

Abstract
Dynamic processes are inherent properties of proteins and are crucial for a wide range of biological functions. To address how changes in protein sequence and structure affect dynamic processes, a quantitative comparison of microsecond-to-microsecond time scale conformational changes, measured by solution NMR spectroscopy, within homologous mesophilic and thermophilic ribonuclease H (RNase H) enzymes is presented. Kinetic transitions between the observed major state (high population) and alternate (low population) conformational state(s) of the substrate-binding handle region in RNase H from the mesophile Escherichia coli (ecRNH) and thermophile Thermus thermophilus (ttRNH) occur with similar kinetic exchange rate constants, but the difference in stability between exchanging conformers is smaller in ttRNH compared to ecRNH. The altered thermodynamic equilibrium between kinetically exchanging conformers in the thermophile is recapitulated in ecRNH by the insertion of a Gly residue within a putative hinge between α-helices B and C. This Gly insertion is conserved among thermophilic RNases H, and allows the formation of additional intrahelical hydrogen bonds. A Gly residue inserted between α-helices B and C appears to relieve unfavorable interactions in the transition state and alternate conformer(s) and represents an important adaptation to adjust conformational changes within RNase H for activity at high temperatures.
Keywords: nuclear magnetic resonance, ribonuclease H, protein dynamics, thermal stability
Protein function relies on a combination of time-average static structure and time-dependent conformational fluctuations. Intrinsic dynamic properties influence many biological processes, including folding, ligand binding, catalysis, and allosterism (Wand 2001; Akke 2002; Benkovic and Hammes-Schiffer 2003; Kern and Zuiderweg 2003; Palmer 2004; Tousignant and Pelletier 2004; Kay 2005). Dynamic processes have been shown to be (partially) rate-limiting for catalysis, and differences in the time scale of motion are directly correlated with differences in the catalytic efficiency of homologous enzymes (Williams and McDermott 1995; Kohen and Klinman 2000; Codreanu et al. 2002; Cole and Loria 2002; Wolf-Watz et al. 2004). Several recent studies have highlighted the importance of individual amino acid residues in modulating dynamic processes within proteins (Eisenmesser et al. 2005; Kovrigin and Loria 2006). However, detailed mechanisms by which local interactions within the protein structure control the kinetic and thermodynamic properties of dynamic processes remain largely unknown.
Ribonucleases H (RNases H, EC 3.1.26.4) from the mesophile Escherichia coli (ecRNH) and thermophile Thermus thermophilus (ttRNH) share significant amino acid sequence and structural similarity, but display differences in thermal stability and catalytic activity. Typical of other mesophilic and thermophilic proteins (Jaenicke and Böhm 1998; Kumar et al. 2001; Vieille and Zeikus 2001), ttRNH is more resistant to thermal (and chemical) denaturation (Kanaya and Itaya 1992; Hollien and Marqusee 1999a,b) and is significantly less catalytically active than ecRNH at mesophilic temperatures (Kanaya and Itaya 1992). One source for these differences may stem from altered dynamic properties between these enzymes: Several regions display localized differences in dynamics on the picosecond-to-nanosecond (ps–ns) and microsecond-to-millisecond (μs–ms) time scales (Butterwick et al. 2004).
Chemical exchange line broadening is observed for nuclear spins in a highly basic loop between α-helices C (αC) and D, termed the handle (see Fig. 1), in both ecRNH and ttRNH (Mandel et al. 1995, 1996; Kroenke et al. 1998; Yamasaki et al. 1998; Butterwick et al. 2004), suggesting that μs–ms time scale conformational changes in this region are an integral property of both enzymes. Residues in αC and the handle are important for RNA:DNA hybrid recognition (Kanaya et al. 1991), although they are not essential for catalytic activity (Keck and Marqusee 1995). Nuclear magnetic resonance (NMR) spectroscopy and circular dichroism studies suggest that the handle changes conformation with respect to the catalytic domain upon complex formation (Oda et al. 1993; Iwai et al. 1996). The structural details of these transitions remain elusive; however, based on chemical shift changes upon RNA:DNA hybrid titration and models of the substrate-bound complex, residues between αB and αC have been suggested as a possible hinge in ecRNH (Oda et al. 1993). This region contains a single amino acid insertion in ttRNH relative to ecRNH: Gly85 is in a left-handed helical conformation (backbone dihedral angles ϕ = 70° and ψ = −9°), and allows an additional hydrogen bond between the Gly85 carboxyl and Gly89 amide, forming the N-terminal cap of αC (Ishikawa et al. 1993d).
Figure 1.

Sequence and structural alignments of RNases H. (A) ClustalW (Thompson et al. 1994) multiple sequence alignment of RNase H from the mesophilic bacterium E. coli (37°C) and several thermophilic bacteria: Chlorobium tepidum (47°C), Moorella thermoacetica (58°C), T. thermophilus (70°C), and Thermoanaerobacter tengcongensis (75°C). The optimal growth temperature for each organism is indicated in parentheses. Conserved active site and substrate-binding residues are highlighted in blue; the inserted Gly is shown in red. α-Helical (αA–αE) and β-strand (β1–β5) secondary structural elements and the handle in WT ecRNH are indicated above the alignment. (B) Structural superposition of WT ecRNH (blue, PDB 2RN2), WT ttRNH (orange, PDB 1RIL), and iG80b ecRNH (gray, PDB 1GOA) using backbone atoms along core helices αA and αD. Structural elements from (A) and the N and C termini are indicated. Ribbon diagrams for this and subsequent figures were drawn using MOLMOL (Koradi et al. 1996).
Using NMR spectroscopy, a quantitative comparison of the μs–ms time scale dynamics of ecRNH and ttRNH is presented. Extending previous results (Butterwick et al. 2004), backbone amide 15N R 1ρ relaxation dispersion experiments show that conformational changes within the handle of both enzymes occur with similar kinetic exchange rate constants at 300 K. Analysis of the relaxation dispersion suggests an altered thermodynamic equilibrium among exchanging conformers exists between ecRNH and ttRNH. Reciprocal Gly insertion/deletion mutations within the putative αB/αC hinge are used to probe the role of this residue in modulating conformational changes within RNases H. Insertion of Gly80b into ecRNH between Gln80 and Trp81 (iG80b) or deletion of Gly85 from ttRNH (dG85) significantly alters μs–ms time scale dynamic processes compared to the wild-type (WT) enzymes. 15N R 1ρ dispersion experiments suggest that the thermodynamic distribution observed for WT ttRNH is largely recapitulated in the iG80b ecRNH mutant. Thus, the Gly insertion represents an important contribution in the adaptation of conformational changes within RNase H for activity at high temperatures.
