

Abstract
Site-2 proteases (S2Ps) form a large family of membrane-embedded metalloproteases that participate in cellular signaling pathways through sequential cleavage of membrane-tethered substrates. Using sequence similarity searches, we extend the S2P family to include remote homologs that help define a conserved structural core consisting of three predicted transmembrane helices with traditional metalloprotease functional motifs and a previously unrecognized motif (GxxxN/S/G). S2P relatives were identified in genomes from Bacteria, Archaea, and Eukaryota including protists, plants, fungi, and animals. The diverse S2P homologs divide into several groups that differ in various inserted domains and transmembrane helices. Mammalian S2P proteases belong to the major ubiquitous group and contain a PDZ domain. Sequence and structural analysis of the PDZ domain support its mediating the sequential cleavage of membrane-tethered substrates. Finally, conserved genomic neighborhoods of S2P homologs allow functional predictions for PDZ-containing transmembrane proteases in extra-cytoplasmic stress response and lipid metabolism.
Keywords: site-2 protease, regulated intramembrane proteolysis, phylogenetic analysis, domain organization, motif recognition, functional prediction
Regulated intramembrane proteolysis (Rip) is a novel mechanism of signal transduction common to all forms of life (Brown et al. 2000). In the process of Rip, membrane-embedded proteases cleave transmembrane-spanning helical (TMH) segments of signaling proteins to release soluble effectors capable of eliciting various biological responses. Although such hydrolysis occurs within the hydrophobic environment of the lipid bilayer, the catalytic residues responsible for this cleavage resemble those used by soluble proteases and comprise recognizable motifs in predicted TMH regions of the membrane-embedded proteases. Several different families of proteases catalyze Rip: including presenilin, signal peptide peptidase, rhomboid, and site 2 protease (S2P) (Urban and Freeman 2002). Members of each family contain multiple predicted TMH segments and characteristic protease motifs. For both the rhomboids (serine protease) and the presenilins (aspartic protease), these conserved sequence features have allowed identification of diverse families of polytopic transmembrane serine and aspartic proteases (Grigorenko et al. 2002; Ponting et al. 2002; Koonin et al. 2003). Similarly, amino acid sequence comparisons of membrane-embedded S2P metalloproteases have established conserved functional motifs (HExxH and DG) (Lewis and Thomas 1999; Rudner et al. 1999) common to soluble metalloproteases (Makarova and Grishin 1999).
Characterized roles for metazoan S2Ps include cleaving different membrane-tethered transcription factors in response to protein (Ye et al. 2000b) and lipid (Sakai et al. 1996; Rawson et al. 1997) composition in the endoplasmic reticulum (ER). Low levels of membrane cholesterol trigger sterol regulatory element-binding protein (SREBP) release that ultimately activates cholesterol biosynthesis in mammalian cells (Sakai et al. 1996); while low levels of phosphatidylethanolamine signal an analogous S2P cascade in Drosophila activating fatty acid biosynthesis (Dobrosotskaya et al. 2002; Seegmiller et al. 2002). When unfolded proteins accumulate in the ER, the same cascade releases the N-terminal activation domain of a different membrane-bound transcription factor, ATF6 (Ye et al. 2000b). These metazoan signaling cascades require a two-step cleavage mediated sequentially by a membrane-anchored serine protease (S1P) and then by S2P (Ye et al. 2000b).
Functional roles for several bacterial S2P homologs have also been described. For example, Escherichia coli RseP performs the second site cleavage of a membrane-associated transcription factor regulator, RseA, with the first site cleavage mediated by periplasmic serine protease DegS (Alba et al. 2002). This RseA cleavage cascade initiates a bacterial extracytoplasmic stress response analogous to that of the mammalian ER stress response. An Enterococcus faecalis S2P homolog (eep) enhances production of an octapeptide sex pheromone (cAD1) through cleavage of a pheromone precursor (An et al. 1999). Finally, cleavage of Bacillus subtilis pro-σk factor by an S2P homolog (SpoIVFB) activates transcription of genes involved in sporulation (Yu and Kroos 2000). However, this signaling cascade does not appear to require sequential cleavage of the pro-σk substrate. Instead, the upstream serine peptidase (SpoIVB) cleaves a SpoIVFB-interacting protein, SpoIVFA, which leads to release of SpoIVFB inhibition allowing cleavage of pro-σk (Dong and Cutting 2003, 2004; Zhou and Kroos 2004).
Despite several examples of characterized S2P-mediated Rip cascades, questions remain as to the mechanism of S2P-catalyzed hydrolysis within the membrane bilayer, the recognition of various different membrane-tethered S2P substrates, and the evolutionary origins and functional constraints governing this unusual signaling mechanism. The nearly ubiquitous presence of the S2P family among all forms of life suggests the presence of an S2P-like signaling cascade in the last common ancestor. To address this hypothesis and to help answer some of these questions we set out to identify and classify all homologous S2P sequences within a functional and evolutionary context.
Results and Discussion
Sequence conservation in the S2P family suggests a compact three-TMH structural core
Using conserved S2P sequence elements as queries, exhaustive PSI-BLAST searches identified a large number of homologs (334 unique sequences ranging in length from 56 [fragment] to 1127 residues) present in all major phylogenetic lineages. Identified sequences possess common elements likely to play general roles in S2P structure and function. Alignment of these elements reveals conserved hydrophobicity patterns (Fig. 1, yellow highlights) that define three predicted TMH segment boundaries (TMH1–TMH3) and characteristic motifs that outline typical metalloprotease functional residues (Fig. 1, black highlights). Two previously identified motifs (Motif 1 HExxH and Motif 3 DG; Lewis and Thomas 1999) form the zinc-binding site, including a catalytic glutamic acid (E) and two metal-coordinating histidines (H) from one motif and a third metal-coordinating aspartic acid (D) from the other motif (Hooper 1994; Rawson et al. 1997). The motifs fall within predicted TMH boundaries: HExxH in TMH1, GxxxN/S/G in TMH2, and NxxPx xxxDG in TMH3 (Fig. 1). Additional less-conserved sequence elements common to S2P homologs include a short hydrophobic span between TMH1 and TMH2 and a TMH predicted to follow TMH3.
Figure 1.

