

Abstract
A large fraction of the Mycobacterium tuberculosis genome codes for proteins of unknown function. We here report the structure of one of these proteins, Rv0130, solved to a resolution of 1.8 å. The Rv0130 monomer features a single hotdog fold composed of a highly curved β-sheet on top of a long and a short α-helix. Two monomers in turn pack to form a double-hotdog-folded homodimer, similar to a large group of enzymes that use thiol esters as substrates. Rv0130 was found to contain a highly conserved R-specific hydratase motif buried deeply between the two monomers. Our biochemical studies show that the protein is able to hydrate a short trans-2-enoyl-coenzyme A moiety with a k cat of 1.1 × 102 sec−1. The importance of the side chains of D40 and H45 for hydratase activity is demonstrated by site-directed mutagenesis. In contrast to many hotdog-folded proteins, a proline residue distorts the central helix of Rv0130. This distortion allows the creation of a long, curved tunnel, similar to the substrate-binding channels of long-chain eukaryotic hydratase 2 enzymes.
Keywords: Rv0130, Mycobacterium tuberculosis, hydratase, hotdog fold, crystal structure
Mycobacterium tuberculosis, the causative agent of tuberculosis (TB), is responsible for over two million deaths every year and is estimated to infect one-third of the world's population (World Health Organization, www.who.org). Although effective drugs exist, the treatment is long and difficult. The cell wall of M. tuberculosis is rich in unusual polysaccharides, lipids, and glycolipids, making it hard to penetrate for drugs and other small molecules (Imaeda et al. 1968; Jarlier and Nikaido 1990). The bacteria can also survive for many years in a dormant state within activated macrophages, and later be reactivated (Dye et al. 1999; O'Regan and Joyce-Brady 2001). This phenomenon has made TB the major cause of death in HIV-infected individuals. In addition, the recent emergence of multidrug-resistant strains makes the need for new drugs compelling. The availability of the complete genome sequence of M. tuberculosis has greatly aided efforts toward understanding the biology of this bacterium (Cole et al. 1998). The ∼4000 gene products were reclassified into 11 functional groups in a study by Camus et al. (2002), in which a putative function could be assigned to 52% of the sequences. However, only a small number of these have been experimentally characterized.
Rv0130 is a 16-kDa protein from M. tuberculosis H37Rv, listed as a conserved hypothetical protein of unknown function in the TubercuList server of the Pasteur Institute (http://genolist.pasteur.fr/TubercuList/). Castell et al. (2005) recently predicted Rv0130 to share a single hotdog fold (SHD) with a large group of enzymes that use thiol esters as substrates. The hotdog fold encompasses a large, highly curved, antiparallel β-sheet wrapped around a long α-helix. This fold was originally observed in the Escherichia coli β-hydroxydecanoyl thiol ester dehydrase (Leesong et al. 1996) and later in the Pseudomonas sp. strain CBS-3 hydroxybenzoyl coenzyme A (CoA) thioesterase (Benning et al. 1998). Many hotdog-folded proteins associate side-by-side to form functional homodimers (Leesong et al. 1996; Benning et al. 1998; Hisano et al. 2003). A number of structures with a double hotdog fold (DHD), similar to the quaternary structure of SHD dimers, have also been described: an E. coli thioesterase II (Li et al. 2000), a Candida tropicalis hydratase 2 (Koski et al. 2004), and a protein of unknown function from M. tuberculosis (Castell et al. 2005).
Results
The structure of Rv0130 was solved by single-wavelength anomalous dispersion (Hendrickson 1991) using selenomethionine-substituted (SeMet) protein. Data collection and refinement statistics are given in Table 1. As previously predicted (Castell et al. 2005), Rv0130 belongs to a family of single hotdog domain proteins (SHDs). The SHD-type structure of Rv0130 is composed of a curved five-stranded antiparallel β-sheet (ordered 1-3-4-5-2) that wraps around the central so called hotdog helix, α4 (numbering according to Hisano et al. 2003). In addition to the minimal hotdog fold, Rv0130 has a short β-strand and α-helix at the N terminus, β0 and α0, arranged such that the strand is aligned parallel to the loop connecting the hotdog helix and β2. The asymmetric unit contains two Rv0130 molecules packed around a noncrystallographic twofold axis, creating a homodimer with an extended 10-stranded sheet (Fig. 1). The high curvature of the sheet is achieved by β-bulges at residues V18/D22, Y89/N92/P97, and V113 in β1, β2, and β3, respectively. In β4 and β5, gradual changes in (φ, ψ) angles of successive residues result in a similar deformation (Chothia and Janin 1982). Four of the strands are connected via type I (β4–β5), type I′ (β3–β4), and type II (β2–β3) β-turns (Venkatachalam 1968; Laskowski et al. 2005). Rv0130 has a well defined lid (Castell et al. 2005) formed by the loop linking β1 and the N terminus of the hotdog helix. The lid consists of a three-turn α-helix, α1, that helps to form the dimer interface, and a pair of single-turn α-helices, α2 and α3. Each of the two lids of the dimer packs on the N-terminal region of the respective neighboring hotdog helix. A number of electrostatic interactions stabilize these sections; E30-R72 connects α1 and the hotdog helix, D36-H41 supports the first loop segment of the lid, while D47-R50 stabilizes α3. The 4.5-turn hotdog helix is distorted with a distinct 25° kink at its center, P71. This change of direction enables the formation of an additional strand, β1b, on the concave side of the molecule, creating a two-stranded sheet with the N-terminal strand β0. Two salt bridges support the sheet; E5-K81 connects the strands together, whereas R2-E16 links this smaller parallel sheet to the large antiparallel sheet.
Table 1.
Data collection and refinement statistics

Figure 1.

Overall structure and topology. (A) The Rv0130 monomer, colored red to blue from N- to C-terminal, forms a homodimer with a neighboring molecule (gold). (B) Topology diagram. The secondary structure elements are numbered according to Hisano et al. (2003). The additional β-strand and α-helix at the N terminus of Rv0130 are called β0 and α0, respectively, and the additional β-strand between the hotdog helix (α4) and β2 is called β1b.
