

Abstract
The phosphoenolpyruvate-dependent carbohydrate transport system (PTS) couples uptake with phosphorylation of a variety of carbohydrates in prokaryotes. In this multienzyme complex, the enzyme II (EII), a carbohydrate-specific permease, is constituted of two cytoplasmic domains, IIA and IIB, and a transmembrane channel IIC domain. Among the five families of EIIs identified in Escherichia coli, the galactitol-specific transporter (IIgat) belongs to the glucitol family and is structurally the least well-characterized. Here, we used nuclear magnetic resonance (NMR) spectroscopy to solve the three-dimensional structure of the IIB subunit (GatB). GatB consists of a central four-stranded parallel β-sheet flanked by α-helices on both sides; the active site cysteine of GatB is located at the beginning of an unstructured loop between β1 and α1 that folds into a P-loop-like structure. This structural arrangement shows similarities with other IIB subunits but also with mammalian low molecular weight protein tyrosine phosphatases (LMW PTPase) and arsenate reductase (ArsC). An NMR titration was performed to identify the GatA-interacting residues.
Keywords: phosphotransferase system, GatB, structure determination, NMR, galactitol
In Gram-negative species, polar solutes and charged molecules have a very low rate of passive flux across the lipid bilayer that makes up the cytoplasmic membrane. Many of these are transported by proteins embedded in the lipid bilayer of the cytoplasmic membrane that bind specific molecules and carry them into or out of the cell (Nikaido and Saier 1992). Three different transport mechanisms are present: (1) facilitated diffusion, which is not a widely used strategy in prokaryotes; (2) active transport driven by ATP or ion-gradient; and (3) group translocation in which the substrate is metabolized as it is transported. An example of the latter metabolism is the phosphoenolpyruvate-dependent phosphotransferase system (PTS) (Meadow et al. 1990; Siebold et al. 2001; Tchieu et al. 2001). The phosphoryl group of phosphoenolpyruvate (PEP) is transferred sequentially through the two general cytosolic proteins, enzyme I (EI) and histidine carrier protein (HPr), to a sugar-specific enzyme II (EII) complex that catalyzes phosphorylation and import of a carbohydrate (mostly hexoses or hexitols). This system is also involved in a signaling cascade that regulates transport, metabolism, and chemotaxis through phosphorylation/dephosphorylation of other proteins or allosteric regulation via protein/protein interactions (Postma et al. 1993; Titgemeyer and Hillen 2002).
On the basis of their amino acid sequence similarity, the different EII's have been divided into five main families: glucose (glc)/sucrose (scr), mannitol (mtl)/fructose (fru), lactose (lac)/cellobiose (cel), mannose (man)/sorbose (sor), and glucitol (gut)/galactitol (gat) (Tchieu et al. 2001). The sequence identity between EII's within one family is at least 25% while the similarities between members of different families are very low mainly confined to a few motifs (Postma et al. 1993). The galactitol (dulcitol)-specific transporter of Escherichia coli (enzyme IIgat complex) consists of three polypeptide chains two cytoplasmic chains, GatA (IIAgat) and GatB (IIBgat), and a transmembrane protein GatC (IICgat) (Nobelmann and Lengeler 1996). GatA and GatC show only limited sequence similarity to the IIA and IIC proteins of the mannitol/fructose permease, while GatB shows ∼45% similarity to the lactose/cellobiose-specific IIB subunit.
Structural information on the IIA and IIB domains are now available for almost all members of the five EII families and each of them shows completely different folds (Siebold et al. 2001). For the central IIB domains of EII's, they are all characterized by α/β structures. Also, several protein–protein complexes from the PTS pathways have been elucidated: EI and HPr (Garrett et al. 1999), HPr and IIAglc (Wang et al. 2000), HPr and IIAmtl (Cornilescu et al. 2002), and IIAglc and IIBglc (Cai et al. 2003). In general, the protein interface requires only minor conformational changes between the free and the bound states. These studies also reveal that the binding surface on each protein is similar for all its partners from PTS (Gemmecker et al. 1997; Peterkofsky et al. 2001) as well as non-PTS carbohydrate transport (Hurley et al. 1993). Here, we describe the NMR structure of the IIB enzyme from the glucitol/galactitol family and its interaction with its partner GatA.
