

Abstract
Patterns of alternation of hydrophobic and polar residues are a profound aspect of amino acid sequences, but a feature not easily interpreted for soluble proteins. Here we report statistics of hydrophobicity patterns in proteins of known structure in a current protein database as compared with results from earlier, more limited structure sets. Previous studies indicated that long hydrophobic runs, common in membrane proteins, are underrepresented in soluble proteins. Long runs of hydrophobic residues remain significantly underrepresented in soluble proteins, with none longer than 16 residues observed. These long runs most commonly occur as buried α helices, with extended hydrophobic strands less common. Avoiding aggregation of partially folded intermediates during intracellular folding remains a viable explanation for the rarity of long hydrophobic runs in soluble proteins. Comparison between database editions reveals robustness of statistics on aqueous proteins despite an approximately twofold increase in nonredundant sequences. The expanded database does now allow us to explain several deviations of hydrophobicity statistics from models of random sequence in terms of requirements of specific secondary structure elements. Comparison to prior membrane-bound protein sequences, however, shows significant qualitative changes, with the average hydrophobicity and frequency of long runs of hydrophobic residues noticeably increasing between the database editions. These results suggest that the aqueous proteins of solved structure may represent an essentially complete sample of the universe of aqueous sequences, while the membrane proteins of known structure are not yet representative of the universe of membrane-associated proteins, even by relatively simple measures of hydrophobic patterns.
Keywords: hydrophobicity, statistics, sequence database, protein structure, bioinformatics
The presence of both hydrophobic and polar residues interspersed through polypeptides chains is one of the most general features of amino acid sequences encoded by genes (Chothia 1984; White 1994). Protein scientists understand this in some general sense; for proteins soluble in aqueous solution the removal of hydrophobic residues from water and their interaction in a buried core is a driving force for chain folding, while the presence of polar amino acids is necessary for the formation of the surface interface between protein and solvent. For integral membrane proteins, the organization of hydrophobic and polar residues differ, with long hydrophobic stretches folding within the apolar lipid environment but polar residues required for stretches of the sequences that are solvent exposed in the cytosolic or extracellular environments. Except for a few specialized cases, such as the heptad repeats directing chains into the coiled-coil fold (O’Shea et al. 1991; Cohen and Parry 1994), few simple patterns unambiguously controlling chain folds are evident in sequences of globular aqueous proteins.
Statistical analyses of patterns of hydrophobicity in protein sequences and structures nonetheless have a long and continuing history (White 1994; Broome and Hecht 2000). Pioneering work in the area came from studies of residue conservation in specific protein families, examining hydrophobicity as one of many factors that appeared to constrain amino acid choice in different structural environments (Lesk and Chothia 1980, 1982; Chothia and Lesk 1982). More recent studies have focused specifically on simplified models of hydrophobicity. For example, White and Jacobs (1990) began studies of hydrophobic/polar (HP) run-length distributions in order to test for randomness of protein sequence hydrophobicities, concluding that most protein sequences are individually indistinguishable from random sequences. Vazquez et al. (1993) examined short HP patterns, finding several significantly favored or suppressed in real protein sequences. Strait and Dewey (1996) examined HP sequences from an information theoretic perspective, showing that they contain much less information than random sequences and therefore exhibit biases for particular arrangements of hydrophobic and hydrophilic residues. Hecht and collaborators (West and Hecht 1995; Xiong et al. 1995; Broome and Hecht 2000) have extensively examined the influence of short or periodic sequence patterns, including HP alternations, in protein structures, finding that such patterns are major determinants of secondary structure and that alternating patterns are specifically disfavored in protein sequences.
In recent years it has become clear that some of the information in amino acid sequences is used to determine the conformations of folding intermediates, which may differ from native states, or to avoid off-pathway aggregation (Goldenberg et al. 1983; Mitraki et al. 1991; King et al. 1996). In considering whether some sequences might be important for folding processes, Istrail et al. (1999) carried out a lattice simulation of the influence of amino acid sequence on propensity of chains to aggregate, building on an approach developed by Gupta et al. (1998). An exhaustive search of the sequence space was carried out for short chains of length 16 with a simple HP model. The results showed that hydrophobic runs increased the chance that folding intermediates would aggregate, preventing them from reaching the native state. Subsequently, statistical tests on protein sequences in a database of proteins of known structure revealed that proteins soluble in aqueous buffers did indeed have a statistically significant elevation of alternations between hydrophobic and hydrophilic residues, as well as preferences for or aversions to specific lengths of blocks of consecutive hydrophobic residues (Schwartz et al. 2001). The statistical significance of the results suggested that they represented general features of amino acid sequences of extant proteins. Simplified simulation models have continued to yield insights into optimal folding conditions and the competition between productive folding and oligomerization and different forms of ordered and disordered aggregation (Gupta et al. 1999; Smith and Hall 2001; Nguyen and Hall 2002; Jang et al. 2004a,b).
In the few years since the prior database study was conducted, there has been an explosive growth in the volume of biological data available. While this growth has been most dramatic with respect to genome sequences (Benson et al. 2005), the volume of protein structure data has also been increasing exponentially with time (Berman et al. 2002a,b). One can reasonably ask whether these databases are undergoing merely quantitative changes in size or are becoming qualitatively different over time. That is, are the proteins being added today similar to those already present? By extension, are the databases in the present forms (or their form five years from now or five years hence) representative of the full range of the proteome, or do they reflect biases in our ability to populate them? We can ask such questions by examining how the character of the databases is changing over time. Hydrophobic/hydrophilic statistics provide an excellent platform for conducting such a comparative analysis. They are simple enough to be easily reproduced and robust to small changes in methodology for individual data sets. Yet they are sufficiently discriminating to show broad changes in sequence properties between distinct data sets.
In this study, we expand on the prior work on analysis of hydrophobicity patterns in proteins by the application of relatively simple measures to evolving databases over time. Specifically, we build on our prior analysis of hydrophobic block lengths and HP alternation statistics to determine how changes in database contents have altered these statistics over time, what new analyses the improved databases permit, and what conclusions can be drawn from them about the changing character of the space of known protein structures.
