the common carboxyl-terminal domain: transactivation is mediated by the conserved amino-terminal domain. Proc. Natl Acad. Sci. USA. 1989;86:510–514. doi: 10.1073/pnas.86.2.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mok YK, de Prat Gay G, Butler PJ, Bycroft M. Equilibrium dissociation and unfolding of the dimeric human papillomavirus strain-16 E2 DNA-binding domain. Protein Sci. 1996;5:310–319. doi: 10.1002/pro.5560050215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McBride AA, Romanczuk H, Howley PM. The papillomavirus E2 regulatory proteins. J. Biol. Chem. 1991;266:18411–18414. [PubMed] [Google Scholar]
- 12.Blakaj DM, Kattamuri C, Khrapunov S, Hegde RS, Brenowitz M. Indirect readout of DNA sequence by papillomavirus E2 proteins depends upon net cation uptake. J. Mol. Biol. 2006;358:224–240. doi: 10.1016/j.jmb.2006.01.093. [DOI] [PubMed] [Google Scholar]
- 13.Thain A, Webster K, Emery D, Clarke AR, Gaston K. DNA binding and bending by the human papillomavirus type 16 E2 protein. Recognition of an extended binding site. J. Biol. Chem. 1997;272:8236–8242. doi: 10.1074/jbc.272.13.8236. [DOI] [PubMed] [Google Scholar]
- 14.Thain A, Jenkins O, Clarke AR, Gaston K. CpG methylation directly inhibits binding of the human papillomavirus type 16 E2 protein to specific DNA sequences. J. Virol. 1996;70:7233–7235. doi: 10.1128/jvi.70.10.7233-7235.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanders CM, Maitland NJ. Kinetic and equilibrium binding studies of the human papillomavirus type-16 transcription regulatory protein E2 interacting with core enhancer elements. Nucleic Acids Res. 1994;22:4890–4897. doi: 10.1093/nar/22.23.4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Xi Z, Hegde RS, Shakked Z, Crothers DM. Predicting indirect readout effects in protein-DNA interactions. Proc. Natl Acad. Sci. USA. 2004;101:8337–8341. doi: 10.1073/pnas.0402319101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dell G, Wilkinson KW, Tranter R, Parish J, Leo Brady R, Gaston K. Comparison of the structure and DNA-binding properties of the E2 proteins from an oncogenic and a non-oncogenic human papillomavirus. J. Mol. Biol. 2003;334:979–991. doi: 10.1016/j.jmb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 18.Hines CS, Meghoo C, Shetty S, Biburger M, Brenowitz M, Hegde RS. DNA structure and flexibility in the sequence-specific binding of papillomavirus E2 proteins. J. Mol. Biol. 1998;276:809–818. doi: 10.1006/jmbi.1997.1578. [DOI] [PubMed] [Google Scholar]
- 19.Kim SS, Tam JK, Wang AF, Hegde RS. The structural basis of DNA target discrimination by papillomavirus E2 proteins. J. Biol. Chem. 2000;275:31245–31254. doi: 10.1074/jbc.M004541200. [DOI] [PubMed] [Google Scholar]
- 20.Alexander KA, Phelps WC. A fluorescence anisotropy study of DNA binding by HPV-11 E2C protein: a hierarchy of E2-binding sites. Biochemistry. 1996;35:9864–9872. doi: 10.1021/bi960447d. [DOI] [PubMed] [Google Scholar]
- 21.Falconi M, Santolamazza A, Eliseo T, de Prat Gay G, Cicero DO, Desideri A. Molecular dynamics of the DNA-binding domain of the papillomavirus E2 transcriptional regulator uncover differential properties for DNA target accommodation. FEBS J. 2007;274:2385–2395. doi: 10.1111/j.1742-4658.2007.05773.x. [DOI] [PubMed] [Google Scholar]
- 22.Cicero DO, Nadra AD, Eliseo T, Dellarole M, Paci M, de Prat Gay G. Structural and thermodynamic basis for the enhanced transcriptional control by the human papillomavirus strain-16 E2 protein. Biochemistry. 2006;45:6551–6560. doi: 10.1021/bi060123h. [DOI] [PubMed] [Google Scholar]
