

Abstract
Origami is the Japanese art of folding a piece of paper into complex shapes and forms. Much like origami of paper, Nature has used conserved protein folds to engineer proteins for a particular task. An example of a protein family, which has been used by Nature numerous times, is the thioredoxin superfamily. Proteins in the thioredoxin superfamily are all structured with a β-sheet core surrounded with α-helices, and most contain a canonical CXXC motif. The remarkable feature of these proteins is that the link between them is the fold; however, their reactivity is different for each member due to small variations in this general fold as well as their active site. This review attempts to unravel the minute differences within this protein family, and it also demonstrates the ingenuity of Nature to use a conserved fold to generate a diverse collection of proteins to perform a number of different biochemical tasks.
Keywords: thioredoxin, protein folds, glutaredoxin, glutathione S-transferase, Dsb, alkylhydroperoxidase, protein engineering
Origami is the ancient Japanese art of paper folding—the simple movement of paper into various shapes and forms some of startling complexity (see Fig. 1). Nature has taken polypeptides and also folded them into very complicated structures. Unlike folded paper, proteins are capable of performing countless numbers of reactions, essential to life. Also, unlike paper origami, protein origami does not necessarily engineer completely new folds, but rather usually alters existing folds slightly to make that protein adopt new roles. Nature currently uses numerous folds. Extrapolating back to the origin of life, it seems likely that all proteins were derived from one or a very limited number of primordial protein folds. These folds then diverged from each other so much that there is often no detectable resemblance that can be found between protein families. However, by close examination of one protein family, we can see how Nature has adapted one particular fold to perform various functions. We will focus on the protein thioredoxin and its adaptable fold.
Figure 1.

Japanese origami. Paper is carefully folded into complex angles using different parts of the paper incorporated with delicate creases. Manipulation of the paper results in a functional representation of an object of shape. In this case, paper was folded to create a replica of a dinosaur. Proteins are also folded with careful creases, creating a functionally active biomolecule.
Thioredoxin is a small ubiquitous protein that reduces protein disulfides, protects cells from oxidative damage, and is important for maintaining the cytoplasm in a reducing state. Thioredoxin remains folded at temperatures up to 80°C. The high degree of stability conferred by the thioredoxin scaffold may explain why it has been adopted by so many proteins for their function. In Escherichia coli, proteins related to thioredoxin include the glutaredoxins, the disulfide bond oxidoreductases, glutathione S-transferases, and alkylhydroperoxidases. One important characteristic of the fold is the presence of a Cys-X-X-Cys motif at the active site, where X is any amino acid. The cysteines can reversibly form a disulfide bond, allowing thioredoxin-related molecules to participate in disulfide exchange reactions. Here we attempt to provide a glimpse at the art of protein structure and function and how it relates to the prototypical protein, thioredoxin. The evolution of thioredoxin via gene duplication events and mutation has altered the function of many members in the thioredoxin superfamily. Although the function may have changed, many thioredoxins and thioredoxin-like proteins have retained key structural features that make them similar. In this review, we will explore the changes each thioredoxin-like protein has changed by manipulating its active site, changing its oligomeric state, and adding new domains to accommodate a particular reactivity. There is vast literature on thioredoxin-related proteins, and to limit the scope of this review, we have chosen to focus on the analysis of the thioredoxin superfamily in the model organism E. coli. Though limited, this focus has the advantage of detailed analyses and experimentation of many thioredoxin-related proteins having been performed in this organism, and E. coli possesses at least one member of all major families of thioredoxin-like proteins.
Thioredoxin
Thioredoxin is the founding member of the thioredoxin family. It is a 12-kDa protein that is involved in many reactions including reducing improper disulfides that have formed in the cytosol, donating reductive equivalents to ribonucleotide reductase, and being an indicator of the intracellular redox status. Thioredoxin is reduced by thioredoxin reductase, a flavoenzyme that transfers reducing equivalents from pyridine nucleotide NADPH to thioredoxin (Holmgren 1985, 1989, 2000).
The function of thioredoxin has been implicated in numerous pathways; principally, it provides a protective role against many different types of damaging stresses. In most organisms, thioredoxin participates as a hydrogen donor to ribonucleotide reductase, a key enzyme in the generation of deoxyribonucleotides for DNA synthesis (Holmgren 1976). Thioredoxin also maintains reductive homeostasis in the cytosol by reducing incorrect disulfides that have formed in proteins, thus returning these proteins to their native state. Many antioxidant proteins such as the peroxiredoxins, another group in the thioredoxin superfamily, require the reductive action of thioredoxin to remove potentially damaging peroxides (Cha et al. 1995).
The structures of thioredoxin of both the oxidized and reduced protein were solved by NMR (Jeng et al. 1994). The structure of thioredoxin is comprised of five β-strands located in the core of the protein, which are encircled by four α-helices (Holmgren et al. 1975) (see Fig. 2). The active-site motif in thioredoxin includes a conserved four amino acid motif C-P-G-C, which is located at the C terminus of β2 and starts in the beginning of an α-helix (Holmgren et al. 1975). These two cysteines reversibly form a disulfide as part of thioredoxin's catalytic mechanism.
Figure 2.
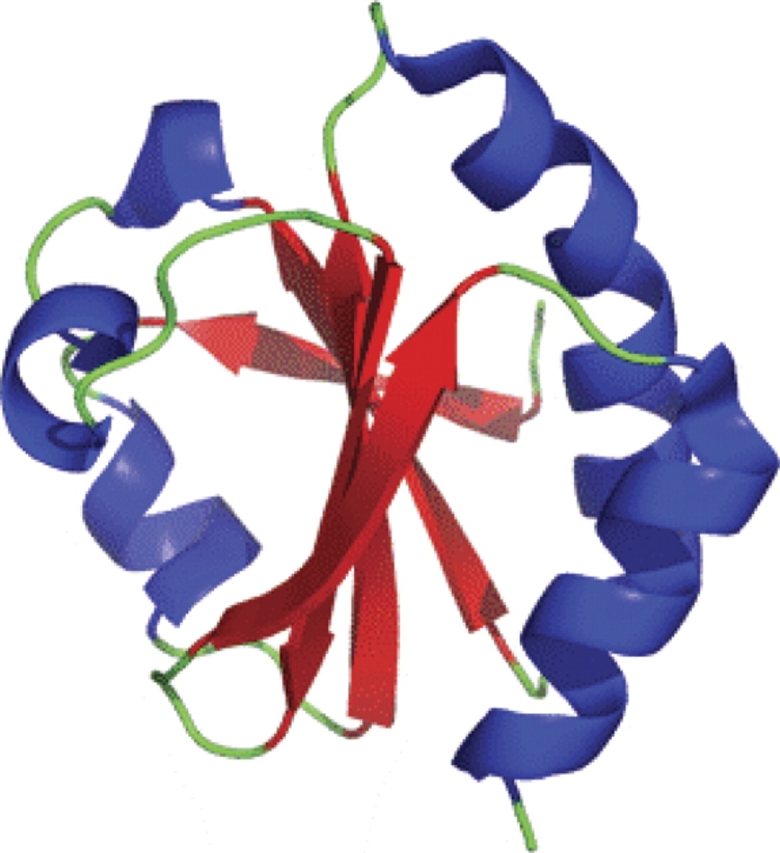
Structure of oxidized thioredoxin. The thioredoxin fold, named for the founding member of the family thioredoxin, is a protein fold consisting of four β-sheets surrounded by three α-helices. The thioredoxin family of protein is typically involved in redox active reactions and usually contains a C-X-X-C motif, where X is any amino acid.