Results
We examined the hydrophobic character of amino acid sequences corresponding to proteins of known structure. We used nonredundant sequences from two versions of the ASTRAL compendium (Brenner et al. 2000), a collection of amino acid sequences corresponding to proteins of known structure that have been assigned structural classes by the SCOP hierarchy (Murzin et al. 1995). One set was extracted from SCOP version 1.48 (released December 1999), which was used in the prior work (Schwartz et al. 2001), and a more recent set was extracted from SCOP version 1.65 (released December 2003). Sequences were then separated into an aqueous and a membrane-associated subset based on SCOP classifications. For the recent SCOP release, the aqueous proteins were further subdivided by SCOP structural classes to extract all-α, all-β, interspersed α and β, and disjoint α and β structures. Residues were converted into hydrophobic (H) or polar (P), yielding collections of HP sequences. Sequence sets were characterized by the fraction of hydrophobic residues in each set (pH), histograms of the number of sequences exhibiting a given fraction of hydrophobic residues (per-sequence hydrophobic residue fractions), the frequency of alternations between hydrophobic and hydrophilic residues (HP or PH patterns in the sequences), and the frequencies of occurrence of maximal runs of consecutive hydrophobic residues. Significance of the alternation and run-length statistics was assessed in comparison to a null hypothesis of random residue orders. The details of the data curation and statistical testing are described in Materials and Methods.
Some general trends are worth noting before the detailed discussion of the data. Overall, the data show a clear bias toward elevated numbers of alternations and reduced numbers of long blocks of hydrophobic residues in aqueous proteins relative to the null hypothesis. These features appear qualitatively nearly identical between the two database versions. The basic character of the two databases in terms of fraction of hydrophobic residues and distributions of these residues among sequences are likewise well conserved. Membrane proteins do not show significant deviations from the null hypothesis in numbers of alternations, although they do show significant elevation in frequencies of many long blocks. The membrane proteins exhibit notable changes in the frequencies of long blocks, the overall hydrophobicity of the sequence set, and the distributions of per-sequence hydrophobicities between the two database editions.
Table 1 shows a comparison of the data available in the prior study to that available in the current study. The number of nonredundant amino acid sequences of known structure available has approximately doubled for both aqueous and membrane-associated proteins between the v. 1.48 and v. 1.65 databases. Hydrophobicity, as measured by the percentage of residues denoted hydrophobic by the definition in Materials and Methods, is nearly unchanged between the two versions for aqueous proteins (45.1% vs. 45.4%), although it has increased somewhat for membrane-associated proteins (47.9% vs. 50.4%).
Table 1.
Statistics on database contents between prior and current work
| SCOP v.1.48 | SCOP v.1.65 | |
| Total sequences | 9,912 | 20,619 |
| Total domains | 22,140 | 54,745 |
| Nonredundant aqueous sequences | 2,753 (45.1% hydrophobic) | 5,385 (45.4% hydrophobic) |
| Nonredundant membrane sequences | 59 (47.9% hydrophobic) | 118 (50.4% hydrophobic) |
We can further test the notion that the set of membrane proteins has qualitatively changed over time to a greater degree than has the set of aqueous proteins by examining per-sequence hydrophobicities over time. Figure 1 shows histograms of the number of sequences with a given fraction of hydrophobic residues for both database versions. Figure 1A reveals little qualitative change between the two database versions for the aqueous proteins. Figure 1B, however, shows a notable shift toward more hydrophobic sequences for the membrane-bound proteins.
Figure 1.

Histograms of per-sequence hydrophobicity content between the two database versions. (A) Histogram for aqueous proteins. (B) Histogram for membrane-associated proteins. Aqueous proteins are plotted with a histogram bin of 1% and membrane-associated proteins with a bin of 10% due to the smaller number of membrane sequences.
Table 2 summarizes the results of alternation analyses for all data sets. Analysis of aqueous proteins reveals similar patterns between the v. 1.48 and v. 1.65 data sets. The percent elevation of alternations is unchanged at 2.22% between the data sets. The Z-score has, however, increased from 14.9 to 21.1 due to the larger number of residues examined. An alternate definition of hydrophobicity based on ability of amino acids to facilitate membrane insertion (Hessa et al. 2005) yields qualitatively similar results. The elevation of alternations is slightly increased as a percentage of expectation, but slightly decreased in significance, most likely because the alternate definition classifies fewer residues as hydrophobic and thereby yields lower total counts. Figure 2 shows the hydrophobic block length statistics, again revealing a strong similarity between the two data sets. The pattern of elevated and suppressed block lengths is nearly identical between the two except at the longest block lengths, where small counts make the numbers unreliable. The larger size of the recent database leads to a narrower ±3 standard deviation region, allowing us to attribute significance to more individual data points than was previously possible.
Table 2.
Alternation statistics by data set
| Aqueous (v. 1.48) | Aqueous (v. 1.65) | Membrane (v. 1.48) | Membrane (v. 1.65) | All-α | All-β | Interspersed α/β | Disjoint α/β | |
| Aexp | 232,541 | 466,081 | 6144 | 11,884 | 74,094 | 92,773 | 170,777 | 95,726 |
| Aobs | 237,716 | 476,405 | 6242 | 11,807 | 76,015 | 95,300 | 174,424 | 98,377 |
| % diff. | +2.22% | +2.22% | +1.60% | −0.65% | +2.59% | +2.72% | +2.14% | +2.77% |
| Z-score | +14.9 | +21.1 | +1.77 | −1.01 | +9.86 | +11.5 | +12.4 | +11.9 |
| Aexp | 193,165 | 390,216 | 5055 | 10,266 | 61,985 | 76,214 | 143,882 | 80,431 |
| Aobs | 197,856 | 400,071 | 5087 | 10,137 | 64,085 | 78,130 | 146,898 | 82,670 |
| % diff. | +2.43% | +2.53% | +0.62% | −1.25% | +3.34% | +2.51% | +2.10% | +2.78% |
| Z-score | +12.2 | +18.2 | +0.51 | −1.51 | +9.71 | +7.83 | +9.24 | +9.11 |
For each data set, the table lists the expected number of H/P alternations (Aexp ), the observed number (Aobs ), the percent difference, and the corresponding Z-score (standard deviations above the mean). The upper rows were derived from our primary definition of hydrophobic residues (Ala, Phe, Ile, Leu, Met, Pro, Val, Trp, and Tyr), while the lower rows were derived from the Hessa et al. (2005) definition (Cys, Ile, Leu, Met, Phe, and Val).