- 23.Ferreiro DU, Dellarole M, Nadra AD, de Prat Gay G. Free energy contributions to direct readout of a DNA sequence. J. Biol. Chem. 2005;280:32480–32484. doi: 10.1074/jbc.M505706200. [DOI] [PubMed] [Google Scholar]
- 24.Nadra AD, Eliseo T, Mok YK, Almeida CL, Bycroft M, Paci M, de Prat Gay G, Cicero DO. Solution structure of the HPV-16 E2 DNA binding domain, a transcriptional regulator with a dimeric beta-barrel fold. J. Biomol. NMR. 2004;30:211–214. doi: 10.1023/b:jnmr.0000048942.96866.76. [DOI] [PubMed] [Google Scholar]
- 25.Ferreiro DU, de Prat Gay G. A protein-DNA binding mechanism proceeds through multi-state or two-state parallel pathways. J. Mol. Biol. 2003;331:89–99. doi: 10.1016/s0022-2836(03)00720-4. [DOI] [PubMed] [Google Scholar]
- 26.Ferreiro DU, Lima LM, Nadra AD, Alonso LG, Goldbaum FA, de Prat Gay G. Distinctive cognate sequence discrimination, bound DNA conformation, and binding modes in the E2 C-terminal domains from prototype human and bovine papillomaviruses. Biochemistry. 2000;39:14692–14701. doi: 10.1021/bi001694r. [DOI] [PubMed] [Google Scholar]
- 27.Newhouse CD, Silverstein SJ. Orientation of a novel DNA binding site affects human papillomavirus-mediated transcription and replication. J. Virol. 2001;75:1722–1735. doi: 10.1128/JVI.75.4.1722-1735.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hooley E, Fairweather V, Clarke AR, Gaston K, Brady RL. The recognition of local DNA conformation by the human papillomavirus type 6 E2 protein. Nucleic Acids Res. 2006;34:3897–3908. doi: 10.1093/nar/gkl466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muller F, Giroglou T, Sapp M. Characterization of the DNA-binding activity of the E1 and E2 proteins and the E1/E2 complex of human papillomavirus type 33. J. Gen. Virol. 1997;78(Pt 4):911–915. doi: 10.1099/0022-1317-78-4-911. [DOI] [PubMed] [Google Scholar]
- 30.Rozenberg H, Rabinovich D, Frolow F, Hegde RS, Shakked Z. Structural code for DNA recognition revealed in crystal structures of papillomavirus E2-DNA targets [see comments] Proc. Natl Acad. Sci. USA. 1998;95:15194–15199. doi: 10.1073/pnas.95.26.15194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hizver J, Rozenberg H, Frolow F, Rabinovich D, Shakked Z. DNA bending by an adenine – thymine tract and its role in gene regulation. Proc. Natl Acad. Sci. USA. 2001;98:8490–8495. doi: 10.1073/pnas.151247298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hegde RS, Androphy EJ. Crystal structure of the E2 DNA-binding domain from human papillomavirus type 16: implications for its DNA binding-site selection mechanism. J. Mol. Biol. 1998;284:1479–1489. doi: 10.1006/jmbi.1998.2260. [DOI] [PubMed] [Google Scholar]
- 33.Bedrosian CL, Bastia D. The DNA-binding domain of HPV-16 E2 protein interaction with the viral enhancer: protein-induced DNA bending and role of the nonconserved core sequence in binding site affinity. Virology. 1990;174:557–575. doi: 10.1016/0042-6822(90)90109-5. [DOI] [PubMed] [Google Scholar]
- 34.Zimmerman JM, Maher LJ., III Solution measurement of DNA curvature in papillomavirus E2 binding sites. Nucleic Acids Res. 2003;31:5134–5139. doi: 10.1093/nar/gkg697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Byun KS, Beveridge DL. Molecular dynamics simulations of papilloma virus E2 DNA sequences: dynamical models for oligonucleotide structures in solution. Biopolymers. 2004;73:369–379. doi: 10.1002/bip.10527. [DOI] [PubMed] [Google Scholar]