Results and Discussion
Overall fold
The structure of the GatB (IIBgat) protein is composed of a central four-stranded parallel open twisted β-sheet, which is flanked by three α-helices (α1, α3, α4) on the concave side and one (α2) on the convex side (Fig. 1B). GatB has a well-defined tertiary structure except for the two loops between β1 and α1 and between β3 and β4 that both lack secondary structures (Fig. 1A). This correlates with the small number of medium and long range NOEs for these portions of the molecule and with low hNOEs values, especially for the loop β1-α1 (Supplemental data). This latter loop corresponds to the active site loop containing the phosphorylation site on residue Cys9 (Fig. 1A). The root-mean-square atomic deviation (RMSD), which is 0.90 Å for the entire protein (residues 2–94), drops to 0.58 Å when the two loops β1–α1 and β3–β4 are excluded. As expected, the quality of the Ramachandran plot is lower for these loops (Table 1). The geometric and energetic statistics for the 20 final GatB structures are given in Table 1.
Figure 1.

Structure of GatB. (A) Stereopair view of the 20 lowest energy structures superimposed on the backbone atoms (N, Cα, C atoms) of residues 2–9, 14–58, and 66–94. (B) Ribbon representation of the average structure. Position of the phosphorylation site Cys9, the termini, and secondary structures are labeled.
Table 1.
Structural statistics of the 20 best structures of GatB

Comparison with cellobiose-specific enzyme IIB and other proteins
A search for protein structures with folds similar to GatB was performed using the DALI server (Holm and Sander 1994). The DALI search revealed 43 candidates with Z-scores above 5.0. Structural comparison with other IIB enzymes suggests that GatB is very close to the IIB domain of the cellobiose- and mannitol-specific enzymes (Fig. 2B) although no significant sequence identity is apparent (Fig. 2A). One can notice that, except for the α-helix between β3 and β4, all the other secondary structures are conserved. Two other proteins that share the same reaction mechanism involving a phosphocysteine intermediate had high DALI Z-scores. These are the bovine low molecular weight phosphotyrosyl phosphatase (LMW PTPase) (Zhang et al. 1997) and the arsenate reductase (ArsC) from Staphylococcus aureus (Zegers et al. 2001) with Z-scores of 6.4 and 5.8, respectively. Along with IIBcel and IIBmtl, these are all members of the PTPase I superfamily. We therefore suggest including GatB in this superfamily of proteins, which all exhibit a very similar motif comprising the central β-sheets and the first α-helix. As suggested by van Montfort et al. (1997), the macrodipole of this first helix stabilizes the phosphocysteine intermediate.
Figure 2.
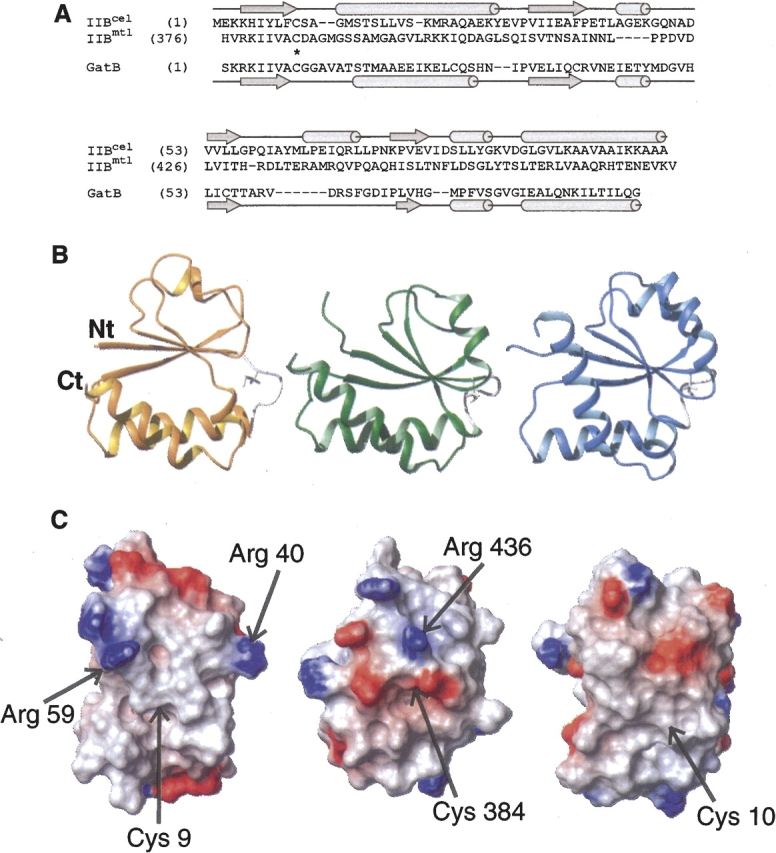
(A) Sequence alignment of GatB with IIBcel and IIBmtl. Sequences were aligned according to their secondary structures (at the top for IIBcel and IIBmtl, and at the bottom for GatB). Gaps and the active residue Cys9 are indicated on sequences with dashes and asterisks, respectively. (B,C) Structural comparison of GatB (left), IIBmtl (middle; PDB code 1VKR) (Legler et al. 2004), and IIBcel (right; PDB code 1IIB) (van Montfort et al. 1997). The IIBmtl and IIBcel structures differ from GatB by an RMSD of ∼2.5 Å. (B) Ribbon representation. The side chain of the active site Cys is shown in white. (C) Potential map of the surfaces around the active sites, calculated with MOLMOL. The surfaces are color coded, with red indicating net negative electrostatic potential and blue indicating positive potential. An arrow shows the position of the cysteine. The orientations of molecules in B and C are related by a 90° rotation along a vertical axis.