Figure 2.

Hydrophobic block statistics for aqueous proteins, comparing v. 1.48 (A) and v. 1.65 (B) databases.
While our primary interest is the broad trends, it is also informative to ask about outliers in the data. In particular, although long hydrophobic blocks are rarer than would be expected by chance, they do nonetheless occur and we can ask whether there are any recurring patterns to their occurrence. Table 3 lists all proteins with hydrophobic blocks of length >11. Of nine such blocks, four consist of α helices largely or completely buried in the protein interior and two consist of buried β strands and connected turns. Three of the blocks are surface-exposed, including two surface helices and one exposed β strand and turn. Adding in an additional 17 proteins with length 11 blocks (Supplementary Table S1) results in 13 largely or completely buried helices, seven largely or completely buried strands, one buried loop region, three surface helices, and two surface strands. Note, however, that these identifications are based on solved structures that are often truncated forms of the proteins or may be lacking normal binding partners; it is possible that some hydrophobic blocks identified here as exposed are, in fact, buried in the biologically active molecules. At least one exposed strand, though, appears to be genuinely solvent-exposed. This strand corresponds to the proline-rich “fin” of phage PRD receptor-binding protein P2, which is proposed to be involved in recognition and attachment to the target receptor (Xu et al. 2003). Figure 3 provides examples of how long blocks are typically accommodated in protein structures, showing a mostly buried helix (Fig. 3A), a buried strand (Fig. 3B), and the unusual projecting PRD receptor block (Fig. 3C).
Table 3.
Aqueous proteins exhibiting long hydrophobic blocks
| Block Length | PDB ID | Name | Reference | Notes |
| 12 | 1LJ8 | Mannitol 2-Dehydrogenase (P. fluroescens) | Kavanagh et al. 2002 | Buried helix |
| 12 | 1UBY | Farnesyl Pyrophosphate Synthetase (G. gallus) | Tarshis et al. 1996 | Buried helix |
| 12 | 1QJ5 | 7,8 Diaminopelargonic Acid Synthase (E. coli) | Kack et al. 1999 | Buried strand and turn |
| 12 | 1ALN | Cytidine Deaminase (E. coli) | Xiang et al. 1996 | Partially buried helix |
| 13 | 1GWI | A3(2) Cyp154C1 (S. coelicolor) | Podust et al. 2003 | Partially buried helix |
| 13 | 1DD3 | Ribosomal Protein L12 (T. maritima) | Wahl et al. 2000 | Surface helix |
| 13 | 1GUQ | Galactose-1-phosphate uridylyltransferase (E. coli) | Thoden et al. 1997 | Buried strand and turn |
| 14 | 1N7V | Receptor Binding Protein P2 (phage PRD1) | Xu et al. 2003 | Surface strand and turn |
| 16 | 1TCA | Lipase B (C. albicans) | Uppenberg et al. 1994 | Surface helix |
The table shows proteins with hydrophobic blocks of at least 12 amino acids, describes the structural nature of the run, and provides a reference to the structure in question.
Figure 3.

Examples of aqueous proteins with unusually long hydrophobic blocks. Images show cartoons of three representative protein structures, with long hydrophobic blocks highlighted in red. (A) Cytidine deaminase, an example of a protein with a hydrophobic block occurring as a partially buried helix; (B) 7,8 diaminopelargonic acid synthase, an example of a protein with a hydrophobic block occurring as a buried strand and turn region; (C) phage PRD1 receptor binding protein P2, illustrating an unusual surface-exposed example of a long hydrophobic block. Prolines in the P2 block are highlighted.
For membrane-associated proteins, the hydrophobicity patterns are somewhat different than they are for the aqueous proteins. The number of alternations shifted from a 1.60% elevation to a −0.65% suppression of alternations relative to expectation. In neither case is the result statistically significant, though (Z-score +1.77 vs. −1.01). Application of the alternate Hessa et al. (2005) hydrophobicity definition again yields qualitatively similar results, although fewer observed alternations relative to expectation for both database editions. Figure 4 illustrates block length statistics for the membrane proteins. The ±3 standard deviation region is narrowed for the recent data, as it was with the aqueous proteins. While previously almost all data points were individually insignificant, the plot now shows a significant suppression of most short block lengths. The elevation of long block frequencies is also comparatively more pronounced now both in the significance and the absolute frequencies with which very long hydrophobic blocks are observed. Table 4 lists the 10 membrane proteins containing the longest hydrophobic blocks, all of length at least 14. Visual inspection showed nine of the 10 to occur within transmembrane helices, with the remaining one (from a V. cholerae ABC transporter) unknown because it occurs in a portion of sequence missing from the solved structure.
Figure 4.

Hydrophobic block statistics for membrane proteins, comparing v. 1.48 (A) and v. 1.65 (B) databases.
Table 4.
Membrane proteins exhibiting long hydrophobic blocks
| Block Length | PDB ID | Name | Reference |
| 14 (×2) | 1EZV | Mitochondrial cytochrome b subunit (S. cerevisiae) | Hunte et al. 2000 |
| 14 | 1PF4 | Multidrug resistance ABC transporter MsbA (V. cholerae) | Chang 2003 |
| 14 | 1FFT | Cytochrome O ubiquinol oxidase, subunit II (E. coli) | Abramson et al. 2000 |
| 14 | 1OCR | Mitochondrial cytochrome c oxidase, subunit I (B. taurus) | Yoshikawa et al. 1998 |
| 14 +15 | 1EHK | Bacterial ba3 type cytochrome c oxidase subunit I (T. thermophilus) | Soulimane et al. 2000 |
| 16 | 1MSL | Gated mechanosensitive channel (M. tuberculosis) | Chang et al. 1998 |
| 16 | 1JBO | Subunit XII of photosystem I reaction centre, PsaM (S. elongatus) | Jordan et al. 2001 |
| 16 | 1M56 | Bacterial aa3 type cytochrome c oxidase subunit IV (R. sphaeroides) | Svensson-Ek et al. 2002 |
The table shows proteins with hydrophobic blocks of at least 14 amino acids, describes the structural nature of the run, and provides a reference to the structure in question. All long hydrophobic runs listed are parts of transmembrane helices, with the possible exception of the length 14 block of 1PF4, which was in a portion of sequence absent from the solved structure.