- 36.Rohs R, Sklenar H, Shakked Z. Structural and energetic origins of sequence-specific DNA bending: Monte Carlo simulations of papillomavirus E2-DNA binding sites. Structure. 2005;13:1499–1509. doi: 10.1016/j.str.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 37.Remm M, Brain R, Jenkins JR. The E2 binding sites determine the efficiency of replication for the origin of human papillomavirus type 18. Nucleic Acids Res. 1992;20:6015–6021. doi: 10.1093/nar/20.22.6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiang CM, Dong G, Broker TR, Chow LT. Control of human papillomavirus type 11 origin of replication by the E2 family of transcription regulatory proteins. J. Virol. 1992;66:5224–5231. doi: 10.1128/jvi.66.9.5224-5231.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sverdrup F, Khan SA. Two E2 binding sites alone are sufficient to function as the minimal origin of replication of human papillomavirus type 18 DNA. J. Virol. 1995;69:1319–1323. doi: 10.1128/jvi.69.2.1319-1323.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu JZ, Sun YN, Rose RC, Bonnez W, McCance DJ. Two E2 binding sites (E2BS) alone or one E2BS plus an A/T-rich region are minimal requirements for the replication of the human papillomavirus type 11 origin. J. Virol. 1993;67:7131–7139. doi: 10.1128/jvi.67.12.7131-7139.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stubenrauch F, Lim HB, Laimins LA. Differential requirements for conserved E2 binding sites in the life cycle of oncogenic human papillomavirus type 31. J. Virol. 1998;72:1071–1077. doi: 10.1128/jvi.72.2.1071-1077.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Demeret C, Le Moal M, Yaniv M, Thierry F. Control of HPV 18 DNA replication by cellular and viral transcription factors. Nucleic Acids Res. 1995;23:4777–4784. doi: 10.1093/nar/23.23.4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong G, Broker TR, Chow LT. Human papillomavirus type 11 E2 proteins repress the homologous E6 promoter by interfering with the binding of host transcription factors to adjacent elements. J. Virol. 1994;68:1115–1127. doi: 10.1128/jvi.68.2.1115-1127.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steger G, Corbach S. Dose-dependent regulation of the early promoter of human papillomavirus type 18 by the viral E2 protein. J. Virol. 1997;71:50–58. doi: 10.1128/jvi.71.1.50-58.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rapp B, Pawellek A, Kraetzer F, Schaefer M, May C, Purdie K, Grassmann K, Iftner T. Cell-type-specific separate regulation of the E6 and E7 promoters of human papillomavirus type 6a by the viral transcription factor E2. J. Virol. 1997;71:6956–6966. doi: 10.1128/jvi.71.9.6956-6966.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thierry F, Howley PM. Functional analysis of E2-mediated repression of the HPV18 P105 promoter. New Biol. 1991;3:90–100. [PubMed] [Google Scholar]
- 47.Demeret C, Desaintes C, Yaniv M, Thierry F. Different mechanisms contribute to the E2-mediated transcriptional repression of human papillomavirus type 18 viral oncogenes. J. Virol. 1997;71:9343–9349. doi: 10.1128/jvi.71.12.9343-9349.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soeda E, Ferran MC, Baker CC, McBride AA. Virology. 2006. Repression of HPV16 early region transcription by the E2 protein. [DOI] [PubMed] [Google Scholar]
- 49.Kim K, Garner-Hamrick PA, Fisher C, Lee D, Lambert PF. Methylation patterns of papillomavirus DNA, its influence on E2 function, and implications in viral infection. J. Virol. 2003;77:12450–12459. doi: 10.1128/JVI.77.23.12450-12459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosl F, Arab A, Klevenz B, zur Hausen H. The effect of DNA methylation on gene regulation of human papillomaviruses. J. Gen. Virol. 1993;74(Pt 5):791–801. doi: 10.1099/0022-1317-74-5-791. [DOI] [PubMed] [Google Scholar]