The two chains of the Rv0130 dimer are very similar and can be superimposed with a root-mean-square distance (RMSD) of 0.45 Å over 149 CA atoms. The subunits have a highly complementary shape, burying a total solvent accessible area of 3300 Å2. Three major regions of quaternary interactions govern this interaction: α1 and α1′ pack side-by-side and share mainly hydrophobic interactions (the prime indicating an element of the adjacent polypeptide chain). The hotdog helices interact via a hydrophobic patch created by F64/L67/L70 and F64′/L67′/L70′ whereas β2 and β2′ shares a number of hydrogen bonds on the convex side, as well as some hydrophobic interactions on the concave side of the extended sheet. The bulge on this strand, N92, is at the dimer interface, forcing a local breakdown of the noncrystallographic symmetry at the carbonyl oxygen. For similar reasons, the neighboring side chain of L91 takes on differing conformations in the two subunits of the dimer.
Similarity to other structures
The DALI server (Holm and Sander 1994) identified a number of structures similar to Rv0130 (Table 2). The most closely related enzymes are an SHD (R)-specific enoyl-CoA hydratase (PhaJ) (PDB code 1IQ6; Hisano et al. 2003), an SHD protein of unknown function from Archaeoglobus fulgius (PDB code 1Q6W; A.A. Fedorov, E.V. Fedorov, R. Thirumuruhan, and S.C. Almo, unpubl.), and Rv0216, a DHD of unknown function from M. tuberculosis (PDB code 2BI0; Castell et al. 2005). The structures of two DHD hydratase 2 enzymes (PDB code 1PN2; PDB code 1S9C; Koski et al. 2004, 2005), two SHD dehydratases (FabZ) (PDB code 1U1Z; PDB code 1Z6B; Kimber et al. 2004; Kostrewa et al. 2005), and a SHD dehydratase-isomerase (FabA) (PDB code 1MKA; Leesong et al. 1996) are more distantly related. The extended β-sheet as well as the first part of the hotdog helix of the Rv0130 dimer are both well conserved in all these proteins. However, due to the broken Rv0130 hotdog helix, the last part of the helix and the small, two-stranded sheet do not have any counterparts in any of the known structures (Fig. 2A). The N-terminal domains of the two eukaryotic DHD enzymes, though, feature a similar distortion where the hotdog helix is broken into two by a short random coil. The change of direction of Rv0130s hotdog helix also makes it more solvent-exposed compared with some of the other structures. The solvent-exposed residues at the C terminus of the hotdog helix are polar, in contrast to the hotdog helix of the dehydratase-isomerase 1MKA, which only features two polar residues over a 19-residue stretch. The unusual N-terminal strand and helix of Rv0130 are both absent in the SHDs 1IQ6 and 1Q6W. In 1MKA, 1U1Z, and 1Z6B, however, the N-terminal segment is significantly longer and folds over to create two strands and a short α-helix. The lid section is conserved in the SHD hydratase as well as in the M. tuberculosis Rv0216. In the DHD hydratases, the lid section is only well conserved in the C-terminal SHD domain where it packs on top of the broken hotdog helix of the first domain to form the active site. In the dehydratase and the dehydratase-isomerase, the lid is much reduced and replaced by a short loop that lacks defined secondary structure.
Table 2.
Structure alignment, DALI, and DALILite statistics (Holm and Sander 1994)

Figure 2.
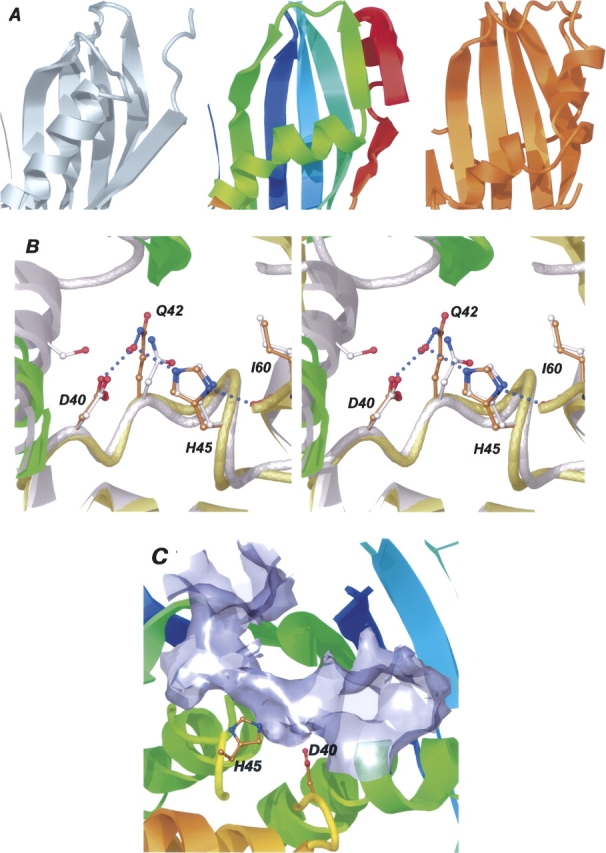
Close-up views of Rv0130. (A) A number of amino acid changes, including a proline residue (P71), create a kink in the hotdog helix of Rv0130 (middle) and enable the formation of a two-stranded parallel β-sheet, not present in the A. caviae SHD R-hydratase (left), or the C. tropicalis DHD hydratase 2 (right). (B) A comparison of Rv0130 and the R-specific enoyl hydratase 1IQ6 (gray) reveals two well conserved active site residues, D40 and H45. The hydrogen bonding network surrounding these is also well conserved, with one exception; a hydrogen bond between the catalytic Asp of 1IQ6 and a residue of the neighboring subunit (S62′) is missing in Rv0130. (C) The broken hotdog helix creates a long, curved substrate-binding tunnel housing the active site residues D40 and H45.
The connections between β-strands in the hotdog fold-family of structures show a great deal of variation (Castell et al. 2005). However, the distinctive β2–β3 loop, packing under the β4–β5 reverse turn, is structurally conserved in all family members. The loop between β3 and β4 is significantly longer in the N-terminal domain of Rv0216. The same loop is shorter in the C-terminal domain of Rv0216, while it is severely truncated in the C-terminal domains of 1PN2 and 1S9C.