The expanded database has allowed us to perform new analyses on individual SCOP structural classes. The results are described in Table 2 and Figure 5. All four classes examined (all-α, all-β, interspersed α/β, and disjoint α+β) show significant elevations of alternations relative to expectation. All four plots of hydrophobic block frequencies (Fig. 5A–D) show qualitatively similar patterns of elevated numbers of short blocks and suppressed numbers of long blocks relative to expectation. The individual block lengths observed to be especially elevated or suppressed differ somewhat from one class to another, however, with differences particularly pronounced between the all-α and all-β class.
Figure 5.
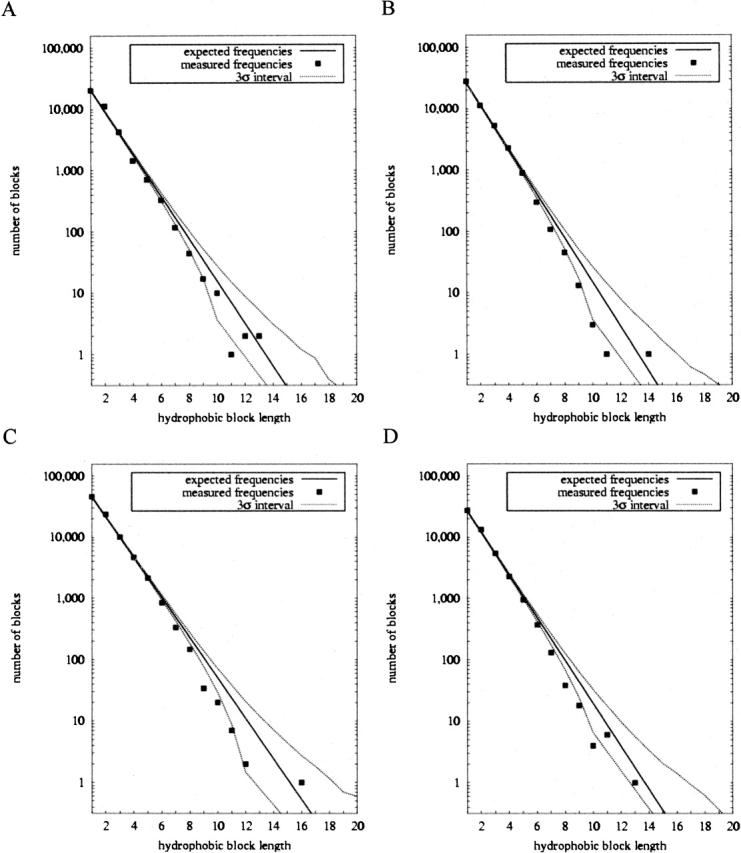
Hydrophobic block statistics for structural classes. (A) All-α proteins. (B) All-β proteins. (C) Interspersed α and β proteins. (D) Disjoint α and β proteins.
Table 5 examines the issue of class-specific block length patterns in more detail, showing how many surplus or missing blocks above expectations are found for block lengths 1 through 7 for the aqueous proteins as a whole and for the all-α and all-β proteins separately. For aqueous proteins as a whole, the total surplus of alternations is explained by a surplus of blocks of lengths 1, 2, and 3, with the length 2 surplus accounting for a large majority of the excess alternations. The examination of all-α and all-β structures shows more subtle patterns of length-specific hydrophobic block surpluses. All-α proteins have a clear suppression of length 1 blocks (isolated hydrophobic residues), a slight elevation of length 3 blocks, but a very large elevation of length 2 blocks. All-β proteins have a deficit of length 2 blocks and small increases of length 3 and length 4 blocks, but a large increase of length 1 blocks.
Table 5.
Surplus hydrophobic blocks by run length
| Total Aqueous | All-α | All-β | |
| 1 | +1,277 | − 340.9 | +1,407 |
| 2 | +5,586 | +1,974 | − 195.4 |
| 3 | +528.6 | +73.9 | +287.9 |
| 4 | − 536.8 | − 447.7 | +100.6 |
| 5 | − 440.4 | − 143.1 | − 69.2 |
| 6 | − 232.2 | − 57.7 | − 118.9 |
| 7 | − 183.6 | − 56.0 | − 74.7 |
The table shows the total number of hydrophobic blocks above expectations found for lengths 1 through 7 for the aqueous proteins as whole, for all-α proteins alone, and for all-β proteins alone.
Discussion
Features of the hydrophobic runs in proteins of known structure
One possible reason for the biases we see in hydrophobic block length frequencies is the propensity of certain HP patterns in particular secondary structures. Previous studies have shown a strong preference for particular HP patterns (Vazquez et al. 1993) or, similarly, hydrophobic periodicities (Hennetin et al. 2003), in particular secondary structures. The most obvious reason for such patterns is a general preference for amphipathic secondary structure elements, as such elements will often have one solvent exposed and one buried surface. This hypothesis would provide a clear explanation for the elevation of length 2 and length 3 blocks in all-α structures and the elevation of length 1 blocks in all-β structures we observe above (Table 5). The elevation of length 3 blocks in all-β structures may also fit this pattern. The elevation of length 4 blocks in all-β structures does not fit the pattern, but might be explained by internally buried β strands that favor hydrophobic surfaces on both sides of the strand.
Preferences for patterns consistent with particular secondary structure elements may explain an apparent contradiction between our conclusion that HP alternations are favored in aqueous proteins and the conclusion of Hecht and colleagues (West and Hecht 1995; Xiong et al. 1995; Broome and Hecht 2000) that alternating HP patterns are specifically disfavored. Alternations are indeed more frequent than would be expected by chance, but this is primarily due to an elevation in blocks of length 2, not to repeated consecutive alternations.
While long hydrophobic runs are significantly rarer than would be expected by chance, they can occur in some outlier sequences (Table 3; Fig 3). These sequences are usually buried internally to the folded protein, generally as α helices, but sometimes as β strands. Even when external, though, they may preferentially correspond to binding interfaces. We can therefore conclude that mechanisms do exist for protecting long hydrophobic blocks both during and after folding, even if selection against such sequences is strongly evident in the proteome in general. The longest hydrophobic runs found in the membrane sequences occurred within transmembrane helices in all cases that could be definitively verified. Given the small size of the available membrane-protein database, though, we cannot definitively conclude that long hydrophobic runs will not be found in cytosolic regions of membrane-associated proteins.