Results
Gly insertion is conserved among thermophilic RNases H
Sequence alignments for mesophilic and thermophilic RNases H highlight several amino acid substitutions that may confer thermal stability or alter catalytic activity. More than 100 bacterial RNase H sequences with a conserved handle are found in the Swiss-Prot database, including four from thermophilic organisms (Fig. 1A). In addition to several amino acid substitutions known to promote increased thermal stability in ecRNH, for example H62P and V74L (Kimura et al. 1992b; Ishikawa et al. 1993b), the Gly insertion between αB and αC is conserved in thermophilic RNases H. Furthermore, this insertion is largely absent from mesophilic sequences: Only RNase H enzymes from mesophiles Lactobacillus plantarum and Deinococcus radiodurans contain this insertion.
Structural and functional consequences of Gly insertion
Although the Gly insertion is conserved in thermophilic RNases H, insertion of Gly80b into ecRNH provides only a moderate increase in thermal stability of 0.1 kJ/mol (Kimura et al. 1992b). However, this insertion leads to a drastic decrease in catalytic efficiency, increasing the binding affinity by 50-fold and decreasing the catalytic rate constant (k cat) by 10-fold at 30°C (Ishikawa et al. 1993c), such that the activity of iG80b ecRNH is similar to the activity of ttRNH (Kanaya and Itaya 1992). Structurally, Gly80b is nicely accommodated between αB and αC, and adopts a similar left-handed helical conformation (ϕ = 52°, ψ = 46°) as Gly85 in ttRNH (Fig. 1B). In addition to the expected N-terminal cap hydrogen bond between Gly80b and Asn84, another hydrogen bond is present between the Ile78 carboxyl and Gly80b amide forming the C-terminal cap of αB (Ishikawa et al. 1993c). The additional Gly also shifts the Gly77 carboxyl–Ile82 amide hydrogen bond to Gly77–Trp81. Given the minimal structural perturbations and marginal stabilization induced by the Gly80b insertion, the drastic decrease in activity may be due to modification of dynamic processes involving the substrate-binding handle.
Backbone amide 1H and 15N resonances for the iG80b ecRNH and dG85 ttRNH mutant enzymes were assigned by comparison to WT spectra complemented with an HMQC-NOESY-HSQC spectrum recorded at 300 K. Chemical shift differences between WT and mutant spectra are small outside the site of mutation, suggesting that insertion or removal of the Gly residue does not significantly alter the protein structure. In addition, NOESY cross-peaks are observed between backbone amide protons separated by one or two residues throughout both αB and αC, confirming the helical nature of these regions (data not shown). The largest 15N chemical shift differences between WT and iG80b ecRNH are for residues Thr79 and Ile82: δiG80b−δWT = 4.94 ppm and −3.77 ppm, respectively. These shift changes are the correct sign and magnitude as expected for gain of a hydrogen bond involving the Ile78 carboxyl and switch of the hydrogen bond involving the Ile82 amide (Xu and Case 2002).
Interestingly, the equivalent residues also exhibit similar 15N chemical shift differences between WT and dG85 ttRNH, but with the opposite sign: δdG85−δWT = −4.78 ppm for Thr83 and 3.89 ppm for Leu87. Chemical shift changes for residues in αB, αC and the handle between WT and mutant ecRNH and ttRNH are negatively correlated overall (Fig. 2A). Not only are the changes in chemical shift similar in magnitude for these proteins, but the iG80b ecRNH chemical shifts resemble WT ttRNH, and dG85 ttRNH shifts resemble WT ecRNH (Fig. 2B). Thus, the hydrogen bonding pattern and backbone geometry is similar between WT ttRNH and iG80b ecRNH and between WT ecRNH and dG85 ttRNH. These results are in slight disagreement with the crystal structure of WT ttRNH (Fig. 2C) (Ishikawa et al. 1993d): Based on chemical shift changes, hydrogen bonds would be expected between carboxyls of Phe82 and Gly77 and amides of Gly85 and Leu87. These interactions appear to be present in solution and slight packing rearrangements may prevent these hydrogen bonds from being recognized in the crystalline state.
Figure 2.
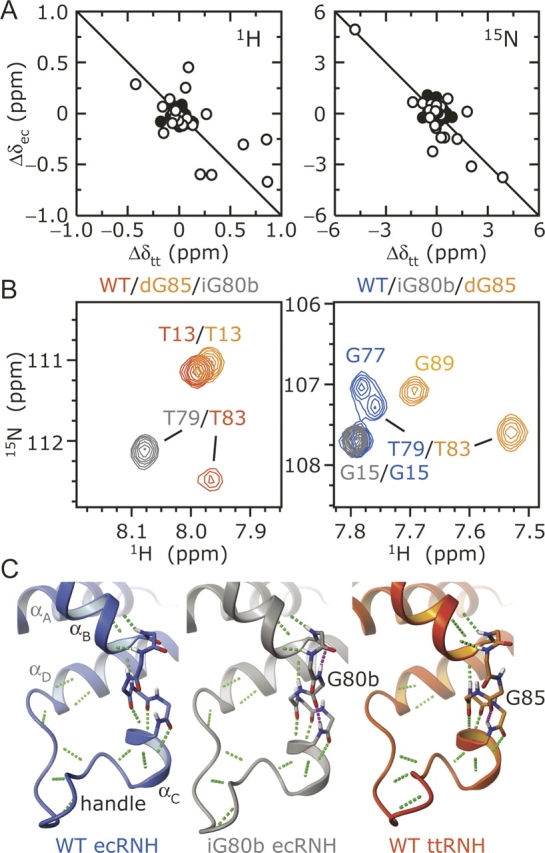
Structural consequences of the Gly insertion. (A) Chemical shift changes between WT and iG80b ecRNH (Δδec) and WT and dG85 ttRNH (Δδtt) are compared as Δδ = δmutant−δWT for backbone amide (left) 1H and (right) 15N nuclei. Residues in αB, αC and the handle (70–100 in ecRNH) are indicated by open circles; the correlation coefficient for these residues are −0.63 for 1H and −0.84 for 15N. (B) Overlay of 1H–15N correlation spectra for iG80b ecRNH and dG85 ttRNH with (left) WT ttRNH and (right) WT ecRNH highlighting the positions of Thr79 (ecRNH) and Thr83 (ttRNH). (C) Main-chain hydrogen bonds in the αB/αC hinge and handle are shown in green for WT ecRNH (blue, PDB 2RN2), iG80b ecRNH (gray, PDB 1GOA), and WT ttRNH (orange, PDB 1RIL). Additional hydrogen bonds seen in the crystal structures of iG80b ecRNH and WT ttRNH are shown in magenta.