Different periplasmic sugar-binding proteins were also found to be structurally close to GatB. This is the case for the d-ribose-, galactose-, and l-arabinose-binding proteins with Z-scores of 5.5, 5.1, and 5.1, respectively. These proteins are characterized by two similar structural domains with the sugar binding site located close to the first loop between β1 and α1, as in GatB, in a deep cleft between the two domains.
Finally, GatB appears remarkably similar to the fold of CheY (Z-score of 6.7), a protein that functions in the chemotaxis signal transduction pathway of E. coli (Armitage 1999). This transduction system is linked to the PTS through EI, which in its unphosphorylated state binds and inhibits the chemotaxis kinase CheA to control CheY phosphorylation in response to changes in sugar transport and metabolism (Grebe and Stock 1998). Structural similarities were found between different members of each system, such as the CheY-binding domain of CheA and HPr (McEvoy et al. 1996), and the N-terminal phosphorylation site domain (P1) of CheA that interacts with CheY (Mourey et al. 2001). These considerations support the idea of an evolutionary relationship between the PTS and chemotaxis pathways.
The active site and the phosphotransfer reaction
As for most IIB enzymes, the phosphorylation site on GatB is located on a cysteine residue (Cys9) at the N-terminal end of the first loop. This residue receives the phosphoryl group from GatA and transfers it to the galactitol when bound at the catalytic site of GatC. In the PTPase superfamily, the cysteine is part of an anion-binding motif, referred to as the P-loop (Evans et al. 1996). This loop shares a common Cys-X5-Arg-(Ser/Thr) motif in members of the PTPase family. In the case of IIBmtl, IIBcel, and GatB, the P-loop lacks the arginine residue (Fig. 2A). This arginine was proposed to contribute to phosphate binding and to promote stabilization of the negatively charged thiolate (Barford 1995). Quantitative differences in electrostatic interactions occurring with the active-site residues were more recently studied for different PTPases (Dillet et al. 2000). Similarities were found around the P-loop and the nearby α-helix dipoles, while differences were mainly observed for residues outside the active site.
Comparing the active-site loop of these three IIB enzymes, a very different electrostatic charge distribution is observed (Fig. 2C). For IIBcel, which shows no charges near the catalytic cysteine, it was suggested that residues located on IIA and IIC were sufficient to stabilize the two phosphotransfer transition states, IIA-IIB and IIB-IIC (van Montfort et al. 1997). IIBmtl protein has a number of negatively-charged residues but also one positive residue, Arg346. Finally, GatB exhibits a very hydrophobic environment around this site, plus two positively-charged residues (Arg40 and Arg59). In the IIBglc subunit, Arg38 and Arg40 were found to stabilize the penta-coordinated transition state of the phosphoryl transfer reaction or the phosphoryl group covalently bound to the cysteine (Eberstadt et al. 1996). They could also participate in the transfer of the phosphate to galactitol. Thus, even if the fold of GatB is very close to the IIBcel and IIBmtl proteins, the activation and stabilization of the catalytic cysteine appears to be closer to that of the IIBglc family. This explains the observation that different families of EII proteins cannot complement each other. Only IIAman from E. coli was able to be functionally replaced by IIAlev from B. subtilis in a mannose-specific system (Schauder et al. 1998) since both belong to the same family.