Very long α helices are common in proteins of eukaryotes, for example, in the coiled coils of tropomyosins, myosins, and intermediate filament proteins. We are not aware of any intrinsic structural features that would limit the length of α helices within soluble proteins. During the folding of soluble proteins, partially folded intermediates that have not fully buried their hydrophobic surfaces are particularly prone to competing aggregation reactions (Mitraki and King 1989; Speed et al. 1996). One of the functions of the ubiquitous chaperonin proteins is to prevent these off-pathway aggregation reactions (Frydman et al. 1994; Frydman and Hartl 1996). A significant body of experimental evidence identifies hydrophobic regions of the partially folded substrates as the binding sites for the chaperonins (Buchner et al. 1991; Wang et al. 1999). We suspect that this aggregation hazard to partly folded intermediates leads to selection against very long hydrophobic runs within soluble proteins (Istrail et al. 1999).
Interestingly, several studies conducted since our original database analysis have confirmed a link between HP patterns and propensity for aggregation. While the hypothesis of such a link was the original motivation behind our prior work, we could only speculate that the effects we measured statistically in protein sequences were in fact related to aggregation propensity of real protein sequences. López de la Paz and Serrano (2004) and Ventura et al. (2004) have since shown that small HP patterns can serve as “aggregation seeds,” promoting aggregation in otherwise disparate proteins. DuBay et al. (2004) examined several possible predictors of aggregation propensity based on HP sequence and found that the presence of specific HP patterns was second only to total hydrophobicity in its effectiveness at predicting aggregation. These experiments provide direct evidence for the argument of our prior work that amino acid sequence must be treated not merely as a “structural code” encoding a protein’s native state but also as a “folding code” providing additional information on how to get to and remain in the native state while avoiding pitfalls such as aggregation.
Use of alternation statistics to assess the status of the protein databases
While the expanded databases did allow us to reach some conclusions we could not before, some of the most interesting conclusions of this study derive from a comparison between the older and the newer data sets. Results for aqueous proteins have essentially been static since our original study. While the size of the database has approximately doubled, increasing the significance of some statistical tests, the overall hydrophobicity and the relative magnitude of the elevation in alternations and elevation or suppression of various block lengths is nearly unchanged. We can thus conclude that there has been no significant qualitative change in the structural databases of aqueous proteins, at least insofar as the simple measures we apply can detect. It is therefore likely that a fairly accurate representation of the full set of aqueous protein structures has been present in the Protein Data Bank for several years. Such a conclusion is necessarily tentative when drawn from only two database editions, though, and should be re-evaluated as the databases continue to grow.
The qualitative results of our analyses of alternation statistics are robust to a significant change in hydrophobicity definition. Hessa et al. (2005), though, raise the possibility that the effective hydrophobicity of a given residue can vary depending on its position in the sequence. Further comparative analyses of statistics on more complex hydrophobicity patterns than were applied here—such as the HP strings considered in some prior sequence database studies (Vasquez et al 1993; West and Hecht 1995; Broome and Hecht 2000)—might be revealing about more subtle changes in the databases over time.
The membrane proteins, on the other hand, have substantially changed in character between the studies. While they also experienced approximately a twofold increase in the number of nonredundant sequences in the database, they have also seen a noticeable increase in hydrophobicity (Fig. 1; Table 1), a decrease in the relative number of alternations (Table 2), and an increase in the counts of long hydrophobic blocks (Fig. 2) relative to expectations. Although we must be cautious in drawing conclusions, given the small number of nonhomologous membrane-associated protein structures available, we believe that this result likely reflects improvements in the technologies required to generate the structures of membrane-associated proteins. In particular, the field of membrane protein crystallization is comparatively young (Garavito and Rosenbusch 1980; Michel and Oesterhelt 1980; Ozawa et al. 1980a,b) and continues to see great advances in solubilizing, reconstituting, and crystallizing membrane proteins. For recent reviews, see Seddon et al. (2004) and Nollert (2005). As it is becoming feasible to solve “harder” membrane-associated protein structures, the database is becoming more representative, shifting toward proteins that are more hydrophobic, larger, and more prone to long blocks of hydrophobic residues. We would therefore suggest that one should be more cautious in drawing conclusions about the nature of membrane protein structures in general from those already known than one must be in drawing conclusions about aqueous protein structures from the current examples. It may be productive in the future to monitor the databases by these and other statistics to better judge when and by what measures they reach an apparently stable state and can thus be considered reliable samples of the universe of protein structures.
Materials and methods
Data collection and curation
We obtained sequences from the ASTRAL compendium (Brenner et al. 2000), a collection of amino acid sequences corresponding to proteins of known structure that have been assigned structural classes by the SCOP hierarchy (Murzin et al. 1995). Analysis was based on two data sets: sequences extracted from SCOP version 1.48, which was used in the prior work (Schwartz et al. 2001), and a more recent set extracted from SCOP version 1.65. Due to a change in the Astral policies, the older data set contained sequences determined to be nonredundant at a 50% similarity level and the newer data set sequences nonredundant at a 40% similarity level. We believe this change in protocol is unlikely to introduce any systematic biases that would affect the results of the present study. In each case, we removed all sequences containing unknown amino acids and we converted the remaining amino acid sequences into hydrophobic-polar (HP) sequences by defining the residues Ala, Ile, Leu, Met, Phe, Pro, Trp, Tyr, and Val as hydrophobic and the remainder as polar. The prior work showed that the statistics analyzed are robust to small changes in the membership of these classes. In order to verify robustness to larger changes, though, we repeated a portion of the database analysis using a recent hydrophobicity definition based on ability of amino acids to facilitate membrane insertion (Hessa et al. 2005). This “biological” hydrophobicity definition identified a notably different set of hydrophobic residues: Cys, Ile, Leu, Met, Phe, and Val.
