

Abstract
The lymphocyte function-associated antigen-1 (LFA-1) binding of a unique class of small-molecule antagonists as represented by compound 3 was analyzed in comparison to that of soluble intercellular adhesion molecule-1 (sICAM-1) and A-286982, which respectively define direct and allosteric competitive binding sites within LFA-1’s inserted (I) domain. All three molecules antagonized LFA-1 binding to ICAM-1-Immunoglobulin G fusion (ICAM-1-Ig) in a competition ELISA, but only compound 3 and sICAM-1 inhibited the binding of a fluorescein-labeled analog of compound 3 to LFA-1. Compound 3 and sICAM-1 displayed classical direct competitive binding behavior with ICAM-1. Their antagonism of LFA-1 was surmountable by both ICAM-1-Ig and a fluorescein-labeled compound 3 analog. The competition of both sICAM-1 and compound 3 with ICAM-1-Ig for LFA-1 resulted in equivalent and linear Schild plots with slopes of 1.24 and 1.26, respectively. Cross-linking studies with a photoactivated analog of compound 3 localized the high-affinity small-molecule binding site to the N-terminal 507 amino acid segment of the α chain of LFA-1, a region that includes the I domain. In addition, cells transfected with a variant of LFA-1 lacking this I domain showed no significant binding of a fluorescein-labeled analog of compound 3 or ICAM-1-Ig. These results demonstrate that compound 3 inhibits the LFA-1/ICAM-1 binding interaction in a directly competitive manner by binding to a high-affinity site on LFA-1. This binding site overlaps with the ICAM-1 binding site on the α subunit of LFA-1, which has previously been localized to the I domain.
Keywords: LFA-1, ICAM-1, antagonist, competitive binding, photoaffinity labeling, Schild analysis
Lymphocyte function-associated antigen-1 (LFA-1) is an integrin that is expressed on the surfaces of all leukocytes as a heterodimercomposed of the αL (CD11a) and β2 (CD18) subunits (Hynes 1992; Gahmberg 1997; Van Kooyk and Figdor 1997). Its primary ligand, intercellular adhesion molecule-1 (ICAM-1), is a member of the immunoglobulin (Ig) protein superfamily containing five Ig-like domains, the first of which is involved in binding to LFA-1 (Marlin and Springer 1987). ICAM-1 is found on the surfaces of endothelial and epithelial cells, including keratinocytes, as well as on leukocytes and fibroblasts, and is up-regulated at sites of inflammation. The LFA-1/ICAM-1 interaction is central to the adhesion of lymphocytes to the vascular endothelium and their subsequent extravasation into the surrounding tissue as part of normal immune function, and is thought to play a role in the pathogenesis of inflammatory disease conditions such as psoriasis, rheumatoid arthritis, and transplant rejection (Yusuf-Makagiansar et al. 2002). Human and animal studies with antibodies directed against LFA-1 or ICAM-1 have demonstrated that the LFA-1/ICAM-1 interaction is a viable target for therapeutic intervention (Gottlieb et al. 2000; Liu 2001a).
The regions of both molecules that are involved in the binding interaction have been characterized by antibody binding, mutagenesis, and crystallographic studies. ICAM-1 has been found to bind to LFA-1’s inserted domain (I domain), a stretch of ~200 amino acids in the N-terminal β propeller region of the LFA-1 α chain (Huang and Springer 1995; Shimaoka et al. 2003a; Fig. 1). The amino acid residues—L205, E241, T243, and K263—within this domain, which define its ICAM-1 binding surface, are proximal to the divalent cation within the metal ion-dependent adhesion site (MIDAS) (Edwards et al. 1995, 1998). The I domain has been stably expressed as a fragment of LFA-1 and shown to bind the first domain of ICAM-1, albeit with significantly reduced affinity (Randi and Hogg 1994; Knorr and Dustin 1997) in the millimolar range (Shimaoka et al. 2001). Residues E34, K39, M64, Y66, N68, and Q73 from the first domain of ICAM-1 have been identified by mutagenesis as critical to LFA-1 binding, and shown to present a complementary binding surface to the LFA-1 I domain (Fisher et al. 1997; Shimaoka et al. 2003a).
Figure 1.
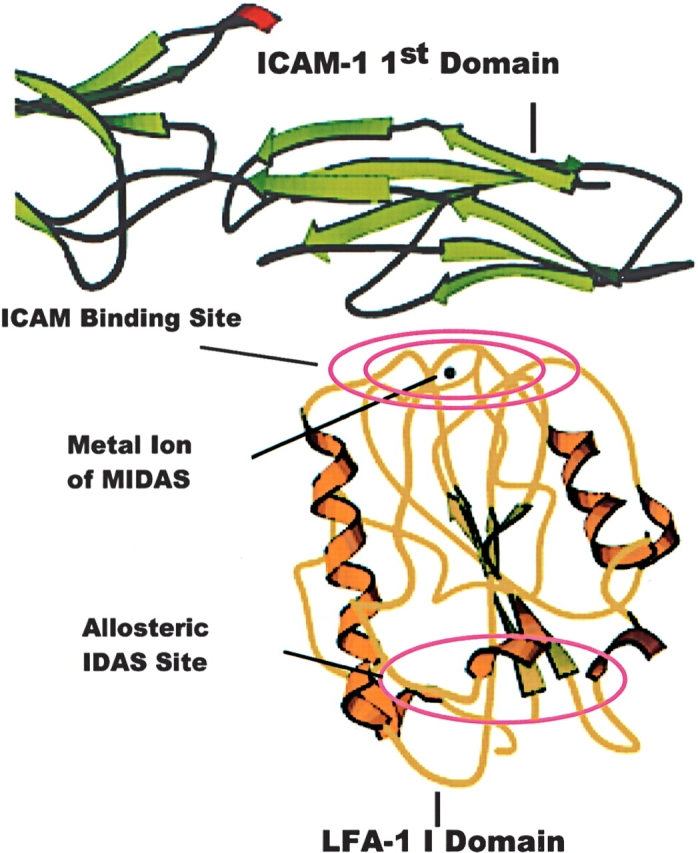
Structure of the I domain of LFA-1 in complex with ICAM-1. The I domain of LFA-1 (shown in yellow) in complex with ICAM-1 (shown in green) taken from the X-ray structure of Shimaoka et al. (2003a), deposited with the Protein Data Bank as PDB 1MQ8. ICAM-1, is seen to bind to the I domain via the magnesium ion of the MIDAS motif. The binding site of allosteric antagonists is on the opposite face of the I domain.