Alignment and phylogeny of S2P core sequences from representative genomes. Sequences are labeled according to gi number, and species abbreviations are as follows: Bh, Bacillus halodurans; Tm, Thermotoga maritima; Tp, Treponema pallidum; Dr, Deinococcus radiodurans; Ss, Synechocystis sp.; Ml, Mycobacterium leprae; Tv, Thermoplasma volcanium; Hl, Halobacterium sp.; Mt, Methanothermobacter thermautotrophicus; Pa, Pyrobaculum aerophilum; At, Arabidopsis thaliana; Ce, Caenorhabditis elegans; Hs, Homo sapiens; Dm, Drosophila melanogaster; Pf, Plasmodium falciparum; Gl, Giardia lamblia. Sequence labels are colored according to superkingdom (bacteria, black; archaea, red; eukaryota, blue). TMH predictions by HMMTOP (Tusnady and Simon 2001) are shown above the alignment (H, TMH; i, intracellular; o, extracellular) with TMH motifs labeled above. Residues are highlighted according to chemical property conservation (hydrophobic, yellow; small, gray; charged/polar, black) and italicized (in Hl15790880 and At15235376) to mark omitted sequence insertions. In the unrooted tree to the left of the alignment, green circles denote internal tree nodes with at least 70% local bootstrap support. Reliably identified subfamilies are outlined with boxes, with the three ubiquitous ones colored gray. Four major groups of S2P subfamilies are labeled using Roman numerals and subfamily taxonomy designations where applicable (E, eukaryotic; B, bacterial; A, archaeal). Domain architectures are indicated with brackets to the right of the tree, including TMH segments (rectangles: dark blue, core TMH; gray, additional common TMH; white, extra nonconserved TMH), PDZ domain (pink hexagon), CBS domain (green oval), cys-rich insert (yellow triangle), and various unknown soluble domains (colored squares).
Combining these conservations with the results of individual S2P TMH topology predictions suggests a model of transmembrane topology consistent with proteolytic cleavage of a TMH substrate (Fig. 2A). This model places the two functional S2P motifs (HExxH) and (DG) toward the cytosolic side of the membrane, defining the spatial proximity of the active site. Such an orientation is compatible with the defined S2P substrate SREBP cleavage site positioned near the cytosolic portion of the membrane (Duncan et al. 1998). The topological prediction also corresponds to experimental data for S2P homologs. For the bacterial proteins RseP and SpoIVFB, fusion experiments with an enzymatic indicator place the sequence N-terminal to TMH3 in the periplasm (corresponds to the lumen in Fig. 2) and the sequences N-terminal to TMH2 and following TMH3 in the cytosol (Green and Cutting 2000: Kanehara et al. 2001). Experimental evidence mapping glycosylation sites for eukaryotic S2P places the sequences N-terminal to TMH1 and following TMH2 in the lumen (Zelenski et al. 1999).
Figure 2.

Model of S2P TMH membrane topology and active site. (A) Transmembrane topology of conserved S2P core TMH segments (predicted TMH1-TMH3, boxed in dark gray) and its SREBP substrate (predicted TMH, boxed in light gray) is indicated with the cytosol and lumen labeled above and below the membrane, respectively (bounded by double horizontal lines). Circles located within the boxed segments represent amino acid residues ordered consecutively (connected by solid lines) from the N terminus to the C terminus (labeled accordingly), with various insertions (indicated by dotted lines). The triangle indicates the placement of the PDZ domain present in some family members. Conserved residues required for catalysis are highlighted in black and labeled according to conserved residue type. Positions corresponding to small residue conservations are highlighted in gray and labeled according to conserved residue type. (B) A stereoview produced with Molscript (Esnouf 1999) depicts a 3D model of the S2P active site based on the placement of active site residues from a number of structurally divergent metalloenzymes (thermolysin 2tlx, mitochondrial processing peptidase 1hr8, peptide deformylase 1Bs8, and leishmanolysin 1lml). Portions of conserved TMH segments (white helixes) were modeled with an HExxH motif-containing metalloprotease helix (residues 256–270 from 1lml) and a proline-containing TMH helix (residues A18–A33 from 1xqf). Side chains of conserved S2P residues required for catalysis (black) and of other conserved S2P residues (gray) are depicted with bond representation, and the zinc ligand is illustrated with partially transparent CPK. The backbone of a peptide substrate (from 1hr8) is shown as ball-and-stick and indicates the potential relative placement of the SREBP peptide substrate.
Potential functional roles for conserved S2P sequence elements
Structures of several different soluble HExxH-motif-containing enzymes have been determined, including various metalloproteases, mitochondrial processing peptidase, and peptide deformylase. Although these proteins belong to different folds, they share convergent structural and functional features that may also extend to the integral membrane S2P enzymes. In each of these structures, the characteristic motif (HExxH or HxxEH) forms an α-helix that positions two metal ion-coordinating residues (His residues of the HExxH motif) near a catalytic acid (Glu from the HExxH motif) and a third ion-coordinating residue contributed by different structural elements. Considering the relative placement of active site residues from these structurally dissimilar metalloprotease examples, a three-dimensional model (Fig. 2B) of the S2P active site emerges (see Materials and Methods). By analogy to soluble HExxH-motif-containing enzymes, whose catalytic acids polarize a metal-bound water molecule for nucleophilic attack of an extended peptide substrate (Hangauer et al. 1984; Makarova and Grishin 1999), the S2P catalytic Glu may polarize a metal-bound water molecule to cleave an extended and therefore accessible TMH substrate peptide bond. Accordingly, the S2P homolog RseP requires residues with low helical propensity in its substrate TMH (Akiyama et al. 2004).
Candidate sequences that may dictate S2P substrate binding include the conserved TMH2 motif (GpxxN/S/G) and the conserved TMH3 motif (NxxPxxxxDG, Fig. 2). The conserved residues in the TMH2 are consistent with a previously defined TMH helix-packing motif, GxxxG (Russ and Engelman 2000). Thus in S2P, TMH2 may dictate specific associations important for binding substrates and/or for proper assembly of protease active site residues. The conserved Asn of the TMH3 (NxxPxxxxDG) motif should be positioned near the ion-coordinating Asp of the same motif, being separated by one turn of the helix (Fig. 2B). Interestingly, Pro residues in TMH segments often play important structural and functional roles by forming kinks where the backbone carbonyls at positions (i-3) and (i-4) are free to form hydrogen bonds (Sansom and Weinstein 2000). Together with the conserved Asn side chain, such backbone carbonyls could provide a hydrogen bond network within the membrane important for positioning an active site water molecule, for binding substrate, or for orienting the TMH segments containing active site residues.
