

Abstract
MRG15 is a transcription factor expressed in a variety of human tissues, and its orthologs have been found in many other eukaryotes which constitute the MRG protein family. It plays a vital role in embryonic development and cell proliferation, and is involved in cellular senescence. The C-terminal part of MRG15 forms a conserved MRG domain which is involved in interactions with the tumor suppressor protein retinoblastoma and a nucleoprotein PAM14 during transcriptional regulation. We report here the characterization of the interaction between the MRG domain of human MRG15 and PAM14 using both yeast two-hybrid and in vitro binding assays based on the crystal structure of the MRG domain. The MRG domain is predominantly hydrophobic, and consists of mainly α-helices that are arranged in a three-layer sandwich topology. The hydrophobic core is stabilized by interactions among a number of conserved hydrophobic residues. The molecular surface is largely hydrophobic, but contains a few hydrophilic patches. Structure-based site-directed mutagenesis studies identified key residues involved in the binding of PAM14. Structural and biochemical data together demonstrate that the PAM14 binding site is consisted of residues Ile160, Leu168, Val169, Trp172, Tyr235, Val268, and Arg269 of MRG15, which form a shallow hydrophobic pocket to interact with the N-terminal 50 residues of PAM14 through primarily hydrophobic interactions. These results provide the molecular basis for the interaction between the MRG domain and PAM14, and reveal insights into the potential biological function of MRG15 in transcription regulation and chromatin remodeling.
Keywords: chromatin remodeling, MRG15, MRG domain, PAM14, protein–protein interaction, transcription regulation
Cellular senescence or replicative senescence is a state of irreversible growth arrest reached by normal cells as a result of replicative exhaustion in culture, and is considered to be an ideal model system for studying the mechanisms of aging and tumor suppression at the cellular level (for review, see Tominaga et al. 2002). Mortality factor on chromosome 4 (MORF4) was first identified in humans as a transcription factor that has the ability to induce replicative senescence in a subset of immortal human cell lines (Bertram et al. 1999). Subsequently, several additional MORF4-related genes were also identified, including MRG15 (M ORF4-related gene on chromosomes 15), MRGX (MORF4-related gene on chromosome X), and four pseudogenes (MRG1, MRG4, MRG5, and MRG11) (Bertram et al. 1999; Bertram and Pereira-Smith 2001). Unlike MORF4, however, MRG15 and MRGX cannot induce replicative senescence when they are introduced into immortal human cell lines. Among these proteins, human MRG15 has drawn more attention because it is ubiquitously expressed in a wide variety of human tissues, and its orthologs have been identified in many eukaryotes from humans to yeasts, which constitute a MRG protein family (Marin and Baker 2000; Bertram and Pereira-Smith 2001).
MRG15 is a transcription factor of 37 kDa, and consists of 323 amino acid residues. It comprises a putative chromo domain at the N terminus (MRG15N, residues 1–85) and a conserved MRG domain at the C terminus (MRG15C, residues 151–323), which are linked together by a flexible region (about residues 86–150) (Bertram and Pereira-Smith 2001). MRG15, MRGX, and MORF4 share a common C-terminal part but a different N-terminal part. MORF4 is equivalent to MRG15C with 96% sequence identity; MRGX has 89% sequence identity with MRG15, but does not contain the N-terminal two-thirds of MRG15N. These differences are likely to be the basis for their functional differences. The exact function of the putative chromo domain of MRG15 is not understood. Nevertheless, biochemical and structural data have shown that proteins containing chromo domains are involved in protein–protein interactions and play important roles in chromatin remodeling that can activate or repress the transcription of a large number of genes (Ball et al. 1997; Brehm et al. 1998; Cavalli and Paro 1998; Akhtar et al. 2000; Jones et al. 2000; Bannister et al. 2001; Lachner et al. 2001; Nielsen et al. 2001). The MRG domain is highly conserved among all MRG proteins, and was predicted to contain several conserved sequence motifs, including a nuclear localization signal, a helix-loop-helix, and a leucine zipper that are commonly found in many transcription factors (Bertram and Pereira-Smith 2001).
Recently, the biological function of MRG15 has been gradually elucidated. Cell biological and biochemical data have shown that, in addition to its involvement in cellular senescence, MRG15 also plays a vital role in embryonic development and cell proliferation. Knockout of MRG15 in mice is embryonic lethal, and exhibits developmental delay (Tominaga et al. 2005). This function of MRG15 is suggested to be most likely through its participation in two nucleoprotein complexes, MAF1 and MAF2 (MRG15-associated factors 1 and 2, respectively), which are involved in chromatin remodeling and transcription regulation (Pardo et al. 2002; Tominaga et al. 2005). MRG15 is shown to interact with hMOF (human male absent on first) in MAF2 through the N-terminal chromo domain (Leung et al. 2001), and with the tumor suppressor protein retinoblastoma (Rb) and the nuclear protein PAM14 (protein associated with MRG15 of 14 kDa) in MAF1, the histone deacetylase (HDAC) containing transcription corepressor mSin3A, and the plant homeodomain zinc finger protein Pf1 through the C-terminal MRG domain (Leung et al. 2001; Yochum and Ayer 2002). In addition, MRG15 is found to be associated with a mammalian TRRAP/Tip60 histone acetyltransferase (HAT) complex through protein MRGBP (MRG15/MRGX binding protein) (Cai et al. 2003). Several MRG15 homologs in other species are also found to be components of multisubunit HAT/HDAC complexes that are involved in transcription regulation through chromatin remodeling (Gorman et al. 1995; Smith et al. 2000; Eisen et al. 2001; Fujita et al. 2002; Nakayama et al. 2003; Morales et al. 2004, 2005; Reid et al. 2004). However, the exact functions of MRG15 and its homologs in these complexes and the underlying molecular mechanisms are still unknown.
Here, we report the crystal structure of the MRG domain of human MRG15 (MRG15C, residues 151–323) at 2.2 Å resolution and the characterization of its interaction with PAM14. After the structure determination and during the functional studies of MRG15C, a crystal structure of MRG15C was reported earlier this year (Bowman et al. 2006). MRG15C was shown to exhibit a structural homology with a DNA-interacting domain of the tyrosine site-specific recombinases XerD, λ integrase, and Cre. Site-directed mutagenesis studies showed that both Val169 and Asn215 are involved in the binding of PAM14. However, it is not clear how PAM14 can interact with the two residues simultaneously because they are located on opposite faces of MRG15C (Bowman et al. 2006). Our MRG15C structure is in agreement with the reported structure. It is composed of mainly α-helices, and is predominantly hydrophobic. The hydrophobic core is stabilized by interactions among a number of conserved hydrophobic residues. The molecular surface is largely hydrophobic, but contains a few hydrophilic patches. Structure-based mutagenesis studies and in vitro binding assays demonstrate that a shallow hydrophobic pocket consisting of residues Ile160, Leu168, Val169, Trp172, Tyr235, Val268, and Arg269 interacts directly with PAM14. Truncation experiments indicate that the N-terminal 50 residues of PAM14 are responsible for its binding with MRG15C. These results further reveal the molecular basis of the interaction between MRG15C and PAM14 and provide insights into the potential biological functions of MRG15 and other MRG proteins in cell development and cellular senescence via chromatin remodeling.