The standard redox protein of thioredoxin is E°′ = −270 mV, indicating that this protein prefers to be in the oxidized, disulfide bonded state (Aslund et al. 1997). The N-terminal cysteine in the thioredoxin motif has a pKa value of ∼7.5, and the C-terminal cysteine has a pKa of ∼9.2. The N-terminal cysteine's pKa of thioredoxin is significantly lower than that of free cysteine due to the hydrogen bonding network created by Asp26 and Lys57, thereby allowing the C-terminal cysteine to have a lower pKa (Kallis and Holmgren 1980; Holmgren and Fagerstedt 1982; Dyson et al. 1991, 1997; Chivers et al. 1997). This low pKa enables thioredoxin to act as a nucleophile and attack disulfide bonds within proteins. The nucleophilic attack on a protein disulfide results in a mixed disulfide between thioredoxin and its target protein. The resolution of this disulfide is carried out by the C-terminal cysteine, resulting in a reduced protein substrate and an oxidized thioredoxin (Holmgren 1995). Thioredoxin thus acts to reduce proteins. The cycle is kept catalytic via thioredoxin reductase and the pyridine nucleotide NADPH (Lennon and Williams 1996). The NMR structures for both reduced and oxidized forms of thioredoxin share similar overall structure with small variations, particularly around the active-site motif (Dyson et al. 1988, 1989, 1991; Katti et al. 1990).
Thioredoxin itself has been very well studied and is the subject of a number of reviews (Holmgren 1985, 1995; Nakamura 2005), and it will not be further discussed here.
Glutaredoxins
The glutaredoxins are distantly related to thioredoxin, and operate via a similar mechanism. They also assist in maintaining the reductive nature of the cytoplasm. The active site of glutaredoxins is nearly always a conserved C-P-Y-C motif (Shi et al. 1999). Glutaredoxins provide hydrogens to the protein ribonucleotide reductase (RNR), which is an essential protein in the synthesis of deoxyribonucleotides. Glutaredoxins also participate in the reduction of dehydroascorbate to ascorbate, the reactivation of DNA binding for some transcription factors, and in the maintenance of HIV-1 protease (Kelley and Bushweller 1998). Maintenance of glutaredoxin in its reduced state is facilitated by reduced glutathione in the cytoplasm. The reduced glutathione is recycled from the oxidized state to the reduced state via glutathione reductase (gor), which in turn is reduced by pyridine nucleotide.
Glutaredoxins were first discovered in 1976 by Arne Holmgren (Holmgren 1976). Glutaredoxin in E. coli has four orthologs: Grx1, Grx2, Grx3, and Grx4. These proteins share the classic thioredoxin/glutaredoxin fold, which is characterized by four β-sheets surrounded by three α-helices (Eklund et al. 1992) (Figs. 3, 4). These proteins participate in a variety of reactions in E. coli by providing hydrogens to ribonucleotide reductase and PAPS reductase as well as the detoxification of many toxic organic molecules.
Figure 3.

Ribbon structures of representative members for the thioredoxin family. Members of the thioredoxin family have a characteristic fold with β-sheets surrounded by α-helices.
Figure 4.

Typical architecture of thioredoxin-like proteins. The figure displays the two-dimensional representation of the thioredoxin fold in representative proteins in the thioredoxin super family. β-Sheets are drawn as arrows and α-helices are drawn in boxes. A typical thioredoxin fold is shown where two parallel β-sheets are intervened by an α-helix. In addition, between the N-terminal and C-terminal portion is an α-helix that connects these two domains. Helices that are not part of the thioredoxin fold are drawn colored in white.
Glutaredoxins have been generalized into three different isoforms. The first isoform characteristic of Grx1 and Grx3 from E. coli has a typical thioredoxin fold as well as a CXXC motif. Grx1 and Grx3 are functionally similar because they both function as reductive enzymes and provide reducing equivalents to proteins (Bushweller et al. 1994; Aslund et al. 1996). The second isoform of glutaredoxins is the glutathione S-transferase-like proteins. These are much larger, ∼24 kDa; in E. coli, the representative is Grx2. Grx2 has an N-terminal glutaredoxin domain with a catalytically active CXXC motif and a C-terminal α-helical domain (Vlamis-Gardikas et al. 1997). These proteins are involved in cellular detoxification of many toxic compounds. Grx4, a very recently discovered E. coli member of the last group of glutaredoxins, are monothiol glutaredoxins that contain a CXFX motif (Fernandes et al. 2005). In eukaryotes they are principally involved in iron–sulfur cluster assembly, oxidative stress response, and other cellular processes (Rodriguez-Manzaneque et al. 1999).
Grx1 was first discovered as a GSH-dependent hydrogen donor to the enzyme ribonucleotide reductase (Holmgren 1976). In vivo it was shown to be 10-fold less in concentration than its counterpart thioredoxin, but also had a 10-fold less K m for ribonucleotide reductase (RNR) (Holmgren 1979b). Grx1 has been shown to be the primary reductant to RNR, and is also an alternate reductant to methionine sulfoxide reductase (Holmgren 1979a,b). Grx1 also is a stress induced protein (Stewart et al. 1998). Grx1 is under the control of OxyR, a potent transcription factor that senses oxidative stress induced by chemicals such as hydrogen peroxide (Tao 1997). OxyR must be glutathionylated to be active in inducing the transcriptions of target genes. Grx1 functions to deglutathionylate OxyR. This system is autoregulatory where Grx1 regulates OxyR activity, and OxyR regulates expression of the gene for Grx1, grxA (Tao 1997). Grx1 is not essential for cell viability in E. coli, suggesting the existence of alternate reducing agents (Zheng et al. 1998).
One such alternative reductant, Grx2, was discovered by looking for suppressors in a ΔgrxA strain. Grx2 is much larger in size than the other three orthologs of glutaredoxin in E. coli, and its structure more closely resembles glutathione S-transferase than glutaredoxin 1, 2, and 4. However, Grx2 does have an N-terminal glutaredoxin domain whose structure is similar to the other glutaredoxins, including a C-P-Y-C active site. Grx2 also contains a C-terminal α-helical domain. Grx2 does not reduce ribonucleotide reductase, and is not a reductant for PAPS reductase, but has an extremely high catalytic activity in resolving the mixed disulfide between β-hydroxyethyl disulfide (HED) and GSH. This suggests that Grx2 participates mainly in monothiol reactions that remove glutathionylation from proteins. Grx2 is regulated by the stationary phase sigma factor (RpoS) and guanosine-3′, 5′-tetraphosphate (ppGpp). These two factors control the regulation of genes in stationary phase of growth, and the expression of Grx2 under these conditions might contribute to the overall defense of organic peroxides and hydrogen peroxides (Aslund et al. 1994; Vlamis-Gardikas et al. 1997; Lillig et al. 1999).
Grx3 was also discovered in a suppressor screen using a ΔgrxA strain. It resembles Grx1 both in size and in structure. Grx3 has ∼33% sequence identity to Grx1, contains a glutaredoxin-like fold, and has the C-P-Y-C redox active motif. Grx3 has ∼5% the catalytic rate of Grx1 for ribonucleotide reductase, and Grx3 cannot reduce PAPS reductase. Deletion of this gene caused increased sensitivity to oxidizing agents such as menadione and cumene hydroperoxide, but it is still unclear what the in vivo role of Grx3 is and how it is regulated (Aslund et al. 1994, 1996; Lillig et al. 1999).