Many therapeutic indications for LFA-1 antagonists require chronic therapy; therefore, small-molecule inhibitors of the LFA-1/ICAM-1 interaction are an attractive alternative to current antibody therapeutics as they have the potential for oral administration as well as a lowered cost of goods. Consequently, several groups are developing small-molecule inhibitors of this interaction (Kallen et al. 1999; Kelly et al. 1999; Frenette 2001; Liu 2001b; Liu et al. 2001; Weitz-Schmidt et al. 2001; Gadek et al. 2002; Welzenbach et al. 2002). The binding site on LFA-1 has been identified for at least one class of these inhibitors, and is localized to a crevice in the I domain distal to the MIDAS between the central β-sheet and the α7 helix (Liu et al. 2001). This site has been termed the I domain allosteric site (IDAS) (Huth et al. 2000; Fig. 1). Inhibitors which bind at the IDAS do not directly compete with ICAM-1 for binding to LFA-1, but are believed to exert an allosteric effect on the I domain, which prevents the conversion of LFA-1 to a high-affinity or an activated conformation (Kallen et al. 1999; Huth et al. 2000; Liu 2001b; Lu et al. 2001).
We have discovered a class of small-molecule LFA-1 antagonists that are based on ICAM-1’s LFA-1 binding epitope and thus possess structural features in common with this epitope and distinct from the compounds known to bind the IDAS (Gadek et al. 2002). This class of small molecules has been shown to exhibit potent activities both in vitro and in vivo. The detailed structure–activity relationships of these antagonists, including the modulation of selectivity for LFA-1 over the closely related integrin, MAC-1 (αMβ2; CD11b/ CD18), are the subject of studies reported elsewhere (Keating et al. 2000; Gadek et al. 2002; Burdick et al. 2003, 2004). One of these small molecules, compound 3 (Fig. 2), has low nanomolar potency in blocking the binding of ICAM-1 to LFA-1, which translates into inhibition of a mixed lymphocyte reaction at low micromolar concentrations and efficacy in a lymphocyte-mediated model of murine contact hypersensitivity (Gadek et al. 2002). Modeling of these compounds onto the structure of ICAM-1 suggests that they bind to LFA-1 in a manner similar to that of ICAM-1. Consequently, these compounds would be expected to bind to LFA-1 in the ICAM-1 binding site and to be direct competitive inhibitors of ICAM-1 binding. Conversely, the investigators in recent publications (Welzenbach et al. 2002; Shimaoka et al. 2003b; Salas et al. 2004; Yang et al. 2004) concluded that two members of this new class of antagonists (compounds 3 and 4) (Fig. 2) and a related Roche compound (XVA143) bind to the β2 subunit I-like domain and interact with the β propeller region of the α subunit of LFA-1 to inhibit ICAM-1 binding by an allosteric mechanism. Accordingly, the investigators have defined these compounds as α/β I-like allosteric antagonists. The structural evidence cited for these conclusions include (1) studies of changes in antibody binding to LFA-1 induced by small molecules, (2) the stabilization of the LFA-1 heterodimer by small molecules under sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) denaturing conditions, (3) failure to demonstrate small-molecule inhibition of ICAM-1 binding to LFA-1 mutants shown to have altered conformational properties, and (4) failure to demonstrate small-molecule binding to the isolated αL subunit I domain. With regard to the antibody and SDS PAGE experiments in particular, these observed effects were indirect studies of the binding of the small molecules to LFA-1, which were not directly linked to the binding of ICAM-1 by LFA-1 nor to their potent inhibition of ICAM-1 binding.
Figure 2.

Structures of compounds 1–4, A-286982, and 5.
In contrast to these studies, we have undertaken a direct comparison of the binding of ICAM-1 and the small-molecule antagonists represented by compounds 3 and 4. These studies seek to define the small-molecule binding sites(s) and to link their mechanism of LFA-1/ ICAM-1 binding antagonism to their potent inhibition of lymphocyte function both in vitro and in vivo (Gadek et al. 2002). These experiments include (1) competition experiments utilizing full-length wild-type LFA-1 comparing the binding of compound 3, sICAM-1 (the extra-cellular domains of LFA-1’s native ligand and a competitive LFA-1/ICAM-1 inhibitor), and A-286982 (an allosteric LFA-1/ICAM-1 inhibitor known to bind to the IDAS) (Liu et al. 2001); (2) binding studies of this class of small molecules and ICAM-1 with a LFA-1 mutant; and (3) chemical cross-linking studies. The results of these experiments demonstrate that this unique class of antagonists inhibits ICAM-1 binding to LFA-1 by direct competition for a common high-affinity binding site on LFA-1. This ICAM-1 binding site has previously been localized to include the MIDAS motif within the I domain of the LFA-1 α subunit (Shimaoka et al. 2003a).
Results
Dependence of ligand affinities on divalent cations
Divalent cations play a critical role in integrin/ligand binding, and their presence is essential in experimental investigations of these interactions (Hynes 1992; Humphries 1996). The affinities of ICAM-1-Ig and compounds 1, 3, and 4 for LFA-1 under two sets of commonly used divalent cation conditions were measured using fluorescence polarization. The affinity of compound 1 for LFA-1 was first measured in a direct binding assay, and then the affinities of ICAM-1-Ig and compounds 3 and 4 for LFA-1 were measured in competition with compound 1 for LFA-1 (Fig. 2; Table 1). The affinity of the A-286982, which binds to the IDAS, was not measured, as it does not compete with compound 1 for binding to LFA-1 (see below). Similar changes in the affinities of compounds 1, 3, and 4 for LFA-1 were measured under the different cation conditions as for ICAM-1-Ig. The small-molecule affinities increase at least 10-fold in the presence of MnCl2 over those measured in CaCl2 and MgCl2. These small molecules do not bind to LFA-1 in the absence of divalent cations (data not shown). Similarly, the binding affinities of the soluble protein, ICAM-1-Ig, for LFA-1 in solution, as measured by the same method, in the presence of MnCl2, is at least fourfold better than the affinity in the presence of CaCl2 and MgCl2. This observation is consistent with previous reports (Marlin and Springer 1987; Dransfield et al. 1992; Woska et al. 1998). Thus, unlike the classes of LFA-1 antagonists, including A-286982, that are known to bind to the IDAS region of the I domain (Huth et al. 2000; Liu et al. 2001) and are reported to bind to LFA-1 in a cation-independent manner (Welzenbach et al. 2002), both ICAM-1-Ig and the class of LFA-1 antagonists represented by compounds 1–4 share a divalent cation sensitivity for LFA-1 binding (Table 1). Consequently, in order to investigate the mechanism of inhibition by small-molecule antagonists of LFA-1/ ICAM-1 binding, all binding assays reported herein were performed under similar conditions, in the presence of MnCl2, which is known to maximize the binding of both ICAM-1 and these cation-sensitive antagonists.
