

Abstract
Cytolethal distending toxin (CDT) secreted by Actinobacillus actinomycetemcomitans induces cell cycle arrest of cultured cells in the G2 phase. The crystal structure of the natural form of A. actinomycetemcomitans DCT (aCDT) has been determined at 2.4 Å resolution. aCDT is a heterotrimer consisting of the three subunits, aCdtA, aCdtB, and aCdtC. Two crystallographically independent aCDTs form a dimer through interactions of the aCdtB subunits. The primary structure of aCDT has 94.3% identity with that of Haemophilus ducreyi CDT (hCDT), and the structure of aCDT is quite similar to that of hCDT reconstituted from the three subunits determined recently. However, the molecular packings in the crystal structures of aCDT and hCDT are quite different. A careful analysis of molecular packing suggests that variation of the amino acid residues in a nonconserved loop (181TSSPSSPERRGY192 of aCdtB and 181NSSSSPPERRVY192 of hCdtB) is responsible for the different oligomerization of very similar CDTs. The loop of aCdtB has a conformation to form a dimer, while the loop conformation of hCdtB prevents hCDT from forming a dimer. Although dimerization of aCDT does not affect toxic activity, it changes the stability of protein. aCDT rapidly aggregates and loses toxic activity in the absence of sucrose in a buffered solution, while hCDT is stable and retains toxic activity. Another analysis of crystal structures of aCDT and hCDT suggests that the receptor contact area is in the deep groove between CdtA and CdtC, and the characteristic “aromatic patch” on CdtA.
Keywords: Cytolethal distending toxin, CDT, Actinobacillus actinomycetemcomitans, crystal structure, oligomerization, stability and toxic activity
Cytolethal distending toxin (CDT) is a bacterial toxin that induces cell cycle arrest of cultured cells in the G2 phase. It has been identified in several pathogenic bacteria including Campylobacter spp. (Johnson and Lior 1988a,b), Escherichia coli (Peres et al. 1997), Shigella dysenteriae (Okuda et al. 1995), Haemophilus ducreyi (Cope et al. 1997), Actinobacillus actinomycetemcomitans (Sugai et al. 1998), and Helicobacter hepaticus (Young et al. 2000). Cells incubated with CDT show a doubling in size of the cytoplasm and the nucleus, followed by cell death (Peres et al. 1997; Sugai et al. 1998; Whitehouse et al. 1998; Deng et al. 2001).
CDT is composed of three subunits, CdtA, CdtB, and CdtC, which form a tripartite complex (Saiki et al. 2001, 2004; Lara-Tejero and Gala’n 2002). CdtA and CdtC are required for the delivery of CdtB, the active subunit (Lara-Tejero and Gala’n 2001; Deng and Hansen 2003; Lee et al. 2003; Shenker et al. 2004). The mechanism of action of this toxin is reasonably well understood. On delivery into host cells by CdtA and CdtC, the active subunit CdtB is transported to the nucleus and causes DNA damage (Elwell and Dreyfus 2000; Lara-Tejero and Gala’n 2000). CdtB has an amino acid sequence similar to the DNase I family of proteins. Indeed, E. coli CdtB and hCdtB have been demonstrated to possess nicking activity toward purified plasmid in vitro (Elwell and Dreyfus 2000; Nešić et al. 2004). In CDT-treated cells, Thr 14 and Tyr 15 in CDC2 were found to be phosphorylated, while these residues were not phosphorylated in uninfected cells, suggesting that CdtB directly damages chromosomal DNA, which results in destruction of the phosphorylation/dephosphorylation cascade (Elwell and Dreyfus 2000).
The recent report of the Surgeon General has estimated that in the United States, severe periodontal disease affects 14% of adults aged 45–54 and 23% of adults aged 65–74 (Office of the Surgeon General 2000). Periodontitis is the result of the response of the periodontium to the presence of certain members of the oral microbiota. One of the most severe forms of periodontal disease is localized aggressive periodontitis (LAP). A. actinomycetemcomitans is one of the key organisms responsible for the pathogenesis of LAP (Slots and Ting 1999). A. actinomycetemcomitans secretes a large number of proteins (Kirby et al. 1995) including leukotoxin (Lally et al. 1999; Narayan et al. 2002), CDT (Sugai et al. 1998; Mayer et al. 1999), chaperonin 60 with potent leukocyte-activating and bone-resorbing activities (Kirby et al. 1995). This organism also produces proteins that inhibit eukaryotic cell cycle progression and proteins and peptides that can induce distinct forms of proinflammatory cytokine networks (Henderson et al. 2002). CDT secreted from A. actinomycetemcomitans has been reported to show a variety of activities. In KB (Saiki et al. 2001), HeLa (Sugai et al. 1998), and HEp-2 (Akifusa et al. 2001) cells, the toxin causes cell distension and cell cycle arrest in the G2 phase, and eventually elicits cell death. The toxin also blocks the proliferation of T lymphocytes to induce immunosuppression (Shenker et al. 1999). One ofthe characteristic features of aCDT is its extreme instability, its propensity to aggregate, and its loses of toxic activity in the absence of sucrose in buffered solutions. An attempt to determine the degree of oligomerization by gel filtration failed because the holotoxin (heterotrimer aCDT) adsorbed to the rapid molecular-sieve chromatography HPLC column, even when the running buffer contained sucrose (Saiki et al. 2004).
A crystal structure of Haemophilus ducreyi CDT (hCDT) reconstituted from the individually purified recombinant subunits (CdtA, CdtB, and CdtC) has been determined, and the assembly of three subunits has been elucidated (Nešić et al. 2004). The DNA binding site on the CdtB subunit and the nicking active site have been proposed on the basis of the structure.
Here we present the crystal structure of naturally formed aCDT from A. actinomycetemcomitans. From the crystal structures of aCDT and hCDT, the instability of aCDT is rationally explained and receptor recognition site is predicted.
Results and Discussion
N-terminal deletion (AΔ19–47CDT) effect
The wild-type A. actinomycetemcomitans CDT (aCDT) purified from an E. coli ER2566 expression system contains two different sizes of CdtA; one starts at residue 59, and the other starts at residue 60 (Saiki et al. 2004). Wild-type aCDT has not been crystallized. On the other hand, a mutant aCDT (AΔ19–47CDT) purified from an E. coli ER2566 expression system used in this study has a single size of CdtA (starts at residue 53), and has been crystallized. The wild-type and mutated aCDT have the same level of toxic activity. The N-terminal of CdtB and CdtC are also truncated at residues 22 and 20, respectively, suggesting that the N-terminal sections of CdtA, CdtB, and CdtC of aCDT are quite susceptible to protease digestion, and these truncated residues are not important for the toxic activity. Similarly, the N-terminal sections of CdtA, CdtB, and CdtC of Haemophilus ducreyi CDT (hCDT) were truncated at residues 17, 22, and 20, respectively (Nešić et al. 2004).