Results
Structure of MRG15C
The crystal structure of MRG15C was solved using the MAD method (Table 1; Fig. 1). There are two MRG15C molecules (A and B) in the asymmetric unit related by a twofold NCS axis. The final model contains residues A156–A203 and A212–A320 of subunit A, residues B156–B204 and B212–B321 of subunit B, and 236 water molecules. The N-terminal five residues (151–155) in both subunits, residues A204–A211, and residues B205–B211 were undefined in the electron density maps. The MRG15C structure reported here is almost identical to that by Bowman et al. (2006). It has a triangular shape of size ∼52 Å × 28 Å × 25 Å, and consists of six long α-helices (α1, α2, α3, α4, α6, and α7), three short α-helices (α5, α8, and α9), and two short β-strands (β1 and β2) (Fig. 1). Helices α1, α2, α3, α6, and α7 form a five-helix bundle and are arranged in a three-layer sandwich topology: helix α1 forms the bottom layer; helices α2 and α3 form the top layer and are in parallel orientation as helix α1; and helices α6 and α7 form the middle layer and are in approximately orthogonal orientation as helix α1. The bottom and top layers are linked together by a short β-strand (β1). Helices α4 and α5 are intercalated between the middle and top layers and cap the helical bundle from one end. The C-terminal region has a relatively extended structure containing two short α-helices (α8 and α9) and a short β-strand (β2), and flanks one side of the helical bundle. MRG15C is predominantly hydrophobic, and the scaffold of the hydrophobic core is stabilized by interactions between a number of highly conserved hydrophobic residues, including Phe225, Leu233–Leu234–Tyr235, Leu267, Leu287–Leu288, Phe294, and Phe306 (Fig. 1D). In addition, a cluster of strictly conserved, charged residues Asp171, Glu238, His262, and Arg265 form a network of salt bridges and hydrogen bonds that also contribute to the stability of the structure.
Table 1.
Summary of diffraction data and structure refinement statistics

Figure 1.
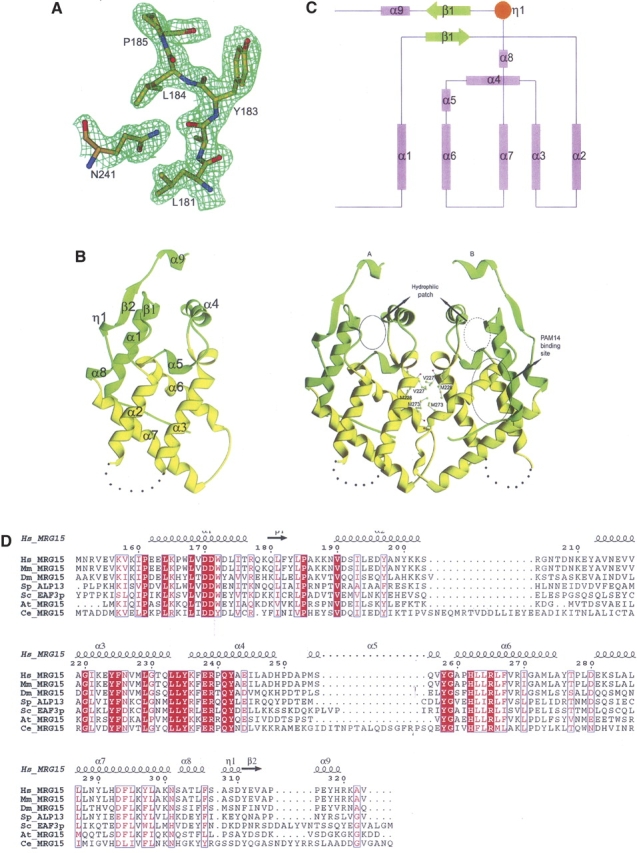
Structure of MRG15C. (A) A representative SIGMMA-weighted 2F o–F c map (1σ contour level) in the C-terminal region of one MRG15C subunit at 2.2 Å resolution. The final coordinates of the structure are shown as a ball-and-stick model. (B) Ribbon diagrams showing the overall structure of MRG15C in monomer (left panel) and homodimer (right panel). The disordered region connecting helices α2 and α3 is indicated with dashed lines. The conserved residues involved in formation of the hydrophobic dimer interface are shown with side chains. The PAM14 binding site and a hydrophilic patch near the C-terminal region are indicated by circles. (C) Secondary structure topology of MRG15C. α-Helices are shown as cylinders; β-strands as arrows. (D) Sequence comparison of the MRG domains between human MRG15 and its homologs. Hs_MRG15, human MRG15; MmMRG15, Mus musculus MRG15; Dm_MRG15, Drosophila melanogaster MRG15; Sp_ALP13, Schizosaccharomyces pombe altered polarity protein 13; Sc_EAF3p, Saccharomyces cerevisiae Esa1p associated factor 3 protein; At_MRG15, Arabidopsis thaliana MRG15; and Ce_MRG15, Caenorhabditis elegans MRG15. Strictly conserved residues are highlighted in shaded red boxes and conserved residues in open red boxes. The secondary structure of the MRG domain of human MRG15 is placed on top of the alignment.
The two MRG15C molecules in the asymmetric unit form a homodimer in a head-to-head fashion (Fig. 1B), which is in agreement with the results of the dynamic light-scattering analysis showing that MRG15C exists as a homodimer in solution as well (data not shown). The dimer interface buries 1097 Å2 (or 12%) of the surface area of each subunit, indicating a moderate subunit–subunit interaction. The dimer interface is mainly formed by helix α3, the C-terminal part of helix α6, the α6–α7 connecting loop, the N-terminal part of helix α7, and a small portion of helix α4 from each subunit. The interface contacts are predominantly hydrophobic and involve only a few hydrogen-bonding interactions. The conserved residues Val227, Met228, and Met273 form the core of the interface (Fig. 1B). The overall structures of the two subunits are very similar (RMSD of 0.92 Å). Conformational differences are only observed in a few surface-exposed regions, including residues 212–219, 246–252, and 271–291. One marked difference is located at region of residues 275–280, which forms part of helix α6 with well-defined electron density in subunit A, but adopts a distorted helical geometry with slightly poorer electron density in subunit B. Tyr-A276 is positioned on the surface and points its side chain toward the solvent, while Tyr-B276 is embedded in a hydrophobic pocket of subunit A and its side-chain hydroxyl group forms a hydrogen bond with the main-chain amide group of Ala-A221. There is a hydrophobic cavity near the strictly conserved Gly-B220 at the dimer interface; a large spherical residual electron density was located in the pocket and was tentatively assigned as a water molecule. In subunit A, this pocket is occupied by Tyr-B276. It is unclear whether this hydrophobic cavity has any biological implication.