Activity measurements
Based on the structural similarity to the enoyl hydratases, a spectroscopic assay (Moskowitz and Merrick 1969; Fukui et al. 1998) was used to investigate hydratase activity. Wild-type protein was found to hydrate crotonoyl-CoA (2-butenoyl-CoA) to hydroxybutyryl-CoA. The enzyme exhibits normal Michaelis-Menten kinetics for the hydratase reaction. For the wild-type enzyme, the K M value was 55 μM, V max was 1.6 × 10−7 M/sec, and k cat was 1.1 × 102 sec−1. The corresponding values for the mutant D40N were 40 μM, 4.4 × 10−8 M/sec, and 0.057 sec−1, respectively. No kinetic values could be determined for the H45Q mutant. In the reverse reaction, with hydroxybutyryl-CoA as substrate, Rv0130 showed limited activity, but no kinetic values could be determined.
Active site
The SHD hydratase, the dehydratase, and the dehydratase-isomerase all feature two active sites located on either side of the homodimer. The DHD hydratases, in turn, exhibit only one. Each of these catalytic sites is situated at equivalent positions in a deep cavity or a tunnel, formed by the dimer or domain interface. The residues responsible for the hydration of a trans-2-enoyl-CoA substrate in both bacterial and eukaryotic R-specific hydratases have been identified through site-directed mutagenesis (Hisano et al. 2003; Koski et al. 2004). In the dehydratase-isomerase, parts of the catalytic machinery have been targeted by a mechanism-based suicide inhibitor (Helmkamp and Bloch 1969; Leesong et al. 1996). The hydratase active site consists of a pair of histidyl/aspartyl residues located at a long loop in the lid region. In the dehydratase-isomerase FabA, a structurally equivalent histidine residue, situated in the small lid section, together with an aspartyl residue from the neighboring subunit, has been implicated in the dehydration and isomerization performed by the enzyme. FabZ, catalyzing only the dehydration reaction, features an almost identical active site, but with the aspartyl side chain exchanged for a glutamyl residue (Kimber et al. 2004; Kostrewa et al. 2005).
A superposition of the lid sections of Rv0130, 1IQ6, 1Q6W, 1PN2, and 1S9C reveals a well conserved active site. In Rv0130, the side chains of H45 and D40 show a close fit to the His-Asp pair of the hydratases (Fig. 2B). The hydrogen-bonding network surrounding these residues also exhibits some conservation; the equivalent of H45 forms a short hydrogen bond to a backbone oxygen of an isoleucine (I60) in all hydratase structures. D40 of Rv0130 is positioned by two conserved hydrogen bonds, to a backbone amino group two residues ahead and to Q42 via a water molecule. In 1IQ6 the counterpart of D40 also forms a hydrogen bond to a serine residue (S62′) and in 1Q6W to an aspartyl side chain (D80′). 1IQ6 and 1PN2 have an asparagine residue instead of Rv0130 Q42, whereas 1Q6W has a phenylalanine. Comparing Rv0130 with the dehydratases 1U1Z/1Z6B and the dehydratase-isomerase 1MKA, one finds that, in spite of the different location of the active site residues, the catalytically relevant nitrogen and oxygen atoms of the His-Asp and His-Glu pairs can be well superimposed.
Although the putative catalytic residues of Rv0130 and the enoyl-hydratases are very similar, the shapes of the cavities that contain them are different. The Aeromonas caviae 1IQ6 features a deep but closed substrate-binding pocket. The depth of this pocket is restricted by the hotdog helix of the adjacent subunit, limiting substrates to C4–C6 enoyl-CoAs (Fukui et al. 1998; Tsuge et al. 2003a). 1Q6W, in contrast, has a long tunnel, going across the loop connecting the hotdog helix with strand β2 and on through the neighboring subunit. However, no functional information has been published about this protein. Due to the distortion of the N-terminal hotdog helix, the two DHD hydratases also exhibit a long curved tunnel passing the active site (Koski et al. 2004, 2005). However, compared with 1Q6W, this cavity is solely created by the DHD-domain interface. The 25° kink in the central helix of Rv0130 results in a similar, long potential substrate-binding tunnel (Fig. 2C). The mouth of the tunnel is created by the splaying apart of the central β2 strands of the extended sheet. Two β-bulges, caused by P97 and by Y89′, on either side of the mouth, together with F56 in the lid section and P99 on β2, form a ring of residues that line the opening of the tunnel. Both the bulges and the two residues are conserved in all SHD and DHD hydratase structures. Other residues around the mouth, such as A98, K138, I87′, and R148′, are less well conserved. Further into the tunnel I60 and I44, lining one side of the potential catalytic histidine, have leucine/isoleucine counterparts in all hydratase structures. On the opposite side, G63 is positioned at the N terminus of the hotdog helix. Any other residue at this position would seriously limit the space within the active site cavity. This residue is also conserved in most hotdog structures. H62, stacking against the potential catalytic H45, is retained in all hydratases but 1PN2. The smallest constriction of the tunnel is between G63, H45, I87′, and N88′, after which it expands to a large opening, created by the neighboring subunit. This cavity is formed as a result of the distorted hotdog helix and the long connection to strand β2. In the structures lacking the hotdog proline, the length from the beginning of the helix to the beginning of β2 correlates well with the presence of a closed crevice, like in 1IQ6, or of an open-ended tunnel, like in the eukaryotic hydratases (Table 3).
Table 3.
Comparison of the amino acid length between the beginning of the hotdog helix and the conserved β-bulge on β2

In Rv0130, the low end of the tunnel is deeper and less hydrophobic than the DHD hydratases. A subset of the hydratase family seems to feature a similar proline residue in the hotdog helix as Rv0130. However, neither the sequences of the C-terminal part of the hotdog helix nor the small two-stranded β-sheet are well conserved within this subset. Consequently, the residues lining the lower part of the tunnel (A86, V147, V127, Q74, Y149, and Y78) are only conserved in a few actino- and proteobacteria that are closely related to M. tuberculosis.