Degree of oligomerization of aCDT holotoxin in solution
An attempt to determine the molecular mass in solution by a gel filtration failed (Saiki et al. 2004). We tried to determine the molecular size in solution by using a light-scattering apparatus. The obtained molecular size is relatively large (~ 310 Å in size), suggesting that monomeric aCDT is not present in a sucrose-containing buffer solution. As will be discussed below, the crystal structure indicates aCDT forms an elongated dimer (~ 140 Å in the maximum length). From the light-scattering data and the crystallographic data, aCDT is most likely a dimer or larger oligomer in solution.
Overall structure of aCDT
The crystal structure of recombinant aCDT composed of CdtA(53–223), CdtB(23–283), and CdtC(21–186) was determined by X-ray diffraction analysis at 2.4 Å resolution. The crystallographic refinement parameters (Table 1), final (2Fo − Fc) maps, and conformational analysis by PROCHECK (Laskowski et al. 1993) suggest that the crystal structure of aCDT has been determined with acceptable statistics. The crystallographic asymmetric unit contains two holotoxins (aCDT-1 and aCDT-2), which are related by a noncrystallographic twofold symmetry to form a dimer (Fig. 1). The light-scattering experiment also suggested that aCDT forms a oligomer in the presence of sucrose (10%). Since the structures of aCDT-1 and aCDT-2 are identical within experimental error (CA RMSD < 0.05 Å), the structure of aCDT-1 is used for the following descriptions. Amino acid residues and loops with a prime belong to aCDT-2.
Table 1.
Crystallographic statistics
| Space group, P212121; Unit cell dimension a = 115.65, b = 117.46, c = 123.36 Å ; Mr = 74,516; number of holotoxins in the unit cell = 8; VM =2.81 Å3; percentage of solvent content, 56%. | |
| Resolution (Å) | 2.4 |
| Total observations | 1,826,944 |
| Unique reflections | 66,570 |
| Completeness (%) | 97.2 |
| Rsyma outer shellc | 0.068 (0.240) |
| Protein nonhydrogen atoms | 9195 |
| Solvent molecules (H2O) | 277 |
| Resolution range (Å) | 20.0–2.4 |
| Total reflection used in Rcryst | 66,570 |
| Total reflection used in Rfree | 6746 |
| Rcrystb (outer shell)c | 0.221 (0.243) |
| Rfree (outer shell)c | 0.251 (0.279) |
| RMSD of bond distances (Å) | 0.010 |
| RMSD of bond angles (°) | 1.4 |
| RMSD of torsion angles (°) | 22.5 |
| Most favored region (%) of Ramachandran plot | 92.3 |
| Additional allowed region (%) of Ramachandran plot | 7.7 |
aRsym = ∑h∑i |Ihi − 〈Ih〉| / ∑h∑i|Ihi|.
bRcryst = ∑| Fo − Fc | / ∑|Fo|.
c Outer shell = 2.4–2.5 Å resolution.
Figure 1.
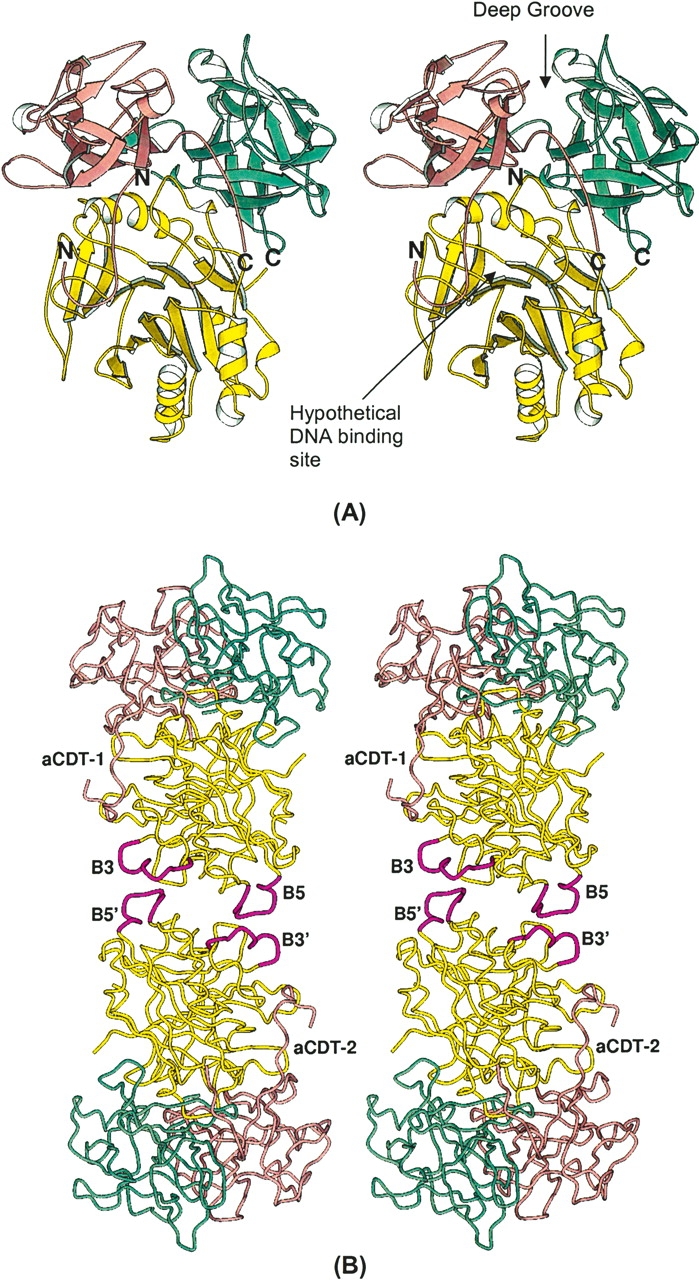
(A) Heterotrimeric aCDT. The CdtA, CdtB, and CdtC subunits are shown by aquamarine, yellow, and light pink colors, respectively. (B) An elongated dimeric aCDT. Loop B3 and B5 of aCdtB are shown with thicker coils in magenta. The detail interactions between loop B3 and B5 are illustrated in Figure 5A.