Table 1.
Cation dependence of the affinities of small-molecule antagonists for LFA-1
| Antagonist | Divalent cations | Ki (nM) | Kd (nM) |
| Compound 3 | CaCl2 + MgCl2 | 95 | |
| MnCl2 | 3.2 | ||
| Compound 4 | CaCl2 + MgCl2 | 6.0 | |
| MnCl2 | 0.58 | ||
| ICAM-1-Iga | CaCl2 + MgCl2 | ~2700 | |
| MnCl2 | ~600 | ||
| Compound 1 | CaCl2 + MgCl2 | 24 | |
| MnCl2 | 0.77 |
a Estimated Ki values for ICAM-1-Ig are based on incomplete inhibition curves, with 40% inhibition in CaCl2 + MgCl2 and 62% inhibition in MnCl2 at the two LFA-1 concentrations and maximum (5.3 μM) ICAM-1-Ig concentration used in the experiments.
Antagonist competition in the LFA-1/ICAM-1 and LFA-1/small-molecule ELISAs
To investigate the ability of compounds 2A and 3, A-286982, and sICAM-1 to inhibit the binding of ICAM-1-Ig to LFA-1, the antagonists were titrated into the LFA-1/ICAM-1 ELISA. Typical competition curves for these inhibitors in the ELISA are shown in Figure 3A. Compound 3 potently inhibited the binding of ICAM-1-Ig to LFA-1 with a 2-nM IC50. Compound 2A, an analog of compound 3, inhibited binding but with an ~10-fold higher IC50 value. A-286982 and sICAM-1 inhibited ICAM-1-Ig binding to LFA-1 but with IC50 values that were >100-fold that of compound 3.
Figure 3.

Antagonist competition by compounds 2A, 3, A-286982, and sICAM-1 in the LFA-1/ICAM-1 and LFA-1/small-molecule ELISAs. 1/5 serial dilutions of compound 3 (•), compound 2A (▴), A-286982 (♦), and sICAM-1 (▾) were incubated with either ICAM-1-Ig (A) or compound 2B (B) on plates containing captured LFA-1. The data shown are the average of two plates from a single experiment and are representative of several independent measurements. The solid lines are the fits of the data. The IC50 values (nM) are provided in the legends.
The ability of these same compounds to inhibit the binding of an FITC-labeled small-molecule antagonist, compound 2B, to LFA-1 was also examined (Fig. 3B). The potencies of compounds 2A and 3 and soluble ICAM-1 as inhibitors of compound 2B binding paralleled their potencies as inhibitors of ICAM-1-Ig binding. Compound 3, compound 2A and sICAM-1 inhibited the binding of compound 2B to LFA-1 with IC50 values of 3, 56, and 1200 nM, respectively. A-286982 did not inhibit, but rather enhanced, the binding of compound 2B to LFA-1, as indicated by the transient increase in the absorbance values, reaching a maximal effect at ~4 μM before decreasing.
The evaluation of IC50 values in the LFA-1/small molecule and LFA-1/ICAM-1 ELISAs was extended to a larger set of compounds, including a group of kistrin-derived peptides and small molecules representing the evolution of this class of LFA-1 small-molecule antagonists (Gadek et al. 2002). As shown in Figure 4, there is a good correlation (R = 0.94) between the IC50 values for competition in each of the two binding assays for this diverse set of compounds, including sICAM-1, compounds 2A and 3, across five log units of potency. The common trend in potencies between the two antagonist competition ELISAs with ICAM-1-Ig and compound 2B as ligands reveals that each compound disrupts the binding of both ICAM-1 and small-molecule ligands in a mechanistically similar fashion. This parallel in potency of inhibition is expected if ICAM-1-Ig and compound 2B are binding to the same site on LFA-1 (Wong et al. 1998).
Figure 4.

Correlation of IC50 values from antagonist competition in the LFA-1/ICAM-1 and LFA-1/small-molecule ELISAs. The IC50 values of a diverse group of compounds (four peptides, five small molecules, and sICAM-1) in competition with compound 2B are plotted against the IC50 values determined in competition with ICAM-1-Ig for binding to LFA-1. The slope of the plot is 0.964, y-intercept is 0.237, and R = 0.940. Each data point is the average of IC50 values from two plates.