The C. tropicalis 1PN2 has been described in complex with its enzymatic product, (3R)-hydroxydecanoyl-CoA (3RHDC) (PDB code 1PN4; Koski et al. 2004). A superposition of Rv0130 and 1PN4 reveals that the pantetheine and acyl chain moieties of 3RHDC can be fitted easily into the putative substrate-binding cleft. An active site water molecule, coordinated by D40, Q42, and H45, occupies the same position as the (3R)-hydroxyl group of the superimposed ligand. This water is equivalent to W6 of 1PN2, which has been proposed to be the source of both the proton and the hydroxyl group added to the CoA-associated acyl thioester substrate of the enzyme (Koski et al. 2004). The ADP-phosphate, bound on the surface of the C. tropicalis enzyme, does not fit Rv0130 without conformational changes. However, the opening of the tunnel contains several side chains that might be involved in interactions with such a substrate. These include the conserved β-bulges (caused by P97 and Y89′), as well as four charged residues (K93, R95, K138, and R148′) that might form salt-bridges to phosphate groups of a CoA moiety.
Substrate specificity of related proteins
The most similar structure to Rv0130, the R-specific enoyl hydratase 1IQ6 (PhaJ) from A. caviae, has been implicated in the production of short-chain polyhydroxyalkanoates (PHAs) by converting enoyl-CoA intermediates from the fatty acid β-oxidation pathway to (R)-3-hydroxyacyl-CoAs. These in turn are polymerized by PHA synthase (PhaC) (Fukui and Doi 1997; Fukui et al. 1998). PHAs are polymers of (R)-hydroxyalkanoic acids that many bacteria accumulate under periods of nutritional imbalance. The insoluble PHA polyesters can later serve as a carbon source during starvation (Madison and Huisman 1999; Sudesh et al. 2000). Tsuge et al. (2003b) recently cloned and characterized four R-hydratases from Pseudomonas aeruginosa involved in PHA biosynthesis, PhaJ1–PhaJ4. PhaJ1 was found to be active only on short-chain enoyl-CoAs (C4–C6), whereas PhaJ2, PhaJ3, and PhaJ4 were able to hydrate longer (C8–C12) enoyl-CoA chains. Comparing these enzymes with Rv0130, one finds that PhaJ1 seems to feature a SHD-type structure with a nondistorted hotdog helix followed by a short link to strand β2, indicating a substrate-binding pocket instead of a tunnel. PhaJ2 and PhaJ3, on the other hand, are DHD-type enzymes with an extended connection between the N-terminal hotdog helix and β2, similar to the eukaryotic hydratase 2 structures (Fig. 3; Table 3). The last protein of the study, PhaJ4, seems to exhibit SHD topology with a proline insertion in the hotdog helix (P70). This might be indicative of a substrate-binding tunnel similar to Rv0130, which in turn would correlate well with the experimental substrate specificity.
Figure 3.
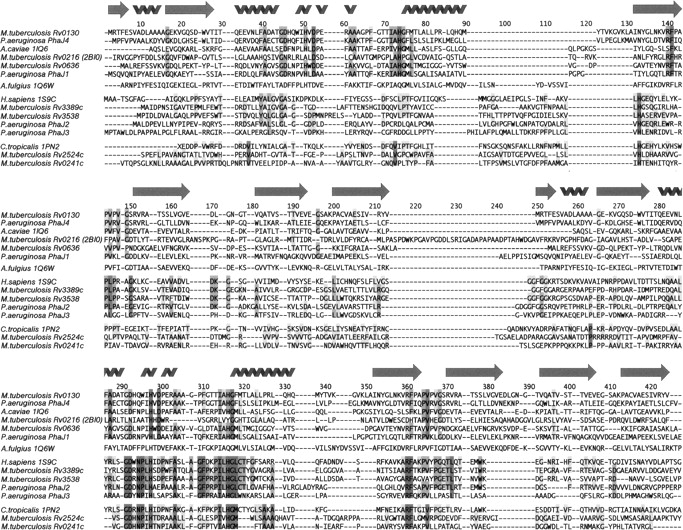
Structure-based sequence alignment. The alignment is grouped according to the closest related structures. The SHD-type proteins are represented twice to enable comparison with the N- and C-terminal domains of the DHD structures.
Related M. tuberculosis proteins
Castell et al. (2005) recently predicted the M. tuberculosis genome to contain at least five DHD- and three SHD-type proteins. Several of these are likely to be hydratase/dehydratase-related enzymes, featuring minor variations of the so called hydratase 2 motif ([YF]-X(1,2)-[LVIG]-[STGC]-G-D-X-N-P-[LIV]-H-X(5)-[AS]) (Qin et al. 2000). In Rv0130, A37 and, in particular, Q42 and W43 deviate from this pattern. Rv0636, predicted by Castell et al. (2005) to be a SHD-folded protein, matches the hydratase 2 motif exactly. This protein is of particular interest since it has been shown to be essential for M. tuberculosis growth both in vitro (Sassetti et al. 2003) and in vivo (Sassetti and Rubin 2003). Rv0636 lacks the proline residue observed in the hotdog helix of Rv0130. The loop between β2 and the hotdog helix is also much reduced compared with both Rv0130 and 1Q6W. We therefore predict a putative Rv0636 homodimer to contain a substrate-binding pocket instead of an open tunnel (Table 3; Fig. 3). Although the active site of Rv0636 is well retained, the mouth of the cavity formed by β2 shows less conservation. The four potential M. tuberculosis DHD proteins that share the hydratase motif (Rv3389c, Rv3538, Rv2524c, and Rv0241c) are more similar to the eukaryotic hydratase 2 enzymes. In these proteins, the N-terminal hotdog helix as well as the following loop seem to be much longer compared with Rv0130 (Table 3).
Discussion
As expected, Rv0130 forms a single hotdog fold (Castell et al. 2005) with a large, highly curved β-sheet that wraps around a long, central α-helix. Like many SHD proteins, two Rv0130 molecules pack side-by-side to form a homodimer with an extended sheet. Rv0130 features a highly conserved R-specific hydratase motif (Qin et al. 2000), buried deeply between the two subunits of the dimer. Indeed, our biochemical studies show that Rv0130 is able to hydrate a trans-2-enoyl-CoA moiety with a k cat of 1.1 × 102 sec−1 and a K M of 55 μM. The two potential active site residues, D40 and H45, were confirmed to be important for this catalytic activity by site-directed mutagenesis. The active site also shares some similarity with the P. aeruginosa and the P. falciparum dehydratases, as well as the E. coli dehydratase-isomerase, indicating the catalytic relationship between the hydratase motif and the dehydratase/dehydratase-isomerase active sites.