The aCDT holotoxin looks like a “fat stem mushroom”, in which the subunit CdtA and CdtC form a “cap” and attach on the end of subunit CdtB (“fat stem”). Subunits CdtA and CdtC have similar structures and have three sets of β-sheets composed of four or three β-strands, respectively, which consist of a pseudo threefold symmetric trefoil of β-strands (Supplemental Fig. 1A,B). These structures are homologous to the B-chain repeats of the plant toxin, ricin (Montfort et al. 1987). All cysteine residues in aCDT are present in these two subunits, four in CdtA and four in CdtC. A previous study using nonreducing SDS-PAGE indicated that CdtA and CdtC contain disulfide bonds (Saiki et al. 2004). Indeed, the two pairs of cysteine residues in each subunit are found. These cysteine pairs are C137–C150 (2.5 Å) and C179–C198 (2.5 Å) in CdtA, and C96–C107 (3.0 Å) and C135–C149 (2.6 Å) in CdtC. In the aCDT structure, CdtA and CdtC bind at one end of CdtB, contacting each other (Fig. 1A). The three subunit assembly of CDT is quite similar to the repeat arrangement in the ricin B-chain and its interaction with the catalytic active subunit (A-chain) (Montfort et al. 1987), although CdtA and CdtC cannot be superimposed as a rigid unit over the ricin repeats (Supplemental Fig. 2). A characteristic long and deep groove (~20 × 10 Å long and 5–8 Å deep) is formed from structural components of both CdtA and CdtC (Fig. 1). The other interesting feature is a large aromatic patch consisting of nine bulky aromatic side chains (“aromatic patch”), which passes through the middle of the surface of CdtA (Fig. 2). These two characteristic surface elements on CdtA and CdtC are directed away from CdtB. Similar characteristic structural features are also observed in the two lectin repeats in the ricin B-chain. The function of these two characteristic elements is likely to be receptor binding to initiate endocytosis of the complex (Lara-Tejero and Gala’n 2001).
Figure 2.

The characteristic “aromatic patch” seen in the aCdtA and hCdtA structures, which might have contacts with the receptor on the target cell. The aCdtA (aquamarine) and hCdtA (light pink) structures superimpose, and the aromatic residues are illustrated. Except for residue 189, all aromatic residues are superimposable.
The structure of CdtB has a pseudo twofold symmetry, and possesses the DNase I family elements. The two β-sheets composed of six and seven β-strands, respectively, face each other to form a core β-sandwich packed between outer α-helices and loops on each side of the sandwich. In each β-sheet, the two central β-strands are parallel to each other, and the others are an antiparallel arrangement. There is a large cleft at the C-terminal ends of the parallel β-strands, which is covered by the N-terminal 13 amino acid residues of CdtC extending away from the globular domain (Fig. 1A). The cleft is hypothesized to be the DNA binding site, and has the essential DNA binding residues R117, R144, and H160 (Nešić et al. 2004). The core structure of CdtB is quite similar to those of DNase I, HAP1 (human DNA repair endonuclease) (Gorman et al. 1997), and ExoIII (exonuclease III of E. coli) (Mol et al. 1995).
A comparison of aCDT and hCDT holotoxins
aCDT used in this study is produced and assembled in E. coli (i.e., the holotoxin is expressed in an E. coli). Recently, hCDT has been reconstituted from the individually purified recombinant CdtA(18–223), CtdB(23–283), and CdtC(21–186) subunits, and its crystal structure has been determined (Nešić et al. 2004). Except for the N terminus length of the CdtA subunit, the natural form aCDT and reconstituted form hCDT have the same number of the amino acid residues, and have similar polypeptide folds in their crystal structures. The CA RMSD between aCDT and hCDT is 0.71 Å, suggesting that the hCDT used in the recent crystal structure was reconstituted correctly.
Differences between the aCDT and hCDT structures induced by nonconserved amino acid residues
The amino acid sequences of aCDT and hCDT have a great deal of similarity (91.5% identity for CdtA, 97.0% identity for CdtB, and 93.5% identity for CdtC) (Table 2). Interestingly, the catalytic subunit CdtB has the least nonconserved sites among the three subunits, and the amino acid residues in its hypothetical DNA binding cleft are completely conserved between aCDT and hCDT (Fig. 3). All of the nonconserved sites are located on the surface of the protein except for residue 118 (I/M) of CdtA, suggesting that most variations have little effect on the tertiary and quaternary structures of the toxin. However, there are a few significant differences between the aCDT and hCDT structures, and some of them are in the nonconserved residue regions (Table 3; Fig. 3). aCDT is rapidly aggregated and loses toxic activity in solution, while hCDT is stable. Some of the variations of the surface residues could be related to the stability of the individual holotoxins.
Table 2.
The nonconserved sites seen in aCDT and hCDT
| Subunit | Nonconserved sites (aCDT/hCDT) |
| CdtA | 7(G/S), 13(L/S), 23(S/N), 41(T/I), 43(F/P), 52(A/T), 53(S/P), 64(K/G), 65(-/P), 67(G/R), 70(S/L), 118(I/M), 138(I/V), 149(T/I), 187(P/T), 189(Y/F), 197(T/V), 205(L/S) |
| CdtB | 8(N/S), 13(T/V), 181(T/N), 184(P/S), 186(S/P), 191(G/V), 205(V/A), 209(A/V) |
| CdtC | 11(S/I), 15(T/A), 17(T/A), 51(I/V), 100(P/L), 102(E/K), 110(I/T), 136(M/I), 145(R/K), 151(I/G), 177(R/I), 181(G/R) |
Figure 3.

A view showing the large CA RMSD regions and the nonconserved sites. The large CA RMSD regions CdtA, CdtB, and CdtC are indicated by thick coils with red, magenta, and blue, respectively, and are listed in Table 3. The nonconserved sites are indicated by black dots with residue numbers and are listed in Table 2. The “aromatic patch“ residues are shown by red dots.
Table 3.
Relatively large RMSD regions between aCDT and hCDT structures
| Range | No. of residues | RMSD (Å) | Mean B (Å2) of aCDT | Mean B (Å2) of hCDT | |
| CdtA | 71–223 | 153 | 0.32a | 9.2 | 13 |
| A1 | 57–70 | 14 | — | Disordered | 16 |
| A2 | 120–124 | 5 | 0.63 | 22 | 21 |
| A3 | 151–154 | 4 | 0.74 | 16 | 22 |
| A4 | 161 | 1 | 0.72 | 9.5 | 12 |
| A5 | 189 | 1 | 1.00 | 8.9 | 14 |
| CdtB | 23–283 | 258 | 0.30a | 7.8 | 16 |
| B1 | 55 | 1 | 0.72 | 13 | 17 |
| B2 | 82–83 | 2 | 0.81 | 35 | 47 |
| B3 | 131–143 | 13 | 3.67 | 7.9 | 20 |
| B4 | 171 | 1 | 0.79 | 4.9 | 16 |
| B5 | 181–191 | 11 | >1.10b | 8.8 | 34 |
| B6 | 212–215 | 4 | 0.97 | 22 | 31 |
| B7 | 246 | 1 | 0.99 | 5.9 | 23 |
| B8 | 263–264 | 2 | 0.75 | 4.1 | 12 |
| CdtC | 25–178 | 154 | 0.33a | 16 | 25 |
| C1 | 54 | 1 | 0.74 | 23 | 23 |
| C2 | 92–93 | 2 | 0.86 | 37 | 18 |
| C3 | 99–105 | 7 | 2.84 | 37 | 18 |
| C4 | 150–151 | 2 | 1.06 | 27 | 20 |
a RMSDs are calculated excluding the regions with large RMSDs.
b Residues 183–185 of hCdtB were disordered, and were not included in the RMSD calculation. Therefore, the RMSD of residue 181–191 should be >1.10 Å.