S2P phylogenetic distribution suggests an ancient origin
S2P sequence homologs are present in completed genomes of archaea, bacteria, and eukaryotes. Among the eukaryotes, major groups such as plants (containing multiple S2P-like sequences), animals, some fungi (genomes of the phylum Basidiomycota), and some protists (Plasmodium and Giardia) possess S2P sequences. Most archaeal genomes contain multiple S2P sequences, while bacterial genomes contain variable numbers of S2P sequences (from one sequence identified in most species to up to seven different sequences identified in Bacillus subtilis). A few bacterial species, mostly the ones with reduced genome sizes (Bifidobacterium longum, Buchnera aphidicola strains, Mycoplasma pneumoniae, Mycoplasma pulmonis, and Ureoplasma urealyticum), lack S2P homologs altogether. The nearly ubiquitous presence of identified S2P sequences suggests its presence in a common ancestor to all species. To better understand the S2P origin and to identify potential S2P subfamilies, we analyzed the phylogeny of identified S2P sequences from a diverse range of species (including four Archaea, six Bacteria, and six Eukaryota).
The evolutionary tree of S2P proteins reveals well-defined subfamilies (Fig. 1, boxes) that agree with previously identified groupings (Lewis and Thomas 1999). Although phylogenetic analysis defines these subfamilies reliably, the precise branching order between them and the placement of the highly divergent sequence Gi29249997 (Fig. 1) remain less clear (bootstrap values <70%). Despite these ambiguities, three of the subfamilies follow the tree of life (i.e., they form bacterial, archaeal, and eukaryotic groups) and contain at least one sequence from each representative genome (with the exception of protists). Given this distribution and the fact that all other subfamilies contain sequences from a limited set of organisms, we unite these three subfamilies into a single Group I (IB, bacterial sequences, and IA/E, archaeal and eukaryotic sequences in Fig. 1) that probably stems from a common ancestor of all organisms.
In the general organization of this ubiquitous Group I, archaeal sequences more closely resemble eukaryotic sequences than the bacterial ones. The plant sequences, however, are present among both eukaryotic sequences (gi|15235376) and bacterial sequences (gi|18390484 and gi|18402981). The two bacterial-like plant sequences group together with cyanobacterial S2P and are predicted to localize to the chloroplast membrane (Emanuelsson et al. 2000), consistent with a genome fusion (Rivera and Lake 2004) or endosymbiotic (Margulis 1970) hypothesis of eukaryotic cells stemming from a union of archaea (forming nucleocytoplasm) and bacteria (forming plastids).
Members of the ubiquitous Group I display similar domain architectures, including the presence of a variable number of PDZ domains (Kanehara et al. 2001) inserted between TMH2 and TMH3, a short hydrophobic sequence stretch between TMH1 and TMH2, and a potential TMH following TMH3 (Fig. 1). Several sequence subsets contain additional distinguishing features. A small bacterial subgroup, to which the characterized Enterococcus faecalis eep belongs, includes an insert that may constitute a soluble domain (~75 residues) C-terminal to the short hydrophobic span between TMH1 and TMH2. The Group I archaeal and eukaryotic subfamilies possess two potential TMH segments N-terminal to the conserved core, and the eukaryotic subfamily contains a cysteine-rich sequence (~80 residues) inserted in the PDZ domain.
In addition to the PDZ-containing Group I sequences, most archaeal genomes and some bacterial genomes include multiple S2P sequence copies that lack a PDZ domain. These sequences cluster into four additional subfamilies (Fig. 1, white boxes). We combined two of these subfamilies, one archaeal and one bacterial (which also includes a eukaryotic sequence from Plasmodium) into a single group (Group II, Fig. 1) based on protein architectures devoid of additional domains and consistent branching patterns in phylogenetic trees. Although Group II sequences are not ubiquitous, their distribution across a somewhat diverse set of organisms may represent the remains of an ancient duplication. Archaeal sequences with various scattered bacterial (and bacterial-derived plant) representatives form the remaining two groups. Group III (Fig. 1) sequences have acquired a C-terminal CBS domain and include the functionally characterized SpoIVFB protein, while previously unidentified Group IV sequences represent the most distantly related S2P homologs. Group IV includes a conserved N-terminal extension that may form a soluble domain.
The nearly universal presence of S2P-like sequences in present-day genomes suggests that the original gene product performed a fundamental function that has evolved to convey an advantage to cells today. S2P cascades contribute to a wide variety of cellular processes (pheromone production [An et al. 1999]; polarity determination [Chen et al. 2005]; sporulation [Yu and Kroos 2000]; and various stress responses [Sakai et al. 1996; Rawson et al. 1997; Ye et al. 2000b; Alba et al. 2002; Dobrosotskaya et al. 2002; Seegmiller et al. 2002]). The best characterized S2P signaling pathway responds to cholesterol content in membranes (Sakai et al. 1996; Dobrosotskaya et al. 2002; Seegmiller et al. 2002), a function innovated by and specific to eukaryotic cells. In turn, archaeal membranes display unique lipid compositions compared to the more similar lipids found in eukaryotic and bacterial membranes. These trends contrast with the S2P phylogenetic distribution, suggesting a non-lipid-based primary influence on its sequence evolution such as interactions with protein substrates or with regulatory domains. Considering S2P substrate diversity and its apparent lack of cleavage specificity (TMH substrates with helix-breaking residues), a significant requirement for cells to regulate S2P activity provides a plausible mechanism for its observed phylogeny. Other characterized S2P-mediated Rip cascades include the bacterial and eukaryotic stress responses to unfolded proteins. Perhaps these more conserved pathways embody early S2P-like sequence function, suggesting that stress-induced S2P-mediated Rip constitutes an early mechanism of cellular signaling.
The S2P PDZ domain
The S2P Group I sequences include insertions between TMH2 and TMH3 identified as a PDZ domain in the bacterial sequence RseP (Kanehara et al. 2001). The archaeal insertions are also confidently identified as PDZ domains (e.g., 10−5 E-value for gi|14520453 with HMM search; Bateman et al. 1999) generating reliable mappings to their closest evolutionary structures (HtrA-like serine proteases 1lcy [Li et al. 2002] or 1ky9 [Krojer et al. 2002]). Eukaryotic PDZ domain identification is less confident (E value 0.74 for gi|19922044 with HMM search; Bateman et al. 1999), and a reliable mapping for these sequences is limited (β-strand b through α-helix B, Fig. 3) due to a cys-rich insertion. However, consensus structure prediction confidence scores (from 3D-Jury; Ginalski and Rychlewski 2003a) support the PDZ domain assignment, and secondary structural predictions, hydrophobicity patterns, and amino acid conservation help place the cys-rich insertion within the alignment (Fig. 3).