Another characteristic of the P-loop is its flexibility. In the 1H-15N HSQC spectrum of GatB, the signals of residues Cys9 to Gly11 are broadened, which is likely the result of conformational flexibility. Similar conformational rearrangements were observed for ArsC (Messens et al. 2002), LMW PTPase (Zhang et al. 1997), and IIBcel (Ab et al. 1997). When they are not bound to their ligand, the P-loop of these molecules is flexible, but it adopts a more rigid conformation when bound to ligand. Similarly, phosphorylation of Cys35 on IIBglc leads to an increase of the amide cross-peak intensities of three residues located in the active site probably due to the formation of hydrogen bonds with the phosphoryl group (Gemmecker et al. 1997). Interestingly, a second mobile loop was observed in GatB between β3 and β4. This is unique with regard to the other members of the PTPase family which usually present a well-ordered α-helix at that place.
NMR study of the GatB/GatA complex
The binding site of the GatB protein upon complex formation was identified by NMR chemical shift perturbation analysis (Fig. 3; Supplemental Material). Residues near the active site Cys9 and neighboring loops had the largest chemical shift changes upon GatA addition. These results identify loop 1 (between β1 and α1), the N terminus of α1, loop 3 (between β2 and α2), loop 5 (between β3 and β4), and α3 as the primary GatA binding sites. Complex formation was in fast exchange on the NMR time-scale (data not shown). From line shape analysis of exchange broadened resonances such as Thr15, we estimate that the lifetime of the complex is on the order of 2–5 msec at 298 K. The binding affinity was measured by isothermal titration calorimetry to be 14 μM.
Figure 3.

(A) Bar plot of chemical shift deviations between free and bound forms. Analysis of ligand-induced shifts (in ppm) was performed by using a weighted chemical shift index: Δ = [(δ 1H)2 + (0.2 * δ 15N)2]1/2 where δ 1H and δ 15N are the chemical shift differences of the amide proton and nitrogen (Shuker et al. 1996). An asterisk (on residue Met73) indicates the presence of severe broadening during the NMR titration that prevented quantification of the chemical shift change. The location of secondary structural elements is shown. (B) Distribution of chemical shift changes on the structure of GatB. The N and C termini and the active-site residue, Cys9, are indicated.
In conclusion, we have determined the structure of GatB and found that it is structurally similar to IIBmtl and IIBcel, with the presence of a P-loop-like structure, despite low sequence similarity. Differences in the electrostatic map around the catalytic site suggest a completely different mode of action for both heterodimerization with the IIA and/or IIC subunits and for the phosphoryl transfer to the carbohydrate. Structural analyses of GatA alone and in complex with its immediate upstream (HPr) and downstream (GatB) partners are currently in progress in order to better understand the atomic mechanisms that govern phosphoryl transfer between the different PTS proteins.
Materials and methods
Cloning, expression, preparation, and purification of recombinant proteins GatA and GatB
Protein expression constructs include an N-terminal His8 tag and a thrombin cleavage site. GatA was cloned from O157 strain E. coli using standard PCR amplification and constructed with the primers 5′ Nde1 and 3′ BamH1. The PCR product was ligated into pET15b (Novagen). GatB was ligated into a pHISTEV vector using BamH1-EcoR1 restriction sites.
Transformed BL21 (DE3) E. coli were grown at 37°C in either Luria Bertani (LB) or M9 minimal medium for preparing 15N- and 15N/13C-labeled protein. Protein expression was induced at 30°C for 4 h by addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). After lysis, the cleared lysate was applied to Ni2+-loaded chelating sepharose (Amersham Pharmacia Biotech) and the His-tagged proteins were eluted with imidazole using standard protocols. After dialysis in 100 mM sodium phosphate buffer (pH 6.8), 75 mM NaCl, 75 mM KCl, and 2 mM dithio-1,4-threitol (DTT), proteins were purified to homogeneity by a further gel-filtration step (Hiload 16/60 Superdex 75 column, Amersham Pharmacia Biotech). The purity of the final products was verified on SDS-polyacrylamide gel electrophoresis (stained with Coomassie Blue) and a single protein band was obtained. The N-terminal His-tag was cleaved from GatA, but not GatB, which becomes unstable when the His-tag is removed, by treatment for 24 h at room temperature with thrombin (Amersham Pharmacia Biotech) at 1 unit per milligram of fusion protein. Benzamidine sepharose was used to remove thrombin. The extra N-terminal residues in GatB were numbered −19 to −1. About 3–4 mg of labeled protein was obtained per liter of culture. The molecular weights of both proteins were confirmed by mass spectrometry.