There are 12 sites that have nonconserved residues in the CdtA(53–223) structure (Table 2; Fig. 3). The most variable region is from residue 63 to 70 (63SKNGQVSP70 vs. 63SGPNRQVLP70) (A1 in Fig. 3). The N terminus of aCdtA is disordered up to residue 70 (i.e., residues 53–70 are disordered), indicating that these residues are not strongly associated with the main body. On the other hand, the residues 18–56 of hCdtA are also disordered but the residues 57–70 form an ordered “U” shape, and are inserted into the deep groove between the CdtA and CdtC subunits of a neighboring hCDT (Fig. 4B). The other site is from residue 186 to 190 (186SPTYY190 vs. 186STTFY190) (A5 in Fig. 3), which forms a loop located on the edge of the deep groove between the CdtA and CdtC subunits. This loop is also the end of the “aromatic patch” on the CdtA surface. The CA and side chain of Y189 of aCdtA are significantly moved from those of F189 of hCdtA (Fig. 2). Actually, the deviations of A1 and A5 are strongly related. In the hCDT crystal structure, the “U” shaped A1 section is inserted into the deep groove of the neighboring hCDT and interacts with loop A5. On the other hand, there is no such interaction in the aCDT structure because the A1 section is disordered.
Figure 4.
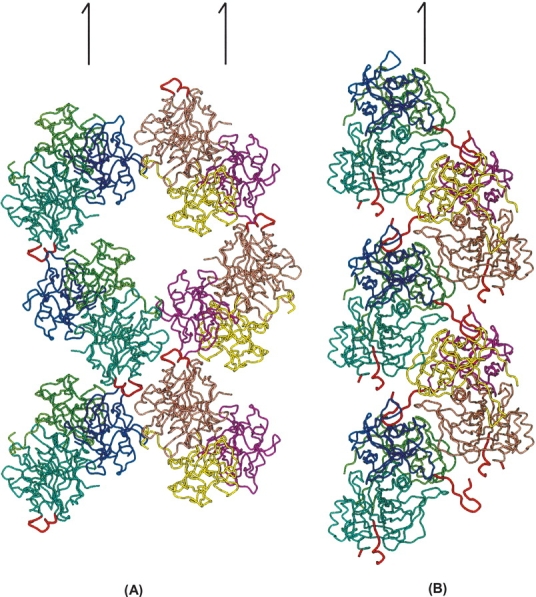
The basic molecular interactions seen in the aCDT and hCDT crystal structures. The significant difference in molecular packing schemes is mainly due to a different degree of oligomerization (i.e., dimeric aCDT vs. monomeric hCDT). (A) Two molecular chains, aCDT-1 chain (blue base color) and aCDT-2 (orange base color), along 21 axes parallel to the crystallographic b form a two-dimensional net sheet structure. It is noted that the crystallographically independent aCDT-1 and aCDT-2 form a dimer around the noncrystallographic twofold symmetry, and two additional aCDTs contact the dimer connection section to form a “cross” structure. (B) Two molecular chains (blue and orange base colors) in the hCDT crystal are related by a 21 axis parallel to the crystallographic b axis. The two chains packed tightly and the N-terminal section of hCdtA is inserted into the deep groove of the 21 related hCDT, and loop B5 is exposed in solvent region and is disordered. Characteristic N-terminal section of hCDT and essentially different loop B5 are illustrated by red coils. Half arrows indicate crystallographic 21 axes parallel to b axis.
There are six sites that have nonconserved residues in the CdtB(23–283) structure. The most variable region is from residue 181 to 192 (181TSSPSSPERRGY192 vs. 181NSSSSPPERRVY192) (loop B5 in Fig. 3). This region of hCdtB is partially disordered (183SSS185 is completely disordered) and is not involved in any intermolecular contact, whereas the same region of aCdtB has a well-defined structure (mean B value is 8.8 Å2) and has strong intermolecular interaction with 135HSDSSVL141 (loop B3 in Fig. 3) of CdtB-2 (dimer partner). The CA RMSDs of the two regions are the largest in the CdtB structure (3.67 Å for loop B3 and >1.10 Å for loop B5). The other interesting difference is seen in residues 263LN264 (B8 in Fig. 3). These residues in both aCdtB and hCdtB have relatively small B values. Residues 263LN264 are located on the intersurface between CdtA and CdtB, and make contact with the nonconserved residue 118 (I/M) of CdtA. The relatively large CA RMSD of 263LN264 is apparently due to the variation of residue 118 (I/M) of CdtA.
There are nine sites that have nonconserved residues in the CdtC(23–178) structure. The region from residue 98 to 106 has two nonconserved residues (98FPGEGTTD106 vs. 98FLGKGTTD106) (C3 in Fig. 3), forms a loop, and has the largest CA RMSD, 2.84 Å in the CdtC structure. The loop in aCdtC is slightly disordered (mean B = 37 Å2), but the same loop in hCdtC has an ordered structure (mean B = 18 Å2), although the loops of aCdtC and hCdtC are not involved in intermolecular contacts. The large CA RMSD and the different B-values of the loops are most likely due to the unusual variation of residue 101. Residues 101 in aCdtC and hCdtC are Glu (E) and Lys (K), respectively, which have opposite charges. The other large CA RMSD is seen around the residue 151, where aCdtC and hCdtC have different amino acid residues (Ile vs. Gly).
Without exception, the sites (64[K/G] and 67[G/R] in CdtA, 191[G/V] in CdtB, 151[I/G] in CdtC), in which Gly residues are replaced with other amino acid residues, have relatively large CA RMSDs through the CDT structures. This observation is consistent with the fact that Gly is able to have various φ and ψ angles, whereas those angles of other amino acids are limited. In connection to the Gly mutation, the “aromatic patch” was mutated with Gly, in order to examine its role of the toxic activity. Quadruple W91G, W98G, Y100G, and Y102A mutations of hCdtA destroyed the hCDT activity toward HeLa cells (Nešić et al. 2004). However, as described above, the result of the quadruple mutations is not reliable because replacement of Trp or Tyr with Gly most likely changes the backbone conformation since the φ and ψ angles of Gly are less restricted than those of the other amino acid residues.