Grx4 is the only monothiol glutaredoxin in E. coli, and was shown to be essential (Gerdes et al. 2003). Grx4 differs from the other orthologs in that it lacks activity toward classic glutaredoxin substrates such as HED, insulin, and others (Fernandes et al. 2005). It was also shown that Grx4 can be a substrate for thioredoxin reductase, and its glutathionylation status is dependent on Grx1 (Fernandes et al. 2005). Recently, Fladvad et al. (2005) solved the NMR solution structure of Grx4 and detailed its monothiol mechanism.
The catalytic mechanism of the glutaredoxins can be classified as being dithiol or monothiol reduction (see Fig. 5 below). In the dithiol mechanism, the N-terminal cysteine in the CXXC has an extremely low pKa and is deprotonated, exposing a thiolate anion. The manipulation of the pKa of the N-terminal cysteine is a common theme in thioredoxin-like proteins including glutaredoxins. An intricate network of hydrogen bonds secures the thiolate and allows it to be catalytically active. A common theme in glutaredoxins is the incorporation of a proline in the first position of the CXXC motif and an aromatic residue at the second position; Grx4 also contains the aromatic residue phenyalanine in a conserved CXFX motif, which also participates in hydrogen bonding to lower the pKa of the N-terminal cysteine (Foloppe et al. 2001; Fladvad et al. 2005). The proline incorporation results in a kink that displaces the N-terminal cysteine into a position that can be hydrogen bonded by the amide proton from the aromatic residue (Foloppe et al. 2001; Fladvad et al. 2005). These modifications contrast the active site with thioredoxin.
Figure 5.

Mechanism of dithiol and monothiol reactivities of glutaredoxin. (A) (1) Reduced glutaredoxin first seeks a substrate that has a disulfide bond; (2) with a reduced cysteine, glutaredoxin attacks the disulfide in the target protein resulting in a mixed disulfide; (3–4) with a deprotonated C-terminal cysteine, glutaredoxin attacks the mixed disulfide, which results in a reduced substrate protein and a completely oxidized glutaredoxin; (5) Glutaredoxin is then reduced to return back into its active form. (B) (1) Glutaredoxin starts off as a reduced species and attacks a protein that is glutathionylated on a cysteine. (2) Glutaredoxin becomes glutathionylated and the substrate protein becomes reduced with a free cystine. (3) A reduced glutathione molecule then attacks the mixed disulfide in glutaredoxin. (4) Glutaredoxin returns back into its reduced conformation and oxidized glutathione disulfide is formed.
The resulting stabilized thiolate anion can then initiate a nucleophilic attack on a sulfur atom of a target disulfide bridge. The glutaredoxin has now formed a mixed disulfide with a target protein; this causes the second cysteine in the CXXC motif to become deprotonated and attack the N-terminal cysteine to resolve the mixed disulfide. This dithiol reaction results in a reduced target protein and an oxidized glutaredoxin (see Fig. 5A). The dithiol reaction is very similar to the one carried out by thioredoxin; however, glutaredoxin can also participate in monothiol reductions in concert with GSH. The principal purpose of the monothiol reaction is to deglutathionylate proteins. This release of the GSH from proteins is first initiated by the N-terminal cysteine of the CXXC motif, which attacks the protein–GSH complex. In addition, the N-terminal cysteine of the CXXC also has a low pKa, which allows the sulfur to serve as a good leaving group in a monothiol reaction. Furthermore, Glutaredoxin has a high affinity site that only interacts with the GSH moiety and not the protein. GSH binding groove is formed in part by Lys8 and Asp67, which provide favorable ionic interactions between GSH and glutaredoxin (Foloppe and Nilsson 2004). The addition of this site to the thioredoxin scaffold is one of the key things that allows glutaredoxins to specifically react with glutaredoxin. These residues are conserved in the glutaredoxins, but are not present in the rest of the thioredoxin superfamily, which explains why proteins such as thioredoxin and DsbA cannot efficiently participate in monothiol reactions involving GSH.
Glutaredoxin catalyzes the movement of GSH from the target protein to the glutaredoxin, creating a glutaredoxin–GSH conjugate. The conjugate is later displaced by another GSH (Bushweller et al. 1994). The resulting reaction results in the deglutathionylation of proteins, the formation of oxidized glutathione, and the release of glutaredoxin in its reduced state (see Fig. 5B).
The Dsb disulfide oxidoreductases
The conditions faced in the extracellular world are harsh, imposing a need for proteins to have extra stability. Disulfide bonds are one modification proteins utilize to retain their tertiary structure even in harsh environments. The disulfide oxidoreductases (Dsb) are proteins that catalyze disulfide bond formation in secreted proteins. In addition, if incorrect disulfide bonds form within a protein, another thioredoxin-like oxidoreductase called DsbC can isomerize the incorrect disulfides to the correct form (Kadokura et al. 2003).
DsbA
DsbA, a 21-kDa thioredoxin-like oxidoreductase, acts as the direct donor of disulfide bonds to secreted proteins (Bardwell et al. 1991). DsbA is the primary oxidant in the E. coli periplasm. Deletion of this gene results in the failure to oxidize a variety of secreted proteins, their instability, and degradation (Hiniker and Bardwell 2004). The resulting phenotypes include sensitivity to the reductant dithiothreitol, loss of motility due to a misfolded flagellar motor protein, FlgI, metal sensitivity, and an increased susceptibility to M13 phage due to incorrect F pilus formation (Dailey and Berg 1993; Missiakas et al. 1993; Stafford et al. 1999). While the Δdsb mutant is not lethal, the activities associated with DsbA are important to the viability of the cell.
The molecular mechanism of DsbA is well studied (see Fig. 6). DsbA is composed of two domains, a thioredoxin-like domain and an all α-helical domain missing in thioredoxin. Thioredoxin shares only about 10% sequence identity with the thioredoxin-like domain of DsbA. The active site of DsbA is a conserved CPHC motif located in the thioredoxin domain (Martin et al. 1993) (Fig. 2). These two cysteines form a highly oxidizing disulfide bond, which is donated to substrate proteins. The standard redox potential of DsbA is −122 mV, which makes DsbA one of the most oxidizing disulfide oxidoreductases known, a key requirement for DsbA function (Zapun et al. 1993).
Figure 6.

Mechanism of disulfide formation and isomerization in the bacterial periplasm. (A) Disulfide bonds are generated by the Dsb family of proteins in the bacterial periplasm. First, DsbA, a thioredoxin-like protein, has a catalytic disulfide and seeks to donate its disulfide to newly translocated proteins, resulting in a reduced DsbA and oxidized substrate protein. For the system to be catalytic, a membrane protein DsbB oxidizes DsbA. DsbB gains its oxidizing equivalents from the electron transport chain with the final electron acceptor oxygen. (B) DsbA is a potent disulfide oxidase and has the potential to incorporate incorrect disulfides. Another pathway exists to correct incorrect disulfides formed by DsbA or another exogenous disulfide source. DsbC is a thioredoxin-like protein that has reduced cysteines. These reduced cysteines accept a disulfide from a substrate protein. Maintaining DsbC in the reduced form is accomplished by the membrane protein DsbD. DsbD gains its reducing equivalents from the cytosolic protein thioredoxin, which ultimately gains its reduced equivalents from NADPH.
The strongly oxidizing nature of DsbA is in part due to the stabilization of the Cys30 thiolate anion by the partial positive charge on the helix dipole of helix α1, hydrogen bond contacts to His32, and stabilization by hydrogen bonding to either Sγ or HN of Cys33 (Grauschopf et al. 1995). These interactions most likely provide a hydrogen bond network that lowers the pKa of Cys30 to ∼3.5. This is much lower than both the pKa of free cysteine and the pKa or thioredoxin. Thus, Cys30 almost always exists as a thiolate in vivo (Grauschopf et al. 1995; Guddat et al. 1998).