NMR spectroscopy
GatB was prepared in the concentration range of 0.8–1.0 mM in 100 mM sodium phosphate (pH 6.8), 75 mM NaCl, 75 mM KCl, 1 mM dithio-1,4-threitol (DTT), and 1 mM NaN3. NMR experiments were performed at 298 K and 303 K on a Bruker 600 MHz AVANCE spectrometer equipped with a triple-resonance inverse cryoprobe head. NMR spectra were processed using XWIN-NMR (Bruker Biospin) and NMRPipe software (Delaglio et al. 1995), and analyzed with Sparky (T.D. Goddard and D.G. Kneller, Sparky 3, University of California, San Francisco).
Sequential backbone assignments were accomplished for GatB using 3D HNCACB (Wittekind and Mueller 1993), CBCA(CO)NH (Grzesiek and Bax 1992a) and HNCO (Grzesiek and Bax 1992b) experiments. Only the backbone amides of Met(−19)–His(−8) segment were not assigned due to exchange broadening. The majority of aliphatic side-chain proton and carbon resonances were obtained using HCCH TOCSY (Kay et al. 1993) while aromatic side-chain protons were assigned from the 2D homonuclear NOESY (τm = 100 msec) and the 1H-13C HSQC recorded in D2O. The assigned chemical shift lists have been deposited in the BioMagResBank (http://www.bmrb.wisc.edu) under accession number 6259. To measure the heteronuclear 15N{1H} nuclear Overhauser effect, two-dimensional spectra were recorded with and without NOE enhancement by 5 sec of 1H presaturation (Peng and Wagner 1994). 1H-15N residual dipolar couplings (RDC) were determined from comparison of the 1 J NH splittings of 15N-labeled GatB in an isotropic medium and an aligned medium containing ∼4 mg of Pf1 phage/mL using the IPAP-HSQC pulse sequence (Ottiger et al. 1998).
Structure calculations of GatB
NOE restraints were obtained from three different NOESY spectra (τm = 100 msec): (1) a 15N-edited three-dimensional NOESY experiment, (2) the 1H-13C region of a 3D 13C/15N-edited NOESY (Sattler et al. 1995), and (3) a two-dimensional homonuclear NOESY spectrum. The two latter spectra were recorded in D2O. Interproton distances were defined as previously described (Volpon et al. 2003). The distribution of restraints by range is summarized in Table 1.
Structures were calculated using standard protocols in CNS version 1.1 (Brunger et al. 1998) with the ensemble of the distance and dihedral restraints described in Table 1. In the final round of calculations, CNS was extended to incorporate 82 RDC restraints for further refinement in torsion angle space. The magnitudes of the axial (Da) and rhombic (R) components of the alignment tensor were determined to be 13 and 0.45, respectively, using standard methods (Clore et al. 1998a,b). Twenty lowest energy structures were selected from the final 100 calculations. None had NOE and dihedral angle violations of >0.5 Å and >5°, respectively. Structural statistics are summarized in Table 1. PROCHECK was used to assess the quality of the structures (Laskowski et al. 1993). Figures were generated using MOLMOL (Koradi et al. 1996). Coordinates have been deposited in the Protein Data Bank (http://www.rcsb.org/pdb/) under accession code 1TVM.
NMR titrations
The titration of labeled GatB (∼0.5 mM) with unlabeled GatA was monitored by a series of two-dimensional 1H-15N HSQC spectra. Solutions were mixed to yield molar ratios (unlabeled/labeled protein) of 0, 0.25, 0.5, 0.75, 1, 1.5, 2, and 3. Assignments of the HSQC signals of 15N-labeled GatB in the complex were made by tracing the fast-exchanging peaks during the titration.
Isothermal titration calorimetry (ITC)
Experiments were carried out on a MicroCal VP-ITC titration calorimeter (MicroCal Inc.) at 15°C in NMR buffer with 0.14 mM GatB and injections of 1.27 mM GatA.