Variation of loop B5 changes the oligomerization and stability of aCDT and hCDT
A dynamic light-scattering experiment suggests that aCDT forms a dimer in solution while hCDT is a monomer in solution (Nešić et al. 2004). Nonconservative amino acid residues between aCDT and hCDT do not change the tertiary and quaternary structures, but do significantly alter their molecular packing in the crystals. The nonconservative residues in aCDT and hCDT, which participate in intermolecular interactions in the crystal structures of aCDT and hCDT are: aCdtA: P187, Y189; hCdtA: G64, P65, R67, T187, F189; aCdtB: P184, S186; and hCdtB: V191.
Since residues 187 and 189 in CdtA are involved in intermolecular interactions in both aCDT and hCDT crystal structures, as will be discussed below, these residues might be involved in receptor recognition. Variations of the rest of residues (residues 64[K/G], 65[-/P], 67[G/R] in CdtA, and 184[P/S], 186[S/P], 191[G/V] in CdtB) apparently account for the molecular packing differences. As shown in Figure 4, these residues belong in the N terminus of CdtA (A1) and loop B5 in CdtB, and are involved in determining basic molecular packing in the crystals. The two molecular chains of aCDT (aCDT-1 chain and aCDT-2 chain) along the b-axis form a two-dimensional net structure with large solvent pools (VM = 2.81 Å3), whereas the two molecular chains of hCDT related by a 21 symmetry are packed tightly (VM = 2.35 Å3) and form a unbranched chain structure. This packing difference is apparently caused by variations of the amino acid residues in loop B5 (181TSSPSSPERRGY192 vs. 181NSSSSPPERRVY192) of CdtB. Loop B5 of aCdtB interacts with loop B3′ (residues 135–141) of the other aCDT-2, and consequently, the crystallographically independent aCDT-1 and aCDT-2 form a twofold dimer through interactions of their aCdtB subunits (Fig. 1B). Although there is no direct H-bond between loop B5 and B3′, two water molecules trapped between loop B5 and B3′ mediate H-bonds between the loops (Fig. 5A). On the other hand, loop B5 of hCdtB is exposed to the solvent region and is disordered. Loop B5 is composed of mostly hydrophilic amino acid residues, and variation of amino acid residues in the loop does not drastically change the polarity of the loop, suggesting that loop B5 changes its conformation by variation of nonconserved amino acid residues. When the nonconserved residues 181, 184, 186, and 191 in the aCdtB structure are replaced with those of hCdtB, respectively, without changing the backbone conformation, obvious problems are seen in the residues 181(T/N) and 186(S/P). For example, variation of residue 181(T/N) requires a significant shift of the CA position because N181 has a longer side chain than T181, and both T181 and N181 form similar H-bonds with W194 and a bound water molecule (Fig. 5B). Actually, deviations of the φ and ψ angles between aCdtB and hCdtB are significantly increased from residue 181 to 191 (The RMSD of φ and ψ angles in residues 181 and 187–191 is 35.0°, whereas that in residues 171–180 and 192–201 is 4.0°). These evidences indicate that variations of the nonconserved residues in loop B5 change the backbone conformation significantly, and consequently, loop B5 of aCdtB has a favorable conformation to make intermolecular contact with loop B3′ of aCdtB-2, whereas loop B5 of hCdtB increases its mobility.
Figure 5.
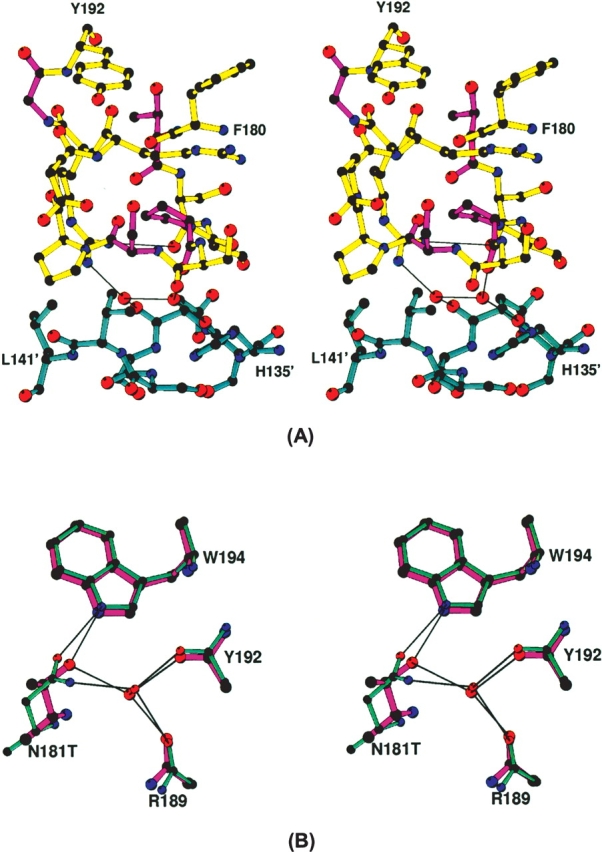
(A) A detailed interaction between loop B5 (yellow) of aCDT-1 and B3′ (cyan) of aCDT-2. The nonconserved residues T181, P184, S186, and G191 are illustrated with magenta colored bonds. H-bonds through the bound two water molecules are illustrated by thin lines. (B) A superimposed view of aCdtB (magenta) and hCdtB (aqua-marine) showing the CA shift by variation of T/N at residue 181, and similar H-bonding. H-bonds are shown by thin lines.
In the two-dimensional net structure of aCDT, two aCDT dimers contact pointing their CdtA to the connection section of third dimer from both sides, so that three aCDT dimers form a “cross” structure around the noncrystallographic twofold symmetry (Fig. 4A). From the crystal structure, sucrose might prevent the “cross” formation by coating the aCdt surface. In conclusion, variations of four amino acid residues in loop B5 (181TSSPSSPERRGY192 vs. 181NSSSSPPERRVY192) change the conformation of the loop, and consequently, change the intermolecular interactions of aCDT and hCDT drastically. The dimeric aCDTs relatively rapidly form a “cross” structure and lose toxic activity, whereas hCDT stays a monomer and retains toxic activity.
The toxic activities of aCDT and hCDT against KB and HeLa, respectively, are nearly the same (0.75 ng/mL vs. 1 ng/mL) (Nešić et al. 2004; Saiki et al. 2004). However, in the absence of sucrose aCDT aggregates and loses toxic activity. Consequently, aCDT is much less toxic than hCDT in a normal environment(i.e., low carbohydrate environment). A. actinomycetemcomitans is present in normal bacterial flora of the mouth of a healthy individual and secretes aCDT to protect themselves from bacterium and virus attacks. In general, bacteria and viruses grow slowly in a low carbohydrate (such as glucose, maltose, sucrose) environment in the mouth, and aCDT is quickly aggregated and loses the toxic activity. On the other hand, bacteria and viruses grow rapidly in a high carbohydrate environment, and aCDT stays the active form and suppresses the bacterium and virus growth in the mouth. Also, high aCDT activity might cause periodontal diseases (Tan et al. 2002; Leung et al. 2005).