In contrast to most bacteria, M. tuberculosis uses two types of systems for fatty acid synthesis, FAS I and FAS II. FAS I is a large multifunctional enzyme, commonly found in eukaryotes, containing all the necessary functions to perform de novo fatty acid synthesis. The FAS II system, on the other hand, contains several dissociated enzymes, each catalyzing a separate reaction (for recent review, see Takayama et al. 2005). Both FAS I and FAS II systems include a dehydration step, which in FAS II is catalyzed by a β-hydroxyacyl-ACP dehydratase, FabZ, which still remains to be identified in M. tuberculosis. In FAS I we have suggested that the dehydratase activity of M. tuberculosis FAS I is localized in a DHD (Castell et al. 2005) with a well conserved hydratase motif at the active site (Qin et al. 2000). In a recent major advance in our understanding of this system, Ban and colleagues have described the mammalian and fungal FAS I structures at 4.5 and 5.0 Å resolution, respectively (Jenni et al. 2006; Maier et al. 2006). In both systems, they localize the dehydratase domain to a DHD of “about twice the expected size” (Maier et al. 2006), confirming our hypothesis. It is becoming clear, therefore, that the hydratase motif is able to shift the reaction equilibrium toward either hydration or dehydration. Based on this catalytic ambiguity and the data describing M. tuberculosis growth in vitro and in vivo (Sassetti and Rubin 2003; Sassetti et al. 2003), Castell et al. (2005) suggested Rv0636 to be the missing FabZ of M. tuberculosis. Interestingly, the classical dehydratase/dehydratase-isomerases also exhibit some catalytic ambiguity; both FabZ and FabA possess substantial hydratase activity in vitro (Heath and Rock 1995, 1996; Sharma et al. 2003). In vivo, the FAS II enoyl-ACP reductase FabI pulls the equilibrium toward dehydration by reducing the enoyl-ACP chain produced by FabZ/FabA to acyl-ACP (Heath and Rock 1995; Sharma et al. 2003). Thus only a low dehydratase activity might be required from a putative hydratase-motif-containing enzyme to act in fatty acid elongation FAS II.
Compared with the previously known structures, the packing of the C-terminal portion of the Rv0130 hotdog helix is altered by a number of changes, including a proline residue situated in the middle of the helix. This modification opens up the dimer interface and creates a long tunnel housing the active site. In the eukaryotic hydratase 2 enzymes, a similar structural feature enables the processing of long chain CoA-thioesters (Koski et al. 2004, 2005). Koski and colleagues argue that in their structure(s) the long loop between the hotdog helix and β2 is highly flexible, adapting to accommodate a long fatty acid substrate. In Rv0130, the end of the tunnel seems to be more rigid, defined by the C-terminal half of the hotdog helix and by the small β-sheet. It is likely that Rv0130 also is able to act on long fatty acid enoyl chains, but with different substrate specificity. In the known 3D structures, the length of the hotdog helix and connecting loop to β2 correlates with the presence of an active site cavity or tunnel. Of the four R-hydratases from P. aeruginosa, PhaJ1 is predicted to feature a substrate-binding pocket, whereas PhaJ2, PhaJ3, and PhaJ4 may feature substrate-binding tunnels. These pocket/tunnel predictions correlate well with the reported substrate specificities. Applying the same reasoning to the five other hydratase/dehydratase proteins from M. tuberculosis that Castell et al. (2005) predicted to exhibit SHD/DHD-type structures, it is suggested that a Rv0636 homodimer features a substrate-binding pocket, while the Rv3389c, Rv3538, Rv2524c, and Rv0241 DHDs have substrate-binding tunnels. However, since FAS II FabZ has a preference to act on long-chain fatty acid derivatives (Takayama et al. 2005), it will most likely require a substrate-binding tunnel, thus making Rv0636 a less likely candidate. On the other hand, the sequence variation of the C-terminal portion of the hotdog helix and the following loop is quite extensive in the large family of R-specific enoyl hydratases, making sequence to structure predictions difficult. One example of how acyl chain length specificity can depend on small sequence changes was reported by Tsuge et al. (2003a), who was able to alter the substrate specificity of the A. caviae hydratase 1IQ6 from C4–C6 to C4–C10 by a single point mutation. Thus, the precise relation between R-hydratase sequence and substrate specificity still requires detailed crystallographic analysis.
Materials and methods
Cloning, expression, and purification
The open reading frame coding for rv0130 was amplified by PCR from cosmid MTCI5 (Philipp et al. 1996) of M. tuberculosis, strain H37Rv, using the primers 5′-TACTGCCGACCAGAGGAGAG (forward) and 5′-ATTCTGACCGCGAGTTGC (reverse). In a second PCR reaction an N-terminal six-histidine tag was introduced using the forward primer 5′-ATGGCTCATCATCATCATCATCATGGTCGCACCTTCGAGTCGGTCG. The high fidelity polymerase PfuUltra (Stratagene) was used for the amplifications. After addition of a 3′ A-overhang by incubation with Taq polymerase (New England Biolabs), the DNA fragment was ligated to the pCR T7 TOPO vector (Invitrogen). Cloning was performed in E. coli TOP10 (Invitrogen), and the correctness of the isolated gene was verified by DNA sequence analysis. Expression was performed in E. coli BL21(DE3) (Invitrogen). The cells were cultured in LB media at 37°C. At OD600 = 0.5–1.0 the temperature was lowered to 24°C and expression of the target gene induced with 0.1 mM IPTG overnight. After harvesting by centrifugation the cell pellet was resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 10% [v/v] glycerol, 0.5% [v/v] Triton X-100 at pH 8.0) with 1 mg/mL lysozyme, 0.01 mg/mL RNase A, and 0.02 mg/mL DNase I and lysed using a One Shot Cell disruptor (Constant Systems Ltd.). The soluble fraction was incubated with 0.7 mL pre-equilibrated Ni-NTA agarose (Qiagen) slurry at 4°C for 1 h then packed into a column. After washing with 20 column volumes of buffer (20 mM imidazole, 50 mM NaH2PO4, 300 mM NaCl, 10% [v/v] glycerol at pH 8.0), the protein was eluted with five column volumes of 250 mM imidazole in the same buffer. Fractions containing the protein were pooled and further purified on a HiLoad 16/60 Superdex 75 prep grade column (GE Healthcare) equilibrated in 20 mM Tris (pH 7.5), 150 mM NaCl, 10% glycerol.