Antagonist modulation of ligand binding in LFA-1/ ICAM-1 and LFA-1/small-molecule ELISAs
To further investigate the mode of binding of compound 3 and related antagonists to LFA-1, the effects of compound 3, A-286982, and sICAM-1 on the binding curves of ICAM-1-Ig and compound 2B to LFA-1 were evaluated (Pratt and Taylor 1990; Fig. 5). If an antagonist inhibits through direct competition with the ligand of interest, then there should be a nonsaturable rightward shift of the ligand binding curves to higher apparent EC50 values with increasing antagonist concentration and no reduction in the maximal binding of the ligand (Pratt and Taylor 1990; Matthews 1993, Kenakin 1997; Lutz and Kenakin 1999). Inhibition will be surmountable but will require increasing amounts of ligand in the presence of increasing concentrations of a direct competitive inhibitor (Gaddum et al. 1955). In contrast, an allosteric inhibitor may alter the ligand binding curves by causing a reduction in maximal binding or saturation in the rightward shifts of the curves (Matthews 1993; Lutz and Kenakin 1999). As shown in Figure 5A, the presence of increasing concentrations of sICAM-1 clearly shifted the ICAM-1-Ig binding curves rightward to higher EC50 values. Additionally, the same maximal extent of binding of ICAM-1-Ig to LFA-1 was observed in the presence and absence of sICAM-1, as expected when two molecular forms of the same natural ligand are competing directly for binding to a single site on a receptor (Pratt and Taylor 1990; Matthews 1993; Kenakin 1997; Lutz and Kenakin 1999). Similarly, increasing concentrations of compound 3 also shifted the binding of ICAM-1-Ig to higher EC50 values with minimal variation in maximal ICAM-1-Ig binding (Fig. 5C). Although the rightward shifts in the ligand binding curves in the presence of a competitive antagonist are typically parallel, this is not always the case (Coultrap et al. 1999). The nonparallel slopes for the LFA-1/ICAM-1-Ig binding curves in the presence and absence of compound 3 may be due to an inability to attain complete equilibrium under the heterogeneous ligand binding ELISA conditions with this compound. In the LFA-1/compound 2B format of the ligand binding ELISA, increasing concentrations of compound 3 also clearly shifted the compound 2B binding curves to higher EC50 values with no reduction in maximal binding (Fig. 5D). Increasing concentrations of sICAM-1 also showed a similar effect (Fig. 5B), although the extent of the shift in the curves was limited by the maximum achievable concentration of sICAM-1 at 2.7 μM. Thus, the effects of both sICAM-1 and compound 3 on ICAM-1-Ig and compound 2B binding to LFA-1 are characteristic of direct competition as described above.The effect of A-286982 on ICAM-1-Ig and compound 2B binding to the receptor was clearly different (Fig. 5E,F). In the LFA-1/ICAM-1 ELISA, the ICAM-1-Ig curves were shifted rightward to higher EC50 values; however, the maximum binding of ICAM-1-Ig to LFA-1 decreased considerably with increasing concentrations of A-286982. The reduction in maximal binding and rightward shift of the ligand binding curves with increasing A-286982 concentration are reflective of allosteric inhibition as described above. A-286982 causes reductions in both ligand affinity and binding capacity (Matthews 1993; Lutz and Kenakin 1999); this demonstrates that A-286982 is an insurmountable antagonist of ICAM-1-Ig binding. In contrast, in the LFA/small-molecule ELISA, the presence of A-286982 at micromolar concentrations shifted the compound 2B binding curves to lower EC50 values and appeared to enhance the binding of compound 2B to LFA-1 (Fig. 5F). Thus, as observed by different assay methods (Figs. 3B, 5F), the presence of A-286982 resulted in a modest but reproducible concentration-dependent enhancement of the binding of compound 2B to LFA-1. The contrasting effects of A-286982 on compound 2B and ICAM-1-Ig binding may be due to the known allosteric effect of the compound binding to the IDAS site on LFA-1. It has been suggested that small molecules that bind at the IDAS disrupt the hydrophobic regulatory pocket and prevent a downward movement of the C-terminal helix, locking the α subunit in a conformation with low affinity for ICAM-1 (Liu 2001b; Liu et al. 2001; Lu et al. 2001). These conformational changes may impact ICAM-1 binding differently than compound 2B or compound 3 binding due to the larger contact surface for ICAM-1 on LFA-1. It is conceivable that the resulting conformational change upon A-286982 binding causes a decrease in the Kd for compound 2B, and therefore increased binding in the ELISAs at the subsaturating ligand concentrations used. This was substantiated as an increase of >45% in the affinity of compound 1 for LFA-1 in the presence of 1 μM A-286982 (data not shown). The A-286982 binding data serve as an illustrative control for allosteric effects on small molecule and protein ligand binding to LFA-1 in the binding experiments used in this study.
Figure 5.
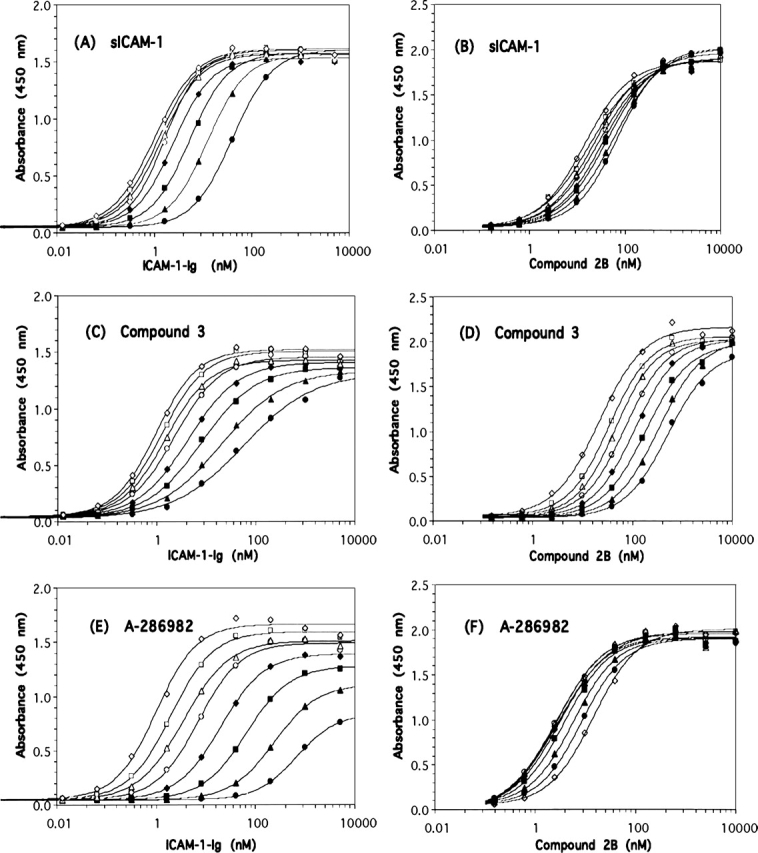
Effect of antagonists on ligand binding in the LFA-1/ICAM-1 and LFA-1/small-molecule ELISAs. Titration of ICAM-1-Ig (A,C,E) or compound 2B (B,D,F) in the absence (⋄) or the presence of antagonist in the LFA-1/ICAM-1 and LFA-1/small-molecule ELISAs. The antagonists were added in twofold dilutions starting at 2.4 (A) and 2.7 (B) μM sICAM-1, 0.040 (C) and 0.10 (D) μM compound 3, and 20 (E) and 50 (F) μM A-286982. The order of antagonist concentrations was □(lowest added antagonist concentration), ▵, ○, ♦, ▪, ▴, and • (highest antagonist concentration). The fits of the data are shown as the solid lines. The data shown are from one plate and representative of a minimum of two experiments. Note that A-286982 (F) resulted in increased binding of compound 2B to LFA-1.