The MRG domain is a functionally uncharacterized protein domain. Searches for structural homologs of MRG15C in the PDB database using the DALI server (Holm and Sander 1997) revealed 20 candidates with a Z-score >2.0, among which six are >3.0, three >4.0, and one >5.0. However, these candidates share very poor sequence identity (<17%) with MRG15C. Three of those structures have a Z-score >3.0 and a RMSD <4.0 Å with MRG15C, which are recombinase XerD domain 1 (PDB code 1A0P, Z = 5.4, and RMSD of 3.8 Å for 78 Cα atoms) (Subramanya et al. 1997), RNA polymerase II transcription initiation factor D TAF(II)250 subdomain double bromodomain module (PDB code 1EQF-A, Z = 3.6, and RMSD of 3.5 Å for 99 Cα atoms) (Jacobson et al. 2000), and Escherichia coli sugar phosphotransferase system enzyme I fragment (PDB code 1ZYM-A, Z = 3.5, and RMSD of 3.4 Å for 89 Cα atoms) (Liao et al. 1996). These structures are orthogonal bundles or up–down bundles of mainly α-helices. However, the topology of MRG15C is different from that of those structures. It was suggested that MRG15C has a structural homology with the DNA binding domains of three recombinases (XerD, λ integrase, and Cre) based on a match of four α-helices (Bowman et al. 2006). However, we found that the numbers of matched residues between them are very limited, and the connection and direction of the two matched α-helix hairpins in MRG15C are different from those in the DNA binding domains of XerD, λ integrase, and Cre. Preliminary biochemical data also suggest that MRG15C does not bind to either single-stranded or double-stranded DNA by itself (J. Zhou, P. Zhang, and J. Ding, unpubl.).
The N-terminal region of PAM14 is responsible for its interaction with MRG15C
Previous biochemical data showed that PAM14 interacts with the C-terminal region of MRG15 in the MAF1 complex (Bertram et al. 1999; Leung et al. 2001). To confirm the direct interaction between MRG15 and PAM14, we performed the yeast two-hybrid experiments in which PAM14 and its fragments (PAM14N, PAM14M, and PAM14C) were used as preys and MRG15, MRG15C, and MRG15C2 (a truncated form of MRG15C in which the N-terminal 12 residues of MRG15C were deleted) as baits. Our results confirm that MRG15 interacts directly with PAM14 and MRG15C is the domain interacting with PAM14, whereas MRG15N has no interaction with PAM14 (Fig. 2).
Figure 2.

Yeast two-hybrid assay of the interaction between MRG15 and PAM14. MRG15, MRG15N, MRG15C, and MRG15C2 were cotransformed with pB42AD as the baits, and PAM14 and its fragments (PAM14N, PAM14M, and PAM14C) were cotransformed with pGilda as the preys. The pSH17-4 plasmid was used as the positive control and the pRFHM1 plasmid as the negative control for transcriptional activation. (A) Yeast two-hybrid analyses of pGilda-MRG15s (MRG15C and MRG15) with the empty prey plasmid pB42AD and pB42AD-PAM14s (PAM14N, PAM14M, PAM14C, and PAM14) with the empty bait plasmid pGilda. The baits or preys alone cannot activate the reporter markers by themselves. (B) Yeast two-hybrid analyses to determine the interactions of MRG15, MRG15N, MRG15C, and MRG15C2 with PAM14 and its fragments (PAM14N, PAM14M, and PAM14C). (Left to right) Full-length MRG15 (residues 1–323), MRG15N (residues 1–150), MRG15C (residues 151–323), and MRG15C2 (residues 173–323). Both MRG15 and MRG15C have direct interactions with PAM14 and PAM14N at comparable levels, whereas MRG15N and MRG15C2 have no interaction with PAM14 or its fragments.
PAM14 is a nuclear protein of 127 residues with unknown biological function, and is predicted to consist of three long α-helices and several coiled coils. To identify which region of PAM14 is required for its interaction with MRG15 or MRG15C, we cloned, expressed, and purified three fragments of PAM14 based on secondary structure prediction, each corresponding to one long α-helix—the N-terminal part (residues 1–50, PAM14N), the middle part (residues 49–84, PAM14M), and the C-terminal part (residues 80–127, PAM14C), respectively—and examined their binding affinity with MRG15 and MRG15C using both yeast two-hybrid assay and in vitro binding assay. The yeast two-hybrid assay results showed that similar to PAM14, PAM14N can interact with MRG15 and MRG15C, whereas PAM14M and PAM14C have no interaction with MRG15 or MRG15C (Fig. 2B). These results are confirmed by the in vitro binding assay results showing that both PAM14 and PAM14N can bind with MRG15C effectively, while PAM14M and PAM14C cannot (Fig. 3A). These data indicate that only the N-terminal part of PAM14 is responsible for its interaction with MRG15C, while the other regions of PAM14 are not involved in the interaction.
Figure 3.
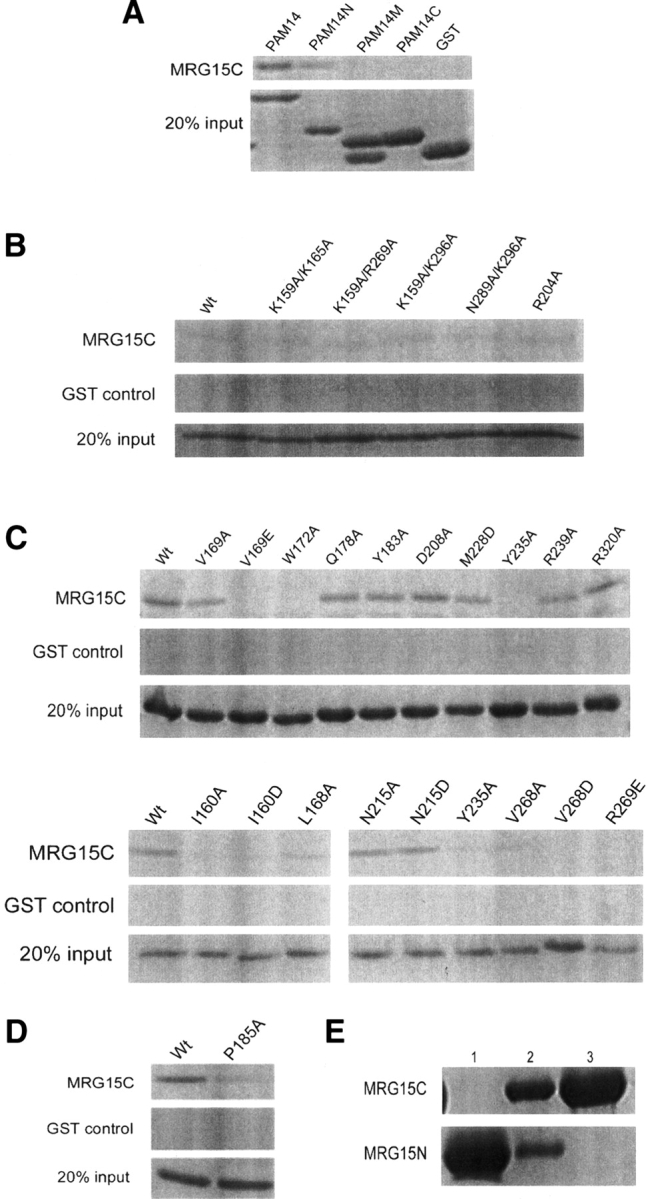