Selenomethionine (SeMet) was introduced into the protein by metabolic inhibition (van Duyne et al. 1993). The SeMet protein was expressed in BL21(DE3) cells (Invitrogen) grown in minimal media supplemented with SeMet, lysine, threonine, phenylalanine, leucine, isoleucine, and valine. Expression was done at room temperature overnight. The purification protocol for the SeMet protein was the same as for the native protein but with 10 mM β-mercaptoethanol in all buffers to prevent selenomethionine oxidation.
Mutagenesis
Mutagenesis was performed with the QuikChange site-directed mutagenesis kit (Stratagene). Temperature cycling was done according to the manual with the rv0130-containing pCR T7 plasmid as template. The two mutagenic primers D40N (5′-CCGACGCAACGGGTAACCACCAGTGGATCCA-3′) and antisense D40N (5′-TGGATCCACTGGTGGTTACCCGTTGCGTCGG-3′) were used for mutation of Asp40 to Asn and the primer pair H45Q (5′-GATCACCAGTGGATCCAAGTCGACCCGGAAC-3′) and antisense H45Q (5′-GTTCCGGGTCGACTTGGATCCACTGGTGATC-3′) for mutation of His45 to Gln. After digestion of parental DNA template with DpnI, the DNA was transformed into Top10 supercompetent cells. Mutations were verified with sequence analysis.
Enzymatic activity
The hydratase activity was measured spectrophotometrically in an assay that monitors the decrease in absorbance at 263 nm when crotonoyl CoA is hydrated to 3-hydroxybutyryl-CoA (Moskowitz and Merrick 1969; Fukui et al. 1998). The reaction was started by addition of enzyme (0.15 pmol of the wild-type enzyme) to varying concentrations (5–100 μM) of crotonoyl-CoA in 50 mM Tris (pH 8.0) at 30°C. The wild-type enzyme was freshly diluted from a 0.15 mM solution before each measurement. For the D40N mutant 0.77 nmol of enzyme, from a 0.31 mM solution, was added to each experiment and for the H45Q mutant up to 3.4 pmol were tested on 25 and 100 μM substrate. In order to get consistent results, the wild-type enzyme had to be diluted just before the reaction was started. The D40N mutant was not diluted but even here a correlation was seen with the freshness of the enzyme and a new aliquot of protein was taken from the storage at −80°C the same day as the measurement was done. The reactions were carried out in a total volume of 1 mL in a quartz cuvette with a 1-cm light path and readings were taken every second. The ɛ263 of the enoyl-thioester bond is taken to be 6.7 × 103 M−1 cm−1 (Stern et al. 1956). The kinetic constants were determined by measuring the reaction rate during the first 2 min of the reaction. Kinetic constants were calculated from Hanes-Woolf plots.
Crystallization
Purified SeMet protein was crystallized by the vapor diffusion method (McPherson 1982). The best crystals were grown at room temperature by mixing 0.5 μL of 2 mg/mL protein solution, still in its purification buffer, with 0.5 μL of 0.1 M HEPES (pH 7.5), 2% PEG 400, and 2 M ammonium sulfate. Crystals in sizes ∼0.07 × 0.05 × 0.05 mm3 could be harvested within 3–4 d.
X-ray data collection and processing
Before flash freezing in liquid nitrogen, Rv0130 crystals were transferred to a second drop containing mother liquor with 20% PEG400 added as a cryo-protectant. X-ray diffraction data were collected at ESRF beamline ID14:3 from a single SeMet crystal above the Se-absorption edge to a resolution of 1.7 Å. The data were indexed, integrated, and scaled using the HKL suite (Otwinowski and Minor 1997) and were found to belong to spacegroup P21212 with unit cell parameters a = 58.7 Å, b = 108.7 Å, and c = 50.4 Å, assuming two molecules in the asymmetric unit give a Matthews coefficient of 2.5 Å3/Da (Matthews 1968), which corresponds to a solvent content of 50.6%. Data collection statistics are given in Table 1.
Four selenium atoms were located using SHELXD (Schneider and Sheldrick 2002) and verified using MLPHARE (Otwinowski 1991). Phase estimates were calculated by SHARP (de la Fortelle and Bricogne 1997). The NCS operators could be identified using O (Jones et al. 1991) and refined with Imp (Jones 1992; Kleywegt et al. 2001). Subsequent twofold averaging, solvent flattening, and histogram matching in DM (Cowtan and Main 1998) improved maps considerably. The ARP/wARP suite (Perrakis 1997) was used for automatic main- and side-chain tracing. Crystallographic refinement was done in REFMAC5 (Murshudov 1997) against the SeMet data to a resolution of 1.8 Å. After each cycle of refinement, the σA-weighted |2Fobs| − |Fcalc| and |Fobs| − |Fcalc| maps (Read 1986) were used for further model rebuilding in O (Jones et al. 1991). Water molecules were added using ARP/wARP (Perrakis 1997) and O, in peaks (>2σ) of the |2Fobs| − |Fcalc| electron density map. Final refinement statistics are given in Table 1. Structural alignments were made using the Lsq tools of O, and structure-based sequence alignments were made using the INDONESIA package (D. Madsen, P. Johansson, and G.J. Kleywegt, unpubl.). Molecular figures were made in O and rendered in Molray (Harris and Jones 2001). Accessible surface area calculations were performed by PDBsum (Laskowski et al. 2005). Coordinates and structure factors of Rv0130 have been deposited at the Protein Data Bank with codes 2C2I and R2C2ISF, respectively.
Acknowledgments
Financial support was received from Swedish Foundation for Strategic Research (SSF), the Swedish Natural Science Research Council, and European Commission programs SPINE (QLG2-CT-2002–00988) and NM4TB (LSHP-CT-2005-018923). Total DNA of M. tuberculosis, strain H37Rv, was a gift from Dr. Stewart Cole (Institut Pasteur, Paris, France).
Footnotes
Reprint requests to: Kristina Bäckbro, Department of Cell and Molecular Biology, Uppsala University, Biomedical Center, Box 596, SE-751 24 Uppsala, Sweden; e-mail: nina@xray.bmc.uu.se; fax: +46-18-530396.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062309306.