Schild analysis can be also used to investigate whether a compound inhibits ligand binding through direct competition for a single binding site (Pratt and Taylor 1990; Matthews 1993; Kenakin 1997; Coultrap et al. 1999; Lutz and Kenakin 1999). This model is based upon the assumptions that equiactive responses in an assay are the result of equivalent occupancy of receptor by ligand and that maximal binding is unchanged by the presence of antagonist. In a Schild analysis, the dose ratio is the ratio of the EC50 values in the presence and the absence of antagonist and is a measure of the ligand concentrations leading to equiactive responses. This dose ratio was determined for each concentration of antagonist, and the Schild regressions were plotted as shown in Figure 6. A linear response with a slope of 1 in a Schild regression indicates that inhibition by an antagonist is directly competitive and reversible (Kenakin 1997; Lutz and Kenakin 1999). The Schild analysis would yield a nonlinear relationship and/or a slope that deviates significantly from 1 in the case of an allosteric inhibitor that does not result in a reduction of maximal binding (Kenakin 1997; Lutz and Kenakin 1999). The Schild regressions for both sICAM-1 and compound 3 were indeed linear (Fig. 6), with comparable slopes of 1.26 and 1.24, respectively. Although the Schild analysis requires a linear regression with a slope close to 1 to demonstrate direct competitive inhibition, there is no guidance in the extensive literature as to what range of Schild values are acceptable. Slopes of 1.24 and 1.26 fall within the bounds of many published Schild values used to support competitive binding conclusions, and therefore, these slope values are not considered significantly different than 1. The linearity of the regression plots and the similarity in slopes of the relationships are consistent with binding of ligand (ICAM-1-Ig) and both antagonists (sICAM-1 and compound 3) to the same site in a similar manner.
Figure 6.
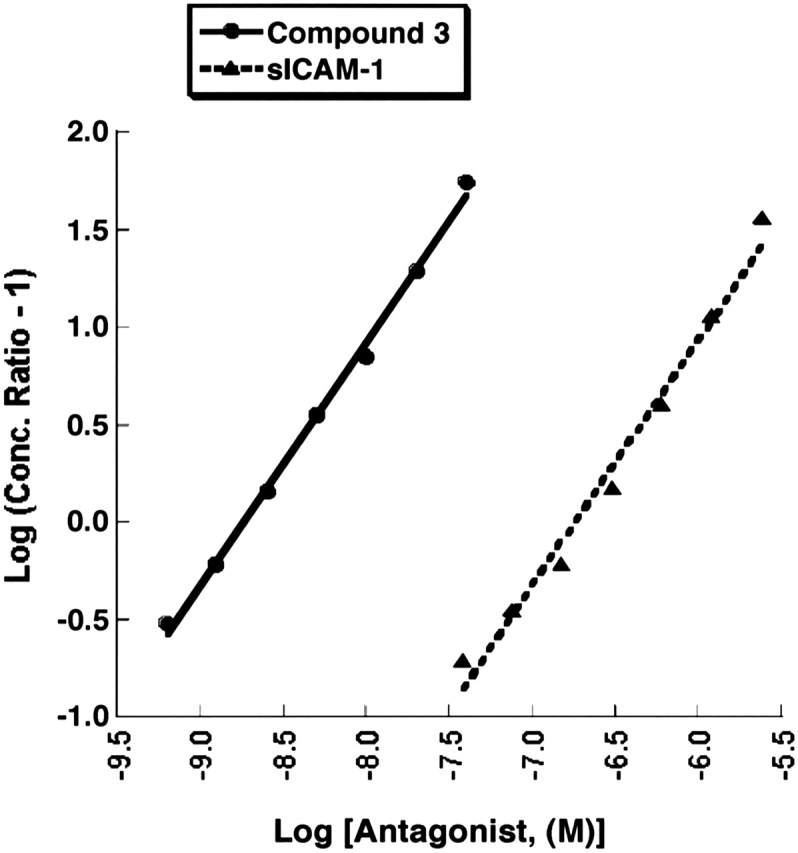
Schild regressions of sICAM-1 and compound 3 antagonism. Schild regressions of s-ICAM-1 (▴) and compound 3 (•) antagonism in the LFA-1/ICAM-1 ligand binding ELISA are plotted from the data in Figure 5, A and C, respectively. The slope of the plot for compound 3 is 1.24 with a y-intercept of 10.9 and R = 0.99832. The slope of the sICAM-1 plot is 1.26, y-intercept is 8.51, and R = 0.99131.
Cross-linking of compound 5 to the α L subunit of LFA-1
To identify the binding site of our small-molecule antagonists, compound 5, a tritium-labeled, photoactivatable analog of compound 3 was bound to LFA-1 and then photo-cross-linked. To maximize specific, high-affinity cross-linking, it was necessary to gel filter the samples to remove unbound or weakly bound compound 5 prior to irradiation (Fig. 7, cf. lanes e and f, g and h). In the absence of gel filtration, substantial low-affinity cross-linking to BSA was observed, whereas none occurred with the gel filtered samples (data not shown). This is consistent with our previously observed binding of compound 3 and related compounds to BSA with low micromolar affinities (S. Keating, L. Stefanich, K. Clark, and M. Beresini, unpubl.). Again, in the absence of gel filtration, there was significant cross-linking of compound 5 to LFA-1 α subunit, β subunit, and hetero-dimer (the band at ~200,000). This cross-linking could be reduced or eliminated when gel filtration was used. Under the latter conditions, compound 5 specifically cross-linked only to the αL subunit (Fig. 7, lanes c,g). Moreover, the presence of compound 3 during the incubation substantially reduced the incorporation of tritium into the αL subunit (Fig. 7, cf. lanes e and g). Similarly, in the presence of compound 3, there was a slight reduction of tritium incorporation into the αL subunit, β2 subunit, and heterodimer in the absence of gel filtration (Fig. 7, cf. lanes f and h). No cross-linking of compound 5 occurred when gel filtered samples of the isolated, structurally intact αL or β2 subunits were used (data not shown). Thus, the high-affinity binding site necessary to cross-link after gel filtration is provided by the intact LFA-1 heterodimer. The absence of a high-affinity site in the isolated αL subunit is consistent with a previous study demonstrating lack of interaction of XVA143 with the isolated I domain (Welzenbach et al. 2002).
Figure 7.