Once DsbA transfers its disulfide to proteins, in order to be catalytic it needs to be reoxidized. DsbB acts to reoxidize DsbA; it removes electrons from DsbA and transfers them to the respiratory chain. In aerobic conditions, DsbB reduces ubiquinone, and ultimately reduces oxygen. In anaerobic conditions, the electron acceptor is menaquinone, which transfers the electrons to nitrate, nitrite, and fumarate (Kobayashi et al. 1997; Bader et al. 1999, 2000; Xie et al. 2002) (see Fig. 6).
DsbC
DsbA is a very powerful oxidant. If a protein has more than two cysteine residues, then DsbA has the potential to form incorrect disulfide bonds. DsbC acts to isomerize incorrectly oxidized proteins. DsbC is a 23.5-kDa protein with two domains, a C-terminal thioredoxin-like domain and an N-terminal dimerization domain (McCarthy et al. 2000). The protein forms a V-shaped dimer with an uncharged cleft that is thought to be involved in peptide binding (McCarthy et al. 2000; Bader et al. 2001; Collet et al. 2002). It is thought that DsbC functions by detecting hydrophobic patches on misfolded proteins via its uncharged cleft, once bound; DsbC's reduced cysteines probe for disulfides in the misfolded protein. Cys98, the nucleophilic cysteine in DsbC, attacks a substrate disulfide and forms a mixed disulfide between DsbC and the substrate protein. The mixed disulfide can be resolved in one of two ways. The mixed disulfide could be resolved when the substrate protein's reduced cysteine attacks the mixed disulfide, creating a new disulfide bond in the target protein and returning DsbC back into its reduced state. Alternatively, the second active site cysteine from DsbC could attack the mixed disulfide, forming an oxidized DsbC and a reduced substrate protein (see Fig. 5). This process would cause the target substrate protein to become reduced, and could allow DsbA to reoxidize that protein to the correct conformation. Both forms of DsbC action require that DsbC be maintained in the reduced state, and this requires DsbD (Walker and Gilbert 1997; Darby et al. 1998a,b) (see Fig. 6).
In contrast, in order to function as an oxidase in vivo, DsbA needs to be kept oxidized. This means that there needs to be a barrier that separates the isomerase pathway from the oxidase pathway. Otherwise, DsbA would oxidize and therefore inactivate DsbC, and DsbC would reduce and therefore inactivate DsbA. Bader et al. (2001) probed the nature of this barrier by the selection of DsbC mutants that could rescue DsbA function in a ΔdsbA strain. The mutations in DsbC that allowed DsbC to function as an oxidase disrupted dimerization and resulted in DsbC monomers. These DsbC mutations allowed DsbC to be oxidized by DsbB, suggesting that the dimeric form of DsbC cannot be oxidized by DsbB. DsbC can thus function as a disulfide oxidase when the dimerization domain is mutated to form monomers. In addition, Sergatori et al. (2006) has also shown that a deletion in the α-helical linker between the N-terminal dimerization domain and the C-terminal thioredoxin domain also causes the dimeric DsbC to function as a substrate for DsbB. This illustrates how simply the overall function of a thioredoxin related protein can be changed by mutagenesis, from that of an isomerase to an oxidase. It also points out the important role of the conjugant reoxidants and reductases have in dictating the overall function of thioredoxin related proteins. Closely related to DsbC in both sequence and structure is the DsbG protein, which is also thought to function as a disulfide isomerase (Andersen et al. 1997).
DsbD is a 59-kDa membrane protein found in E. coli that reduces both DsbC and DsbG. DsbD is composed of three domains: DsbDα is a thioredoxin-like domain, DsbDγ resembles an immunoglobulin-like fold, and DsbDβ is a membrane-bound domain that contains eight transmembrane segments (Goulding et al. 2002). Each domain contains a pair of cysteines postulated to be involved in the reduction of DsbC and DsbG. The mechanism of moving reducing equivalents from the cytoplasm to the periplasm has been shown to work in concert with the thioredoxin system. The proposed mechanism starts with reduced thioredoxin docking to the β-domain and reducing the cysteine pair located in that subunit. This subunit subsequently reduces the γ-domain, which then reduces the α-domain. The α-domain then reduces oxidized DsbC and DsbG in a disulfide cascade reaction that involves multiple thiol–disulfide exchange reactions. This achieves a net flow of reducing equivalents from the cytoplasm to the periplasm (Rietsch et al. 1996, 1997; Collet et al. 2002). Since thioredoxin-like proteins function primarily as thiol–disulfide oxidoreductases, it is not surprising that a thioredoxin-like protein be involved in this cascade, but it is remarkable that the disulfide isomerization pathway consists of no less than four thioredoxin related proteins: thioredoxin itself, DsbD, DsbC, and DsbG. The disulfide cascade within this pathway appears to be controlled by ever increasing redox potentials that allow the disulfides to fall down an energy gradient.
Alkyl hydroperoxidases
Reactive oxygen species (ROS, ROOH, H2O2, O2 −, and OH·) are generated in the cell by a variety of cellular processes that include incomplete reduction of molecular oxygen by the respiration chain, phagocytosis, and redox cycling of xenobiotics (Cha et al. 1995). These reactive oxygen species can be extremely detrimental by oxidizing, thereby critically damaging many cellular components. They can oxidize proteins resulting in denaturation, and can cause oxidation of nucleotides in DNA and thus mutagenesis. A class of thioredoxin-like proteins called the peroxiredoxins (PRX) or the alkylhydroperoxidases (AHP) provide a line of defense against inorganic and organic peroxides. In E. coli, examples of these proteins are the thiol-dependent peroxidase and alkylhydroperoxide reductase (Bieger and Essen 2001). In general, these proteins reduce hydroperoxides by having a cysteine react with a peroxide moiety. The peroxide moiety is reduced to a less harmful alcohol and the cysteine is oxidized to form a sulfenic acid intermediate. The sulfenic acid intermediate is then resolved by another protein to reform the thiol. Alternatively, the sulfenic acid intermediate reacts with another cysteine in the protein causing the formation of a disulfide bond and water. The disulfide bond is then reduced by a thiol reductase such as thioredoxin, returning the peroxiredoxin or alklhydroperoxidase back into its reduced catalytically active thiol form (Cha et al. 1995; Poole 1996; Wood et al. 2003) (see Fig. 7).
Figure 7.

Generalized mechanisms of atypical 2-Cys, typical 2-Cys, and 1-Cys peroxiredoxins. (A) In the typical 2-Cys peroxiredoxins, a homodimer, where one monomer has the active peroxidatic site cysteine and the other having the resolving cysteine. The peroxidatic becomes oxidized to a sulfenic acid, thereby reducing hydrogen peroxide to water. The resolving cysteine now attacks the peroxidatic cysteine to form an intersubunit disulfide bond. The disulfide is reduced from cytosolic reductants such as thioredoxin. (B) In the atypical 2-Cys peroxiredoxins, the mechanism of action is very similar to the typical 2-Cys peroxiredoxins except that both the peroxidatic and the resolving cysteines are both on the same monomer, thereby eliminating the need for two protein monomers. (C) The 1-Cys peroxiredoxins function differently than the 2-Cys peroxiredoxins. These peroxiredoxins interact with the peroxide directly and form a sulfenic acid intermediate on the peroxidatic cysteine. The 2-Cys peroxiredoxins then interact with a thiol reducing source such as thioredoxin or glutaredoxin and become reduced, releasing water and regenerating the reduced 1-Cys peroxiredoxin. (Derived from Wood et al. 2003.)