Abbreviations: CoA, coenzyme A; DHD, double hotdog; LALI RMSD, root-mean-square distance; SeMet, seleno-methionine; SHD, single hotdog; TB, tuberculosis.
References
- Benning, M.M., Wesenberg, G., Liu, R., Taylor, K.L., Dunaway-Mariano, D., Holden, H.M. 1998. The three-dimensional structure of 4-hydroxybenzoyl-CoA thioesterase from Pseudomonas sp. strain CBS-3. J. Biol. Chem. 273: 33572–33579. [DOI] [PubMed] [Google Scholar]
- Camus, J.C., Pryor, M.J., Médigue, C., Cole, S.T. 2002. Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiol. 148: 2967–2973. [DOI] [PubMed] [Google Scholar]
- Castell, A., Johansson, P., Unge, T., Jones, T.A., Bäckbro, K. 2005. Rv0216, a conserved hypothetical protein from Mycobacterium tuberculosis that is essential for bacterial survival during infection, has a double hotdog-fold. Protein Sci. 14: 1850–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothia, C. and Janin, J. 1982. Orthogonal packing of β-pleated sheets in proteins. Biochemistry 21: 3955–3965. [DOI] [PubMed] [Google Scholar]
- Cole, S.T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., Gordon, S.V., Eiglmeier, K., Gas, S., Barry, C.E. III et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537–544. [DOI] [PubMed] [Google Scholar]
- Cowtan, K. and Main, P. 1998. Miscellaneous algorithms for density modification. Acta Crystallogr. D Biol. Crystallogr. 54: 487–493. [DOI] [PubMed] [Google Scholar]
- de la Fortelle, E. and Bricogne, G. 1997. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol. 276: 472–494. [DOI] [PubMed] [Google Scholar]
- Dye, C., Scheele, S., Dolin, P., Pathania, V., Raviglione, M.C. 1999. Consensus statement. Global burden of tuberculosis: Estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA 282: 677–686. [DOI] [PubMed] [Google Scholar]
- Engh, R. and Huber, R. 1991. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr. A 47: 392–400. [Google Scholar]
- Fukui, T. and Doi, Y. 1997. Cloning and analysis of the poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) biosynthesis genes of Aeromonas caviae . J. Bacteriol. 179: 4821–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui, T., Shiomi, N., Doi, Y. 1998. Expression and characterization of (R)-specific enoyl coenzyme A hydratase involved in polyhydroxyalkanoate biosynthesis by Aeromonas caviae . J. Bacteriol. 180: 667–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, M. and Jones, T.A. 2001. Molray—A web interface between O and the POV-Ray ray tracer. Acta Crystallogr. D Biol. Crystallogr. 57: 1201–1203. [DOI] [PubMed] [Google Scholar]
- Heath, R.J. and Rock, C.O. 1995. Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli . J. Biol. Chem. 270: 26538–26542. [DOI] [PubMed] [Google Scholar]
- Heath, R.J. and Rock, C.O. 1996. Roles of the FabA and FabZ β-hydroxyacyl-acyl carrier protein dehydratases in Escherichia coli fatty acid biosynthesis. J. Biol. Chem. 271: 27795–27801. [DOI] [PubMed] [Google Scholar]
- Helmkamp, G.M. Jr. and Bloch, K. 1969. β-hydroxydecanoyl thioester dehydrase. Studies on molecular structure and active site. J. Biol. Chem. 244: 6014–6022. [PubMed] [Google Scholar]
- Hendrickson, W.A. 1991. Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science 254: 51–58. [DOI] [PubMed] [Google Scholar]
- Hisano, T., Tsuge, T., Fukui, T., Iwata, T., Miki, K., Doi, Y. 2003. Crystal structure of the (R)-specific enoyl-CoA hydratase from Aeromonas caviae involved in polyhydroxyalkanoate biosynthesis. J. Biol. Chem. 278: 617–624. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1994. Searching protein structure databases has come of age. Proteins 19: 165–173. [DOI] [PubMed] [Google Scholar]
- Imaeda, T., Kanetsuna, F., Galindo, B. 1968. Ultrastructure of cell walls of genus. Mycobacterium. J. Ultrastruct. Res. 25: 46–63. [DOI] [PubMed] [Google Scholar]
- Jarlier, V. and Nikaido, H. 1990. Permeability barrier to hydrophilic solutes in Mycobacterium chelonei . J. Bacteriol. 172: 1418–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenni, S., Leibundgut, M., Maier, T., Ban, N. 2006. Architecture of a fungal fatty acid synthase at 5 Å resolution. Science 311: 1263–1267. [DOI] [PubMed] [Google Scholar]
- Jones, T.A. 1992. A, yaap, asap, @#*? A set of averaging programs. In Molecular replacement (ed. Dodson E.J.) . pp. 91–105. SERC Daresbury Laboratory, Warrington, UK.