- 51.Bhattacharjee B, Sengupta S. CpG methylation of HPV 16 LCR at E2 binding site proximal to P97 is associated with cervical cancer in presence of intact E2. Virology. 2006;354:280–285. doi: 10.1016/j.virol.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 52.Wiley DJ, Huh J, Rao JY, Chang C, Goetz M, Poulter M, Masongsong E, Chang CI, Bernard HU. Methylation of human papillomavirus genomes in cells of anal epithelia of HIV-infected men. J. Acquir. Immune Defic. Syndr. (1999) 2005;39:143–151. [PubMed] [Google Scholar]
- 53.Badal S, Badal V, Calleja-Macias IE, Kalantari M, Chuang LS, Li BF, Bernard HU. The human papillomavirus-18 genome is efficiently targeted by cellular DNA methylation. Virology. 2004;324:483–492. doi: 10.1016/j.virol.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 54.Kalantari M, Calleja-Macias IE, Tewari D, Hagmar B, Lie K, Barrera-Saldana HA, Wiley DJ, Bernard HU. Conserved methylation patterns of human papillomavirus type 16 DNA in asymptomatic infection and cervical neoplasia. J. Virol. 2004;78:12762–12772. doi: 10.1128/JVI.78.23.12762-12772.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Badal V, Chuang LS, Tan EH, Badal S, Villa LL, Wheeler CM, Li BF, Bernard HU. CpG methylation of human papillomavirus type 16 DNA in cervical cancer cell lines and in clinical specimens: genomic hypomethylation correlates with carcinogenic progression. J. Virol. 2003;77:6227–6234. doi: 10.1128/JVI.77.11.6227-6234.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ozbun MA, Meyers C. Human papillomavirus type 31b E1 and E2 transcript expression correlates with vegetative viral genome amplification. Virology. 1998;248:218–230. doi: 10.1006/viro.1998.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 2006;110:525–541. doi: 10.1042/CS20050369. [DOI] [PubMed] [Google Scholar]
- 58.Tan S, Leong LE, Walker PA, Bernard H. The human papillomavirus type 16 E2 transacription factor binds with low cooperativity to two flanking sites and represses the E6 promoter through displacement of Sp1 and TFIID. J. Virol. 1994;68:6411–6420. doi: 10.1128/jvi.68.10.6411-6420.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klucevsek K, Wertz M, Lucchi J, Leszczynski A, Moroianu J. Characterization of the nuclear localization signal of high risk HPV16 E2 protein. Virology. 2007;360:191–198. doi: 10.1016/j.virol.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hou SY, Wu SY, Chiang CM. Transcriptional activity among high and low risk human papillomavirus E2 proteins correlates with E2 DNA binding. J. Biol. Chem. 2002;277:45619–45629. doi: 10.1074/jbc.M206829200. [DOI] [PubMed] [Google Scholar]
- 61.Parish JL, Kowalczyk A, Chen HT, Roeder GE, Sessions R, Buckle M, Gaston K. E2 proteins from high- and low-risk human papillomavirus types differ in their ability to bind p53 and induce apoptotic cell death. J. Virol. 2006;80:4580–4590. doi: 10.1128/JVI.80.9.4580-4590.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garcia-Vallve S, Iglesias-Rozas JR, Alonso A, Bravo IG. Different papillomaviruses have different repertoires of transcription factor binding sites: convergence and divergence in the upstream regulatory region. BMC Evol. Biol. 2006;6:20. doi: 10.1186/1471-2148-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Büchen-Osmond C. The Universal Virus Database ICTVdB. Computing in Science & Engineering. 2003;5:16–25. [Google Scholar]
- 65.Tatusova TA, Madden TL. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 1999;174:247–250. doi: 10.1111/j.1574-6968.1999.tb13575.x. [DOI] [PubMed] [Google Scholar]
- 66.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Clamp M, Cuff J, Searle SM, Barton GJ. The Jalview Java alignment editor. Bioinformatics. 2004;20:426–427. doi: 10.1093/bioinformatics/btg430. [DOI] [PubMed] [Google Scholar]