SDS-PAGE analysis of compound 5 cross-linked LFA-1. SDS polyacrylamide gel electrophoresis followed by Coomassie blue staining (lanes a–d) and autoradiography (lanes e–h) was used to visualize LFA-1 αL and β2 subunits after irradiation of LFA-1 following an overnight incubation with 4.1 μM compound 5. Samples in lanes a, c, e, and g were subjected to gel filtration prior to irradiation. Samples in lanes b, d, f, and h did not undergo gel filtration before irradiation. Samples in lanes a, b, e, and f were incubated with compound 5 in the presence of 290 μM compound 3.
The site of cross-linking was further defined by fragmenting the affinity-labeled αL subunit with hydroxylamine, electrophoretically separating the fragments, and then performing N-terminal sequencing on the radio-labeled fragments to determine their locations within the protein sequence. Two sequences were identified, the first starting with residue 1 (sequence found: YNLDVR GARSFS) and the second with residue 30 (sequence found: GVIVGAPGEGNST) (Larson et al. 1989). Both peptides were ~500 amino acids long, as judged by their sizes on SDS-PAGE (50–60 kDa); this fragment size is consistent with the next two predicted cleavage sites (N-G) for hydroxylamine, N507 and N530 (Bornstein 1969; Larson et al. 1989). No label was incorporated into the C-terminal half of the subunit. Attempts to refine the cross-linking site further were not successful. No definable labeled peptides were recoverable after limited digestion of the labeled αL subunit with either cyanogen bromide or Lys-C. The inability to recover labeled peptides in the LFA-1 cross-linked reaction using compound 5 after cyanogen bromide treatment may be due to instability of the cross-linked product under these degradative conditions. A similar benzoyl cross-linking agent was reported to preferentially bind to methionines; however, the cross-linked methionine product was found to be unstable to cyanogen bromide treatment (Kage et al. 1996).
Lack of binding of compound 2B to LFA-1 lacking the I domain
To investigate the role of the I domain in the binding of compound 2B and related analogs to LFA-1, a construct of the αL subunit lacking the I domain, was prepared. The β2 construct alone (mock) or together with the construct lacking the I domain or wild-type αL was transfected into 293 cells, and the binding of compound 2B to the transfected cells was examined (Fig. 8). Compound 2B showed substantial binding to the wild-type αL transfected cells but demonstrated no significant binding to the cells transfected with αL lacking the I domain relative to binding to mock (β2) transfected cells. Transfectants were also tested for their ability to adhere to ICAM-1-Ig, and as expected, the LFA-1 transfected cells lacking the I domain and mock transfectants showed indistinguishable background levels of binding, while the wild-type αL transfected cells showed robust adhesion ( Yalamanchili et al. 2000; Fig. 8B). Evaluation of the binding of a panel of LFA-1 antibodies to the transfected cells indicated that, apart from loss of binding by antibodies that mapped to the I domain, the LFA-1 heterodimer appeared to be intact in the transfected cells lacking the αL I domain (data not shown).
Figure 8.
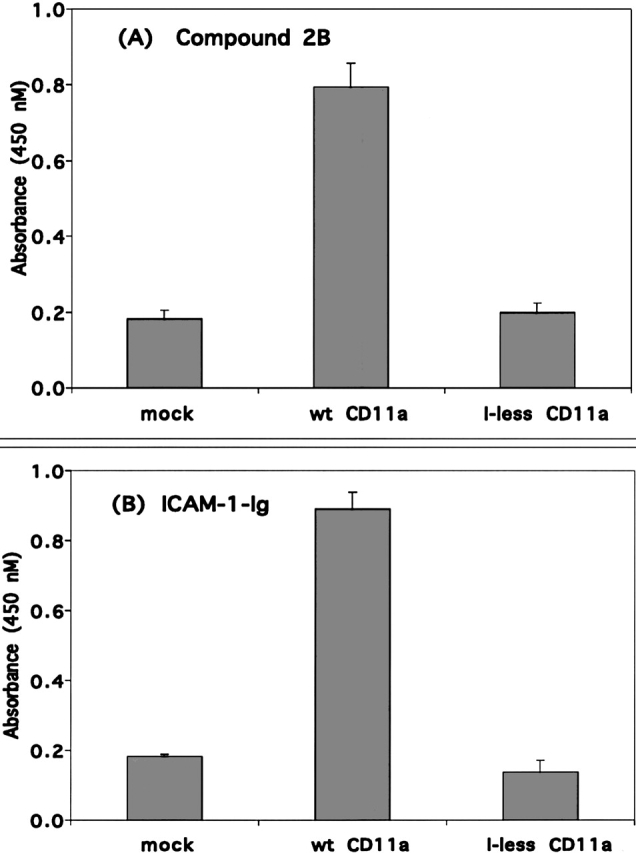
Binding of compound 2B and ICAM-1-Ig to 293 cells expressing wild-type LFA-1 or LFA-1 lacking the I domain. Binding of 1 μM compound 2B (A) or plate bound ICAM-1-Ig (B) to 293 cells cotransfected with β2 and either mock construct, wild-type αL or αL lacking the I domain (I-less). Each condition was carried out in triplicate in three experiments.
Discussion
The data obtained from the experiments described in this report uniformly support the conclusion that compound 3 and related molecules bind to a high-affinity site on LFA-1 that overlaps with the ICAM-1 binding site, which has previously been shown to include the MIDAS motif of the I domain in the αL subunit of LFA-1 (Shimaoka et al. 2003a). In particular, each protein or small-molecule antagonist (i.e., sICAM-I and compound 3) competitively inhibits the binding of both ICAM-1-Ig and compound 2B to LFA-1 under various ELISA assay formats (Fig. 4). Furthermore, the correlation of IC50 values obtained for a set of antagonist compounds across the broad range of structures studied (e.g., proteins, peptides, and small molecules) under both the ICAM-1-Ig and compound 2B formats of these ELISAs demonstrates the similar binding behavior of the small-molecule antagonist compounds, sICAM-1, ICAM-1-Ig, and compound 2B. These binding similarities extend to analogous divalent cation sensitivities in the affinities of compound 3 and ICAM-1-IgG for LFA-1, and suggest a common LFA-1 binding mode. The demonstration that both sICAM-1 and compound 3 behave as surmountable antagonists of the binding of ICAM-1-Ig indicates that the binding of ICAM-1-Ig is excluded by the binding of either sICAM-1 or compound 3, and substantiates a direct competition between compound 3 and ICAM-1 for a common binding site on LFA-1. Additional verification of this direct competition between compound 3 and ICAM-1 is provided by the nearly identical Schild regressions of the ligand binding data for both s-ICAM-1 and compound 3 in competition with ICAM-1-Ig for binding to LFA-1.