Electronic supplemental material
Supplemental material includes 1H-15N heteronuclear NOE data for GatB, number of NOE restraints per residue used for the NMR structure, and average RMSD plots for each residue of the NMR ensemble. It also includes a superposition of 1H-15N HSQC spectra of uniformly 15N-labeled GatB in the presence of GatA at different molar ratios.
Acknowledgments
We thank Pietro Iannuzzi for providing the GatB clone. This work was supported by a Canadian Institutes of Health Research Genomics grant for the Montreal-Kingston Bacterial Structural Genomics Initiative.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Laurent Volpon, Institut de Recherche en Immunologie et Cancérologie, Université de Montréal, Pavillon Marcelle-Coutu, 2950, Chemin Polytechnique, Montreal, Quebec H3T 1J4, Canada; e-mail: laurent.volpon@umontreal.ca; fax: (514) 343-5839.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062337406.
Abbreviations: GatA, IIA domain of the galactitol specific transporter; GatB, IIB domain of the galactitol specific transporter; PTS, phosphotransferase system; EI, enzyme I; HPr, histidine-containing phosphocarrier protein; EII, enzyme II; LMW PTPase, low molecular weight protein tyrosine phosphatase; PEP, phosphoenolpyruvate; TOCSY, total correlation spectroscopy; NOESY, nuclear Overhauser effect spectroscopy; HSQC, heteronuclear single quantum correlation; hNOE, heteronuclear nuclear Overhauser effect; gat, galactitol; cel, cellobiose; glc, glucose; mtl, mannitol; lac, lactose; man, mannose.
References
- Ab, E., Schuurman-Wolters, G., Reizer, J., Saier, M.H., Dijkstra, K., Scheek, R.M., Robillard, G.T. 1997. The NMR side-chain assignments and solution structure of enzyme IIBcellobiose of the phosphoenolpyruvate-dependent phosphotransferase system of Escherichia coli . Protein Sci. 6: 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage, J.P. 1999. Bacterial tactic responses. Adv. Microb. Physiol. 41: 229–289. [DOI] [PubMed] [Google Scholar]
- Barford, D. 1995. Protein phosphatases. Curr. Opin. Struct. Biol. 5: 728–734. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S. et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Cai, M., Williams, D.C. Jr., Wang, G., Lee, B.R., Peterkofsky, A., Clore, G.M. 2003. Solution structure of the phosphoryl transfer complex between the signal-transducing protein IIAGlucose and the cytoplasmic domain of the glucose transporter IICBGlucose of the Escherichia coli glucose phosphotransferase system. J. Biol. Chem. 278: 25191–25206. [DOI] [PubMed] [Google Scholar]
- Clore, G.M., Gronenborn, A.M., Bax, A. 1998a. A robust method for determining the magnitude of the fully asymmetric alignment tensor of oriented macromolecules in the absence of structural information. J. Magn. Reson. 133: 216–221. [DOI] [PubMed] [Google Scholar]
- Clore, G.M., Gronenborn, A.M., Tjandra, N. 1998b. Direct structure refinement against residual dipolar couplings in the presence of rhombicity of unknown magnitude. J. Magn. Reson. 131: 159–162. [DOI] [PubMed] [Google Scholar]
- Cornilescu, G., Delaglio, F., Bax, A. 1999. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13: 289–302. [DOI] [PubMed] [Google Scholar]
- Cornilescu, G., Lee, B.R., Cornilescu, C.C., Wang, G., Peterkofsky, A., Clore, G.M. 2002. Solution structure of the phosphoryl transfer complex between the cytoplasmic A domain of the mannitol transporter IIMannitol and HPr of the Escherichia coli phosphotransferase system. J. Biol. Chem. 277: 42289–42298. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- Dillet, V., Van Etten, R.L., Bashford, D. 2000. Stabilization of charges and protonation states in the active site of the protein tyrosine phosphatases: A computational study. J. Phys. Chem. B 104: 11321–11333. [Google Scholar]