In vitro binding assay of the interaction between MRG15C and PAM14. GST was used as the negative control, which cannot bind to MRG15C. (A) In vitro binding assays of MRG15C with GST-fused PAM14 (residues 1–127) and its three fragments GST-PAM14N (residues 1–50), GST-PAM14M (residues 49–84), and GST-PAM14C (residues 80–127). The results show that MRG15C has interactions with PAM14 and PAM14N, but no interaction with PAM14M and PAM14C. (B) In vitro binding assays of GST-PAM14N with wild-type MRG15C and representative MRG15C mutants containing mutations of positively-charged residues. Shown here are double mutations K159A/K165A, K159A/R269A, K159A/K296A, and N289A/K296A, and single mutation R204A. The results show that these mutations have no obvious effect on the binding affinity of MRG15C with PAM14N compared to wild-type MRG15C. (C) In vitro binding assays of GST-PAM14N with wild-type MRG15C and representative MRG15C mutants containing mutations of both hydrophobic and hydrophilic residues. Shown here are single mutations V169A, V169E, W172A, Q178A, Y183A, D208A, M228D, Y235A, R239A, R320A, I160A, I160D, L168A, N215A, N215D, V268A, V268D, and R269E. The results show that mutation L168A and V169A significantly reduce the binding affinity of MRG15C with PAM14N; mutation I160A, I160D, V169E, W172A, Y235A, V268A, V268D, and R269E completely abolish the binding of PAM14N; and other mutations have no significant impact on the binding of PAM14N. (D) In vitro binding assay of GST-PAM14 with P185A mutant MRG15C. The result shows that mutation P185A substantially decreases the binding of MRG15C with PAM14. (E) In vitro binding assay of MRG15N and MRG15C. (Lane 1) His-tagged MRG15N; (lane 2) mixture of His-tagged MRG15N and GST-tagged MRG15C; (lane 3) mixture of His-tagged MRG15N and GST-tagged MRG15C after being washed three times with a washing buffer. The result shows that MRG15N and MRG15C have no direct interaction with each other.
MRG15C uses a shallow hydrophobic pocket to interact with PAM14
To further determine the binding site of PAM14 on MRG15C, we performed structure-based site-directed mutagenesis studies of MRG15C and examined the binding affinity of these MRG15C mutants with PAM14 by in vitro binding assay. Analysis of the primary sequence of PAM14N reveals that there are 15 negatively-charged, acidic residues out of 50 residues in PAM14N, leading us initially to speculate that the interaction between PAM14 and MRG15C might be mediated through hydrogen-bonding or hydrophilic interactions. In addition, previous biochemical data showed that a MRG15 mutant containing deletion of residues 125–191 could not bind to PAM14 and Rb, suggesting that residues 125–191 of MRG15 may be involved in the binding of PAM14 and Rb (Leung et al. 2001). Our biochemical data show that MRG15C (residues 151–323) itself is sufficient to bind with PAM14, suggesting that the region of residues 125–150 is not involved in PAM14 binding. However, deletion of the N-terminal 12 residues (including helix α1) of MRG15C (MRG15C2, residues 173–323) abolishes its interaction with PAM14 and PAM14N (Fig. 2B), suggesting that the N-terminal region of MRG15C is indispensable in the binding of PAM14. Taking these two considerations into account, we first generated mutations on a number of positively-charged, basic amino acids in or near the N-terminal region of MRG15C, including K157A, K159A, K165A, R269A, and K296A. However, these mutations had no effect on the binding of MRG15C with PAM14N (data not shown). Furthermore, we made several double mutations in this region, including K159A/K165, K159A/R269A, K159A/K296A, and K296A/N289A, which also did not show any obvious effect on the interaction between MRG15C and PAM14N (Fig. 3B). These results imply that the N-terminal region of MRG15C either is not directly involved in PAM14 binding or interacts with PAM14 via a different interaction mechanism.
We then investigated if MRG15C uses other structural element(s) instead of the N-terminal region to interact with PAM14. Analysis of the MRG15C structure indicates that although MRG15C exhibits a largely hydrophobic surface, there is a negatively-charged surface patch at the C-terminal region consisting of several conserved residues (Glu244, Asp248, Glu312, and Glu317) and a strictly conserved Pro185 with a cis-conformation (Fig. 4A). On the opposite face of the molecule, there are several positively-charged, basic residues (Lys236, Arg239, and Arg269) that are highly conserved in all MRG proteins. In addition, the region connecting helices α2 and α3 (residues 204–211), which was disordered in both subunits of the homodimer, contains several conserved hydrophilic residues as well (Lys201, Arg204, and Asp208). The conservation of these residues might imply a possible role in biological function(s), such as protein–protein interaction. Thus, we performed mutagenesis studies on these surface exposed hydrophilic residues and carried out in vitro binding assay. The results showed that mutations of these residues with alanine had no influence on the binding ability of MRG15C with PAM14 (some of the data are shown in Fig. 3B,C). Therefore, these experiments indicate that the interaction between MRG15C and PAM14N is not through hydrophilic residues, instead, it is most likely through hydrophobic residues.
Figure 4.