Figure 3.
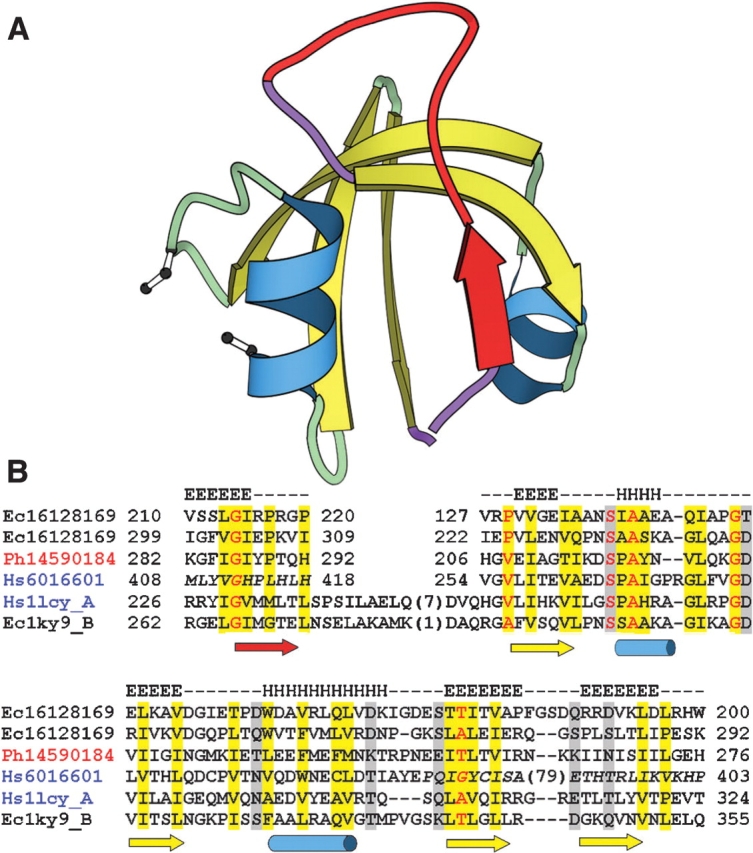
Schematic representation of PDZ domain structure and corresponding alignment. (A) PDZ domain model from HtrA-like serine protease (1lcy) produced with MOLSCRIPT (Esnouf 1999) Helices (blue) and strands (yellow) are colored and numbered according to HtrA-like serine protease topology, with circularly permuted β-strand and corresponding loops colored in red and purple, respectively. Positions of cysteine residues found in eukaryotic S2P PDZ domains that cluster spatially and may form a disulfide bond are depicted with ball-and-stick. (B) Alignment of representative S2P PDZ sequences (labeled according to Fig. 1) with HtrA-like serine protease sequences (labeled with PDB ID). Locations of the secondary structure elements in S2P (gi|16128169, consensus of secondary structure predictions) and HtrA (1lcy) are shown above and below the alignment, respectively. Color shading of sequence labels corresponds to that in Figure 1. Residues are highlighted according to conservation of chemical properties (hydrophobic, yellow; charged/polar, gray) and conserved small residues are red. Italicized sequence is not confidently mapped in the alignment. The residue numbers indicate the corresponding start and end sites.
Completing the S2P PDZ domain sequence mapping requires an alteration to the topological connectivity known as circular permutation, where the predicted C-terminal S2P β-strand structurally corresponds to the N-terminal HtrA PDZ β-strand (red β-strand, Fig. 3). In this alignment (Fig. 3B) a conserved N-terminal β-strand sequence motif (YIGV from 1lcy) corresponds to a conserved motif containing an invariant glycine residue from the C terminus of S2P PDZ (SLGI in gi|16128169). The HtrA-like serine protease PDZ domain represents one of two existing circularly permuted variants of the PDZ fold, where its C-terminal β-strand structurally corresponds to the very N-terminal β-strand found in other PDZ structures. Thus, the PDZ domain found in S2P is predicted to represent a third type of permutation to the basic PDZ fold.
S2P PDZ domains closely resemble those of HtrA-like serine proteases, which include mitochondrial serine protease HtrA2 (1lcy; Li et al. 2002), extracytoplasmic protease DegP (1ky9; Krojer et al. 2002), and DegS protease responsible for site-1 cleavage of RseA in the E. coli S2P cascade (Alba et al. 2002; Walsh et al. 2003). In fact, S2P PDZ sequences from the bacterial or archaeal groups detect HtrA-like serine protease domains before they detect each other in PSI-BLAST searches (bacterial gi|15614983 detects the C-terminal PDZ domain of HtrA-like protease gi|23103613 with the E-value 0.002 before iteration). Such a close relationship between the PDZ domain sequences in S2P and HtrA suggests that their protein products perform similar functions. Generally, PDZ domains bind to a protein substrate C terminus to mediate such processes as substrate recognition or molecular complex tethering (Nourry et al. 2003). The HtrA PDZ domains appear to negatively regulate their corresponding protease active sites (Krojer et al. 2002; Li et al. 2002; Walsh et al. 2003). This inhibition is relieved when the PDZ domain of at least one member of this family, DegS, binds misfolded outer membrane porins, allowing initiation of its cleavage cascade (Walsh et al. 2003).
Similar to this HtrA PDZ domain function, the S2P PDZ domain may regulate protease activity through C-terminal peptide binding. The site created by site-1 cleavage of the S2P substrate provides one candidate for this role, explaining the obligate sequential cleavage observed in two-step proteolytic cascades. Accordingly, S2P topology places the PDZ domain on the extracellular or luminal side of the plasma membrane, proximal to the substrate site-1 cleavage site (Fig. 1). Furthermore, site-1 cleavage of SREBP leaves a C-terminal peptide of the motif RSVL (Ye et al. 2000a), which exemplifies a typical class I PDZ substrate. An identified PDZ signature sequence, GLGF (Nourry et al. 2003) found in the S2P PDZ domains mediates binding to such class I substrates. Experimental data for two bacterial S2P homologs also support this hypothesis. First, loss of PDZ function in RseP results in unregulated substrate cleavage (Kanehara et al. 2003). Second, the substrate (Pro-σk) of an S2P family member that has not retained its PDZ domain (SpoIVFB) is cleaved only once, suggesting a functional correlation between the presence of a PDZ domain and sequential proteolysis. Alternatively, the PDZ domain may bind other members of the S2P cascade.