- Eberstadt, M., Grdadolnik, S.G., Gemmecker, G., Kessler, H., Buhr, A., Erni, B. 1996. Solution structure of the IIB domain of the glucose transporter of Escherichia coli . Biochemistry 35: 11286–11292. [DOI] [PubMed] [Google Scholar]
- Evans, B., Tishmack, P.A., Pokalsky, C., Zhang, M., Van Etten, R.L. 1996. Site-directed mutagenesis, kinetic, and spectroscopic studies of the P-loop residues in a low molecular weight protein tyrosine phosphatase. Biochemistry 35: 13609–13617. [DOI] [PubMed] [Google Scholar]
- Garrett, D.S., Seok, Y.J., Peterkofsky, A., Gronenborn, A.M., Clore, G.M. 1999. Solution structure of the 40,000 Mr phosphoryl transfer complex between the N-terminal domain of enzyme I and HPr. Nat. Struct. Biol. 6: 166–173. [DOI] [PubMed] [Google Scholar]
- Gemmecker, G., Eberstadt, M., Buhr, A., Lanz, R., Grdadolnik, S.G., Kessler, H., Erni, B. 1997. Glucose transporter of Escherichia coli: NMR characterization of the phosphocysteine form of the IIB(Glc) domain and its binding interface with the IIA(Glc) subunit. Biochemistry 36: 7408–7417. [DOI] [PubMed] [Google Scholar]
- Grebe, T.W. and Stock, J. 1998. Bacterial chemotaxis: The five sensors of a bacterium. Curr. Biol. 8: R154–R157. [DOI] [PubMed] [Google Scholar]
- Grzesiek, S. and Bax, A. 1992a. Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J. Am. Chem. Soc. 114: 6291–6293. [Google Scholar]
- ——— 1992b. Improved 3D triple-resonance NMR techniques applied to a 31 kDa protein. J. Magn. Reson. 96: 432–440. [Google Scholar]
- Holm, L. and Sander, C. 1994. The FSSP database of structurally aligned protein fold families. Nucleic Acids Res. 22: 3600–3609. [PMC free article] [PubMed] [Google Scholar]
- Hurley, J.H., Faber, H.R., Worthylake, D., Meadow, N.D., Roseman, S., Pettigrew, D.W., Remington, S.J. 1993. Structure of the regulatory complex of Escherichia coli IIIGlc with glycerol kinase. Science 259: 673–677. [PubMed] [Google Scholar]
- Kay, L.E., Xu, G.Y., Singer, A.U., Muhandiram, D.R., Form-Kay, J.D. 1993. A gradient-enhanced HCCH-TOCSY experiment for recording side-chain 1H and 13C correlations in H2O samples of proteins. J. Magn. Reson. B 101: 333–337. [Google Scholar]
- Koradi, R., Billeter, M., Wuthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14: 51–55. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Legler, P.M., Cai, M., Peterkofsky, A., Clore, G.M. 2004. Three-dimensional solution structure of the cytoplasmic B domain of the mannitol transporter IIMannitol of the Escherichia coli phosphotransferase system. J. Biol. Chem. 279: 39115–39121. [DOI] [PubMed] [Google Scholar]
- McEvoy, M.M., Muhandiram, D.R., Kay, L.E., Dahlquist, F.W. 1996. Structure and dynamics of a CheY-binding domain of the chemotaxis kinase CheA determined by nuclear magnetic resonance spectroscopy. Biochemistry 35: 5633–5640. [DOI] [PubMed] [Google Scholar]
- Meadow, N.D., Fox, D.K., Roseman, S. 1990. The bacterial phosphoenolpyruvate: Glycose phosphotransferase system. Annu. Rev. Biochem. 59: 497–542. [DOI] [PubMed] [Google Scholar]
- Messens, J., Martins, J.C., Brosens, E., Van Belle, K., Jacobs, D.M., Willem, R., Wyns, L. 2002. Kinetics and active site dynamics of Staphylococcus aureus arsenate reductase. J. Biol. Inorg. Chem. 7: 146–156. [DOI] [PubMed] [Google Scholar]
- Mourey, L., Da Re, S., Pedelacq, J.D., Tolstykh, T., Faurie, C., Guillet, V., Stock, J.B., Samama, J.P. 2001. Crystal structure of the CheA histidine phosphotransfer domain that mediates response regulator phosphorylation in bacterial chemotaxis. J. Biol. Chem. 276: 31074–31082. [DOI] [PubMed] [Google Scholar]
- Nikaido, H. and Saier, M.H. Jr. 1992. Transport proteins in bacteria: Common themes in their design. Science 258: 936–942. [DOI] [PubMed] [Google Scholar]