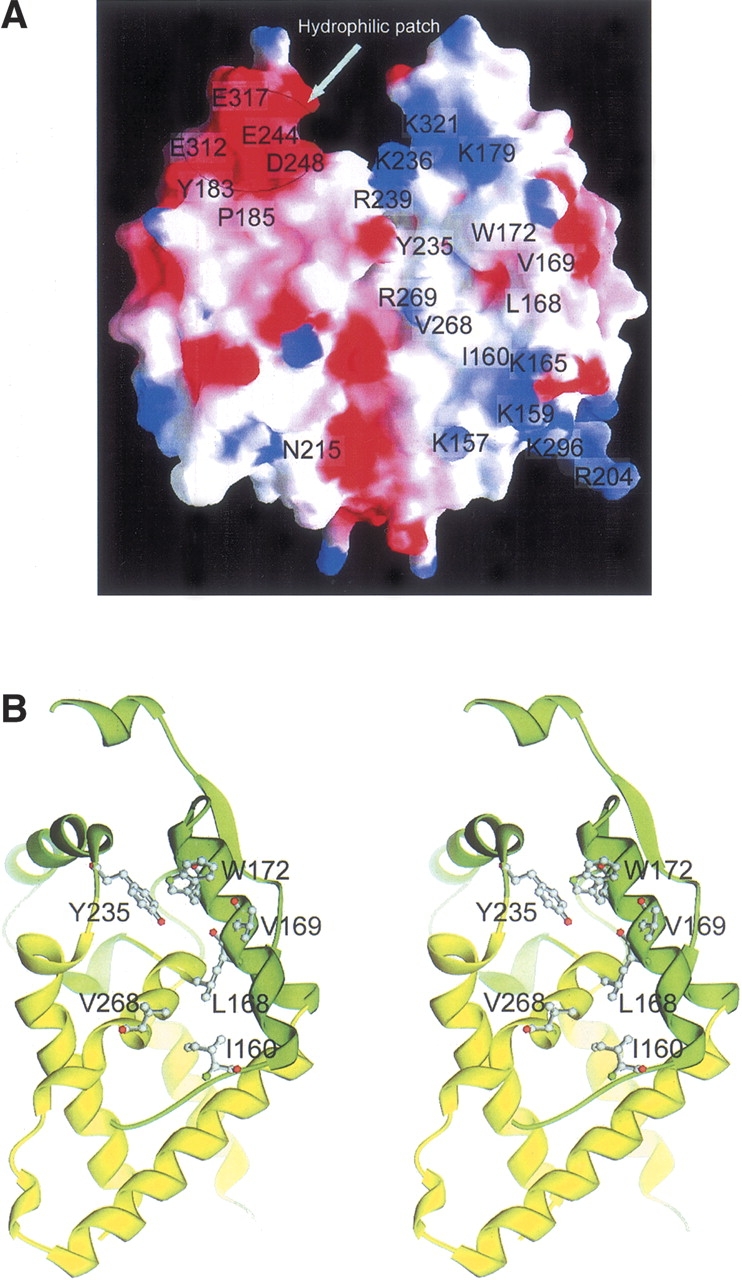
The PAM14 binding site. (A) Electrostatic surface of the dimeric MRG15C. MRG15 is highly hydrophobic but contains a few hydrophilic surface patches. The PAM14 binding site is located in a shallow hydrophobic pocket. A negatively-charged surface patch is located near the C-terminal region on one face of the molecule and several positively-charged patches on the opposite face. The residues forming the hydrophobic pocket and the hydrophilic surface patches are highlighted. (B) A stereoview of the structure of the PAM14 binding site. The PAM14 binding site consists of residues Ile160, Leu168, Val169, Trp172, Tyr235, Val268, and Arg269, which form a shallow hydrophobic pocket on one surface of the molecule. The hydrophobic residues are shown with side chains.
To further delineate the potential PAM14 binding site, we performed mutagenesis studies on a number of surface-exposed hydrophobic residues in the N-terminal region and several other regions of MRG15C. The in vitro binding assay results showed that mutation V169A substantially decreased the binding of MRG15C with PAM14N, and mutation W172A completely disrupted the interaction between MRG15C and PAM14N, whereas other mutations, including Q178A, Y183A, D208A, M228D, R239A, and R320A, had no effect on the interaction (Fig. 3C). Analysis of the MRG15C structure shows that several hydrophobic residues are located very closely to Val169 and Trp172 (α1), including Ile160 and Leu168 of helix α1, Tyr235 of the α3–α4 connecting loop, and Val268 of helix α6, and these residues together form a shallow hydrophobic pocket (Fig. 4), suggesting that in addition to Val169 and Trp172, these residues may also be involved in interaction with PAM14. Indeed, mutation L168A significantly decreased the binding of MRG15C with PAM14N; mutations I160A, I160D, V268A, V268D, and Y235A disrupted the binding of MRG15C with PAM14N (Fig. 3C). Moreover, a highly conserved Arg269 is positioned at the rim of the shallow pocket (Fig. 4A). Although mutation R269A had no significant effect on the binding of MRG15C with PAM14N, mutation R269E abolished the binding of PAM14N, suggesting that the hydrophilic residue(s) nearby the shallow pocket also plays an important role in the binding of PAM14. When full-length PAM14 was used, we had similar results as PAM14N (data not shown). While we were performing those functional studies, Bowman et al. (2006) reported that mutation V169E of MRG15C completely abolished PAM14 binding, and mutation N215R could cause a reduced affinity for PAM14 binding. Thus, we further carried out in vitro binding assays for mutations V169E, N215A, and N215D (Fig. 3C). Our in vitro binding assay data confirmed that mutation V169E completely disrupted the interaction of MRG15C with PAM14N or PAM14. However, in our experiments, mutation N215A or N215D had no obvious impact on the binding affinity of MRG15C with PAM14N or PAM14. Our biochemical results can be explained very well in the structural context. Residues Ile160, Leu168, Val169, Trp172, Tyr235, Val268, and Arg269 constitute a shallow hydrophobic pocket as the binding site for PAM14, and the interaction between MRG15C and PAM14 are mainly hydrophobic. Mutation V169A reduces the size of the hydrophobic side chain, which would cause a mild change in the geometry of the PAM14 binding site, and therefore decreases the binding affinity of MRG15C with PAM14. When Val169 is substituted with Glu, the mutation would not only increase the size of the side chain, but also change the electrostatic property of the PAM14 binding site, leading to the abolishment of the binding ability of MRG15C with PAM14. Similarly, residues Ile160, Leu168, Val268, Try172, and Tyr235 are hydrophobic residues with moderate to large aromatic side chains, and their mutations to Ala or hydrophilic residues (Asp) would significantly alter the geometry of the PAM14 binding site and destabilize the interaction between MRG15C and PAM14, leading to the loss of binding affinity of MRG15C with PAM14. Arg269 is a hydrophilic residue with a large positively-charged side chain. Mutation R269A appears to have a minor impact on the PAM14 binding site, whereas change of Arg269 to Glu would reverse the electrostatic property of the side chain, and therefore would have a drastic impact on the binding of PAM14. Asn215 is located on the opposite face of the protein from that of the hydrophobic pocket involved in interaction with PAM14 or very far away from the hydrophobic pocket on the same face in the dimer, and therefore its mutation should not affect the binding of MRG15C with PAM14. Thus, we are not sure whether the reported reduction in the binding affinity of MRG15C for PAM14 caused by mutation N215R is an experimental artifact or through some unknown mechanism. Taking the structural and biochemical data together, we conclude that the PAM14 binding site of MRG15C consists of mainly Ile160, Leu168, Val169, Trp172, Tyr235, Val268, and Arg269, which form a shallow hydrophobic pocket to interact with the N-terminal 50 residues of PAM14 through primarily hydrophobic interactions.
It is noteworthy that there is a strictly conserved Pro185 in the loop connecting strand β1 to helix α2 with a cis-conformation. This residue is located at the back of the hydrophobic pocket for binding PAM14, and forms part of the negatively-charged surface patch at the C-terminal region. Mutation of Pro185 to Ala resulted in a large fraction of the protein expressed in inclusion bodies with only a small amount in supernatant. The P185A MRG15C mutant had substantially reduced binding affinity with PAM14 compared to the wild-type protein (Fig. 3D). It seems very likely that mutation P185A destabilizes the conformation and structure of the N-terminal region, leading to the decrease of the binding affinity with PAM14.