Functional predictions from genome context
The genomic neighborhoods of bacterial S2P sequences with PDZ domains display remarkable conservation, which can result from functional constraints imposed by neighboring genes (Snel et al. 2000). Accordingly, genome context analysis predicts a number of highly confident (neighborhood STRING score >0.7) functional associations. Identifying genes fall within one of three broad functional categories: lipid metabolism, outer membrane cell envelope biogenesis, and translation. Although precise molecular roles for S2P in these general cellular functions remain elusive, identified gene products could interact directly with S2P as substrates or regulators, or they could be targets of S2P-controlled transcription factors. Consistent with these predictions, eukaryotic S2P regulates lipid metabolism through cleaving its SREBP transcription factor substrate (Rawson 2003), whereas the bacterial S2P homolog RseP regulates lipid A synthesis and other components of cell envelope biogenesis through cleaving its RseA negative transcription factor regulator substrate (Alba and Gross 2004).
A gene encoding 1-deoxy-D-xylulose 5-phosphate (Dxp) reductoisomerase represents the most confidently predicted prokaryotic S2P neighborhood association (STRING 0.896). Dxp reductoisomerase catalyzes the first committed step of the methylerythritol phosphate (MEP) pathway of isoprenoid biosynthesis specific to plant chloroplasts, many pathogenic bacteria, and the malarial parasite Plasmodium falciparum (Lichtenthaler 2000). Two additional genes that encode metabolic enzymes related to isoprenoid metabolism reside in the genomic neighborhood of prokaryotic S2P sequences: undecaprenyl pyrophosphate synthetase (STRING 0.858), which synthesizes a 55-carbon-long chain-product from isopentenyl pyrophosphate (Chang et al. 2004), and 1-hydroxy-2-methyl-2-butenyl 4-diphosphate synthase (medium confidence STRING 0.462), which generates the precursor to isopentenyl pyrophosphate in the MEP pathway of isoprenoid biosynthesis (Lichtenthaler 2000).
Another lipid metabolic enzyme, membrane-bound phosphatidate cytidylyltransferase (CdsA), represents the second-highest predicted S2P functional association (STRING 0.882). The CDP-diacylglycerol reaction product of this enzyme serves as an important branch point intermediate in the glycerophospholipid metabolic pathway common to all organisms, and participates in the phosphatidylinositol signaling system in eukaryotes. CdsA has been identified as a component of the lipogenic response regulated by SREBP in fetal rat lung development (Zhang et al. 2004). Other identified lipid metabolic enzymes include a bacterial fatty acid biosynthesis protein, 3-hydroxymyristoyl/3-hydroxydecanoyl-(acyl carrier protein) dehydratase (STRING 0.439), and an additional glycerophospholipid metabolic enzyme, phosphatidylglycerophosphate synthase (STRING 0.439).
The next highest-scoring S2P functional association is to a nucleotide metabolic enzyme, uridylate kinase (STRING 0.867), whose gene is transcribed in a Lactococcus lactis operon (Wadskov-Hansen et al. 2000) with a gene encoding ribosomal recycling factor, also predicted to associate with S2P (STRING 0.865). Although the functional relationship between these two proteins and S2P remains elusive, several additional proteins that participate in protein translation reside in the conserved S2P genomic neighborhood: translation elongation factor Ts (STRING 0.852), ribosomal protein S2 (STRING 0.839), and prolyl trna synthetase (STRING 0.542). The role of the bacterial S2P homolog RseP in mediating stress response to misfolded envelope polypeptides may dictate such a link between S2P and control of protein translation.
The final set of genes predicted to associate with prokaryotic S2P participates in bacterial cell wall membrane biogenesis. This set includes two outer membrane proteins: protective antigen OMA87 (STRING 0.717) and extracytoplasmic protein folding catalyst ompH (STRING 0.682), and three lipopolysaccharide biosynthetic enzymes: two acyltransferases (LpxA and LpxD, both STRING 0.651) and lipid A-disaccharide synthase (LpxR, STRING 0.615). With the exception of LpxR, each of these genes is transcribed in E. coli by the S2P substrate (RseA)-controlled envelope stress response transcription factor SigmaE (Dartigalongue et al. 2001), supporting the predicted link.
The conserved genomic neighborhoods of prokaryotic PDZ-containing S2P genes imply a role for these enzymes in three broad functional categories: lipid metabolism, cell wall membrane biogenesis, and translation. In support of these predictions, transcription factors regulated by experimentally characterized S2P cascades control genes involved in both lipid metabolism (SREBP) and cell wall membrane biogenesis (SigmaE). These predictions further support the notion that S2P signaling cascades function universally to maintain membrane integrity. Just as the S2P cascade controls cholesterol content in animal membranes, the corresponding bacterial pathway controls lipid A content in outer membranes. Finally, changes in protein translation levels accompany the ER unfolded protein response (Harding et al. 2002). A role for S2P proteolysis in this process has not previously been recognized and is worthy of experimental characterization.
Materials and methods
S2P sequences were detected using transitive PSI-BLAST (Altschul et al. 1997) searches (E-value cutoff 0.0005) against the nonredundant database (nr posted Apr 24, 2003, 1415660 sequences) with query sequence gi|6016601 (amino acid range 143 to 247). Hits were grouped (1 bit per site threshold) and PSI-BLAST searches were initiated from representative sequences of each group. This transitive search procedure was repeated to convergence (three iterations). We identified additional sequences from unfinished eukaryotic genomes (http://www.ncbi.nlm.nih.gov) by performing TBLASTN searches (Altschul et al. 1990) against nucleotide sequence databases translated in all reading frames, using TMH segments with conserved motifs as queries. Potential open reading frames containing consecutive S2P core TMH motifs were considered hits.
To assemble an S2P multiple sequence alignment, identified homologs were first grouped based on BLAST score-based single-linkage clustering (blastclust) (Altschul et al. 1990). Similar groups and outlier sequences were unified based on the presence of additional TMH segments and inserted domains. In such cases, TMH segments were predicted using HMMTOP (Tusnady and Simon 2001) and domains were defined by precomputed CDD links in the Entrez protein database (Marchler-Bauer et al. 2003), by profile HMM search of the PFAM database (Bateman et al. 1999), or by consensus fold recognition (3D-Jury; Ginalski et al. 2003). Multiple sequence alignments for each resulting group were generated using PCMA (Pei et al. 2003). A global multiple sequence alignment of the S2P core was assembled by aligning the conserved TMH motifs.