Receptor contact site
It is believed that CdtA and CdtC recognize the CDT receptor on the target cell and deliver the catalytic active CdtB into the target cell (Deng et al. 2003; Lee et al. 2003; Shenker et al. 2004). The primary sequences of CDTs from various organisms are generally not very conserved. Notable differences are seen in the amino acid sequences of CdtA and CdtC subunits. For example, the percentages of amino acid identity between E. coli CDT and aCDT are 25.9% for CdtA, 48% for CdtB, and 19.2% for CdtC (Pickett and Whitehouse 1999). The nonconserved features found in the CDT subunits (especially in CdtA and CdtC) might show the species–specific adaptation of a CDT toxin to an infection strategy of a CDT-producing bacterium.
Since the surface of CDT, which interacts with the receptor (protein) on the target cells, is believed to be relatively hydrophobic in character, it tends to participate in intermolecular contacts in crystals. A careful examination of the intermolecular contacts in the CDT crystal structures might reveal the receptor contact surfaces of CDT. The two independent aCDT holotoxins (aCDT-1 and aCDT-2) in the crystal structure have identical structure, and have similar intermolecular contacts in the crystal structure (Fig. 6A,B). Although the structure of hCDT is quite similar to that of aCDT, the intermolecular interactions of hCDT are quite different from those of aCDT (Fig. 6C). Most of the differences of the intermolecular contacts are artifacts due to the molecular packing energy. Since aCDT and hCDT have quite similar amino acid sequences and structures, the overall receptor recognition schemes of aCDT and hCDT are expected to be similar rather than drastically different. Therefore, the common intermolecular contact area in the three CDTs (aCDT-1, aCDT-2, and hCDT) are most likely the receptor contact surfaces of aCDT and hCDT. A careful examination shows that there are two common intermolecular contact areas in aCDT and hCDT (Fig. 6). These are: (1) Around residue 141 of CdtA where the end of the “aromatic patch” is. (2) Around residue 187 of CdtA where the other end of the “aromatic patch” and the surface of the deep groove between CdtA and CdtC are.
Figure 6.
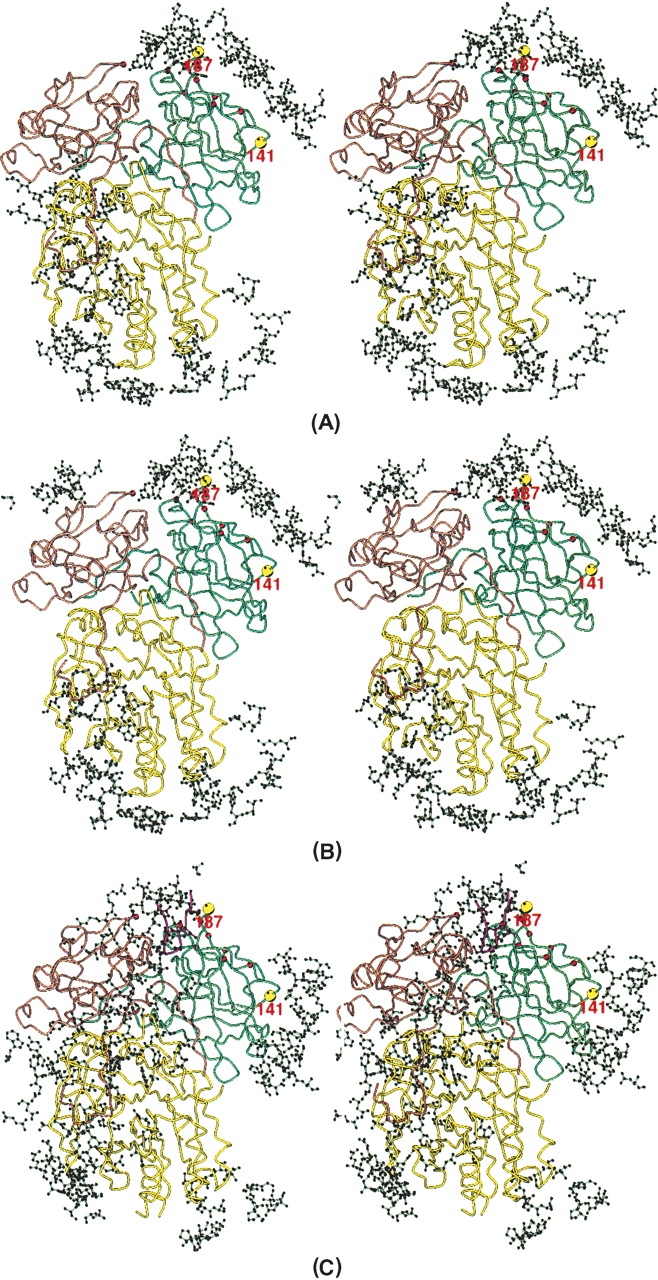
Intermolecular contacts seen in aCDT-1 (A), aCDT-2 (B), and hCDT (C). Amino acid residues that contact holotoxin within 7 Å are illustrated with a ball-and-stick presentation. The common intermolecular contact areas around residues 141 and 187 are shown by large yellow circles.
Interestingly, both the deep groove and the “aromatic patch” predicted to be the receptor contact areas are characteristic structure features of CDT and located close together on CdtA. It has been predicted that the deep groove between CdtA and CdtC is the receptor contact area because the catalytic active CdtB is not transported into target cells without both CdtA and CdtC (Deng and Hansen 2003; Lee et al. 2003; Shenker et al. 2004). It is important that the receptor contact area deduced from the crystal structures of aCDT and hCDT agrees to the predicted receptor contact area deduced from various cloning experiments (Deng et al. 2003; Lee et al. 2003; Shenker et al. 2004). Although insertion of the N-terminal of hCDT into the deep groove of neighboring hCDT is an artifact due to the molecular packing in the crystal, this interaction might be a mimic of receptor–toxin interaction.
Materials and methods
Purification and crystallization procedures
A. actinomycetemcomitans AΔ19–47CDT (aCDT) used in this study is the recombinant toxin produced in E. coli ER2566 (pTYT7-cdtAΔ19–47-B-His-BamHI-C) (Saiki et al. 2004). AΔ19–47CDT was purified from E. coli extracts using a Ni2+-chelating Sepharose Fast Flow column (Amersham Pharmacia Biotech AB) (Saiki et al. 2004). An SDS-PAGE analysis showed a heterotrimer structure composed of CdtA, CdtB, and CdtC in a nearly 1:1:1 stoichiometry. The N-terminal amino acid sequences of CdtA, CdtB, and CdtC indicated that the N-termini were truncated by proteolysis at residues 52, 22, and 20, respectively (Saiki et al. 2004). The purified AΔ19–47CDT showed the same strong CDT activity as that of the wild-type CDT. Both wild-type CDT and AΔ19–47CDT were only stable, and have toxic activity in the presence of sucrose (10% [w/v]) in a buffered solution.