The initial data set was divided into several subclasses. Sequences corresponding to designed proteins were discarded. Those classified by SCOP as “Membrane and Cell Surface Proteins and Peptides” were removed to leave a set of aqueous protein sequences and a set of membrane-associated protein sequences for each SCOP version examined. For the recent SCOP release, the aqueous proteins were further subdivided by SCOP structural class to extract all-α, all-β, interspersed α and β, and disjoint α and β structures. Additional SCOP classes were examined but are omitted from further discussion because they were of insufficient size to obtain informative results. The subdivision by SCOP classes other than the membrane-based was likewise not applied to the original sequence databases, because the smaller database would have made it difficult to derive statistically significant results.
Statistical analysis
The mathematical formulas described in this section were developed for our prior study of hydrophobic/hydrophilic sequences and are described in more detail in that study (Schwartz et al. 2001). Following the previous study, statistics were derived on the frequency of alternations between hydrophobic and hydrophilic residues (HP or PH patterns in the sequences) and the frequencies of occurrence of maximal runs of consecutive hydrophobic residues. For each of the sequence sets derived in the data curation step, we counted the number of alternations and the number of occurrences of maximal hydrophobic runs of each run length. We further measured the fraction of hydrophobic residues in the set, which we denote pH. We also computed histograms of the fraction of hydrophobic residues in each sequence. For the aqueous proteins, we used a bin size of 1% in collecting sequences for the histograms. For membrane proteins, we used a bin size of 10% due to the smaller number of nonredundant sequences available.
Again following the prior work, we compared observed values to a null hypothesis of residues randomly selected with hydrophobic probability pH. For this purpose, we computed the expected number of alternations in each data set. The expected number of alternations in a single sequence of length n is given by:
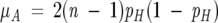 |
The variance in this number is given by:
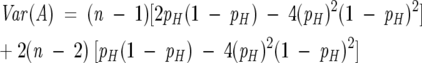 |
The mean and variance for a full sequence set can be determined by summing the above per-sequence formulas over all sequence lengths in the set.
We determined the expected frequency of hydrophobic runs of each length using the function P(n,k,m), the probability that a sequence of length n has exactly k blocks of length exactly m, which is solved for the null hypothesis by the recurrence relation:
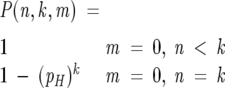 |
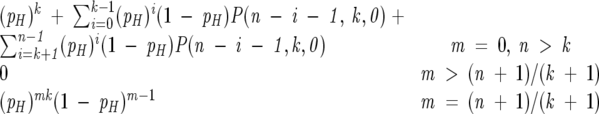 |
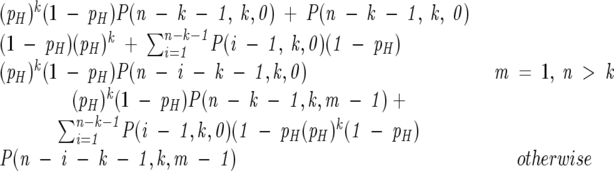 |
We do not have an exact analytical formula for the variance in run-length counts under the null hypothesis, and therefore estimated it for each sequence set by generating 1,000,000 random data sets under the null hypothesis.
Analysis of the structural nature of outliers exhibiting long hydrophobic blocks was performed by visual inspection using Rasmol version 2.7.2.1.1 (Bernstein 2000). Rasmol was also used to prepare the structure images in Figure 3.
Electronic supplemental material
Supplementary Table S1 provides PDB IDs, sources, and descriptions of hydrophobic runs of length >10 found in the aqueous sequences examined.
Acknowledgments
R.S. is supported by NSF award no. 0346981. J.K. is supported by NIH grant no. GM 17980.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051741806.
Supplemental material: see www.proteinscience.org
References
- Abramson, J., Riistama, S., Larsson, G., Jasaitis, A., Svensson-Ek, M., Laakkonen, L., Puustinen, A., Iwata, S., and Wikstrom, M. 2000. The structure of the ubiquinol oxidase from Escherichia coli and its ubiquinone binding site. Nat. Struct. Biol. 7: 910–917. [DOI] [PubMed] [Google Scholar]
- Benson, D.A., Karsch-Mizrachi, I., Lipman, D.J., Ostell, J., and Wheeler, D.L. 2005. GenBank. Nucleic Acids Res. 33: D34–D38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, H.M., Battistuz, T., Bhat, T.N., Bluhm, W.F., Bourne, P.E., Burkhardt, K., Feng, Z., Gilliland, G.L., Iype, L., Jain, S., et al. 2002a. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 58: 899–907. [DOI] [PubMed] [Google Scholar]
- Berman, H.M., Goodsell, D.S., and Bourne, P.E. 2002b. Protein structures: From famine to feast. Am. Sci. 90: 350–359. [Google Scholar]
- Bernstein, H.J. 2000. Recent changes to RasMol, recombining the variants. Trends Biochem. Sci. 25: 453–455. [DOI] [PubMed] [Google Scholar]
- Brenner, S.E., Koehl, P., and Levitt, M. 2000. The ASTRAL compendium for protein structure and sequence analysis. Nucleic Acids Res. 28: 254–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broome, B.M. and Hecht, M.H. 2000. Nature disfavors sequences of alternating polar and non-polar amino acids: Implications for amyloidogenesis. J. Mol. Biol. 296: 961–968. [DOI] [PubMed] [Google Scholar]
- Buchner, J., Schmidt, M., Fuchs, M., Jaenicke, R., Rudolph, R., Schmid, F.X., and Kiefhaber, T. 1991. GroE facilitates refolding of citrate synthase by suppressing aggregation. Biochemistry 30: 1586–1591. [DOI] [PubMed] [Google Scholar]
- Chang, G. 2003. Structure of Msba from Vibrio cholera: A multidrug resistance ABC transporter homolog in a closed conformation. J. Mol. Biol. 330: 419–430. [DOI] [PubMed] [Google Scholar]
- Chang, G., Spencer, R.H., Lee, A.T., Barclay, M.T., and Rees, D.C. 1998. Structure of the MscL homolog from Mycobacterium tuberculosis: A gated mechanosensitive ion channel. Science 282: 2220–2226. [DOI] [PubMed] [Google Scholar]
- Chothia, C. 1984. Principles that determine the structure of proteins. Annu. Rev. Biochem. 53: 537–572. [DOI] [PubMed] [Google Scholar]