Tpx is a 20-kDa peroxiredoxin found in the E. coli periplasm and other pathogenic bacterial species and has been implicated in the reduction of hydrogen peroxide. It was discovered in 1995 by its thiol-dependent antioxidant activity (Cha et al. 1995). Tpx contains two catalytically active cysteines that participate in the reduction of hydrogen peroxide and other organic hydroperoxides. Tpx can efficiently suppress the inactivation of glutamine synthetase via metal catalyzed oxidation, a standard assay in measuring peroxidase activity (Cha et al. 1995). Tpx is reduced by thioredoxin in vivo.
Tpx contains a thioredoxin-like fold consisting of a seven-stranded twisted β-sheet surrounded by four α-helices (Choi et al. 2003). It shares ∼16% sequence identity with thioredoxin. Thr58, Cys61, and Arg133 form a catalytic triad. Thr58 and Arg133 act to lower the pKa of Cys61’s active site sulfur, thus allowing the resulting thiolate to participate in nucleophilic attack of the target hydroperoxide. A lower than normal pKa of the active site cysteine is a common mechanism that thioredoxin-like proteins including thioredoxin, glutaredoxin, DsbA, DsbC, DsbG, and Tpx use to increase their reactivity. The pyrrolidine ring on Pro54 of Tpx acts to limits solvent accessibility as well as peroxide accessibility of Cys61. This may prevent overoxidation of that cysteine from a sulfenic acid intermediate to species such as sulfinic and sulfonic acid. Also observed in the crystal structure was an L-shaped hydrophobic cleft that is formed by the N-terminal β-hairpin, which seems to be able to accommodate long fatty acid hydroperoxides. This N-terminal β-hairpin is absent in other peroxiredoxins, a possible reason why Tpx has high specificity toward alkylhydroperoxides. The structure with the extra N-terminal β-hairpin is uniquely present in Gram-negative bacteria, particularly in pathogenic bacteria (Choi et al. 2003).
AhpR is a multimeric protein composed of AhpC and AhpF, which combine to form a potent alkylhydroperoxidase complex. AhpC is a 21-kDa thioredoxin-like protein that has two cysteine residues. AhpC forms an intersubunit disulfide with another AhpC monomer resulting in a homodimer. This intersubunit disulfide seems to be undergoing reversible reduction during catalysis and the addition of N-ethylmaleimide can irreversibly inhibit AhpC, evidence that this intersubunit disulfide is part of a disulfide exchange mechanism (Wood et al. 2003).
AhpF forms the other component of the AhpR complex. AhpF is a 57-kDa protein that contains an FAD moiety as well as six total cysteines, with one pair forming a disulfide and the other four existing in the reduced form. AhpF has an N-terminal thioredoxin-fold with two cysteines that are involved in thiol–disulfide exchange. It also contains a C terminus that is highly homologous to thioredoxin reductase and is responsible for binding the flavin moiety. Thus, the AhpF enzyme could be thought of as being derived from a fusion of the two main components of the thioredoxin–thioredoxin reductase pathway. The complete AhpF enzyme contains two disulfides, Cys129–Cys132 and Cys345–Cys 348, as well as two thiols in the C terminus (Cys476 and Cys489). The Cys129 and Cys132 in the thioredoxin domain of AhpF are the cysteine pair that directly reduces AhpC as part of AhpC's catalytic cycle. Removal of this domain causes a dramatic decrease in NADH consumption as well as decreased reduction rate of AhpC by AhpF (Poole et al. 2000; Reynolds and Poole 2000; Bieger and Essen 2001; Wood et al. 2001).
Glutathione S-transferases
The glutathione S-transferases are a class of proteins that participate in the addition of glutathione to electrophilic groups of many hydrophobic compounds including organic halides, epoxides, α- and β-unsaturated carbonyls, organic nitrate esters, and organic thiocyanates (Mannervik and Danielson 1988; Coles and Ketterer 1990). The addition of glutathione to these xenobiotic compounds allows the compounds to become more hydrophilic, and tends to make the compounds less reactive to cellular components. Glutathione S-transferases also participate in concert with GSH to detoxify intracellular organic hydroperoxides. In addition, bacterial species including E. coli use glutathionylation as a means to promote degradation of antibiotics, and glutathione S-transferases have also been shown to bind to the antibiotics themselves (Vuilleumier 1997). These examples show that glutathione S-transferases participate in the degradation of toxic compounds imported into the cell. They do so by making them more water soluble and deactivating reactive groups that could potentially cause damage.
Most glutathione S-transferases are homodimers, with each monomer folding into two domains connected via hydrogen bonds. Each monomer in the dimer has two distinct domains, an N-terminal thioredoxin-like domain (amino acids ∼1–80) and a C-terminal domain (amino acids ∼89–201) (using E. coli numbering). The N-terminal domain participates in binding the glutathione moiety via its thioredoxin-like domain while the C-terminal domain contains several hydrophobic α-helices that specifically bind hydrophobic substrates. Thus, similar to DsbC, DsbD, and AhpF, the function of the thioredoxin-like domain has become modified by the covalent addition on another domain. Another example of a thioredoxin-like protein that has an additional domain fused onto it is YbbN, a 33-kDa heat-shock inducible protein of unknown function. This protein consists of an N-terminal thioredoxin domain and a C-terminal glcnac transferase-like domain. In thioredoxin-like proteins the N-terminal cysteine of the CXXC motif is exposed and reactive, but intriguingly, YbbN lacks the N-terminal cysteine of this motif. The protein has no activity in the reduction of insulin's disulfide bonds, nor does it show any chaperone activity when using citrate synthase as a model substrate (J.L. Pan and J.C. Bardwell, unpubl.). YbbN may be an example of a thioredoxin-like fold that is in the process of evolving away from a redox-related function.
The E. coli glutathione S-transferase structure was solved at 2.1 Å bound to glutathione sulfonate, a glutathione analog. The overall topology and structure show that the protein has a similar shape to many other glutathione S-transferases structures. E. coli glutathione S-transferase shares ∼26%–80% sequence identity with other bacterial glutathione S-transferases and <20% sequence identity to the eukaryotic glutathione S-transferases. Mechanistically, the reaction scheme for eukaryotic glutathione S-transferases involves either Tyr5 or Ser11 in the positioning of the glutathione molecule within the N-terminal domain (Nishida et al. 1998). In E. coli glutathione S-transferase, these two residues are unable to participate in hydrogen bonding with glutathione due to spatial constraints. In E. coli, Tyr5, instead, has its side chain directed into the solvent region and Ser11 is almost 7.1 Å away, placing the amino acid far away from the sulfur of the glutathione analog. Thus, Ser11 and Try5 in E. coli are not playing a part in stabilizing the glutathione; instead, Cys10 and His106 are involved. This is an example of how different organisms have used different residues but maintained a certain activity (Nishida et al. 1998).
Glutathione S-transferases are extremely specific in their ability to detoxify aromatic and other hydrophobic compounds. In Burkholderia, bphK is a glutathione S-transferase thought to be involved specifically in the degradation of biphenyls (Vuilleumier 1997). In Sphingomonas paucimobilis the glutathione S-transferases ligE and ligF can actually distinguish between the two different enantiomers of a β-aryl ether model compound, allowing for selective degradation of one product over another (Masai et al. 2003).
Conclusions and laboratory evolution of thioredoxin-like proteins
The thioredoxin-like fold is found in numerous proteins in the model genetic organism E. coli and in other species. This particular fold is seen in predominantly redox active proteins that participate in the reduction or oxidation of disulfide bonds, electron donation, and detoxification. These examples show that enzymatic evolution has used the thioredoxin fold as a scaffold for a number of different purposes. Nature has been seen to maintain catalytically active protein folds and alter them so that they broaden their ability to catalyze a new substrate. These changes occurred as a selection to new substrates, and have led to the evolution of thioredoxin into more specialized proteins that maintain cell homeostasis.