Chemical exchange is more pronounced in the presence of the inserted Gly
Dynamic processes on the μs–ms time scale were identified by the excess contribution to backbone amide 15N transverse relaxation (R 2) from chemical exchange line broadening (R ex). A TROSY-based Hahn-echo experiment (Wang et al. 2003) was used to measure R ex for WT ecRNH and mutant RNases H at a static magnetic field of 14.1 T and a temperature (T) of 300 K (Fig. 3); data for WT ttRNH have been reported previously (Butterwick et al. 2004). In WT ecRNH, two residues show significant R ex (R ex ≥ 2.5 sec−1): Lys60 in the αA/β4 loop and Trp90 in the handle (Mandel et al. 1995, 1996; Kroenke et al. 1998; Yamasaki et al. 1998). WT ttRNH is characterized by an increase in both the number of residues influenced by chemical exchange and the magnitude of R ex. Specifically, Lys80, Thr83, Gly85, and Trp86 surrounding the αB/αC hinge, Trp95 and Arg96 within the handle, and His124 and Val 126 in β5 have significant values of R ex at 300 K (Butterwick et al. 2004).
Figure 3.

Chemical exchange line broadening in WT and mutant RNases H. Backbone amide 15N R ex at 300 K, 14.1 T for WT ecRNH, WT ttRNH, iG80b ecRNH, and dG85 ttRNH are shown. Residues with significant chemical exchange line broadening (R ex ≥ 2.5 sec−1) are indicated by open circles. Vertical dashed lines indicate the sites of mutation. Secondary structural elements are diagrammed at the top of the figure. Data for WT ttRNH are from Butterwick et al. (2004).
Insertion of Gly80b into ecRNH or deletion of Gly85 from ttRNH within the αB/αC hinge significantly alters chemical exchange line broadening. iG80b ecRNH shows an increase in both the number of residues influenced by chemical exchange line broadening and magnitude of R ex for residues within the handle and αB/αC hinge. In particular, Ile82, Arg88, Lys95, Lys99, and Asn100 show significant R ex not seen in the WT enzyme, while Trp90 shows a significant increase in R ex (over 10-fold). Relative to changes in the handle, chemical exchange for Lys60 is only moderately perturbed (decreased by 0.8-fold) in the iG80b ecRNH mutant. Conversely, dG85 ttRNH shows a significant decrease in R ex for residues in both the handle and β5. Specifically, residues near the Gly85 deletion no longer show significant R ex, and Trp95 in the handle and His124 and Val126 in β5 show decreased R ex, relative to the WT enzyme. Importantly, changes in R ex for both mutants are not localized solely to the site of mutation, but are distributed throughout the handle and other regions of the protein, suggesting that μs–ms time scale motions are highly coupled events that affect much of the enzyme.
Temperature dependence of chemical exchange
The temperature dependence of R ex provides information on the time scale and energetics of conformational transitions (Mandel et al. 1996; Evenäs et al. 1999; Butterwick et al. 2004). R ex measurements were repeated at temperatures between 285 K and 310 K for WT ecRNH and mutant RNases H; data for WT ttRNH have been reported previously (Butterwick et al. 2004). Values of R ex increase with decreasing temperature in both WT enzymes, indicating that chemical exchange is intermediate to fast on the chemical shift time scale (Fig. 4A; Mandel et al. 1996; Butterwick et al. 2004). In addition, although R ex is not a kinetic rate constant per se, the temperature dependence of R ex displays Arrhenius behavior over the temperature ranges studied, where the thermophilic enzyme is characterized by a larger “apparent” activation energy barrier (Ê a) (Fig. 4B; Butterwick et al. 2004). At T ≤ 290 K, Ile82 and Asn100 have significant R ex in WT ecRNH, but accurate Ê a could not be determined for these residues (see Materials and Methods).
Figure 4.

Temperature dependence of chemical exchange line broadening. (A) R ex measured at multiple temperatures and 14.1 T and (B) Arrhenius plots are shown for (left) Trp90 in WT and iG80b ecRNH and (right) Trp95 in WT and dG85 ttRNH. Open and closed symbols represent data for WT and mutant RNases H, respectively. Apparent activation energies, Ê a, derived from the slope of the lines in B are listed in Table 1. Square symbols represent data that deviate from a linear Arrhenius relationship, and are not included for determining Ê a. Data for WT ttRNH are from Butterwick et al. (2004).
In a similar manner to the WT enzymes, R ex in both mutants increases with decreasing temperature (Fig. 4A). However, despite the differences in R ex between WT and mutant enzymes, Ê a remain strikingly similar (Fig. 4B; Table 1) This is not true for all residues: His124 and Val126 have depressed Ê a in dG85 ttRNH relative to WT ttRNH. Amide nuclei in these residues are 14–16 Å away from the site of mutation, and backbone nuclei between the site of mutation and these residues are not preferentially broadened. Thus, indirect effects, such as time-dependent aromatic ring current shifts arising from conformational dynamics within the handle, are unlikely to contribute to the broadening of 15N nuclei in the loop, particularly given the low magnetogyric ratio of 15N. Thus, these results suggest coupling of motional processes between the handle and β5 near the active site. Removal of Gly85 from ttRNH likely disrupts this connection, but leaves motion within the handle relatively unperturbed. Although the relatively large uncertainty in Ê a for dG85 ttRNH renders quantitative comparisons difficult, the similarity between Trp95 in WT and dG85 ttRNH suggests that interactions within the handle determine the kinetic and thermodynamic properties of motion. In contrast to the WT proteins, R ex for both mutants deviate from Arrhenius behavior at low temperatures; these data are not included in the determination of Ê a.
Table 1.
Apparent activation energies for WT and mutant RNases H

Relaxation dispersion of backbone amide nuclei
To further characterize the exchange kinetics within WT and mutant RNases H, backbone amide 15N R 2(ω e) relaxation dispersion was measured at a temperature of 300 K for WT ecRNH, WT ttRNH, and iG80b ecRNH using R 1ρ spin relaxation techniques. The magnitude of exchange broadening in the dG85 ttRNH mutant is too small for accurate relaxation dispersion measurements (see Materials and Methods). All dispersion data are well described by a two-state exchange model
 |
characterized by a kinetic exchange rate constant k ex = k AB + k BA, where k AB and k BA are the forward and reverse rate constants between exchanging conformers A and B. On- and off-resonance R 1ρ experiments (Akke and Palmer 1996; Mulder et al. 1998; Massi et al. 2004) were combined to span effective fields of ω e ≈ 102–104 sec−1 at static magnetic fields of 11.7 and 18.8 T. For all exchanging residues in WT ecRNH and ttRNH, R ex(ω e) is barely suppressed at the highest ω e obtained, such that k ex >> 104 sec−1 for residues in both enzymes (Fig. 5; Table 2). In this regime, the individual forward and reverse rate constants and the thermodynamic and structural characteristics of the exchange process cannot be separated and only k ex and ϕ ex = p A p BΔω 2 can be determined, where p A and p B are the fractional populations of the conformers A and B, respectively (p A ≥ p B), and Δω is the chemical shift difference between exchanging conformers (Palmer 2004).
Figure 5.

R 1ρ relaxation dispersion for backbone amide 15N nuclei. R 2(ω e) dispersion results are shown for residues Trp90 and Lys60 in WT and iG80b ecRNH, and Trp95 and Val126 in WT ttRNH at 300 K. Closed and open symbols represent data collected at 11.7 and 18.8 T, respectively.