The hanging-drop vapor-diffusion method was employed for crystallization of aCDT. Crystals were grown in a solution containing 50 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 6.5), 10% 2-methyl-2,4-pentanediol (MPD), 10% sucrose, 4% (w/v) polyethylene glycol molecular weight 4000 (PEG-4000) with a protein concentration of 10 mg/mL in a 22°C incubator. Plate-shaped crystals suitable for X-ray diffraction studies were obtained using a microseeding method.
Data measurement
Diffraction data were collected from cryo-cooled crystals at beamline 19BM at the Advance Photon Source in the Argonne National Laboratory. The crystals (~0.3 × 0.2 × 0.1 mm) diffract up to 2.3 Å, and one set of data was collected at 2.4 Å resolution. The data were processed with the program DENZO/SCALEPACK (Otwinowski and Minor 1997). Data statistics are given in Table 1.
Structure determination
The unit cell dimensions and the space group indicate that an asymmetric unit contains two heterotrimer CDTs. The crystal structure was initially determined by a molecular replacement procedure using a poly-Ser model of hCDT (PDB code: 1SR4). The side chains of the polypeptide chain were built in (2Fo − Fc) maps. The model was refined by the simulated annealing procedures of CNS (Brünger et al. 1998). (2Fo − Fc) and (Fo − Fc) maps showed that the N-termini of CdtA, CdtB, and CdtC started at residues 71, 23, and 25, respectively, indicating that the residues 53–70 of CdtA and residues 21–24 of CdtC are disordered in the crystal structure. During the final refinement stage, well-defined residual electron density peaks in difference maps were assigned to water molecules if peaks were able to bind to the protein molecules with hydrogen bonds. The two CDTs in the asymmetric unit were restrained to have the same structure in order to improve the accuracy of the structure. The final crystallographic R-factor was 0.221 for all data (no sigma cutoff) from 20.0 to 2.4 Å resolution. The free-R for 10% randomly selected data is 0.251.
Molecular size determination in solution by a dynamic light scattering
The molecular size of aCDT in solution was examined by using a dynamic light-scattering apparatus (DynaPro, Protein Solution, Inc.). A solution of aCDT (10 mg/mL), used for the above crystallizations, was diluted in 50 mM Tris/HCl buffer (pH 8.0) containing 10% sucrose to a final concentration of 1 mg/mL. The sample was filtered through a Whatman Anodisc 0.1-μm pore size filter. The dynamic light scatterings from the sample were measured at 25° C. The data were analyzed by the associated program (Dynamics version 5.25.44). The molecular size of aCDT in solution deduced from dynamic light scattering data is ~310 Å. The same experiment was repeated several times, and similar molecular sizes were obtained. The crystal structure indicates that the maximum dimensions of aCDT monomer and elongated dimer is ~75 Å and ~140 Å, respectively. When the 0.01-μm pore size filter was used, we could not get enough light-scattering signals, indicating that aCDT is a dimer or larger oligomer in solution.
Coordinates
The atomic coordinate of aCDT has been deposited with the Broovkhaven Protein Data Bank under accession code 2F2F.
Electronic supplemental material
The supplemental material contains two figures showing the CdtA and CdtC subunits and difference between the CdtA–CdtC of CDT and the B-chain of ricin.
Acknowledgments
We thank Professor Richard H. Himes for a critical reading of this manuscript and very valuable comments. This work has been supported by the American Heart Association Grant 0455454Z, and the National Institutes of Health Grant GM37233. Use of the Argonne National Laboratory Structural Biology Center beamline at the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Energy Research, under Contract No. W-31-109-ENG-38. This publication was made possible by NIH Grant Number P20 RR-17708 from the National Center for Research Resources.
Abbreviations
A. actinomycetemcomitans, Actinobacillus actinomycetemcomitans
CDT, cytolethal distending toxin
aCDT, A. actinomycetemcomitans CDT
hCDT, Haemophilus ducreyi CDT
aCdtA, subunit A of aCDT
aCdtB, subunit B of aCDT
aCdtC, subunit C of aCDT
hCdtA, subunit A of hCDT
hCdtB, subunit B of hCDT
hCdtC, subunit C of hCDT
CA RMSD, root-mean-square-deviation of CA positions between aCDT and hCDT
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051790506.
Supplemental material: see www.proteinscience.org
References
- Akifusa, S., Poole, S., Lewthwaite, J., Henderson, B., and Nair, S.P. 2001. Recombinant Actinobacillus actinomycetemcomitans cytolethal distending toxin proteins are required to interact to inhibit human cell cycle progression and to stimulate human leukocyte cytokine synthesis. Infect. Immun. 69: 5925–5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D54: 905–921. [DOI] [PubMed] [Google Scholar]
- Cope, L., Lumbley, S., Latimer, J., Klesney-Tait, J., Stevens, M., Johnson, L., Purven, M., Munson, R.J., Lagergard, T., Radolf, J., et al. 1997. A diffusible cytotoxin of Haemophilus ducreyi. Proc. Natl. Acad. Sci. 94: 4056–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, K. and Hansen, E.J. 2003. A CdtA–CdtC complex can block killing of HeLa cells by Haemophilus ducreyi cytolethal distending toxin. Infect. Immun. 71: 6633–6640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, K., Latimer, J.L., Lewis, D.A., and Hansen, E.J. 2001. Investigation of the interaction among the components of the cytolethal distending toxin of Haemophilus ducreyi. Biochem. Biophys. Res. Commun. 20: 609–615. [DOI] [PubMed] [Google Scholar]
- Elwell, C.A. and Dreyfus, L. 2000. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol. Microbiol. 37: 952–963. [DOI] [PubMed] [Google Scholar]
- Gorman, M.A., Morera, S., Rothwell, D.G., de La Fortelle, E., Mol, C.D., Tainer, J.A., Hickson, I.D., and Freemont, P.S. 1997. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 16: 6548–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson, B., Wilson, M., Sharp, L., and Ward, J.M. 2002. Actinobacillus actinomycetemcomitans. J. Med. Microbiol. 51: 1013–1020. [DOI] [PubMed] [Google Scholar]