- 68.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vacic V, Iakoucheva LM, Radivojac P. Two Sample Logo: a graphical representation of the differences between two sets of sequence alignments. Bioinformatics. 2006;22:1536–1537. doi: 10.1093/bioinformatics/btl151. [DOI] [PubMed] [Google Scholar]
- 70.Berg OG, von Hippel PH. Selection of DNA binding sites by regulatory proteins. II. The binding specificity of cyclic AMP receptor protein to recognition sites. J. Mol. Biol. 1988;200:709–723. doi: 10.1016/0022-2836(88)90482-2. [DOI] [PubMed] [Google Scholar]
- 71.Berg OG, von Hippel PH. Selection of DNA binding sites by regulatory proteins. Statistical-mechanical theory and application to operators and promoters. J. Mol. Biol. 1987;193:723–750. doi: 10.1016/0022-2836(87)90354-8. [DOI] [PubMed] [Google Scholar]
- 72.Berg OG, von Hippel PH. Selection of DNA binding sites by regulatory proteins. Trends Biochem. Sci. 1988;13:207–211. doi: 10.1016/0968-0004(88)90085-0. [DOI] [PubMed] [Google Scholar]
- 73.Workman CT, Yin Y, Corcoran DL, Ideker T, Stormo GD, Benos PV. enoLOGOS: a versatile web tool for energy normalized sequence logos. Nucleic Acids Res. 2005;33:W389–W392. doi: 10.1093/nar/gki439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.MacQueen JB. Vol. 1. Berkeley: University of California Press; 1967. Proceedings of 5-th Berkeley Symposium on Mathematical Statistics and Probability; pp. 281–297. [Google Scholar]
- 75.Rivals I, Personnaz L, Taing L, Potier MC. Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics. 2007;23:401–407. doi: 10.1093/bioinformatics/btl633. [DOI] [PubMed] [Google Scholar]
- 76.Liang H, Petros AM, Meadows RP, Yoon HS, Egan DA, Walter K, Holzman TF, Robins T, Fesik SW. Solution structure of the DNA-binding domain of a human papillomavirus E2 protein: evidence for flexible DNA-binding regions. Biochemistry. 1996;35:2095–2103. doi: 10.1021/bi951932w. [DOI] [PubMed] [Google Scholar]
- 77.Pyles EA, Lee JC. Escherichia coli cAMP receptor protein-DNA complexes. 2. Structural asymmetry of DNA bending. Biochemistry. 1998;37:5201–5210. doi: 10.1021/bi972451a. [DOI] [PubMed] [Google Scholar]
- 78.Ramirez-Carrozzi V, Kerppola T. Asymmetric recognition of nonconsensus AP-1 sites by Fos-Jun and Jun-Jun influences transcriptional cooperativity with NFAT1. Mol. Cell. Biol. 2003;23:1737–1749. doi: 10.1128/MCB.23.5.1737-1749.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.King DA, Zhang L, Guarente L, Marmorstein R. Structure of a HAP1-DNA complex reveals dramatically asymmetric DNA binding by a homodimeric protein. Nat. Struct. Biol. 1999;6:64–71. doi: 10.1038/4940. [DOI] [PubMed] [Google Scholar]
- 80.Raumann BE, Rould MA, Pabo CO, Sauer RT. DNA recognition by beta-sheets in the Arc repressor-operator crystal structure. Nature. 1994;367:754–757. doi: 10.1038/367754a0. [DOI] [PubMed] [Google Scholar]
- 81.Winston RL, Ehley JA, Baird EE, Dervan PB, Gottesfeld JM. Asymmetric DNA binding by a homodimeric bHLH protein. Biochemistry. 2000;39:9092–9098. doi: 10.1021/bi000947d. [DOI] [PubMed] [Google Scholar]