- Nobelmann, B. and Lengeler, J.W. 1996. Molecular analysis of the gat genes from Escherichia coli and of their roles in galactitol transport and metabolism. J. Bacteriol. 178: 6790–6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottiger, M., Delaglio, F., Bax, A. 1998. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson. 131: 373–378. [DOI] [PubMed] [Google Scholar]
- Peng, J.W. and Wagner, G. 1994. Investigation of protein motions via relaxation measurements. Methods Enzymol. 239: 563–596. [DOI] [PubMed] [Google Scholar]
- Peterkofsky, A., Wang, G., Garrett, D.S., Lee, B.R., Seok, Y.J., Clore, G.M. 2001. Three-dimensional structures of protein-protein complexes in the E. coli PTS. J. Mol. Microbiol. Biotechnol. 3: 347–354. [PubMed] [Google Scholar]
- Postma, P.W., Lengeler, J.W., Jacobson, G.R. 1993. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 57: 543–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler, M., Maurer, M., Schleucher, J., Griesinger, C. 1995. A simultaneous 15N,1H- and 13C,1H–HSQC with sensitivity enhancement and a heteronuclear gradient echo. J. Biomol. NMR 5: 97–102. [DOI] [PubMed] [Google Scholar]
- Schauder, S., Nunn, R.S., Lanz, R., Erni, B., Schirmer, T. 1998. Crystal structure of the IIB subunit of a fructose permease (IIBLev) from Bacillus subtilis . J. Mol. Biol. 276: 591–602. [DOI] [PubMed] [Google Scholar]
- Shuker, S.B., Hajduk, P.J., Meadows, R.P., Fesik, S.W. 1996. Discovering high-affinity ligands for proteins: SAR by NMR. Science 274: 1531–1534. [DOI] [PubMed] [Google Scholar]
- Siebold, C., Flukiger, K., Beutler, R., Erni, B. 2001. Carbohydrate transporters of the bacterial phosphoenolpyruvate:sugar phosphotransferase system (PTS). FEBS Lett. 504: 104–111. [DOI] [PubMed] [Google Scholar]
- Tchieu, J.H., Norris, V., Edwards, J.S., Saier, M.H. Jr. 2001. The complete phosphotranferase system in Escherichia coli . J. Mol. Microbiol. Biotechnol. 3: 329–346. [PubMed] [Google Scholar]
- Titgemeyer, F. and Hillen, W. 2002. Global control of sugar metabolism: A Gram-positive solution. Antonie Van Leeuwenhoek 82: 59–71. [PubMed] [Google Scholar]
- van Montfort, R.L., Pijning, T., Kalk, K.H., Reizer, J., Saier, M.H. Jr., Thunnissen, M.M., Robillard, G.T., Dijkstra, B.W. 1997. The structure of an energy-coupling protein from bacteria, IIBcellobiose, reveals similarity to eukaryotic protein tyrosine phosphatases. Structure 5: 217–225. [DOI] [PubMed] [Google Scholar]
- Volpon, L., Lievre, C., Osborne, M.J., Gandhi, S., Iannuzzi, P., Larocque, R., Cygler, M., Gehring, K., Ekiel, I. 2003. The solution structure of YbcJ from Escherichia coli reveals a recently discovered αL motif involved in RNA binding. J. Bacteriol. 185: 4204–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, G., Louis, J.M., Sondej, M., Seok, Y.J., Peterkofsky, A., Clore, G.M. 2000. Solution structure of the phosphoryl transfer complex between the signal transducing proteins HPr and IIAGlucose of the Escherichia coli phosphoenolpyruvate:sugar phosphotransferase system. EMBO J. 19: 5635–5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittekind, M. and Mueller, L. 1993. HNCACB, a high sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the α and β carbon resonances in proteins. J. Magn. Reson. B 101: 201–205. [Google Scholar]
- Zegers, I., Martins, J.C., Willem, R., Wyns, L., Messens, J. 2001. Arsenate reductase from S. aureus plasmid pI258 is a phosphatase drafted for redox duty. Nat. Struct. Biol. 8: 843–847. [DOI] [PubMed] [Google Scholar]
- Zhang, M., Zhou, M., Van Etten, R.L., Stauffacher, C.V. 1997. Crystal structure of bovine low molecular weight phosphotyrosyl phosphatase complexed with the transition state analog vanadate. Biochemistry 36: 15–23. [DOI] [PubMed] [Google Scholar]