Table 2.
Relaxation dispersion parameters for WT and mutant RNases H at 300 K

In WT ecRNH, R ex(ω e) could be quantified for Lys60 in the αD/β4 loop, and Trp90 and Asn100 in the handle. Residues in the handle were individually fit with similar k ex ≈ 40,000–45,000 sec−1; subsequent global analysis of these residues yielded k ex = (42 ± 8) × 103 sec−1. In agreement with its reduced Ê a, Lys60 exhibited much faster kinetics with k ex = (100 ± 23) × 103 sec−1. Surprisingly, residues in WT ttRNH exhibited very similar exchange kinetics to residues in WT ecRNH. Individual fits of residues Lys80, Phe82, Thr83, Gly85, Trp86, Gly89, and Trp90 near the αB/αC hinge and Trp95, Arg96 and Arg106 in the handle region were characterized by k ex ≈ 30,000–50,000 sec−1. Interestingly, residues His124 and Val126 in β5 were also individually fit with k ex ≈ 40,000 sec−1, suggesting a common motional process affects residues in the handle and near the active site. In a similar manner for WT ecRNH, these residues could be globally described by k ex = (40 ± 4) × 103 sec−1. Thus, the larger values of R ex for residues in WT ttRNH are due to approximately threefold larger ϕ ex values and either the population distribution or chemical shift differences between the exchanging conformers are altered between the two enzymes.
In a similar manner to the WT enzymes, chemical exchange for all residues in iG80b ecRNH are fast on the chemical shift time scale (Fig. 5; Table 2). Residues in αC and the handle region were generally individually fit with slower kinetics than WT ecRNH with k ex ≈ 15,000–40,000 sec−1; subsequent global analysis of Trp81, Ile82, Arg88, Trp90, Thr92, Lys95, Lys99, and Asn100 yielded k ex = (27 ± 2) × 103 sec−1. The reduced k ex does not completely account for the increased R ex relative to WT ecRNH, and residues in iG80b ecRNH are fit with approximately fourfold larger ϕ ex. Relaxation dispersion for Lys60, however, yields k ex = (101 ± 30) × 103 sec−1, which is identical within experimental uncertainties to the value obtained for WT ecRNH. Thus, reduced R ex for Lys60 in iG80b ecRNH compared to WT is due to an ∼0.8-fold reduction in ϕ ex.
Interpreting chemical exchange effects
In the fast exchange regime, only k ex and ϕ ex can be determined from relaxation dispersion measurements; consequently, the underlying energetics and chemical shift changes of the conformational transition cannot be determined. However, ratios of k ex and ϕ ex provide information on the relative changes in the free energy difference between exchanging conformers (ΔΔG) and between transition states (ΔΔG ‡) for two proteins. Assuming that k AB and k BA are described by transition state theory, k AB = k oexp[−ΔG ‡/RT] and k BA = k oexp[−(ΔG ‡−ΔG)/RT], and that site populations follow a Boltzmann distribution, p B/p A = exp[−ΔG/RT], then
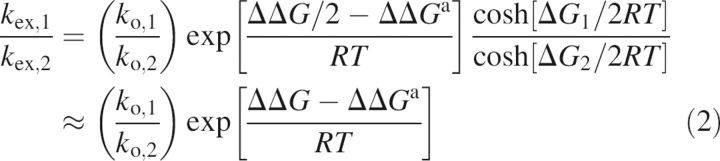 |
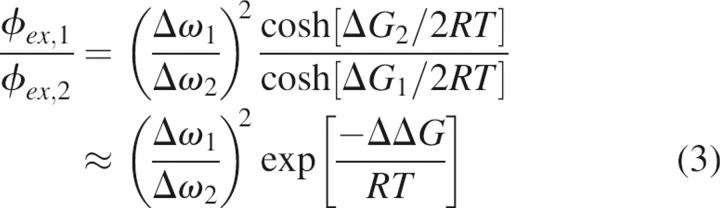 |
where the subscripts 1 and 2 represent the two proteins of interest, k o is the preexponential factor for k AB and k BA, R is the universal gas constant, and ΔG and ΔG ‡ are the energy differences of the minor conformer and transition state relative to the major conformer, respectively, and ΔΔG = ΔG 1 − ΔG 2 and ΔΔG ‡ = ΔG 1 ‡ − ΔG 2 ‡. The second equalities in Equations 2 and 3 arise if p A − p B ≈ 1 for either protein.
Given the small chemical shift changes between WT and iG80b ecRNH outside the site of mutation, the exchanging conformer structures probably are similar for both proteins, such that Δω WT ≈ Δω iG80b. In this case, using R 1ρ results for Trp90 (δN,iG80b − δN,WT = 0.41 ppm), ΔΔG = ΔG iG80b − ΔG WT = −3.6 ± 0.1 kJ/mol and ΔΔG ‡ = ΔG ‡ iG80b − ΔG ‡ WT = −2.5 ± 0.5 kJ/mol at 300 K. Thus, insertion of Gly80b reduces the stability difference between exchanging conformers, increasing p B relative to WT ecRNH (Fig. 6). The decreased stability difference between conformers is slightly larger than the decrease in activation energy, leading to slower kinetics in the mutant enzyme. Virtually identical results are obtained with data from Asn100.
Figure 6.

Summary of conformational changes in RNases H. Energy diagram comparing the relative stabilities and activation energies for exchanging conformers in WT ecRNH (blue), WT ttRNH (orange), and iG80b ecRNH (gray) at 300 K. Diagram is drawn to scale assuming p B = 1% for WT ecRNH and k o = k B T/h, where k B and h are Boltzmann's and Planck's constants, respectively.
The significant amino acid conservation and structural similarity between WT ecRNH and ttRNH also suggest that Δω probably are similar between these enzymes. Using R 1ρ results for Trp90 (ecRNH) and Trp95 (ttRNH) (δN,ttRNH − δN,ecRNH = 1.03 ppm), yields ΔΔG = ΔG WT,Tt − ΔG WT,Ec = −2.7 ± 0.1 kJ/mol and ΔΔG ‡ = ΔG ‡ WT,Tt − ΔG ‡ WT,Ec = −2.6 ± 0.6 kJ/mol at 300 K. Similar exchange kinetics are observed between WT enzymes because ΔΔG ≈ ΔΔG ‡; however, the minor conformer is stabilized relative to the major conformer in the thermophilic enzyme leading to a larger p B (Fig. 6). Most surprisingly, the increase in transition state and minor conformer stabilities seen in WT ttRNH is similar to that conferred by the Gly80b insertion into ecRNH.
Discussion
Conformational changes are ubiquitous in biology. While structural studies have identified numerous cases in which structural changes occur during a biological process, the roles of specific residues and local interactions in controlling thermodynamics and kinetics of structural transitions remain largely uncharacterized. In the present work, NMR spectroscopy was used to characterize μs–ms time scale motional processes in homologous mesophilic and thermophilic RNase H enzymes. Comparison of the conformational dynamic properties of WT and Gly insertion/deletion mutants was used to probe the role of local interactions surrounding a putative hinge in modulating dynamics within the substrate-binding handle.