- 82.Kalodimos CG, Bonvin AM, Salinas RK, Wechselberger R, Boelens R, Kaptein R. Plasticity in protein-DNA recognition: lac repressor interacts with its natural operator 01 through alternative conformations of its DNA-binding domain. EMBO J. 2002;21:2866–2876. doi: 10.1093/emboj/cdf318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sarai A, Takeda Y. Lambda repressor recognizes the approximately 2-fold symmetric half-operator sequences asymmetrically. Proc. Natl Acad. Sci USA. 1989;86:6513–6517. doi: 10.1073/pnas.86.17.6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ensser A, Pfister H. Epidermodysplasia verruciformis associated human papillomaviruses present a subgenus-specific organization of the regulatory genome region. Nucleic Acids Res. 1990;18:3919–3922. doi: 10.1093/nar/18.13.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li R, Knight J, Bream G, Stenlund A, Botchan M. Specific recognition nucleotides and their DNA context determine the affinity of E2 protein for 17 binding sites in the BPV-1 genome. Genes Dev. 1989;3:510–526. doi: 10.1101/gad.3.4.510. [DOI] [PubMed] [Google Scholar]
- 86.Ptashne M. A Genetic Switch - Phage Lambda Revisited. 3rd. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2004. [Google Scholar]
- 87.Brown JH. Breaking symmetry in protein dimers: designs and functions. Protein Sci. 2006;15:1–13. doi: 10.1110/ps.051658406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gloss B, Bernard HU, Seedorf K, Klock G. The upstream regulatory region of the human papilloma virus-16 contains an E2 protein-independent enhancer which is specific for cervical carcinoma cells and regulated by glucocorticoid hormones. EMBO J. 1987;6:3735–3743. doi: 10.1002/j.1460-2075.1987.tb02708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fontaine V, van der Meijden E, de Graaf J, ter Schegget J, Struyk L. A functional NF-kappaB binding site in the human papillomavirus type 16 long control region. Virology. 2000;272:40–49. doi: 10.1006/viro.2000.0363. [DOI] [PubMed] [Google Scholar]
- 90.Thomassin H, Flavin M, Espinas ML, Grange T. Glucocorticoid-induced DNA demethylation and gene memory during development. EMBO J. 2001;20:1974–1983. doi: 10.1093/emboj/20.8.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Matsuo K, Silke J, Georgiev O, Marti P, Giovannini N, Rungger D. An embryonic demethylation mechanism involving binding of transcription factors to replicating DNA. EMBO J. 1998;17:1446–1453. doi: 10.1093/emboj/17.5.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kirillov A, Kistler B, Mostoslavsky R, Cedar H, Wirth T, Bergman Y. A role for nuclear NF-kappaB in B-cell-specific demethylation of the Igkappa locus. Nat. Genet. 1996;13:435–441. doi: 10.1038/ng0895-435. [DOI] [PubMed] [Google Scholar]
- 93.Wu MH, Chan JY, Liu PY, Liu ST, Huang SM. Human papillomavirus E2 protein associates with nuclear receptors to stimulate nuclear receptor- and E2-dependent transcriptional activations in human cervical carcinoma cells. Int. J. Biochem. Cell Biol. 2007;39:413–425. doi: 10.1016/j.biocel.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 94.Bernard HU. Saveria Campo M. Wymondham: Caisreir Academic Press; 2006. Papillomavirus Research: From Natural History to Vaccines and Beyond. Vol. Chapter 2, pp. 11–18. [Google Scholar]
- 95.Stubenrauch F, Leigh IM, Pfister H. E2 represses the late gene promoter of human papillomavirus type 8 at high concentrations by interfering with cellular factors. J. Virol. 1996;70:119–126. doi: 10.1128/jvi.70.1.119-126.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Guido MC, Zamorano R, Garrido-Guerrero E, Gariglio P, Garcia-Carranca A. Early promoters of genital and cutaneous human papillomaviruses are differentially regulated by the bovine papillomavirus type 1 E2 gene product. J. Gen. Virol. 1992;73(Pt 6):1395–1400. doi: 10.1099/0022-1317-73-6-1395. [DOI] [PubMed] [Google Scholar]
- 97.Sichero L, Villa LL. Epidemiological and functional implications of molecular variants of human papillomavirus. Braz. J. Med. Biol. Res. 2006;39:707–717. doi: 10.1590/s0100-879x2006000600002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.