- Johnson, W.M. and Lior, H. 1988a. A new heat-labile cytolethal distending toxin (CLDT) produced by Escherichia coli isolates from clinical material. Microb. Pathog. 4: 103–113. [DOI] [PubMed] [Google Scholar]
- ———. 1988b.Anew heat-labile cytolethal distending toxin (CLDT) produced by Campylobacter spp. Microb. Pathog. 4: 115–126. [DOI] [PubMed] [Google Scholar]
- Kirby, A.C., Meghji, S., Nair, S.P., White, P., Reddi, K., Nishihara, T., Nakashima, K., Willis, A.C., Sim, R., Wilson, M., et al. 1995. The potent bone resorbing mediator of Actinobacillus actinomycetemcomitans is homologous to the molecular chaperone GroEL. J. Clin. Invest. 96: 1185–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lally, E.T., Hill, R.B., Kieba, I.R., and Korostoff, J. 1999. The interaction between RTX toxins and target cells. Trends Microbiol. 7: 356–361. [DOI] [PubMed] [Google Scholar]
- Lara-Tejero, M. and Gala’n, J.E. 2000. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 290: 354–357. [DOI] [PubMed] [Google Scholar]
- ———. 2001. CdtA, CdtB, and CdtC form a tripartite complex that is required for cytolethal distending toxin activity. Infect. Immun. 69: 4358–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2002. Cytolethal distending toxin: Limited damage as a strategy to modulate cellular functions. Trends Microbiol. 10: 147–152. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Lee, R.B., Hassane, D.C., Cottle, D.L., and Pickett, C.L. 2003. Interactions of Campylobacter jejuni cytolethal distending toxin subunits CdtA and CdtC with HeLa cells. Infect. Immun. 71: 4883–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung, W.K., Ngai, V.K., Yau, J.Y., Cheung, B.P., Tsang, P.W., and Corbet, E.F. 2005. Characterization of Actinobacillus actinomycetemcomitans isolated from young Chinese aggressive periodontal patients. J. Periodontal Res. 40: 258–268. [DOI] [PubMed] [Google Scholar]
- Mayer, M.P.A., Bueno, L.A., Hansen, E.J., and DiRienzo, J.M. 1999. Identification of a cytolethal distending toxin gene locus and features of a virulence-associated region in Actinobacillus actinomycetemcomitans. Infect. Immun. 67: 1227–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mol, C.D., Kuo, C.F., Thayer, M.M., Cunningham, R.P., and Tainer, J.A. 1995. Structure and function of the multifunctional DNA-repair enzyme exonuclease III. Nature 374: 381–386. [DOI] [PubMed] [Google Scholar]
- Montfort, W., Villafranca, J.E., Monzingo, A.F., Ernst, S.R., Katzin, B., Rutenber, E., Xuong, N.H., Hamlin, R., and Robertus, J.D. 1987. The three-dimensional structure of ricin at 2.8 A¢ª . J. Biol. Chem. 262: 5398– 5403. [PubMed] [Google Scholar]
- Narayan, S.M., Nagaraja, T.G., Chengappa, M.M., and Stewart, G.C. 2002. Leukotoxins of Gram-negative bacteria. Vet. Microbiol. 84: 337–356. [DOI] [PubMed] [Google Scholar]
- Nešić, D., Hsu, Y., and Stebbins, C.E. 2004. Assembly and function of a bacterial genotoxin. Nature 429: 429–433. [DOI] [PubMed] [Google Scholar]
- Office of the Surgeon General. 2000. Oral Health in America: A report of the Surgeon General US Public Health Service. http://www.surgeongeneral.gov/library/reports.htm.
- Okuda, J., Kurazono, H., and Takeda, Y. 1995. Distribution of the cytolethal distending toxin A gene (cdtA) among species of Shigella and Vibrio, and cloning and sequencing of the cdt gene from Shigella dysenteriae. Microb. Pathog. 18: 167–172. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Peres, S.Y., Marches, O., Daigle, F., Nougayrede, J.P., Herault, F., Tasca, C., De Rycke, J., and Oswald, E. 1997. A new cytolethal distending toxin (CDT) from Escherichia coli producing CNF2 blocks HeLa cell division in G2/M phase. Mol. Microbiol. 24: 1095–1107. [DOI] [PubMed] [Google Scholar]
- Pickett, C.L. and Whitehouse, C.A. 1999. The cytolethal distending toxin family. Trends Microbiol. 7: 292–297. [DOI] [PubMed] [Google Scholar]
- Saiki, K., Konishi, K., Gomi, T., Nishihara, T., and Yoshikawa, M. 2001. Reconstitution and purification of cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Microbiol. Immunol. 45: 497–506. [DOI] [PubMed] [Google Scholar]
- Saiki, K., Gomi, T., and Konishi, K. 2004. Deletion and purification studies to elucidate the structure of the Actinobacillus actinomycetemcomitans cytolethal distending toxin. J. Biochem. (Tokyo) 136: 335–342. [DOI] [PubMed] [Google Scholar]
- Shenker, B.J., McKay, T., Datar, S., Miller, M., Chowhan, R., and Demuth, D. 1999. Actinobacillus actinomycetemcomitans immunosuppressive protein is a member of the family of cytolethal distending toxins capable of causing a G2 arrest in human T cells. J. Immunol. 162: 4773–4780. [PubMed] [Google Scholar]
- Shenker, B.J., Besack, D., McKay, T., Pankoski, L., Zekavat, A., and Demuth, D.R. 2004. Actinobacillus actinomycetemcomitans cytolethal distending toxin (Cdt): Evidence that the holotoxin is composed of three subunits: CdtA, CdtB, and CdtC. J. Immunol. 172: 410–417. [DOI] [PubMed] [Google Scholar]
- Slots, J. and Ting, M. 1999. Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in human periodontal disease: Occurrence and treatment. Periodontol 2000 20: 82–121. [DOI] [PubMed] [Google Scholar]
- Sugai, M., Kawamoto, T., Peres, S.Y., Ueno, Y., Komatsuzawa, H., Fujiwara, F., Suginaka, H., and Oswald, E. 1998. The cell cycle-specific growth-inhibitory factor produced by Actinobacillus actinomycetemcomitans is a cytolethal distending toxin. Infect. Immun. 66: 5008–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, K.S., Song, K.P., and Ong, G. 2002. Cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Occurrence and association with periodontal disease. Periodontal Res. 37: 268–272. [DOI] [PubMed] [Google Scholar]
- Whitehouse, C.A., Balbo, P.B., Pesci, E.C., Cottle, D.L., Mirabito, P.M., and Pickett, C.L. 1998. Campylobacter jejuni cytolethal distending toxin causes a G2-phase cell cycle block. Infect. Immun. 66: 1934–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, V.B., Knox, K.A., and Schauer, D.B. 2000. Cytolethal distending toxin sequence and activity in the enterohepatic pathogen Helicobacter hepaticus. Infect. Immun. 68: 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]