Corroborating evidence for the close proximity of the ICAM-1 and small-molecule antagonist binding sites on LFA-1 can be seen in the common effect of the deletion of the I domain on the binding of both ICAM-1-Ig and compound 2B. Both compound 2B and ICAM-1 were unable to bind to LFA-1 lacking the I domain, the domain in which the ICAM-1 binding site is located. Moreover, the ability of A-286982 to allosterically modify the binding of both ICAM-1-Ig and compound 2B is consistent with a close proximity of their binding sites to the A-286982 binding site in the IDAS motif in the I domain of the LFA-1 α subunit (Liu 2001b; Liu et al. 2001). The selective photochemical cross-linking of compound 5 to the α chain of LFA-1 localizes its binding site to within residues 30–507 of this subunit. All of the findings noted above are consistent with a single high-affinity small-molecule binding site located in the I domain of the α chain of LFA-1.
Close examination of the photochemical cross-linking study performed with a relatively high concentration of compound 5 (4.1 μM) (Fig. 7) affords direct evidence for an additional low-affinity small-molecule binding site on LFA-1. Dramatically different protein and cross-linking patterns are observed in the presence and the absence of gel filtration. When samples are gel filtered to remove unbound and weakly bound molecules prior to irradiation, only high-affinity labeling of the α subunit is observed. However, in the absence of the gel filtration step, irradiation of the complex of compound 5 with LFA-1 results in high-intensity cross-linking to the α subunit and lower intensity cross-linking to a low-affinity binding site in the β subunit whose complex with compound 5 is too weak to survive gel filtration. Under both conditions, the observed cross-linking is partially inhibited by a large excess (290 μM) of compound 3 (Fig. 7, lanes e and g, f and h) demonstating the specific nature of the binding to both sites. Attempts to cross-link compound 5 to either of the isolated α or β subunits failed to afford high-affinity complexes capable of surviving the gel filtration process. Consequently, it appears that the high-affinity competitive binding of the class of compounds represented by compound 3 requires the presence of an intact full length LFA-1 heterodimer. Attempts to capture this binding site in constructs of either of the LFA-1 subunits or the isolated I domain results in diminished affinity of LFA-1 for ICAM-1 and small-molecule analogs of compound 3 (e.g., XVA143) (Shimaoka et al. 2001; Welzenbach et al. 2002). It is particularly interesting to note the presence of a minor LFA-1 heterodimer band that appears in the absence of gel filtration (Fig. 7, band at >200,000 Da). The intensity of the LFA-1 band, as judged by both Coomassie blue staining and autoradiography, is significantly lower than previous reports of the stabilization of LFA-1 by compound 3 under SDS-PAGE suggest (Shimaoka et al. 2003b; see Discussion), but consistent with low-affinity binding to a second site on the β chain that stabilizes the heterodimer. Overall, these cross-linking results indicate that there are two distinct binding sites for this class of LFA-1 small-molecule antagonists.
Recent publications describe a conformational interaction between the αL I domain and the homologous I-like domain in the β2 subunit of LFA-1 and hypothesize that compounds 3 and 4, and a compound from Roche, XVA143, bind to the I-like domain in the β2 subunit and interact with the β-propeller domain of the α subunit at or near Glu310 and inhibit ICAM-1 binding in an allosteric fashion by inhibiting activation of the ICAM-1 binding site in the αL I domain (Welzenbach et al. 2002; Shimaoka et al. 2003b; Salas et al. 2004; Yang et al. 2004). While the data presented in these publications demonstrate that compound 4 and XVA143 are potent high-affinity antagonists of LFA-1/ICAM-1 binding, they also show that these compounds bind to and stabilize LFA-1 lacking the I domain (Shimaoka et al. 2003b). Additional data indicate that at concentrations significantly above their IC50’s for the inhibition of LFA-1/ICAM-1 binding, these compounds bind to and induce conformational changes in the I-like domain in LFA-1’s β2 subunit as detected with specific antibodies, and that XVA143 and ICAM-1 can simultaneously bind to the Glu310Ala mutant of LFA-1 (Yang et al. 2004). The proposed allosteric mechanism involving interdomain communication that derived from these studies is at odds with the direct competition for a single high-affinity binding site on the LFA-1 heterodimer that we have observed between ICAM-1 and the class of antagonists represented by compounds 3 and 4. If the potent inhibitory and immunosuppressive activities of these compounds are a result of their binding at a site in the β subunit distant from the ICAM-1 binding site in the I domain of the α subunit—blocking the relay of an activating conformational signal from the β I-like to the α I domain and causing the I domain to remain in the default low-affinity state—then the inhibition of ICAM-1-Ig binding by compound 3 would neither be expected to be surmountable, nor would it result in a linear Schild regression with a slope comparable to that of sICAM-1. On the contrary, allosteric ICAM inhibition such as this would be expected to exhibit the unsurmountable competition we have observed for A-286982 as a result of the passage of this allostery through the A-286982 binding site in its transmission from the β subunit I-like domain to the α subunit ICAM binding site (Huth et al. 2000; Shimaoka and Springer 2004).
In one of the reports discussed above, the binding of compounds 3 and 4 and XVA143 to wild-type LFA-1 and a deletion mutant lacking the I domain is inferred from a stabilization of the LFA-1 heterodimer by these compounds under the denaturing conditions of SDS-PAGE (Shimaoka et al. 2003b). This is apparently at odds with our result showing that neither ICAM-1 nor FITC-labeled compound 2B bind to LFA-1 lacking the I domain (Fig. 8). However, both of these observations are consistent with the two binding sites noted for the cross-linking above: a high-affinity binding of compound 2B in the α subunit I domain, which is stable enough to detect with an anti-FITC antibody, and a less stable binding site in the β subunit. If compound 2B binds to the I-like domain of the β subunit in the absence of the I domain, its complex with this truncated LFA-1 lacks the stability necessary for detection with an anti-FITC antibody in our studies (Fig. 8). Consequently, compound 2B behaves like ICAM-1 in binding to a high-affinity site on LFA-1, and this binding is abrogated by deletion of the I domain. Furthermore, the appearance of a weak LFA-1 band stabilized by concentrations of the small molecules (Fig. 7) far in excess of their IC50 values for their inhibition of LFA- 1/ICAM-1 binding (≤4 μM vs. 0.002 μM for compound 3), indicates that the stabilization of the LFA-1 hetero-dimer to SDS-PAGE by compound 3 is unrelated to its potent inhibition of ICAM-1 binding to LFA-1. It is clear from published gel stabilization studies (Shimaoka et al. 2003b; Salas et al. 2004; Yang et al. 2004), that the binding site responsible for the stabilization of LFA-1 to SDS-PAGE resides in the I-like domain of the β subunit. It is also clear from the data presented in this paper that this β subunit binding site is not related to the high-affinity binding site in the α subunit, which is responsible for the direct competitive inhibition of ICAM-1 binding. However, the β subunit binding site responsible for LFA-1 stabilization by compound 3 may be the same as the low-affinity β subunit cross-linking site we have observed.