- Chothia, C. and Lesk, A.M. 1982. Evolution of proteins formed by β-sheets. I. Plastocyanin and azurin. J. Mol. Biol. 160: 309–323. [DOI] [PubMed] [Google Scholar]
- Cohen, C. and Parry, D.A. 1994. α-helical coiled coils: More facts and better predictions. Science 263: 488–489. [DOI] [PubMed] [Google Scholar]
- DuBay, K.F., Pawar, A.P., Chiti, F., Zurdo, J., Dobson, C.M., and Vendruscolo, M. 2004. Prediction of the absolute aggregation rates of amyloidogenic polypeptide chains. J. Mol. Biol. 341: 1317–1326. [DOI] [PubMed] [Google Scholar]
- Frydman, J. and Hartl, F.U. 1996. Principles of chaperone-assisted protein folding: Differences between in vitro and in vivo mechanisms. Science 272: 1497–1502. [DOI] [PubMed] [Google Scholar]
- Frydman, J., Nimmesgern, E., Ohtsuka, K., and Hartl, F.U. 1994. Folding of nascent polypeptide chains in a high molecular mass assembly with molecular chaperones. Nature 370: 111–117. [DOI] [PubMed] [Google Scholar]
- Garavito, R.M. and Rosenbusch, J.P. 1980. Three-dimensional crystals of an integral membrane protein: An initial x-ray analysis. J. Cell Biol. 86: 327–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg, D.P., Smith, D.H., and King, J. 1983. Genetic analysis of the folding pathway for the tail spike protein of phage P22. Proc. Natl. Acad. Sci. 80: 7060–7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, P., Hall, C.K., and Voegler, A.C. 1998. Effect of denaturant and protein concentrations upon protein refolding and aggregation: A simple lattice model. Protein Sci. 7: 2642–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1999. Computer simulation of the competition between protein folding and aggregation. Fluid Phase Equil. 160: 87–93. [Google Scholar]
- Hennetin, J., Le, T.K., Canard, L., Colloc’h, N., Mornon, J.P., and Callebaut, I. 2003. Non-intertwined binary patterns of hydrophobic/nonhydrophobic amino acids are considerably better markers of regular secondary structures than nonconstrained patterns. Proteins 51: 236–244. [DOI] [PubMed] [Google Scholar]
- Hessa, T., Kim, H., Bihlmaier, K., Lundin, C., Boekel, J., Andersson, H., Nilsson, I., White, S.H., and von Heijne, G. 2005. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433: 377–381. [DOI] [PubMed] [Google Scholar]
- Hunte, C., Koepke, J., Lange, C., Rossmanith, T., and Michel, H. 2000. Structure at 2.3 Å resolution of the cytochrome bc1 complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Struct. Fold. Des. 8: 669–684. [DOI] [PubMed] [Google Scholar]
- Istrail, S., Schwartz, R., and King, J. 1999. Lattice simulations of aggregation funnels for protein folding. J. Comput. Biol. 6: 143–162. [DOI] [PubMed] [Google Scholar]
- Jang, H., Hall, C.K., and Zhou, Y. 2004a. Assembly and kinetic folding pathways of a tetrameric β-sheet complex: Molecular dynamics simulations on simplified off-lattice protein models. Biophys. J. 86: 31–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2004b. Thermodynamics and stability of a β-sheet complex: Molecular dynamics simulations on simplified off-lattice protein models. Protein Sci. 13: 40–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan, P., Fromme, P., Witt, H.T., Klukas, O., Saenger, W., and Krauss, N. 2001. Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution. Nature 411: 909–917. [DOI] [PubMed] [Google Scholar]
- Kack, H., Sandmark, J., Gibson, K., Schneider, G., and Lindqvist, Y. 1999. Crystal structure of diaminopelargonic acid synthase: Evolutionary relationships between pyridoxal-5′-phosphate-dependent enzymes. J. Mol. Biol. 291: 857–876. [DOI] [PubMed] [Google Scholar]
- Kavanagh, K.L., Klimacek, M., Nidetzky, B., and Wilson, D.K. 2002. Crystal structure of Pseudomonas fluorescens mannitol 2-dehydrogenase binary and ternary complexes. Specificity and catalytic mechanism. J. Biol. Chem. 277: 43433–43442. [DOI] [PubMed] [Google Scholar]
- King, J., Haase-Pettingell, C., Robinson, A., Speed, M.A., and Mitraki, A. 1996. Thermolabile folding intermediates: Inclusion body precursors and chaperonin substrates. FASEB J. 10: 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesk, A.M. and Chothia, C. 1980. How different amino acid sequences determine similar protein structures: The structure and evolutionary dynamics of the globins. J. Mol. Biol. 136: 225–270. [DOI] [PubMed] [Google Scholar]
- ———. 1982. Evolution of proteins formed by β-sheets. II. The core of the immunoglobulin domains. J. Mol. Biol. 160: 325–342. [DOI] [PubMed] [Google Scholar]
- López de la Paz, M. and Serrano, L. 2004. Sequence determinants of amyloid fibril formation. Proc. Natl. Acad. Sci. 101: 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel, H. and Oesterhelt, D. 1980. Three-dimensional crystals of membrane proteins: Bacteriorhodopsin. Proc. Natl. Acad. Sci. 77: 1283–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitraki, A. and King, J. 1989. Protein folding intermediates and inclusion body formation. Bio/Technology 7: 690–697. [Google Scholar]
- Mitraki, A., Fane, B., Haase-Pettingell, C., Sturtevant, J., and King, J. 1991. Global suppression of protein folding defects and inclusion body formation. Science 253: 54–58. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247: 536–540. [DOI] [PubMed] [Google Scholar]
- Nguyen, H.D. and Hall, C.K. 2002. Effect of rate of chemical or thermal renaturation on refolding and aggregation of a simple lattice protein. Biotechnol. Bioeng. 80: 823–834. [DOI] [PubMed] [Google Scholar]
- Nollert, P. 2005. Membrane protein crystallization in amphiphile phases: Practical and theoretical considerations. Prog. Biophys. Mol. Biol. 88: 339–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea, E.K., Klemm, J.D., Kim, P.S., and Alber, T. 1991. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science 254: 539–544. [DOI] [PubMed] [Google Scholar]