- Jones, T.A., Zou, J.Y., Cowan, S.W., Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47: 110–119. [DOI] [PubMed] [Google Scholar]
- Kimber, M.S., Martin, F., Lu, Y., Houston, S., Vedadi, M., Dharamsi, A., Fiebig, K.M., Schmid, M., Rock, C.O. 2004. The structure of (3R)-hydroxyacyl-acyl carrier protein dehydratase (FabZ) from Pseudomonas aeruginosa . J. Biol. Chem. 279: 52593–52602. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G.J. and Jones, T.A. 1996. Phi/psi-chology: Ramachandran revisited. Structure 4: 1395–1400. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G.J., Zou, J.Y., Kjeldgaard, M., Jones, T.A. 2001. Around O. In International tables for crystallography (eds. Rossmann M.G. and Arnold, E.) . pp. 366–367. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- Koski, M.K., Haapalainen, A.M., Hiltunen, J.K., Glumoff, T. 2004. A two-domain structure of one subunit explains unique features of eukaryotic hydratase 2. J. Biol. Chem. 279: 24666–24672. [DOI] [PubMed] [Google Scholar]
- Koski, M.K., Haapalainen, A.M., Hiltunen, J.K., Glumoff, T. 2005. Crystal structure of 2-enoyl-CoA hydratase 2 from human peroxisomal multifunctional enzyme type 2. J. Mol. Biol. 345: 1157–1169. [DOI] [PubMed] [Google Scholar]
- Kostrewa, D., Winkler, F.K., Folkers, G., Scapozza, L., Perozzo, R. 2005. The crystal structure of PfFabZ, the unique β-hydroxyacyl-ACP dehydratase involved in fatty acid biosynthesis of Plasmodium falciparum . Protein Sci. 14: 1570–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, R.A., Chistyakov, V.V., Thornton, J.M. 2005. PDBsum more: New summaries and analyses of the known 3D structures of proteins and nucleic acids. Nucleic Acids Res. 33: D266–D268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leesong, M., Henderson, B.S., Gillig, J.R., Schwab, J.M., Smith, J.L. 1996. Structure of a dehydratase-isomerase from the bacterial pathway for biosynthesis of unsaturated fatty acids: Two catalytic activities in one active site. Structure 4: 253–264. [DOI] [PubMed] [Google Scholar]
- Li, J., Derewenda, U., Dauter, Z., Smith, S., Derewenda, Z.S. 2000. Crystal structure of the Escherichia coli thioesterase II, a homolog of the human Nef binding enzyme. Nat. Struct. Biol. 7: 555–559. [DOI] [PubMed] [Google Scholar]
- Madison, L.L. and Huisman, G.W. 1999. Metabolic engineering of poly(3-hydroxyalkanoates): From DNA to plastic. Microbiol. Mol. Biol. Rev. 63: 21–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier, T., Jenni, S., Ban, N. 2006. Architecture of mammalian fatty acid synthase at 4.5 Å resolution. Science 311: 1258–1262. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33: 491–497. [DOI] [PubMed] [Google Scholar]
- McPherson, A. In Preparation and analysis of protein crystals .1982. John Wiley & Sons, New York.
- Moskowitz, G.J. and Merrick, J.M. 1969. Metabolism of poly-β-hydroxybutyrate. II. Enzymatic synthesis of D-(−)-β hydroxybutyryl coenzyme A by an enoyl hydrase from Rhodospirillum rubrum . Biochemistry 8: 2748–2755. [DOI] [PubMed] [Google Scholar]
- Murshudov, G.N. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- O'Regan, A. and Joyce-Brady, M. 2001. Latent tuberculosis may persist for over 40 years. BMJ 323:–635. [PMC free article] [PubMed]
- Otwinowski, Z. 1991. Maximum likelihood refinement of heavy atom parameters. In CCP4 study weekend. Isomorphous replacement and anomalous scattering (ed. Wolf W.) . pp. 80–86. Daresbury Laboratory, Warrington, UK.
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Perrakis, A. 1997. wARP: Improvement and extension of crystallographic phases by weighted averaging of multiple-refined dummy atomic models. Acta Crystallogr. D Biol. Crystallogr. 53: 448–455. [DOI] [PubMed] [Google Scholar]
- Philipp, W.J., Poulet, S., Eiglmeier, K., Pascopella, L., Balasubramanian, V., Heym, B., Bergh, S., Bloom, B.R., Jacobs, Jr. W.R., Cole, S.T. 1996. An integrated map of the genome of the tubercle bacillus, Mycobacterium tuberculosis H37Rv and comparison with Mycobacterium leprae . Proc. Natl. Acad. Sci. 93: 3132–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, Y.M., Haapalainen, A.M., Kilpeläinen, S.H., Marttila, M.S., Koski, M.K., Glumoff, T., Novikov, D.K., Hiltunen, J.K. 2000. Human peroxisomal multifunctional enzyme type 2. Site-directed mutagenesis studies show the importance of two protic residues for 2-enoyl-CoA hydratase 2 activity. J. Biol. Chem. 275: 4965–4972. [DOI] [PubMed] [Google Scholar]
- Read, R.J. 1986. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. A42: 140–149. [Google Scholar]
- Sassetti, C.M. and Rubin, E.J. 2003. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. 100: 12989–12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti, C.M., Boyd, D.H., Rubin, E.J. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48: 77–84. [DOI] [PubMed] [Google Scholar]
- Schneider, T.R. and Sheldrick, G.M. 2002. Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 58: 1772–1779. [DOI] [PubMed] [Google Scholar]
- Sharma, S.K., Kapoor, M., Ramya, T.N., Kumar, S., Kumar, G., Modak, R., Sharma, S., Surolia, N., Surolia, A. 2003. Identification, characterization, and inhibition of Plasmodium falciparum β-hydroxyacyl-acyl carrier protein dehydratase (FabZ). J. Biol. Chem. 278: 45661–45671. [DOI] [PubMed] [Google Scholar]
- Stern, J.R., del Campillo, A., Raw, I. 1956. Enzymes of fatty acid metabolism. I. General introduction; crystalline crotonase. J. Biol. Chem. 218: 971–983. [PubMed] [Google Scholar]
- Sudesh, K., Fukui, T., Iwata, T., Doi, Y. 2000. Factors affecting the freeze-fracture morphology of in vivo polyhydroxyalkanoate granules. Can. J. Microbiol. 46: 304–311. [PubMed] [Google Scholar]
- Takayama, K., Wang, C., Besra, G.S. 2005. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis . Clin. Microbiol. Rev. 18: 81–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuge, T., Hisano, T., Taguchi, S., Doi, Y. 2003a. Alteration of chain length substrate specificity of Aeromonas caviae R-enantiomer-specific enoyl-coenzyme A hydratase through site-directed mutagenesis. Appl. Environ. Microbiol. 69: 4830–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuge, T., Taguchi, K., Seiichi, T., Doi, Y. 2003b. Molecular characterization and properties of (R)-specific enoyl-CoA hydratases from Pseudomonas aeruginosa: metabolic tools for synthesis of polyhydroxyalkanoates via fatty acid β-oxidation. Int. J. Biol. Macromol. 31: 195–205. [DOI] [PubMed] [Google Scholar]
- van Duyne, G.D., Standaert, R.F., Karplus, P.A., Schreiber, S.L., Clardy, J. 1993. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229: 105–124. [DOI] [PubMed] [Google Scholar]
- Venkatachalam, C.M. 1968. Stereochemical criteria for polypeptides and proteins. V. Conformation of a system of three linked peptide units. Biopolymers 6: 1425–1436. [DOI] [PubMed] [Google Scholar]