Previous biochemical data also showed that residues 284–305 (helix α7) of MRG15, which were predicted to form a leucine zipper, are involved in binding with PAM14 and Rb (Bertram et al. 1999; Bertram and Pereira-Smith 2001; Leung et al. 2001). Analysis of the MRG15C structure indicates that although this region forms a long α-helix (α7), it is not involved in hydrophobic interaction with any other helix to form a typical leucine zipper structure. Most of the conserved leucine residues point their side chains toward the interior of the protein to stabilize the hydrophobic core of the overall structure. Moreover, mutagenesis data show that mutations of two surface exposed hydrophilic residues on this helix (N289A and K296A) had no effect on the PAM14 binding (Fig. 3B). These results suggest that helix α7 is not involved in direct binding of PAM14, consistent with Bowman et al. (2006). The necessity of this region for binding PAM14 is most likely because of its contribution to the formation of the structural scaffold, rather than direct participation in protein–protein interaction.
Discussion
The MRG proteins exist in many eukaryotes from yeast to human, and MRG15, MORF4, and MRGX are typical representatives (Bertram and Pereira-Smith 2001). The MRG domain is highly conserved among all known MRG proteins, underscoring an important functional role (Fig. 1D). Although the exact biological functions of these MRG proteins are yet unclear, the crystal structure of MRG15C, together with biochemical data, has provided the molecular basis for the interaction of MRG15 with PAM14, and revealed some insights into the potential biological function(s) of MRG15 and other MRG proteins. It is unexpected that the interaction of MRG15C with the N-terminal part of PAM14 is mainly through hydrophobic residues. Residues Ile160, Leu168, Val169, Trp172, Tyr235, Val268, and Arg269 of MRG15C form a shallow hydrophobic pocket as the PAM14 binding site, and the hydrophobic interactions dictate the binding affinity for PAM14. Although the N-terminal region of PAM14 contains a large number of acidic residues (15 out 50 residues), they appear not to be the primary residues involved in the interaction. Similarly, there are a number of conserved, charged residues forming several hydrophilic patches on the MRG15C surface, but they are also not involved in PAM14 binding. Nevertheless, since MRG15C and other MRG domains can also interact with other proteins, even though these hydrophilic surface patches or residues are not involved in interaction with PAM14, we cannot exclude the possibility that they might be potential binding sites for or involved in interaction with other proteins. Further biochemical and structural studies of MRG15 in complexes with its protein partners will provide detailed molecular basis for these interactions.
Sequence comparisons show that the MRG domains of MRG15, MRGX, and MORF4 are strongly conserved, and differ only by several amino acids. These varying residues are mostly located on surface exposed regions and, thus appear not affecting the overall structure. Moreover, the MRG domains of other MRG proteins also share very high sequence homology with MRG15C, and contain a number of strictly conserved hydrophobic residues that form the scaffold of the structure (Fig. 1D). Hence, we predict that the MRG domains of MRGX and MORF4 and most likely other MRG proteins would assume a similar structure as that of MRG15C. On the other hand, MRG15, MRGX, and MORF4 show differed functions in cellular senescence, and this variation is very likely due to the differences in the N-terminal domains.
MRG15 is suggested to play an important role in transcription regulation and cell development via chromatin remodeling (Tominaga et al. 2005). MRG15 consists of MRG15N and MRG15C linked together by a flexible region. Biochemical data have shown that MRG15N is involved in interactions with proteins (most likely modified histones) in the HAT/HDAC complexes (Leung et al. 2001), and MRG15C in interactions with several protein partners, including Rb, PAM14, mSin3A, and Pf1 (Leung et al. 2001; Yochum and Ayer 2002). So far, we have been able to express, purify, and crystallize both MRG15N and MRG15C, and solve their individual crystal structures (the crystal structure of MRG15N will be published elsewhere). However, we have not been able to crystallize the full-length MRG15. These results suggest that MRG15N and MRG15C exist as two independent and stable domains that are connected together by a structurally flexible region. Indeed, our in vitro binding assay data show that MRG15N and MRG15C do not interact with each other (Fig. 3E). Based on the biochemical and structural data, we propose that MRG15 functions as an adaptor protein to interact with other proteins in nuclear complexes during chromatin remodeling and transcription regulation. The two domains of MRG15 (MRG15N and MRG15C) interact with different protein partners and play different functional roles, but work in a concerted way. The flexible connecting region may function in modulating the relative conformation of the two domains and facilitating the overall structure and biological function of MRG15.
Materials and methods
Expression and purification of MRG15C
The cDNA corresponding to MRG15C (residues 151–323) was cloned into the pET-28a(+) expression plasmid (Novagen) using the NcoI and XhoI restriction sites. The plasmid was transferred into the E. coli BL21(DE3) strain, and the bacterial cells were grown in LB medium supplemented with 0.1 mg/mL ampicillin at 37°C until OD600 reached ∼0.8. Protein expression was induced by adding isopropyl-β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM at 22°C for about 10 h. The bacterial cells were harvested by centrifugation and resuspended in a lysis buffer (20 mM Tris-HCl at pH 8.0, 500 mM NaCl, 2 mM β-mercaptoethanol, and 1 mM PMSF), and then lysed by sonication on ice. The supernatant was loaded onto an Ni-NTA agarose column (Amersham-Pharmacia) pre-equilibrated with a binding buffer (20 mM Tris-HCl at pH 8.0, 500 mM NaCl, and 1 mM PMSF) and then the resin was washed with a washing buffer (the binding buffer supplemented with 50 mM imidazole) to remove nonspecific binding proteins. The target proteins were eluted with an elution buffer (the binding buffer supplemented with 300 mM imidazole). Further purification was performed using gel-filtration on a Superdex 26/60 (prep grade) column (Amersham-Pharmacia) pre-equilibrated with the binding buffer. The target protein was collected and concentrated to ∼20 mg/mL by ultrafiltration for structural and functional studies. SDS-PAGE analysis of the protein showed a single band with molecular mass of 20 kDa. The purity and homogeneity of the protein were also confirmed with dynamic light-scattering analysis. Selenomethionine (Se-Met)-substituted MRG15C was expressed in the methionine auxotroph E. coli B834(DE3) strain (Novagen) and purified using the same method as for the native protein.
Constructs of the mutant MRG15Cs containing point mutations were generated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). Expression plasmid of a truncated MRG15C containing deletion of the N-terminal 12 residues (MRG15C2, residues 173–323) was generated using the same method as MRG15C. All of these clones were verified by DNA sequencing. Expression and purification of these MRG15C mutants were the same as the wild-type protein described above.
Expression and purification of PAM14
The cDNA corresponding to the full-length PAM14 was cloned into a pGEX-4T1 vector in which a glutathione-S-transferase (GST) tag was fused at the N-termini of the protein. The GST-fused PAM14 was expressed in E. coli BL21(DE3) and purified using affinity chromatography with the glutathione Sepharose 4B column (Amersham Biosciences). To identify the region of PAM14 involved in interaction with MRG15C, the full-length PAM14 was divided into three fragments: PAM14N (the N-terminal residues 1–50), PAM14M (residues 49–84), and PAM14C (the C-terminal residues 80–127). Cloning, expression, and purification of the three PAM14 fragments were the same as the wild-type protein.