Laboratory evolution of the thioredoxin-like fold further emphasizes the flexibility of the fold and helps illustrate how various functions can be acquired by individual members that did not possess these functions prior to the imposed selection. For example, substitution of thioredoxin's active site CGPC to DsbA's active site CPHC can result in a protein that functions very similarly to DsbA. The CXXC sequence can even be thought of as a signature motif that defines the function of the family. Another example of in vitro evolution is the selection of thioredoxin mutants that can compensate for the whole DsbA–DsbB pathway. A mutation from CGPC to CACC in exported versions of thioredoxin was capable of complementing null mutations in the DsbA–DsbB pathway. They did so by acquiring a 2Fe–2S iron–sulfur cluster, and presumably a whole new mechanism of action. This shows that thioredoxin is extremely amenable to mutation, conferring the protein with new catalytic properties and the ability to participate in new redox reactions (Masip et al. 2004).
Ritz et al. (2001) deleted thioredoxin reductase and glutathione oxidoreductase; both of these proteins are required for the maintenance of a reducing cytosol. However, it was discovered that an insertion of a single TCT insertion in the gene ahpC allowed the AhpC protein product to function as a disulfide reductase as opposed to the peroxiredoxin role that it normally participates in within the cell. AhpC has lost its peroxidase activity while gaining a disulfide reductase activity.
These examples show that thioredoxin and thioredoxin-like proteins can evolve, both in function and substrate specificity, with only few amino acid changes in the protein. Although the function and specificity has changed, the thioredoxin fold is still conserved. The observed pliability and adaptation of thioredoxin makes this protein extremely suitable for the construction of novel redox reactions that participate in numerous processes, ranging from protein folding, detoxification, and metabolite synthesis.
Acknowledgments
We thank Jimmy Regeimbal, Stefan Gleiter, and Annie Hiniker for reading over this article. We also thank Arne Holgren for providing his helpful comments. Funding was made available from NIH and HHMI to J.C.A.B. J.L.P. is a Ruth L. Kirchenstein Fellow.
Footnotes
Reprint requests to: James C.A. Bardwell, 830 North University, Rm. 4007, Ann Arbor, MI 48109, USA; e-mail: jbardwel@umich.edu; fax: (313) 647-0884.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062268106.
References
- Andersen, C.L., Matthey-Dupraz, A., Missiakas, D., Raina, S. 1997. A new Escherichia coli gene, dsbG, encodes a periplasmic protein involved in disulphide bond formation, required for recycling DsbA/DsbB and DsbC redox proteins. Mol. Microbiol. 26: 121–132. [DOI] [PubMed] [Google Scholar]
- Aslund, F., Ehn, B., Miranda-Vizuete, A., Pueyo, C., Holmgren, A. 1994. Two additional glutaredoxins exist in Escherichia coli: Glutaredoxin 3 is a hydrogen donor for ribonucleotide reductase in a thioredoxin/glutaredoxin 1 double mutant. Proc. Natl. Acad. Sci 91: 9813–9817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslund, F., Nordstrand, K., Berndt, K.D., Nikkola, M., Bergman, T., Ponstingl, H., Jornvall, H., Otting, G., Holmgren, A. 1996. Glutaredoxin-3 from Escherichia coli: Amino acid sequence, 1H and 15N NMR assignments, and structural analysis. J. Biol. Chem. 271: 6736–6745. [DOI] [PubMed] [Google Scholar]
- Aslund, F., Berndt, K.D., Holmgren, A. 1997. Redox potentials of glutaredoxins and other thiol-disulfide oxidoreductases of the thioredoxin superfamily determined by direct protein–protein redox equilibria. J. Biol. Chem. 272: 30780–30786. [DOI] [PubMed] [Google Scholar]
- Bader, M., Muse, W., Ballou, D.P., Gassner, C., Bardwell, J.C. 1999. Oxidative protein folding is driven by the electron transport system. Cell 98: 217–227. [DOI] [PubMed] [Google Scholar]
- Bader, M.W., Xie, T., Yu, C.A., Bardwell, J.C. 2000. Disulfide bonds are generated by quinone reduction. J. Biol. Chem. 275: 26082–26088. [DOI] [PubMed] [Google Scholar]
- Bader, M.W., Hiniker, A., Regeimbal, J., Goldstone, D., Haebel, P.W., Riemer, J., Metcalf, P., Bardwell, J.C. 2001. Turning a disulfide isomerase into an oxidase: DsbC mutants that imitate DsbA. EMBO J. 20: 1555–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell, J.C., McGovern, K., Beckwith, J. 1991. Identification of a protein required for disulfide bond formation in vivo. Cell 67: 581–589. [DOI] [PubMed] [Google Scholar]
- Bieger, B. and Essen, L.O. 2001. Crystal structure of the catalytic core component of the alkylhydroperoxide reductase AhpF from Escherichia coli . J. Mol. Biol. 307: 1–8. [DOI] [PubMed] [Google Scholar]
- Bushweller, J.H., Billeter, M., Holmgren, A., Wuthrich, K. 1994. The nuclear magnetic resonance solution structure of the mixed disulfide between Escherichia coli glutaredoxin(C14S) and glutathione. J. Mol. Biol. 235: 1585–1597. [DOI] [PubMed] [Google Scholar]
- Cha, M.K., Kim, H.K., Kim, I.H. 1995. Thioredoxin-linked “thiol peroxidase” from periplasmic space of Escherichia coli . J. Biol. Chem. 270: 28635–28641. [DOI] [PubMed] [Google Scholar]
- Chivers, P.T., Prehoda, K.E., Volkman, B.F., Kim, B.M., Markley, J.L., Raines, R.T. 1997. Microscopic pKa values of Escherichia coli thioredoxin. Biochemistry 36: 14985–14991. [DOI] [PubMed] [Google Scholar]
- Choi, J., Choi, S., Cha, M.K., Kim, I.H., Shin, W. 2003. Crystal structure of Escherichia coli thiol peroxidase in the oxidized state: insights into intramolecular disulfide formation and substrate binding in atypical 2-Cys peroxiredoxins. J. Biol. Chem. 278: 49478–49486. [DOI] [PubMed] [Google Scholar]
- Coles, B. and Ketterer, B. 1990. The role of glutathione and glutathione transferases in chemical carcinogenesis. Crit. Rev. Biochem. Mol. Biol. 25: 47–70. [DOI] [PubMed] [Google Scholar]
- Collet, J.F., Riemer, J., Bader, M.W., Bardwell, J.C. 2002. Reconstitution of a disulfide isomerization system. J. Biol. Chem. 277: 26886–26892. [DOI] [PubMed] [Google Scholar]
- Dailey, F.E. and Berg, H.C. 1993. Mutants in disulfide bond formation that disrupt flagellar assembly in Escherichia coli . Proc. Natl. Acad. Sci. 90: 1043–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby, N.J., Penka, E., Vincentelli, R. 1998a. The multi-domain structure of protein disulfide isomerase is essential for high catalytic efficiency. J. Mol. Biol. 276: 239–247. [DOI] [PubMed] [Google Scholar]