A Gly insertion between αB and αC is conserved in thermophilic RNases H that utilize a handle region for RNA:DNA hybrid recognition, suggesting that this residue is important for biological function at high temperatures. This insertion is a focal point for additional intrahelical hydrogen bonds that marginally stabilize the protein. Chemical shifts surrounding this region are similar between WT ttRNH and iG80b ecRNH and between WT ecRNH and dG85 ttRNH, suggesting that interactions within the iG80b ecRNH mutant are more “thermophilic-like” while those in dG85 ttRNH are more “mesophilic-like.” The similarity between WT ttRNH and iG80b ecRNH is further reflected in their reduced catalytic efficiencies relative to WT ecRNH (Kanaya and Itaya 1992; Ishikawa et al. 1993c). dG85 ttRNH, however, does not show significantly increased catalytic activity at low temperatures (to resemble WT ecRNH), possibly due to the uncoupling of conformational changes between the handle and β5 near the active site (J.A. Butterwick and A.G. Palmer III, unpubl.). Thus, while the crystallographic structures of WT ttRNH and iG80b ecRNH display slight differences, both proteins likely incorporate the same additional hydrogen bonds in solution, compared to WT ecRNH and dG85 ttRNH.
Currently, the structural details of alternative conformations sampled in solution and leading to chemical exchange line broadening are unknown. Differences in R ex between WT and mutant RNases H are spread throughout residues in αC and the handle, suggesting that conformational changes within these regions are highly coupled. Large R ex are not observed for most residues, indicating that Δω are small and the local structure in αC and the handle is maintained. Because chemical exchange line broadening is not observed for residues Thr79, Gly80b and Asn84 in iG80b ecRNH, the additional hydrogen bonds most likely are maintained during conformational changes about this region. Residues Trp90 and Asn100 may serve as hinges for motions of the handle: Trp90 is the first and Asn100 is the last residue in the handle, both Trp90 and Asn100 show significant increase in R ex in the iG80b ecRNH mutant, and both Trp90 and Asn100 have left-handed helical backbone dihedral angles in the observed major conformation.
The hypothesis that chemical exchange reflects movement of the entire handle as a whole is supported by the dynamic properties of a thermostabilized ecRNH quintuple G23A/H62P/V74L/K95G/D134H mutant (Yamasaki et al. 1998). Mutation of the left-handed Lys95 to Gly is well accommodated within the handle of ecRNH (Ishikawa et al. 1993a) and significantly stabilizes the protein (Kimura et al. 1992a). While no global changes in dynamics were observed in the quintuple mutant compared to the WT protein, several residues surrounding the sites of mutation experienced a change in dynamics across multiple time scales. Interestingly, an increase in R ex was observed for residues Ile82, Trp90, Lys91, and Asn100 in the quintuple mutant (Yamasaki et al. 1998), although to a lesser extent than seen in iG80b ecRNH. Lys95 is 5–14 Å away from these residues, yet removal of its bulky side chain likely alters the thermodynamics or kinetics of structural transitions across the handle.
The remarkable similarity between the free energy profiles of iG80b ecRNH and WT ttRNH (Fig. 6) suggests that the Gly insertion plays a major role in modulating conformational dynamics of the handle region of RNase H. In iG80b ecRNH, both the transition and minor states are stabilized by a similar extent relative to WT ecRNH. Thus, the Gly insertion does not drastically alter the kinetic rate constants, but rather adjusts the thermodynamic equilibrium between conformers. Lacking a Cβ atom, Gly occupies approximately threefold more Ramachandran space compared to other amino acids. This structural plasticity appears to alleviate unfavorable interactions in the transition state and minor conformer(s) in RNase H.
For RNases H, the kinetics of the chemical exchange process are likely too fast to be rate limiting for enzymatic activity. Previous characterization of the WT proteins using a nonanucleotide hybrid substrate at 30°C yielded k cat ≈ 1.5 and 0.3 sec−1 for WT ecRNH and WT ttRNH, respectively (Kanaya et al. 1990; Kanaya and Itaya 1992). Given the current results, the lower limit for WT ecRNH is p B ≈ 1%, corresponding to k AB ≈ 400 sec−1 and k BA ≈ 41,600 sec−1, which are much faster than k cat. Thus, the altered thermodynamic distribution rather than changes in the kinetic rate constants afforded by the Gly insertion likely represents an important adaptation to fine tune conformational changes within RNase H for activity at high temperatures. The Gly80b insertion into ecRNH does not significantly stabilize the protein; thus, the mutant ecRNH enzyme is unfolded and inactive at the optimal temperature for activity in the wild-type thermophilic ttRNH enzyme (∼343 K) (Kanaya and Itaya 1992). Additional amino acid substitutions, such as H62P and V74L (Kimura et al. 1992b; Ishikawa et al. 1993b), may be required to confer increased thermal stability in order for the effects of the Gly insertion to be seen at higher temperatures.
Insertion of Gly80b into ecRNH has few effects on the ground-state structure outside of the αB/αC hinge region and insignificantly affects overall thermodynamic stability, but renders the structure of the αB/αC hinge region and enzymatic activity more similar to the hinge structure and enzymatic activity observed for WT ttRNH (Ishikawa et al. 1993c). The crystallographically observed structures of apo WT ecRNH, WT ttRNH, and iG80b ecRNH are consistent with the dominant conformations observed in solution by NMR spectroscopy, but these structures appear incapable of productive interactions between the handle and substrate (Iwai et al. 1996). The present results demonstrate that the potential energy surface controlling μs–ms time scale transitions to alternate higher energy conformations also is more similar in iG80b ecRNH to WT ttRNH than WT ecRNH. These observations imply that enzymatic activity of RNases H depends upon the structures and energetics of the alternative conformations that are sampled at equilibrium in solution.
Materials and methods
Sample preparation
Plasmids containing the WT RNase H coding sequences have been described previously (Mandel et al. 1995; Hollien and Marqusee 1999b). For improved solubility, ttRNH contains four Cys mutations (Hollien and Marqusee 1999b): C17A, C45S, C67A, and C154S. Gly insertion (GGT codon) and deletion mutations were created using the Quik-Change protocol (Stratagene). WT and mutant RNases H were expressed in E. coli BL21(DE3) (Stratagene) using M9 minimal media (Sambrook et al. 1989) prepared with 99% (v/v) deuterium oxide and [98% 15N]-ammonium chloride (Cambridge Isotope Laboratories, Inc.) and purified as previously described (Kroenke et al. 1998; Hollien and Marqusee 1999b). 2H incorporation at nonexchangeable proton sites was estimated to be >85% for all samples by mass spectrometry. Final protein concentrations were 1.0 mM for WT and iG80b ecRNH, and 0.3 mM for WT and dG85 ttRNH in 100 mM d 3-sodium acetate at pH 5.5, 10% (v/v) deuterium oxide, 0.02% (w/v) sodium azide, and 3 mM sodium 2,2-dimethyl-2-silapentane-sulfonate (DSS); ecRNH samples also contained 1 mM d 10-dithiothreitol.