- Ozawa, T., Suzuki, H., and Tanaka, M. 1980a. Crystallization of part of the mitochondrial electron transfer chain: Cytochrome c oxidase–cytochrome c complex. Proc. Natl. Acad. Sci. 77: 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa, T., Tanaka, M., and Shimomura, Y. 1980b. Crystallization of the middle part of the mitochondrial electron transfer chain: Cytochrome bc1-cytochrome c complex. Proc. Natl. Acad. Sci. 77: 5084–5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podust, L.M., Kim, Y., Arase, M., Neely, B.A., Beck, B.J., Bach, H., Sherman, D.H., Lamb, D.C., Kelly, S.L., and Waterman, M.R. 2003. The 1.92-Å structure of Streptomyces coelicolor A3(2) CYP154C1. A new monooxygenase that functionalizes macrolide ring systems. J. Biol. Chem. 278: 12214–12221. [DOI] [PubMed] [Google Scholar]
- Schwartz, R., Istrail, S., and King, J. 2001. Frequencies of amino acid strings in globular protein sequences indicate suppression of blocks of consecutive hydrophobic residues. Protein Sci. 10: 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon, A.M., Curnow, P., and Booth, P.J. 2004. Membrane proteins, lipids and detergents: Not just a soap opera. Biochim. Biophys. Acta 1666: 105–117. [DOI] [PubMed] [Google Scholar]
- Smith, A.V. and Hall, C.K. 2001. Protein refolding versus aggregation: Computer simulations on an intermediate-resolution protein model. J. Mol. Biol. 312: 187–202. [DOI] [PubMed] [Google Scholar]
- Soulimane, T., Buse, G., Bourenkov, G.P., Bartunik, H.D., Huber, R., and Than, M.E. 2000. Structure and mechanism of the aberrant ba3-cytochrome c oxidase from Thermus thermophilus. EMBO J. 19: 1766–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speed, M.A., Wang, D.I., and King, J. 1996. Specific aggregation of partially folded polypeptide chains: The molecular basis of inclusion body composition. Nat. Biotechnol. 14: 1283–1287. [DOI] [PubMed] [Google Scholar]
- Strait, B.J. and Dewey, T.G. 1996. The Shannon information entropy of protein sequences. Biophys. J. 71: 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson-Ek, M., Abramson, J., Larsson, G., Tornroth, S., Brezezinski, P., and Iwata, S. 2002. The X-ray crystal structures of wild-type and eq(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J. Mol. Biol. 321: 329–339. [DOI] [PubMed] [Google Scholar]
- Tarshis, L.C., Proteau, P.J., Kellogg, B.A., Sacchettini, J.C., and Poulter, C.D. 1996. Regulation of product chain length by isoprenyl diphosphate synthases. Proc. Natl. Acad. Sci. 93: 15018–15023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoden, J.B., Ruzicka, F.J., Frey, P.A., Rayment, I., and Holden, H.M. 1997. Structural analysis of the H166G site-directed mutant of galactose-1-phosphate uridylyltransferase complexed with either UDP-glucose or UDP-galactose: Detailed description of the nucleotide sugar binding site. Biochemistry 36: 1212–1222. [DOI] [PubMed] [Google Scholar]
- Uppenberg, J., Hansen, M.T., Patkar, S., and Jones, T.A. 1994. The sequence, crystal structure determination and refinement of two crystal forms of lipase B from Candida antarctica. Structure 2: 293–308. [DOI] [PubMed] [Google Scholar]
- Vazquez, S., Thomas, C., Lew, R.A., and Humphreys, R.E. 1993. Favored and suppressed patterns of hydrophobic and nonhydrophobic amino acids in protein sequences. Proc. Natl. Acad. Sci. 90: 9100–9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura, S., Zurdo, J., Narayanan, S., Parreno, M., Mangues, R., Reif, B., Chiti, F., Giannoni, E., Dobson, C.M., Aviles, F.X., et al. 2004. Short amino acid stretches can mediate amyloid formation in globular proteins: The Src homology 3 (SH3) case. Proc. Natl. Acad. Sci. 101: 7258–7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl, M.C., Bourenkov, G.P., Bartunik, H.D., and Huber, R. 2000. Flexibility, conformational diversity and two dimerization modes in complexes of ribosomal protein L12. EMBO J. 19: 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z., Feng, H., Landry, S.J., Maxwell, J., and Gierasch, L.M. 1999. Basis of substrate binding by the chaperonin GroEL. Biochemistry 38: 12537–12546. [DOI] [PubMed] [Google Scholar]
- West, M.W. and Hecht, M.H. 1995. Binary patterning of polar and non-polar amino acids in the sequences and structures of native proteins. Protein Sci. 4: 2032–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, S.H. 1994. Global statistics of protein sequences: Implications for the origin, evolution, and prediction of structure. Annu. Rev. Biophys. Biomol. Struct. 23: 407–439. [DOI] [PubMed] [Google Scholar]
- White, S.H. and Jacobs, R.E. 1990. Statistical distribution of hydrophobic residues along the length of protein chains. Implications for protein folding and evolution. Biophys. J. 57: 911–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, S., Short, S.A., Wolfenden, R., and Carter Jr., C.W. 1996. Cytidine deaminase complexed to 3-deazacytidine: A “valence buffer” in zinc enzyme catalysis. Biochemistry 35: 1335–1341. [DOI] [PubMed] [Google Scholar]
- Xiong, H., Buckwalter, B.L., Shieh, H.M., and Hecht, M.H. 1995. Periodicity of polar and nonpolar amino acids is the major determinant of secondary structure in self-assembling oligomeric peptides. Proc. Natl. Acad. Sci. 92: 6349–6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, L., Benson, S.D., Butcher, S.J., Bamford, D.H., and Burnett, R.M. 2003. The receptor binding protein P2 of PRD1, a virus targeting antibiotic-resistant bacteria, has a novel fold suggesting multiple functions. Structure (Camb.) 11: 309–322. [DOI] [PubMed] [Google Scholar]
- Yoshikawa, S., Shinzawa-Itoh, K., Nakashima, R., Yaono, R., Yamashita, E., Inoue, N., Yao, M., Fei, M.J., Libeu, C.P., Mizushima, T., et al. 1998. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science 280: 1723–1729. [DOI] [PubMed] [Google Scholar]