- Darby, N.J., Raina, S., Creighton, T.E. 1998b. Contributions of substrate binding to the catalytic activity of DsbC. Biochemistry 37: 783–791. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J., Holmgren, A., Wright, P.E. 1988. Structural differences between oxidized and reduced thioredoxin monitored by two-dimensional 1H NMR spectroscopy. FEBS Lett. 228: 254–258. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J., Holmgren, A., Wright, P.E. 1989. Assignment of the proton NMR spectrum of reduced and oxidized thioredoxin: Sequence-specific assignments, secondary structure, and global fold. Biochemistry 28: 7074–7087. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J., Tennant, L.L., Holmgren, A. 1991. Proton-transfer effects in the active-site region of Escherichia coli thioredoxin using two-dimensional 1H NMR. Biochemistry 30: 4262–4268. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J., Jeng, M.F., Tennant, L.L., Slaby, I., Lindell, M., Cui, D.S., Kuprin, S., Holmgren, A. 1997. Effects of buried charged groups on cysteine thiol ionization and reactivity in Escherichia coli thioredoxin: Structural and functional characterization of mutants of Asp 26 and Lys 57. Biochemistry 36: 2622–2636. [DOI] [PubMed] [Google Scholar]
- Eklund, H., Ingelman, M., Soderberg, B.O., Uhlin, T., Nordlund, P., Nikkola, M., Sonnerstam, U., Joelson, T., Petratos, K. 1992. Structure of oxidized bacteriophage T4 glutaredoxin (thioredoxin). Refinement of native and mutant proteins. J. Mol. Biol. 228: 596–618. [DOI] [PubMed] [Google Scholar]
- Fernandes, A.P., Fladvad, M., Berndt, C., Andresen, C., Lillig, C.H., Neubauer, P., Sunnerhagen, M., Holmgren, A., Vlamis-Gardikas, A. 2005. A novel monothiol glutaredoxin (Grx4) from Escherichia coli can serve as a substrate for thioredoxin reductase. J. Biol. Chem. 280: 24544–24552. [DOI] [PubMed] [Google Scholar]
- Fladvad, M., Bellanda, M., Fernandes, A.P., Mammi, S., Vlamis-Gardikas, A., Holmgren, A., Sunnerhagen, M. 2005. Molecular mapping of functionalities in the solution structure of reduced Grx4, a monothiol glutaredoxin from Escherichia coli . J. Biol. Chem. 280: 24553–24561. [DOI] [PubMed] [Google Scholar]
- Foloppe, N. and Nilsson, L. 2004. The glutaredoxin -C-P-Y-C- motif: Influence of peripheral residues. Structure 12: 289–300. [DOI] [PubMed] [Google Scholar]
- Foloppe, N., Sagemark, J., Nordstrand, K., Berndt, K.D., Nilsson, L. 2001. Comparison with functionally related proteins. J. Mol. Biol. . Escherichia coli 310: 449–470. [DOI] [PubMed] [Google Scholar]
- Gerdes, S.Y., Scholle, M.D., Campbell, J.W., Balazsi, G., Ravasz, E., Daugherty, M.D., Somera, A.L., Kyrpides, N.C., Anderson, I., Gelfand, M.S. et al. 2003. Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J. Bacteriol. 185: 5673–5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding, C.W., Sawaya, M.R., Parseghian, A., Lim, V., Eisenberg, D., Missiakas, D. 2002. Thiol-disulfide exchange in an immunoglobulin-like fold: Structure of the N-terminal domain of DsbD. Biochemistry 41: 6920–6927. [DOI] [PubMed] [Google Scholar]
- Grauschopf, U., Winther, J.R., Korber, P., Zander, T., Dallinger, P., Bardwell, J.C. 1995. Why is DsbA such an oxidizing disulfide catalyst? Cell 83: 947–955. [DOI] [PubMed] [Google Scholar]
- Guddat, L.W., Bardwell, J.C., Martin, J.L. 1998. Crystal structures of reduced and oxidized DsbA: Investigation of domain motion and thiolate stabilization. Structure 6: 757–767. [DOI] [PubMed] [Google Scholar]
- Hiniker, A. and Bardwell, J.C. 2004. In vivo substrate specificity of periplasmic disulfide oxidoreductases. J. Biol. Chem. 279: 12967–12973. [DOI] [PubMed] [Google Scholar]
- Holmgren, A. 1976. Hydrogen donor system for Escherichia coli ribonucleoside-diphosphate reductase dependent upon glutathione. Proc. Natl. Acad. Sci. 73: 2275–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren, A. 1979a. Glutathione-dependent synthesis of deoxyribonucleotides. Characterization of the enzymatic mechanism of Escherichia coli glutaredoxin. J. Biol. Chem. 254: 3672–3678. [PubMed] [Google Scholar]
- Holmgren, A. 1979b. Glutathione-dependent synthesis of deoxyribonucleotides. Purification and characterization of glutaredoxin from Escherichia coli . J. Biol. Chem. 254: 3664–3671. [PubMed] [Google Scholar]
- Holmgren, A. 1985. Thioredoxin. Annu. Rev. Biochem. 54: 237–271. [DOI] [PubMed] [Google Scholar]
- Holmgren, A. 1989. Thioredoxin and glutaredoxin systems. J. Biol. Chem. 264: 13963–13966. [PubMed] [Google Scholar]
- Holmgren, A. 1995. Thioredoxin structure and mechanism: Conformational changes on oxidation of the active-site sulfhydryls to a disulfide. Structure 3: 239–243. [DOI] [PubMed] [Google Scholar]
- Holmgren, A. 2000. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2: 811–820. [DOI] [PubMed] [Google Scholar]
- Holmgren, A. and Fagerstedt, M. 1982. The in vivo distribution of oxidized and reduced thioredoxin in Escherichia coli . J. Biol. Chem. 257: 6926–6930. [PubMed] [Google Scholar]
- Holmgren, A., Soderberg, B.O., Eklund, H., Branden, C.I. 1975. Three-dimensional structure of Escherichia coli thioredoxin-S2 to 2.8 Å resolution. Proc. Natl. Acad. Sci. 72: 2305–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeng, M.F., Campbell, A.P., Begley, T., Holmgren, A., Case, D.A., Wright, P.E., Dyson, H.J. 1994. High-resolution solution structures of oxidized and reduced Escherichia coli thioredoxin. Structure 2: 853–868. [DOI] [PubMed] [Google Scholar]
- Kadokura, H., Katzen, F., Beckwith, J. 2003. Protein disulfide bond formation in prokaryotes. Annu. Rev. Biochem. 72: 111–135. [DOI] [PubMed] [Google Scholar]
- Kallis, G.B. and Holmgren, A. 1980. Differential reactivity of the functional sulfhydryl groups of cysteine-32 and cysteine-35 present in the reduced form of thioredoxin from Escherichia coli . J. Biol. Chem. 255: 10261–10265. [PubMed] [Google Scholar]
- Katti, S.K., LeMaster, D.M., Eklund, H. 1990. Crystal structure of thioredoxin from Escherichia coli at 1.68 Å resolution. J. Mol. Biol. 212: 167–184. [DOI] [PubMed] [Google Scholar]
- Kelley, J.J. III and Bushweller, J.H. 1998. 1H, 13C and 15N NMR resonance assignments of vaccinia glutaredoxin-1 in the fully reduced form. J. Biomol. NMR 12: 353–355. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T., Kishigami, S., Sone, M., Inokuchi, H., Mogi, T., Ito, K. 1997. Respiratory chain is required to maintain oxidized states of the DsbA–DsbB disulfide bond formation system in aerobically growing Escherichia coli cells. Proc. Natl. Acad. Sci. 94: 11857–11862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon, B.W. and Williams, C.H. Jr. 1996. Enzyme-monitored turnover of Escherichia coli thioredoxin reductase: Insights for catalysis. Biochemistry 35: 4704–4712. [DOI] [PubMed] [Google Scholar]