NMR spectroscopy
All NMR data were collected at Columbia University (11.7 and 14.1 T) or the New York Structural Biology Center (16.4 and 18.8 T). Data at static magnetic fields of 11.7 and 18.8 T were acquired on Bruker DRX500 and AV800, respectively, equipped with triple-resonance probes and three-axis gradients. Data at static magnetic fields of 14.1 and 16.4 T were acquired on Bruker DRX600 and AV700, respectively, equipped with triple-resonance cryoprobes with Z-axis gradients. For all experiments, the temperature was calibrated using a 100% (v/v) methanol standard (Cavanagh et al. 1996). Data processing and analysis were performed with NMRPipe (Delaglio et al. 1995) and Sparky (T.D. Goddard and D.G. Kneller, Sparky 3, University of California, San Francisco) software, along with in-house written programs.
Resonance assignments for mutant RNases H
Backbone 1H and 15N resonance assignments for iG80b ecRNH and dG85 ttRNH were determined by comparison with WT spectra supplemented by HMQC-NOESY-HSQC spectra (Cavanagh et al. 1996) acquired at a static magnetic field of 16.4 T and a temperature of 300 K. The spectra were recorded using with (t 1 × t 2 × t 3) 48 × 32 × 768 complex points and 2.857 × 2.857 × 10 kHz spectral widths. Assignments, referenced to internal DSS, have been deposited to the BioMagResBank (http://www.bmrb.wisc.edu) under accession codes 7277 (dG85 ttRNH) and 7278 (iG80b ecRNH).
Analysis of chemical exchange line broadening
For residues subject to chemical exchange line broadening, R 2 = R 2° + R ex, where R 2° is the intrinsic transverse relaxation rate constant which is sensitive to motion on the ps–ns time scale and the protein size. A TROSY-based Hahn-echo sequence (Wang et al. 2003) was used to measure R 2° and R ex at a static magnetic field of 14.1 T and temperatures between 285 K and 310 K. This method uses the transverse 15N chemical shift anisotropy/1H–15N dipolar interference rate constant (η xy) to determine R 2° = κη xy, where κ is the trimmed mean R 2/η xy ratio for nonexchanging residues. Values of κ were between 1.31 and 1.47 at all temperatures for all proteins. Analysis of the data with a weighted mean κ = 1.40 does not significantly change the results. Relaxation data were collected in an interleaved manner with (t 1 × t 2) 150 × 1024 complex points and 2.5 × 10 kHz spectral widths. A minimum of four experiments were acquired at each temperature for each protein. The average and standard deviation were taken as the relaxation rate constant and uncertainty, respectively.
Apparent activation energies, Ê a, were determined from the temperature dependence of chemical exchange assuming an Arrhenius relationship for R ex:
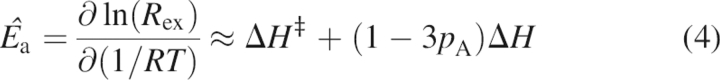 |
where ΔH ‡ is the transition state enthalpy for the forward reaction and ΔH is the difference in enthalpy between the two exchanging states (Mandel et al. 1996; Evenäs et al. 1999; Butterwick et al. 2004). The second equality assumes that exchange is fast (k ex >> Δω). For each residue, Ê a was determined from a linear fit of ln(R ex) versus 1/T using R ex data acquired at a minimum of three temperatures. At low temperatures, deviations from linearity may become apparent if the exchange kinetics slow to the intermediate regime (k ex ≈ Δω); these data are not included in the determination of Ê a.
R1ρ relaxation dispersion analysis
Chemical exchange line broadening is suppressed in the presence of an effective field, ω e. In an R 1ρ experiment, magnetization is spin-locked parallel to ω e by the application of a continuous radiofrequency (rf) field with frequency ω rf and amplitude ω 1. The magnitude of the effective field is ω e 2 = ω 1 2 + ΔΩ2, where ΔΩ = Ωo − ω rf is the observed resonance offset from the applied field. The relaxation rate constant, R 1ρ, for magnetization aligned parallel to ω e is given by
where R 1 is the longitudinal relaxation rate constant, θ = arctan(ω 1/ΔΩ) is the tilt angle of the effective field, and
is the effective transverse relaxation constant where R ex(ω e) is the residual R ex.
R 1 and on-resonance R 1ρ relaxation rate constants were measured at a temperature of 300 K and static magnetic fields of 11.7 and 18.8 T using relaxation time delays between 0 and 1.2 sec and 0 and 100 msec, respectively (Farrow et al. 1994; Massi et al. 2004). Seven to nine unique time points were sampled with three duplicates for estimation of the error in signal intensity. The relaxation rate constants were determined from the best single exponential fit to the data with the error estimated using a jackknife algorithm. Off-resonance R 1ρ relaxation rate constants were measured from pairs of interleaved spectra with relaxation delays of 2 msec and 60–200 msec assuming a single exponential decay (Akke and Palmer 1996; Mulder et al. 1998); errors were estimated based on repeat experiments. For on-resonance experiments, magnetization was aligned using a free precession period of 1/ω 1 (Yamazaki et al. 1994; Massi et al. 2004), while off-resonance experiments utilized 10 msec tanh/tan adiabatic pulses each with a frequency sweep of 15 kHz (Mulder et al. 1998). For all R 1ρ experiments, ω 1 was calibrated using the residual scalar coupling in the presence and absence of the applied field (Palmer et al. 2001).
Analysis of relaxation dispersion data followed a similar protocol previously described (Grey et al. 2006). Initially, R 1 and R 1ρ relaxation data at both static magnetic fields along with R 2° and free precession R ex estimated from TROSY Hahn-echo experiments acquired at 11.7 and 18.8 T were analyzed on a per-residue basis using Equations 5 and 6. F-statistical testing (at α = 0.05 confidence level) was used to select the appropriate functional form for R ex(ω e): either chemical exchange is not present, with R ex(ω e) = 0; or exchange is fast on the chemical shift time scale (k ex >> ω e), with
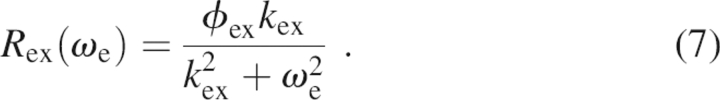 |
Additional R ex(ω e) models that include exchange outside the fast time scale regime (Trott and Palmer 2002), or a three-site exchange mechanism (Grey et al. 2003) did not significantly improve the fitted parameters. Optimization of R 1 and R 2° rate constants and ϕ ex and k ex exchange parameters were accomplished with routines written in Mathematica (v4.1, Wolfram Research, Inc.) utilizing the Levenberg-Marquart algorithm. Off-resonance R 1ρ data were included only if 20° ≤ θ ≤ 65° while on-resonance R 1ρ data required ΔΩ ≤ |0.4ω 1/2π|. Residues characterized by similar k ex values by inspection were subsequently analyzed globally with a single k ex, maintaining individual ϕ ex.