Crystallization and diffraction data collection
Crystallization of MRG15C was carried out using the hanging drop vapor diffusion method. Crystals of both native and Se-Met MRG15C were grown in drops containing equal volumes of the protein solution (20 mg/mL) and the crystallization solution (0.1 M HEPES at pH 7.9, 20% PEG4000, and 5% isopropanol) to approximate dimensions of 0.2 × 0.2 × 0.3 mm3 in 10 d at 20°C. Crystals of MRG15C belong to space group R3 with the cell parameters of a = b = 111.2 Å and c = 87.0 Å. The native diffraction data were collected to 2.2 Å resolution from a flash-cooled crystal at 100 K at beamline BL-6A of Photon Factory, Japan. The multiwavelength anomalous dispersion (MAD) data were collected to 2.6 Å resolution at beamline BL-18B of Photon Factory. The diffraction data were processed, integrated, and scaled together with HKL2000 (Otwinowski and Minor 1997). The statistics of the diffraction data are summarized in Table 1.
Structure determination and refinement
Crystal structure of MRG15C was solved by the MAD method implemented in the program SOLVE (Terwilliger and Berendzen 1999). The MAD phases revealed six Se sites in the asymmetric unit. Since each MRG15C contains four Met residues (including the first residue Met151), the SOLVE results suggest that there are two MRG15C molecules in the asymmetric unit, and the first Met residue is likely disordered. The MAD phases were improved by statistical density modification including solvent flattening and histogram matching using the program RESOLVE (Terwilliger 2001), increasing the overall figure of merit from 0.31 to 0.63 at 2.6 Å resolution. The resulting electron density map had very good quality and was used to build an initial model. RESOLVE automatically built 250 polyalanines out of 334 residues and successfully located most of the secondary structural elements. A complete model was built manually using the program O (Jones et al. 1991). Structure refinement was carried out against the 2.2 Å resolution native data set using the program CNS (Brunger et al. 1998). Strict twofold noncrystallographic symmetry (NCS) restraints were used in the initial stage of refinement but were released in the later stage of refinement. The final structure refinement was performed using the maximum likelihood algorithm implemented in the program REFMAC5 (Murshudov et al. 1997). A bulk solvent correction and a free R-factor monitor (calculated with 5% of randomly chosen reflections) were applied throughout the refinement. The statistics of the structure refinement and the quality of the structure model are summarized in Table 1.
In vitro protein–protein binding assay
For in vitro protein–protein binding assay, the GST-PAM14 protein or its fragments (5 μg) were absorbed onto the glutathione Sepharose 4B beads (10 μL) in a binding buffer (PBS containing 1 mM PMSF) and then 10 μg of the wild-type or mutant MRG15C protein and 50 μg of BSA were added in a total volume of 500 μL. After 2 h at 4°C, the beads were washed four times with 500 μL of a washing buffer (20 mM Tris-HCl at pH 8.0, 1 M NaCl, 1% NP40, 2 mM DTT, 1 mM EDTA, and 1 mM PMSF). The bound proteins were eluted with SDS-sample buffer and analyzed by SDS-PAGE with Coomassie blue staining. Binding assay with the GST protein was used as a negative control.
Yeast two-hybrid assay
The yeast two-hybrid assay was carried out using the yeast HLY819 strain, which was transformed with the Matchmaker LexA two-hybrid system (BD Biosciences Clontech). The pGilda plasmid was used to express the LexA DNA-binding domain in fusion with MRG15C (bait). The pB42AD plasmid was used to express the AD transcriptional activation domain in fusion with PAM14 (prey). pSH17–4, which encodes LexA fused to the activation domain of the yeast activator protein GAL4 and can strongly activate transcription, was used as a positive control plasmid for transcriptional activation. pRFHM1 was used as a negative control plasmid, which encodes LexA fused to the N terminus of the Drosophila protein bicoid and has no ability to activate transcription. The MRG15C:pGilda and PAM14:pB42AD plasmids were cotransformed into the yeast HLY819 strain, which contains two different inducible reporter markers for leucine and β-galactosidase synthesis. The prey plasmid was cotransformed with the bait plasmid by the lithium acetate method and selected on plates lacking uracil, tryptophan, and histidine. These colonies were subsequently streaked on plates lacking uracil, tryptophan, histidine, and leucine, where cell growth indicates an interaction between the bait and prey proteins which activates an inducible reporter marker. The β-galactosidase reporter assays were performed on colonies transferred to nitrocellulose filters (Whatman) to assess the strength of the protein interaction. X-gal was used as a substrate. Colonies (2 mm diameter) on filters were frozen with liquid nitrogen for 1 min and then incubated with X-gal (334 μg/mL) at 30°C.
Protein Data Bank accession code
The structure of the MRG domain of human MRG15 has been deposited with the RCSB Protein Data Bank under accession code 2F5J.
Acknowledgments
We thank the staff members of Photon Factory, Japan, for technique support in data collection, and Qiuhua Huang of Shanghai Institute of Hematology, Rui-Jin Hospital, Shanghai Second Medical University, for providing the MRG15 plasmid. This work was supported by NSFC Grants (30125011 and 30570379), MOST Grants (2002BA711A13, 2004CB520801, and 2004CB720102), and CAS Grant (KSCX1-SW-17).
Footnotes
Reprint requests to: Jianping Ding, State Key Laboratory of Molecular Biology, Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue-Yang Road, Shanghai 200031, China; e-mail: jpding@sibs.ac.cn; fax: 86-21-5492-1116.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062397806.
Abbreviations: MORF4, mortality factor on chromosome 4; MRG15, MORF4-related gene on chromosome 15; MRGX, MORF4-related gene on chromosome X; MRG domain, the C-terminal domain of MRG15; MRG15C, the C-terminal MRG domain of MRG15; MRG15N, the N-terminal chromo domain of MRG15; MAF1 and MAF2, MRG15-associated factor 1 and 2; PAM14, protein associated with MRG15 of 14 kDa; HAT, histone acetyltransferase; HDAC, histone deacetylase.