- Lillig, C.H., Prior, A., Schwenn, J.D., Aslund, F., Ritz, D., Vlamis-Gardikas, A., Holmgren, A. 1999. New thioredoxins and glutaredoxins as electron donors of 3′-phosphoadenylylsulfate reductase. J. Biol. Chem. 274: 7695–7698. [DOI] [PubMed] [Google Scholar]
- Mannervik, B. and Danielson, U.H. 1988. Glutathione transferases—Structure and catalytic activity. CRC Crit. Rev. Biochem. 23: 283–337. [DOI] [PubMed] [Google Scholar]
- Martin, J.L., Bardwell, J.C., Kuriyan, J. 1993. Crystal structure of the DsbA protein required for disulphide bond formation in vivo. Nature 365: 464–468. [DOI] [PubMed] [Google Scholar]
- Masai, E., Ichimura, A., Sato, Y., Miyauchi, K., Katayama, Y., Fukuda, M. 2003. Roles of the enantioselective glutathione S-transferases in cleavage of β-aryl ether. J. Bacteriol. 185: 1768–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masip, L., Pan, J.L., Haldar, S., Penner-Hahn, J.E., DeLisa, M.P., Georgiou, G., Bardwell, J.C., Collet, J.F. 2004. An engineered pathway for the formation of protein disulfide bonds. Science 303: 1185–1189. [DOI] [PubMed] [Google Scholar]
- McCarthy, A.A., Haebel, P.W., Torronen, A., Rybin, V., Baker, E.N., Metcalf, P. 2000. Crystal structure of the protein disulfide bond isomerase, DsbC, from Escherichia coli . Nat. Struct. Biol. 7: 196–199. [DOI] [PubMed] [Google Scholar]
- Missiakas, D., Georgopoulos, C., Raina, S. 1993. Identification and characterization of the Escherichia coli gene dsbB, whose product is involved in the formation of disulfide bonds in vivo. Proc. Natl. Acad. Sci. 90: 7084–7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, H. 2005. Thioredoxin and its related molecules: Update 2005. Antioxid. Redox Signal. 7: 823–828. [DOI] [PubMed] [Google Scholar]
- Nishida, M., Harada, S., Noguchi, S., Satow, Y., Inoue, H., Takahashi, K. 1998. Three-dimensional structure of Escherichia coli glutathione S-transferase complexed with glutathione sulfonate: Catalytic roles of Cys10 and His106. J. Mol. Biol. 281: 135–147. [DOI] [PubMed] [Google Scholar]
- Poole, L.B. 1996. Flavin-dependent alkyl hydroperoxide reductase from Salmonella typhimurium. 2. Cystine disulfides involved in catalysis of peroxide reduction. Biochemistry 35: 65–75. [DOI] [PubMed] [Google Scholar]
- Poole, L.B., Godzik, A., Nayeem, A., Schmitt, J.D. 2000. AhpF can be dissected into two functional units: Tandem repeats of two thioredoxin-like folds in the N-terminus mediate electron transfer from the thioredoxin reductase-like C-terminus to AhpC. Biochemistry 39: 6602–6615. [DOI] [PubMed] [Google Scholar]
- Reynolds, C.M. and Poole, L.B. 2000. Attachment of the N-terminal domain of Salmonella typhimurium AhpF to Escherichia coli thioredoxin reductase confers AhpC reductase activity but does not affect thioredoxin reductase activity. Biochemistry 39: 8859–8869. [DOI] [PubMed] [Google Scholar]
- Rietsch, A., Belin, D., Martin, N., Beckwith, J. 1996. An in vivo pathway for disulfide bond isomerization in Escherichia coli . Proc. Natl. Acad. Sci. 93: 13048–13053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietsch, A., Bessette, P., Georgiou, G., Beckwith, J. 1997. Reduction of the periplasmic disulfide bond isomerase, DsbC, occurs by passage of electrons from cytoplasmic thioredoxin. J. Bacteriol. 179: 6602–6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz, D., Lim, J., Reynolds, C.M., Poole, L.B., Beckwith, J. 2001. Conversion of a peroxiredoxin into a disulfide reductase by a triplet repeat expansion. Science 294: 158–160. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Manzaneque, M.T., Ros, J., Cabiscol, E., Sorribas, A., Herrero, E. 1999. Grx5 glutaredoxin plays a central role in protection against protein oxidative damage in Saccharomyces cerevisiae . Mol. Cell. Biol. 19: 8180–8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segatori, L., Murphy, L., Arredondo, S., Kadokura, H., Gilbert, H., Beckwith, J., Georgiou, G. 2006. Conserved role of the linker α-helix of the bacterial disulfide isomerase DsbC in the avoidance of misoxidation by DsbB. J. Biol . Chem. 281: 4911–4919. [DOI] [PubMed] [Google Scholar]
- Shi, J., Vlamis-Gardikas, A., Aslund, F., Holmgren, A., Rosen, B.P. 1999. Reactivity of glutaredoxins 1, 2, and 3 from Escherichia coli shows that glutaredoxin 2 is the primary hydrogen donor to ArsC-catalyzed arsenate reduction. J. Biol. Chem. 274: 36039–36042. [DOI] [PubMed] [Google Scholar]
- Stafford, S.J., Humphreys, D.P., Lund, P.A. 1999. Mutations in dsbA and dsbB, but not dsbC, lead to an enhanced sensitivity of Escherichia coli to Hg2+and Cd2+ . FEMS Microbiol. Lett. 174: 179–184. [DOI] [PubMed] [Google Scholar]
- Stewart, E.J., Aslund, F., Beckwith, J. 1998. Disulfide bond formation in the Escherichia coli cytoplasm: An in vivo role reversal for the thioredoxins. EMBO J. 17: 5543–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao, K. 1997. oxyR-dependent induction of Escherichia coli grx gene expression by peroxide stress. J. Bacteriol. 179: 5967–5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlamis-Gardikas, A., Aslund, F., Spyrou, G., Bergman, T., Holmgren, A. 1997. Cloning, overexpression, and characterization of glutaredoxin 2, an atypical glutaredoxin from Escherichia coli . J. Biol. Chem. 272: 11236–11243. [DOI] [PubMed] [Google Scholar]
- Vuilleumier, S. 1997. Bacterial glutathione S-transferases: What are they good for? J. Bacteriol. 179: 1431–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, K.W. and Gilbert, H.F. 1997. Scanning and escape during protein–disulfide isomerase-assisted protein folding. J. Biol. Chem. 272: 8845–8848. [DOI] [PubMed] [Google Scholar]
- Wood, Z.A., Poole, L.B., Karplus, P.A. 2001. Structure of intact AhpF reveals a mirrored thioredoxin-like active site and implies large domain rotations during catalysis. Biochemistry 40: 3900–3911. [DOI] [PubMed] [Google Scholar]
- Wood, Z.A., Schroder, E., Robin Harris, J., Poole, L.B. 2003. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 28: 32–40. [DOI] [PubMed] [Google Scholar]
- Xie, T., Yu, L., Bader, M.W., Bardwell, J.C., Yu, C.A. 2002. Identification of the ubiquinone-binding domain in the disulfide catalyst disulfide bond protein B. J. Biol. Chem. 277: 1649–1652. [DOI] [PubMed] [Google Scholar]
- Zapun, A., Bardwell, J.C., Creighton, T.E. 1993. The reactive and destabilizing disulfide bond of DsbA, a protein required for protein disulfide bond formation in vivo. Biochemistry 32: 5083–5092. [DOI] [PubMed] [Google Scholar]
- Zheng, M., Aslund, F., Storz, G. 1998. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science 279: 1718–1721. [DOI] [PubMed] [Google Scholar]