Acknowledgments
We thank members of the Palmer laboratory for helpful discussions, and M. Grey and F. Massi additionally for assistance with R 1ρ measurements and critical reviews of the manuscript. This work was supported by National Institutes of Health (NIH) Grant GM50291 (A.G.P.). A.G.P. is a member of the New York Structural Biology Center supported by NIH Grant GM66354.
Footnotes
Reprint requests to: Arthur G. Palmer III, Department of Biochemistry and Molecular Biophysics, Columbia University, 650 West 168 Street, Black Building 507, New York, NY 10032, USA; e-mail: agp6@columbia.edu; fax: (212) 305-6949.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062398606.
References
- Akke, M. 2002. NMR methods for characterizing microsecond to millisecond dynamics in recognition and catalysis. Curr. Opin. Struct. Biol. 12: 642–647. [DOI] [PubMed] [Google Scholar]
- Akke, M. and Palmer III, A.G. 1996. Monitoring macromolecular motions on microsecond to millisecond time scales by R 1ρ − R 1 constant relaxation time NMR spectroscopy. J. Am. Chem. Soc. 118: 911–912. [Google Scholar]
- Benkovic, S.J. and Hammes-Schiffer, S. 2003. A perspective on enzyme catalysis. Science 301: 1196–1202. [DOI] [PubMed] [Google Scholar]
- Butterwick, J.A., Loria, J.P., Astrof, N.S., Kroenke, C.D., Cole, R., Rance, M., and Palmer III, A.G. 2004. Multiple time scale backbone dynamics of homologous thermophilic and mesophilic ribonuclease HI enzymes. J. Mol. Biol. 339: 855–871. [DOI] [PubMed] [Google Scholar]
- Cavanagh, J., Fairbrother, W.J., Palmer III, A.G., and Skelton, N.J. 1996. Protein NMR spectroscopy: Principles and practice. Academic Press, San Diego, CA.
- Codreanu, S.G., Ladner, J.E., Xiao, G., Stourman, N.V., Hachey, D.L., Gilliland, G.L., and Armstrong, R.N. 2002. Local protein dynamics and catalysis: Detection of segmental motion associated with rate-limiting product release by a glutathione transferase. Biochemistry 41: 15161–15172. [DOI] [PubMed] [Google Scholar]
- Cole, R. and Loria, J.P. 2002. Evidence for flexibility in the function of ribonuclease A. Biochemistry 41: 6072–6081. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- Eisenmesser, E.Z., Millet, O., Labeikovsky, W., Korzhnev, D.M., Wolf-Watz, M., Bosco, D.A., Skalicky, J.J., Kay, L.E., and Kern, D. 2005. Intrinsic dynamics of an enzyme underlies catalysis. Nature 438: 117–121. [DOI] [PubMed] [Google Scholar]
- Evenäs, J., Forsén, S., Malmendal, A., and Akke, M. 1999. Backbone dynamics and energetics of a calmodulin domain mutant exchanging between closed and open conformations. J. Mol. Biol. 289: 603–607. [DOI] [PubMed] [Google Scholar]
- Farrow, N.A., Muhandiram, R., Singer, A.U., Pascal, S.M., Kay, C.M., Gish, G., Shoelson, S.E., Pawson, T., Forman-Kay, J.D., and Kay, L.E. 1994. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33: 5984–6003. [DOI] [PubMed] [Google Scholar]
- Grey, M.J., Wang, C.Y., and Palmer III, A.G. 2003. Disulfide bond isomerization in basic pancreatic trypsin inhibitor: Multisite chemical exchange quantified by CPMG relaxation dispersion and chemical shift modeling. J. Am. Chem. Soc. 125: 14324–14335. [DOI] [PubMed] [Google Scholar]
- Grey, M.J., Tang, Y., Alexov, E., McKnight, C.J., Raleigh, D.P., and Palmer III, A.G. 2006. Characterizing a partially folded intermediate of the villin headpiece domain under non-denaturing conditions: Contribution of His41 to the pH-dependent stability of the N-terminal subdomain. J. Mol. Biol. 355: 1078–1094. [DOI] [PubMed] [Google Scholar]
- Hollien, J. and Marqusee, S. 1999a. Structural distribution of stability in a thermophilic enzyme. Proc. Natl. Acad. Sci. 96: 13674–13678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien, J. and Marqusee, S. 1999b. A thermodynamic comparison of mesophilic and thermophilic ribonucleases H. Biochemistry 38: 3831–3836. [DOI] [PubMed] [Google Scholar]
- Ishikawa, K., Kimura, S., Kanaya, S., Morikawa, K., and Nakamura, H. 1993a. Structural study of mutants of Escherichia coli ribonuclease HI with enhanced thermostability. Protein Eng. 6: 85–91. [DOI] [PubMed] [Google Scholar]
- Ishikawa, K., Nakamura, H., Morikawa, K., and Kanaya, S. 1993b. Stabilization of Escherichia coli ribonuclease HI by cavity-filling mutations within a hydrophobic core. Biochemistry 32: 6171–6178. [PubMed] [Google Scholar]
- Ishikawa, K., Nakamura, H., Morikawa, K., Kimura, S., and Kanaya, S. 1993c. Cooperative stabilization of Escherichia coli ribonuclease HI by insertion of Gly-80b and Gly-77 → Ala substitution. Biochemistry 32: 7136–7142. [DOI] [PubMed] [Google Scholar]
- Ishikawa, K., Okumura, M., Katayanagi, K., Kimura, S., Kanaya, S., Nakamura, H., and Morikawa, K. 1993d. Crystal structure of ribonuclease H from Thermus thermophilus HB8 refined at 2.8 Å resolution. J. Mol. Biol. 230: 529–542. [DOI] [PubMed] [Google Scholar]
- Iwai, S., Wakasa, M., Ohtsuka, E., Kanaya, S., Kidera, A., and Nakamura, H. 1996. Interaction of the basic protrusion of Escherichia coli ribonuclease HI with its substrate. J. Mol. Biol. 263: 699–706. [DOI] [PubMed] [Google Scholar]
- Jaenicke, R. and Böhm, G. 1998. The stability of proteins in extreme environments. Curr. Opin. Struct. Biol. 8: 738–748. [DOI] [PubMed] [Google Scholar]
- Kanaya, S. and Itaya, M. 1992. Expression, purification, and characterization of a recombinant ribonuclease H from Thermus thermophilus HB8. J. Biol. Chem. 267: 10184–10192. [PubMed] [Google Scholar]
- Kanaya, S., Kohara, A., Miura, Y., Sekiguchi, A., Iwai, S., Inoue, H., Ohtsuka, E., and Ikehara, M. 1990. Identification of the amino acid residues involved in an active site of Escherichia coli ribonuclease H by site-directed mutagenesis. J. Biol. Chem. 265: 4615–4621. [PubMed] [Google Scholar]