Phylogenetic trees were constructed using distances estimated with the amino acid transition probability matrix of Jones, Taylor, and Thornton (Jones et al. 1992). Initial tree topologies were built using Njdist from the MOLPHY package (Saitou and Nei 1987; Adachi and Hasegawa 1992) or FITCH from the PHYLIP package with global optimization (−G option) (Felsenstein 1997). Maximum likelihood trees were built using the local rearrangement search of the initial tree topologies (−R option) of the PROTML program in MOLPHY (Adachi and Hasegawa 1992). The reliability of the resulting tree topologies was assessed by resampling of estimated log-likelihood (RELL) method of MOLPHY, and the tree with the highest RELL bootstrap probability was chosen (Kishino et al. 1990). Cladogram-like rooted trees were plotted with drawgram of the PHYLIP package without using branch lengths (Felsenstein 1993).
A three dimensional model of the S2P active site containing the C-terminal portion of two motif-containing TMH segments (TMH1 and TMH3) was generated by assembling pieces of various existing structures. The TMH1 fragment was built with the biopolymer module of the insight II graphics package by replacing residues (256–270 from 1lml) of the leishmanolysin HExxH motif-containing helix with those of the human S2P TMH1 sequence (gi|6016601, residues 163–177). The TMH3 fragment was built similarly by replacing residues (A18–A33 of 1xqf) of a proline-containing TMH segment from the ammonia transporter Amtb with the corresponding human S2P residues (gi|6016601, residues 454–469). The relative positions of conserved residues (NxxPxxxxD) in this TMH3 fragment are supported by comparison to a second proline-containing TMH segment (A49 to A64 of 2a65) from the bacterial Leutaa transporter (superimposes with RMSD 1.71 over all fragment residues, with conserved residue positions oriented similarly). Placement of TMH3 with respect to TMH1 was based on a number of factors. First, the active sites of several structurally diverse metalloenzymes (2tlx, 1hr8, and 1bs8) were superimposed based on the active site metal ion and its coordinating residues. In each case, the HExxH (or HxxEH) motif-containing helix and the third metal ion-coordinating residue occupy similar spatial locations. The TMH3 helix was therefore placed so that the conserved Asp side chain orients towards the metal ion in a similar manner as the superimposed metalloenzymes. Finally, the THH3 helix was rotated so that placement of additional side chains (LxxVxxxxCFxL) roughly corresponds to placement of residues falling within 5Å of the Zn metal or dipeptide product in thermolysin. The peptide substrate (1hr8) or peptide products (2tlx and 1bs8) of these superimposed structures bind in similar extended conformations. We therefore indicate the relative position of a potential S2P substrate using the backbone of the mitochondrial processing peptidase substrate (residues Q16–Q19 from 1hr8).
To generate reliable multiple sequence alignments for S2P PDZ domains and their closest evolutionary structures (HtrA-like serine protease PDZ domains 1lcy [Li et al. 2002] and 1ky9 [Krojer et al. 2002]), close homologs of 1lcy and 1ky9 were collected with PSI-BLAST and aligned to identified S2P PDZ sequences using PCMA (Pei et al. 2003). Sequence conservation pattern, secondary structure prediction, and tertiary fold recognition were carried out using Meta Server (Bujnicki et al. 2001; http://bioinfo.pl/meta/) coupled with 3D-Jury (Ginalski et al. 2003) guided manual alignment adjustments. Representative S2P PDZ sequences (bacterial gi|16128169, archaeal gi|14590184, and eukaryotic gi|6016601) were reliably mapped on HtrA-like PDZ structures using the consensus alignment approach and 3D assessment (Ginalski and Rychlewski 2003b). Sequences were also artificially permuted by placing the C-terminal predicted β-strand region on the N terminus, and submitted to the Meta Server to establish a multiple sequence alignment encompassing the complete PDZ fold.
Genomic neighborhood analysis of S2P was carried out using STRING (Snel et al. 2000) with orthology derived from the COG database (Tatusov et al. 2003). The majority of S2P sequences belong to two different COGs, with one comprised of PDZ-containing sequences (COG0750) and one with SpoIVFB-like sequences (COG1994). Limiting analysis to STRING neighborhood scores, only COG0750 predicted associations with confidence values above 0.6 (scores >0.7 are considered high-confidence; Snel et al. 2000). Broad functional categories of predicted associations were defined using COG database annotations (Tatusov et al. 2003).
Acknowledgments
We thank Dr. Joseph L. Goldstein for critical reading of the manuscript. This work was supported in part by NIH grant GM67165 to N.V.G.
Abbreviations
Rip, regulated intramembrane proteolysis
TMH, transmembrane-spanning helix
S2P, site 2 protease
ER, endoplasmic reticulum
SREBP, sterol regulatory element-binding protein
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051766506.
References
- Adachi, J. and Hasegawa, M. 1992. Molphy: Programs for molecular phylogenetics based on maximum likelihood. In Computer Science Monographs, Institute of Statistical Mathematics, Tokyo.