References
- Akhtar, A., Zink, D., Becker, P.B. 2000. Chromodomains are protein–RNA interaction modules. Nature 407: 405–409. [DOI] [PubMed] [Google Scholar]
- Ball, L.J., Murzina, N.V., Broadhurst, R.W., Raine, A.R., Archer, S.J., Stott, F.J., Murzin, A.G., Singh, P.B., Domaille, P.J., Laue, E.D. 1997. Structure of the chromatin binding (chromo) domain from mouse modifier protein 1. EMBO J. 16: 2473–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister, A.J., Zegerman, P., Partridge, J.F., Miska, E.A., Thomas, J.O., Allshire, R.C., Kouzarides, T. 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410: 120–124. [DOI] [PubMed] [Google Scholar]
- Bertram, M.J. and Pereira-Smith, O.M. 2001. Conservation of the MORF4 related gene family: Identification of a new chromo domain subfamily and novel protein motif. Gene 266: 111–121. [DOI] [PubMed] [Google Scholar]
- Bertram, M.J., Berube, N.G., Hang-Swanson, X., Ran, Q., Leung, J.K., Bryce, S., Spurgers, K., Bick, R.J., Baldini, A., Ning, Y. et al. 1999. Identification of a gene that reverses the immortal phenotype of a subset of cells and is a member of a novel family of transcription factor-like genes. Mol. Cell. Biol. 19: 1479–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman, B.R., Moure, C.M., Kirtane, B.M., Welschhans, R.L., Tominaga, K., Pereira-Smith, O.M., Quiocho, F.A. 2006. Multipurpose MRG domain involved in cell senescence and proliferation exhibits structural homology to a DNA-interacting domain. Structure 14: 151–158. [DOI] [PubMed] [Google Scholar]
- Brehm, A., Miska, E.A., McCance, D.J., Reid, J.L., Bannister, A.J., Kouzarides, T. 1998. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391: 597–601. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S. et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Cai, Y., Jin, J., Tomomori-Sato, C., Sato, S., Sorokina, I., Parmely, T.J., Conaway, R.C., Conaway, J.W. 2003. Identification of new subunits of the multiprotein mammalian TRRAP/TIP60-containing histone acetyltransferase complex. J. Biol. Chem. 278: 42733–42736. [DOI] [PubMed] [Google Scholar]
- Cavalli, G. and Paro, R. 1998. Chromo-domain proteins: Linking chromatin structure to epigenetic regulation. Curr. Opin. Cell Biol. 10: 354–360. [DOI] [PubMed] [Google Scholar]
- Eisen, A., Utley, R.T., Nourani, A., Allard, S., Schmidt, P., Lane, W.S., Lucchesi, J.C., Cote, J. 2001. The yeast NuA4 and Drosophila MSL complexes contain homologous subunits important for transcription regulation. J. Biol. Chem. 276: 3484–3491. [DOI] [PubMed] [Google Scholar]
- Fujita, M., Takasaki, T., Nakajima, N., Kawano, T., Shimura, Y., Sakamoto, H. 2002. MRG-1, a mortality factor-related chromodomain protein, is required maternally for primordial germ cells to initiate mitotic proliferation in C. elegans . Mech. Dev. 114: 61–69. [DOI] [PubMed] [Google Scholar]
- Gorman, M., Franke, A., Baker, B.S. 1995. Molecular characterization of the male-specific lethal-3 gene and investigations of the regulation of dosage compensation in Drosophila . Development 121: 463–475. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1997. Dali/FSSP classification of three-dimensional protein folds. Nucleic Acids Res. 25: 231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson, R.H., Ladurner, A.G., King, D.S., Tjian, R. 2000. Structure and function of a human TAFII250 double bromodomain module. Science 288: 1422–1425. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A47: 110–119. [DOI] [PubMed] [Google Scholar]
- Jones, D.O., Cowell, I.G., Singh, P.B. 2000. Mammalian chromodomain proteins: Their role in genome organisation and expression. Bioessays 22: 124–137. [DOI] [PubMed] [Google Scholar]
- Lachner, M., O'Carroll, D., Rea, S., Mechtler, K., Jenuwein, T. 2001. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410: 116–120. [DOI] [PubMed] [Google Scholar]
- Leung, J.K., Berube, N., Venable, S., Ahmed, S., Timchenko, N., Pereira-Smith, O.M. 2001. MRG15 activates the B-myb promoter through formation of a nuclear complex with the retinoblastoma protein and the novel protein PAM14. J. Biol. Chem. 276: 39171–39178. [DOI] [PubMed] [Google Scholar]
- Liao, D.I., Silverton, E., Seok, Y.J., Lee, B.R., Peterkofsky, A., Davies, D.R. 1996. The first step in sugar transport: Crystal structure of the amino terminal domain of enzyme I of the E. coli PEP:sugar phosphotransferase system and a model of the phosphotransfer complex with HPr. Structure 4: 861–872. [DOI] [PubMed] [Google Scholar]
- Marin, I. and Baker, B.S. 2000. Origin and evolution of the regulatory gene male-specific lethal-3 . Mol. Biol. Evol. 17: 1240–1250. [DOI] [PubMed] [Google Scholar]
- Morales, V., Straub, T., Neumann, M.F., Mengus, G., Akhtar, A., Becker, P.B. 2004. Functional integration of the histone acetyltransferase MOF into the dosage compensation complex. EMBO J. 23: 2258–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales, V., Regnard, C., Izzo, A., Vetter, I., Becker, P.B. 2005. The MRG domain mediates the functional integration of MSL3 into the dosage compensation complex. Mol. Cell. Biol. 25: 5947–5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Nakayama, J., Xiao, G., Noma, K., Malikzay, A., Bjerling, P., Ekwall, K., Kobayashi, R., Grewal, S.I. 2003. Alp13, an MRG family protein, is a component of fission yeast Clr6 histone deacetylase required for genomic integrity. EMBO J. 22: 2776–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, A.L., Oulad-Abdelghani, M., Ortiz, J.A., Remboutsika, E., Chambon, P., Losson, R. 2001. Heterochromatin formation in mammalian cells: Interaction between histones and HP1 proteins. Mol. Cell 7: 729–739. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Pardo, P.S., Leung, J.K., Lucchesi, J.C., Pereira-Smith, O.M. 2002. MRG15, a novel chromodomain protein, is present in two distinct multiprotein complexes involved in transcriptional activation. J. Biol. Chem. 277: 50860–50866. [DOI] [PubMed] [Google Scholar]
- Reid, J.L., Moqtaderi, Z., Struhl, K. 2004. Eaf3 regulates the global pattern of histone acetylation in Saccharomyces cerevisiae . Mol. Cell. Biol. 24: 757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, E.R., Pannuti, A., Gu, W., Steurnagel, A., Cook, R.G., Allis, C.D., Lucchesi, J.C. 2000. The Drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol. Cell. Biol. 20: 312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanya, H.S., Arciszewska, L.K., Baker, R.A., Bird, L.E., Sherratt, D.J., Wigley, D.B. 1997. Crystal structure of the site-specific recombinase, XerD. EMBO J. 16: 5178–5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T.C. 2001. Maximum-likelihood density modification using pattern recognition of structural motifs. Acta Crystallogr. D Biol. Crystallogr. 57: 1755–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T.C. and Berendzen, J. 1999. Evaluation of macromolecular electron-density map quality using the correlation of local r.m.s. density. Acta Crystallogr. D Biol. Crystallogr. 55: 1872–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga, K., Olgun, A., Smith, J.R., Pereira-Smith, O.M. 2002. Genetics of cellular senescence. Mech. Ageing Dev. 123: 927–936. [DOI] [PubMed] [Google Scholar]
- Tominaga, K., Kirtane, B., Jackson, J.G., Ikeno, Y., Ikeda, T., Hawks, C., Smith, J.R., Matzuk, M.M., Pereira-Smith, O.M. 2005. MRG15 regulates embryonic development and cell proliferation. Mol. Cell. Biol. 25: 2924–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yochum, G.S. and Ayer, D.E. 2002. Role for the mortality factors MORF4, MRGX, and MRG15 in transcriptional repression via associations with Pf1, mSin3A, and Transducin-Like Enhancer of Split. Mol. Cell. Biol. 22: 7868–7876. [DOI] [PMC free article] [PubMed] [Google Scholar]