- Kanaya, S., Katsuda-Nakai, C., and Ikehara, M. 1991. Importance of the positive charge cluster in Escherichia coli ribonuclease HI for the effective binding of the substrate. J. Biol. Chem. 266: 11621–11627. [PubMed] [Google Scholar]
- Kay, L.E. 2005. NMR studies of protein structure and dynamics. J. Magn. Reson. 173: 193–207. [DOI] [PubMed] [Google Scholar]
- Keck, J.L. and Marqusee, S. 1995. Substitution of a highly basic helix/loop sequence into the RNase H domain of human immunodeficiency virus reverse transcriptase restores its Mn2+-dependent RNase H activity. Proc. Natl. Acad. Sci. 92: 2740–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern, D. and Zuiderweg, E.R. 2003. The role of dynamics in allosteric regulation. Curr. Opin. Struct. Biol. 13: 748–757. [DOI] [PubMed] [Google Scholar]
- Kimura, S., Kanaya, S., and Nakamura, H. 1992a. Thermostabilization of Escherichia coli ribonuclease HI by replacing left-handed helical Lys95 with Gly or Asn. J. Biol. Chem. 267: 22014–22017. [PubMed] [Google Scholar]
- Kimura, S., Nakamura, H., Hashimoto, T., Oobatake, M., and Kanaya, S. 1992b. Stabilization of Escherichia coli ribonuclease HI by strategic replacement of amino acid residues with those from the thermophilic counterpart. J. Biol. Chem. 267: 21535–21542. [PubMed] [Google Scholar]
- Kohen, A. and Klinman, J.P. 2000. Protein flexibility correlates with degree of hydrogen tunneling in thermophilic and mesophilic alcohol dehydrogenases. J. Am. Chem. Soc. 122: 10738–10739. [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14: 51–55: 29–32. [DOI] [PubMed] [Google Scholar]
- Kovrigin, E.L. and Loria, J.P. 2006. Enzyme dynamics along the reaction coordinate: Critical role of a conserved residue. Biochemistry 45: 2636–2647. [DOI] [PubMed] [Google Scholar]
- Kroenke, C.D., Loria, J.P., Lee, L.K., Rance, M., and Palmer III, A.G. 1998. Longitudinal and transverse 1H−15N dipolar/15N chemical shift anisotropy relaxation interference: Unambiguous determination of rotational diffusion tensors and chemical exchange effects in biological macromolecules. J. Am. Chem. Soc. 120: 7905–7915. [Google Scholar]
- Kumar, S., Tsai, C.J., and Nussinov, R. 2001. Thermodynamic differences among homologous thermophilic and mesophilic proteins. Biochemistry 40: 14152–14165. [DOI] [PubMed] [Google Scholar]
- Mandel, A.M., Akke, M., and Palmer III, A.G. 1995. Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J. Mol. Biol. 246: 144–163. [DOI] [PubMed] [Google Scholar]
- Mandel, A.M., Akke, M., and Palmer III, A.G. 1996. Dynamics of ribonuclease H: Temperature dependence of motions on multiple time scales. Biochemistry 35: 16009–16023. [DOI] [PubMed] [Google Scholar]
- Massi, F., Johnson, E., Wang, C., Rance, M., and Palmer III, A.G. 2004. NMR R 1ρ rotating-frame relaxation with weak radio frequency fields. J. Am. Chem. Soc. 126: 2247–2256. [DOI] [PubMed] [Google Scholar]
- Mulder, F.A.A., de Graaf, R.A., Kaptein, R., and Boelens, R. 1998. An off-resonance rotating frame relaxation experiment for the investigation of macromolecular dynamics using adiabatic rotations. J. Magn. Reson. 131: 351–357. [DOI] [PubMed] [Google Scholar]
- Oda, Y., Iwai, S., Ohtsuka, E., Ishikawa, M., Ikehara, M., and Nakamura, H. 1993. Binding of nucleic acids to E. coli RNase HI observed by NMR and CD spectroscopy. Nucleic Acids Res. 21: 4690–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer III, A.G. 2004. NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 104: 3623–3640. [DOI] [PubMed] [Google Scholar]
- Palmer III, A.G., Kroenke, C.D., and Loria, J.P. 2001. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 339: 204–238. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., Fritsch, E.F., and Maniatis, T. 1989. Molecular cloning: A laboratory manual2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tousignant, A. and Pelletier, J.N. 2004. Protein motions promote catalysis. Chem. Biol. 11: 1037–1042. [DOI] [PubMed] [Google Scholar]
- Trott, O. and Palmer III, A.G. 2002. R 1ρ relaxation outside of the fast-exchange limit. J. Magn. Reson. 154: 157–160. [DOI] [PubMed] [Google Scholar]
- Vieille, C. and Zeikus, G.J. 2001. Hyperthermophilic enzymes: Sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 65: 1–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wand, A.J. 2001. Dynamic activation of protein function: A view emerging from NMR spectroscopy. Nat. Struct. Biol. 8: 926–931. [DOI] [PubMed] [Google Scholar]
- Wang, C., Rance, M., and Palmer III, A.G. 2003. Mapping chemical exchange in proteins with MW > 50 kD. J. Am. Chem. Soc. 125: 8968–8969. [DOI] [PubMed] [Google Scholar]
- Williams, J.C. and McDermott, A.E. 1995. Dynamics of the flexible loop of triosephosphate isomerase: The loop motion is not ligand gated. Biochemistry 34: 8309–8319. [DOI] [PubMed] [Google Scholar]
- Wolf-Watz, M., Thai, V., Henzler-Wildman, K., Hadjipavlou, G., Eisenmesser, E.Z., and Kern, D. 2004. Linkage between dynamics and catalysis in a thermophilic–mesophilic enzyme pair. Nat. Struct. Mol. Biol. 11: 945–949. [DOI] [PubMed] [Google Scholar]
- Xu, X.P. and Case, D.A. 2002. Probing multiple effects on 15N, 13Cα, 13Cβ, and 13C′ chemical shifts in peptides using density functional theory. Biopolymers 65: 408–423. [DOI] [PubMed] [Google Scholar]
- Yamasaki, K., Akasako-Furukawa, A., and Kanaya, S. 1998. Structural stability and internal motions of Escherichia coli ribonuclease HI: 15N relaxation and hydrogen-deuterium exchange analyses. J. Mol. Biol. 277: 707–722. [DOI] [PubMed] [Google Scholar]
- Yamazaki, T., Muhandiram, R., and Kay, L.E. 1994. NMR experiments for the measurement of carbon relaxation properties in highly enriched, uniformly 13C, 15N-labeled proteins: Application to 13Cα carbons. J. Am. Chem. Soc. 116: 8266–8278. [Google Scholar]