- Akiyama, Y., Kanehara, K., and Ito, K. 2004. RseP (YaeL), an Escherichia coli RIP protease, cleaves transmembrane sequences. EMBO J. 23: 4434–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alba, B.M. and Gross, C.A. 2004. Regulation of the Escherichia coli σ-dependent envelope stress response. Mol. Microbiol. 52: 613–619. [DOI] [PubMed] [Google Scholar]
- Alba, B.M., Leeds, J.A., Onufryk, C., Lu, C.Z., and Gross, C.A. 2002. DegS and YaeL participate sequentially in the cleavage of RseA to activate the σ(E)-dependent extracytoplasmic stress response. Genes & Dev. 16: 2156–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An, F.Y., Sulavik, M.C., and Clewell, D.B. 1999. Identification and characterization of a determinant (eep) on the Enterococcus faecalis chromosome that is involved in production of the peptide sex pheromone cAD1. J. Bacteriol. 181: 5915–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman, A., Birney, E., Durbin, R., Eddy, S.R., Finn, R.D., and Sonnhammer, E.L. 1999. Pfam 3.1: 1313 multiple alignments and profile HMMs match the majority of proteins. Nucleic Acids Res. 27: 260–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, M.S., Ye, J., Rawson, R.B., and Goldstein, J.L. 2000. Regulated intramembrane proteolysis: A control mechanism conserved from bacteria to humans. Cell 100: 391–398. [DOI] [PubMed] [Google Scholar]
- Bujnicki, J.M., Elofsson, A., Fischer, D., and Rychlewski, L. 2001. Structure prediction meta server. Bioinformatics 17: 750–751. [DOI] [PubMed] [Google Scholar]
- Chang, S.Y., Ko, T.P., Chen, A.P., Wang, A.H., and Liang, P.H. 2004. Substrate binding mode and reaction mechanism of undecaprenyl pyrophosphate synthase deduced from crystallographic studies. Protein Sci. 13: 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J.C., Viollier, P.H., and Shapiro, L. 2005. A membrane metalloprotease participates in the sequential degradation of a Caulobacter polarity determinant. Mol. Microbiol. 55: 1085–1103. [DOI] [PubMed] [Google Scholar]
- Dartigalongue, C., Missiakas, D., and Raina, S. 2001. Characterization of the Escherichia coli σE regulon. J. Biol. Chem. 276: 20866–20875. [DOI] [PubMed] [Google Scholar]
- Dobrosotskaya, I.Y., Seegmiller, A.C., Brown, M.S., Goldstein, J.L., and Rawson, R.B. 2002. Regulation of SREBP processing and membrane lipid production by phospholipids in Drosophila. Science 296: 879–883. [DOI] [PubMed] [Google Scholar]
- Dong, T.C. and Cutting, S.M. 2003. SpoIVB-mediated cleavage of SpoIVFA could provide the intercellular signal to activate processing of Pro-σK in Bacillus subtilis. Mol. Microbiol. 49: 1425–1434. [DOI] [PubMed] [Google Scholar]
- ———. 2004. The PDZ domain of the SpoIVB transmembrane signaling protein enables cis-trans interactions involving multiple partners leading to the activation of the pro-σK processing complex in Bacillus subtilis. J. Biol. Chem. 279: 43468–43478. [DOI] [PubMed] [Google Scholar]
- Duncan, E.A., Dave, U.P., Sakai, J., Goldstein, J.L., and Brown, M.S. 1998. Second-site cleavage in sterol regulatory element-binding protein occurs at transmembrane junction as determined by cysteine panning. J. Biol. Chem. 273: 17801–17809. [DOI] [PubMed] [Google Scholar]
- Emanuelsson, O., Nielsen, H., Brunak, S., and von Heijne, G. 2000. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300: 1005–1016. [DOI] [PubMed] [Google Scholar]
- Esnouf, R.M. 1999. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr. D Biol. Crystallogr. 55 (Pt 4): 938–940. [DOI] [PubMed] [Google Scholar]
- Felsenstein, J. 1993. PHYLIP (Phylogeny Inference Package), 3.5c ed, Department of Genetics, University of Washington, Seattle.
- ———. 1997. An alternating least squares approach to inferring phylogenies from pairwise distances. Syst. Biol. 46: 101–111. [DOI] [PubMed] [Google Scholar]
- Ginalski, K. and Rychlewski, L. 2003a. Detection of reliable and unexpected protein fold predictions using 3D-Jury. Nucleic Acids Res. 31: 3291–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2003b. Protein structure prediction of CASP5 comparative modeling and fold recognition targets using consensus alignment approach and 3D assessment. Proteins 53 (Suppl. 6): 410–417. [DOI] [PubMed] [Google Scholar]
- Ginalski, K., Elofsson, A., Fischer, D., and Rychlewski, L. 2003. 3D-Jury: A simple approach to improve protein structure predictions. Bioinformatics 19: 1015–1018. [DOI] [PubMed] [Google Scholar]
- Green, D.H. and Cutting, S.M. 2000. Membrane topology of the Bacillus subtilis pro-σ (K) processing complex. J. Bacteriol. 182: 278–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorenko, A.P., Moliaka, Y.K., Korovaitseva, G.I., and Rogaev, E.I. 2002. Novel class of polytopic proteins with domains associated with putative protease activity. Biochemistry (Mosc.) 67: 826–835. [DOI] [PubMed] [Google Scholar]
- Hangauer, D.G., Monzingo, A.F., and Matthews, B.W. 1984. An interactive computer graphics study of thermolysin-catalyzed peptide cleavage and inhibition by N-carboxymethyl dipeptides. Biochemistry 23: 5730–5741. [DOI] [PubMed] [Google Scholar]
- Harding, H.P., Calfon, M., Urano, F., Novoa, I., and Ron, D. 2002. Transcriptional and translational control in the mammalian unfolded protein response. Annu. Rev. Cell Dev. Biol. 18: 575–599. [DOI] [PubMed] [Google Scholar]
- Hooper, N.M. 1994. Families of zinc metalloproteases. FEBS Lett. 354: 1–6. [DOI] [PubMed] [Google Scholar]
- Jones, D.T., Taylor, W.R., and Thornton, J.M. 1992. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8: 275–282. [DOI] [PubMed] [Google Scholar]
- Kanehara, K., Akiyama, Y., and Ito, K. 2001. Characterization of the yaeL gene product and its S2P-protease motifs in Escherichia coli. Gene 281: 71–79. [DOI] [PubMed] [Google Scholar]
- Kanehara, K., Ito, K., and Akiyama, Y. 2003. YaeL proteolysis of RseA is controlled by the PDZ domain of YaeL and a Gln-rich region of RseA. EMBO J. 22: 6389–6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishino, H., Miyata, T., and Hasegawa, M. 1990. Maximum likelihood inference of protein phylogeny and the origin of chloroplasts. J. Mol. Evol. 30: 151–160. [Google Scholar]
- Koonin, E.V., Makarova, K.S., Rogozin, I.B., Davidovic, L., Letellier, M.C., and Pellegrini, L. 2003. The rhomboids: A nearly ubiquitous family of intramembrane serine proteases that probably evolved by multiple ancient horizontal gene transfers. Genome Biol. 4: R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krojer, T., Garrido-Franco, M., Huber, R., Ehrmann, M., and Clausen, T. 2002. Crystal structure of DegP (HtrA) reveals a new protease-chaperone machine. Nature 416: 455–459. [DOI] [PubMed] [Google Scholar]
- Lewis, A.P. and Thomas, P.J. 1999. A novel clan of zinc metallopeptidases with possible intramembrane cleavage properties. Protein Sci. 8: 439–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W., Srinivasula, S.M., Chai, J., Li, P., Wu, J.W., Zhang, Z., Alnemri, E.S., and Shi, Y. 2002. Structural insights into the pro-apoptotic function of mitochondrial serine protease HtrA2/Omi. Nat. Struct. Biol. 9: 436–441. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler, H.K. 2000. Non-mevalonate isoprenoid biosynthesis: Enzymes, genes and inhibitors. Biochem. Soc. Trans. 28: 785–789. [PubMed] [Google Scholar]
- Makarova, K.S. and Grishin, N.V. 1999. Thermolysin and mitochondrial processing peptidase: How far structure-functional convergence goes. Protein Sci. 8: 2537–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer, A., Anderson, J.B., DeWeese-Scott, C., Fedorova, N.D., Geer, L.Y., He, S., Hurwitz, D.I., Jackson, J.D., Jacobs, A.R., Lanczycki, C.J., et al. 2003. CDD: A curated Entrez database of conserved domain alignments. Nucleic Acids Res. 31: 383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulis, L. 1970. Origin of eukaryotic cells: Evidence and research implications for a theory of the origin and evolution of microbial, plant, and animal cells on the Precambrian earth. Yale University Press, New Haven, CT.
- Nourry, C., Grant, S.G., and Borg, J.P. 2003. PDZ domain proteins: Plug and play! Sci STKE 2003: RE7. [DOI] [PubMed]
- Pei, J., Sadreyev, R., and Grishin, N.V. 2003. PCMA: Fast and accurate multiple sequence alignment based on profile consistency. Bioinformatics 19: 427–428. [DOI] [PubMed] [Google Scholar]
- Ponting, C.P., Hutton, M., Nyborg, A., Baker, M., Jansen, K., and Golde, T.E. 2002. Identification of a novel family of presenilin homologues. Hum. Mol. Genet. 11: 1037–1044. [DOI] [PubMed] [Google Scholar]
- Rawson, R.B. 2003. Control of lipid metabolism by regulated intramembrane proteolysis of sterol regulatory element binding proteins (SREBPs). Biochem. Soc. Symp. 70: 221–231. [DOI] [PubMed] [Google Scholar]
- Rawson, R.B., Zelenski, N.G., Nijhawan, D., Ye, J., Sakai, J., Hasan, M.T., Chang, T.Y., Brown, M.S., and Goldstein, J.L. 1997. Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol. Cell 1: 47–57. [DOI] [PubMed] [Google Scholar]
- Rivera, M.C. and Lake, J.A. 2004. The ring of life provides evidence for a genome fusion origin of eukaryotes. Nature 431: 152–155. [DOI] [PubMed] [Google Scholar]
- Rudner, D.Z., Fawcett, P., and Losick, R. 1999. A family of membrane-embedded metalloproteases involved in regulated proteolysis of membrane-associated transcription factors. Proc. Natl. Acad. Sci. 96: 14765–14770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ, W.P. and Engelman, D.M. 2000. The GxxxG motif: A framework for transmembrane helix-helix association. J. Mol. Biol. 296: 911–919. [DOI] [PubMed] [Google Scholar]
- Saitou, N. and Nei, M. 1987. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4: 406–425. [DOI] [PubMed] [Google Scholar]
- Sakai, J., Duncan, E.A., Rawson, R.B., Hua, X., Brown, M.S., and Goldstein, J.L. 1996. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 85: 1037–1046. [DOI] [PubMed] [Google Scholar]
- Sansom, M.S. and Weinstein, H. 2000. Hinges, swivels and switches: The role of prolines in signalling via transmembrane α-helices. Trends Pharmacol. Sci. 21: 445–451. [DOI] [PubMed] [Google Scholar]
- Seegmiller, A.C., Dobrosotskaya, I., Goldstein, J.L., Ho, Y.K., Brown, M.S., and Rawson, R.B. 2002. The SREBP pathway in Drosophila: Regulation by palmitate, not sterols. Dev. Cell 2: 229–238. [DOI] [PubMed] [Google Scholar]
- Snel, B., Lehmann, G., Bork, P., and Huynen, M.A. 2000. STRING: A web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 28: 3442–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov, R.L., Fedorova, N.D., Jackson, J.D., Jacobs, A.R., Kiryutin, B., Koonin, E.V., Krylov, D.M., Mazumder, R., Mekhedov, S.L., Nikolskaya, A.N., et al. 2003. The COG database: An updated version includes eukaryotes. BMC Bioinformatics 4: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusnady, G.E. and Simon, I. 2001. The HMMTOP transmembrane topology prediction server. Bioinformatics 17: 849–850. [DOI] [PubMed] [Google Scholar]
- Urban, S. and Freeman, M. 2002. Intramembrane proteolysis controls diverse signalling pathways throughout evolution. Curr. Opin. Genet Dev. 12: 512–518. [DOI] [PubMed] [Google Scholar]
- Wadskov-Hansen, S.L., Martinussen, J., and Hammer, K. 2000. The pyrH gene of Lactococcus lactis subsp. cremoris encoding UMP kinase is transcribed as part of an operon including the frr1 gene encoding ribosomal recycling factor 1. Gene 241: 157–166. [DOI] [PubMed] [Google Scholar]
- Walsh, N.P., Alba, B.M., Bose, B., Gross, C.A., and Sauer, R.T. 2003. OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell 113: 61–71. [DOI] [PubMed] [Google Scholar]
- Ye, J., Dave, U.P., Grishin, N.V., Goldstein, J.L., and Brown, M.S. 2000a. Asparagine-proline sequence within membrane-spanning segment of SREBP triggers intramembrane cleavage by site-2 protease. Proc. Natl. Acad. Sci. 97: 5123–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, J., Rawson, R.B., Komuro, R., Chen, X., Dave, U.P., Prywes, R., Brown, M.S., and Goldstein, J.L. 2000b. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6: 1355–1364. [DOI] [PubMed] [Google Scholar]
- Yu, Y.T. and Kroos, L. 2000. Evidence that SpoIVFB is a novel type of membrane metalloprotease governing intercompartmental communication during Bacillus subtilis sporulation. J. Bacteriol. 182: 3305–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelenski, N.G., Rawson, R.B., Brown, M.S., and Goldstein, J.L. 1999. Membrane topology of S2P, a protein required for intramembranous cleavage of sterol regulatory element-binding proteins. J. Biol. Chem. 274: 21973–21980. [DOI] [PubMed] [Google Scholar]
- Zhang, F., Pan, T., Nielsen, L.D., and Mason, R.J. 2004. Lipogenesis in fetal rat lung: Importance of C/EBPα, SREBP-1c, and stearoyl-CoA desaturase. Am. J. Respir. Cell Mol. Biol. 30: 174–183. [DOI] [PubMed] [Google Scholar]
- Zhou, R. and Kroos, L. 2004. BofA protein inhibits intramembrane proteolysis of pro-σK in an intercompartmental signaling pathway during Bacillus subtilis sporulation. Proc. Natl. Acad. Sci. 101: 6385–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]