Overall, the cross-linking results we have presented indicate that there are two distinct binding sites for this class of LFA-1 small-molecule antagonists. The first is a high-affinity binding site in the αL subunit of LFA-1 through which the small molecule and LFA-1 form a complex that is stable enough (e.g., Kd < 25 nM) to survive the gel filtration process. It is this small-molecule binding site that has been characterized in the binding experiments reported here as overlapping the ICAM-1 binding site, and that correlates with the potent inhibition of LFA-1/ICAM-1 binding by compounds 3 and 4 (compound 4 IC50 = 1.4 nM), their potent inhibition of LFA-1-induced lymphocyte proliferation (compound 4 IC50 = 3 nM) in vitro, and their inhibition of the immune system’s response in vivo (Gadek et al. 2002). The second site is a lower-affinity binding site (e.g., Kd > 1 μM) in the β subunit, which is involved with stabilization of the LFA-1 heterodimer under SDS-PAGE. This site is more dynamic by nature (i.e., faster off rate) and does not survive the gel filtration/photolysis process. The characteristics of this second low-affinity site are consistent with those of the recently described α/β I-like allosteric antagonist binding site in the I-like domain of the β subunit (Welzenbach et al. 2002; Shimaoka et al. 2003b; Salas et al. 2004; Yang et al. 2004). The low-affinity binding of the ICAM-1 mimetics described herein to the β subunit of LFA-1, presumably to the I-like domain, was unanticipated in their design. This is likely due to the sequence homology between the I and I-like domains, particularly with regard to similarities in MIDAS motifs and their affinities for the carboxylic acid moiety common to this class of antagonists. Given that the β2 family of integrins, including MAC-1, share this subunit, the affinity of compounds for the I-like domain in the β2 subunit must be attenuated to select antagonists which are specific to LFA-1 (Keating et al. 2000). Subsequent reports will describe the structural origins of the selectivity of compounds 3 and 4 and analogs for LFA-1 versus MAC-1.
Taken together, the work described herein substantiates the high-affinity binding of compounds 3 and 4 to LFA-1 in a manner that is similar to that of ICAM-1, at a site overlapping the ICAM-1 binding site involving the MIDAS motif within the I domain of the LFA-1 α subunit (Shimaoka et al. 2003a). This is consistent with their proposed mimicry of the ICAM-1 epitope (Gadek et al. 2002) and inconsistent with the previous conclusion that they function as α/β I-like allosteric antagonists of LFA-1/ICAM-1 (Shimaoka et al. 2003b; Shimaoka and Springer 2004). The binding of these ICAM-1 mimetics to the β2 integrin subunit, albeit with lower affinity, raises the question of whether ICAM-1 itself binds to a second site in the I-like domain (Welzenbach et al. 2002; Shimaoka et al. 2003b; Salas et al. 2004; Shimaoka and Springer 2004; Yang et al. 2004) as part of a feedback mechanism. The different conclusions reached from direct and indirect binding studies conducted in different laboratories with the same compounds highlights the need for a correlation between antagonist binding and target protein function in the formulation of integrin signaling mechanisms.
Materials and methods
Materials
Full-length recombinant human membrane-bound LFA-1 and recombinant human 5-domain ICAM-1-Ig fusion (ICAM-1-Ig) were produced in human 293 cells and purified as described (Fisher et al. 1997; Keating et al. 2000). sICAM-1 (a truncated form of native ICAM-1 without the transmembrane and cytoplasmic domains for ease of use in in vitro assays, but with the intact LFA-1 binding epitope) and MEM-48 were from R&D Systems. Mouse monoclonal anti-human β2 integrin (clone PLM2) was generated using standard procedures (Fisher et al. 1997). Small molecules and peptide antagonists were synthesized as described (Burdick 1999; Liu et al. 2000; Gadek et al. 2002). Compounds 1–5 and A-286982 are shown in Figure 2. Compounds 1, 2A, and 2B are similar to compound 3 but with the addition of linkers to enable conjugation to fluorescein (compounds 1 and 2B; 2A was not conjugated to fluorescein). Fluorescein conjugates were prepared via coupling of an amine functionality with fluorescein-5-isothiocyanate (FITC) (Keating et al. 2000). Additional molecules analyzed include compounds previously described (Gadek et al. 2002) and identified as compounds 1 and 2 (distinct from compounds 1 and 2 in this report), kistrin (Dennis et al. 1990), the non-Kistrin heptapeptides, H2N-CGFDMPC-CO2H and H2N-CGY(m)DMPC-CO2H, cyclic kistrin peptide CRIPRGDMPDDRC and tetra-peptide, H2N-CN(F) PC-CO2H, wherein Y(m) is meta-tyrosine and N(F) is N′-3-phenylpropyl asparagine. Compounds 3 and 4 in this article are identical to compounds 3 and 4 previously described (Gadek et al. 2002). All small-molecule antagonists were stored as 10 mM solutions in 50% DMSO at −20°C. Compound 5 was a gift from Hoffman-La Roche Inc.
Affinity measurements
The affinities of the small molecules for LFA-1 were measured using fluorescence polarization (FP) (Panvera 1995; Lakowicz 1999) in a competitive format with a small-molecule antagonist, compound 1 (Fig. 2), as previously described (Keating et al. 2000). All measurements were performed in buffer containing 50 mM HEPES (pH 7.2), 150 mM NaCl, 0.05% n-octyglucoside and 0.05% bovine γ globulins (BGG) and either 1 mM MnCl2, or 1 mM CaCl2 and 1 mM MgCl2. The affinity of compound 1 for LFA-1 was first measured by addition of 2 nM compound 1 to serial dilutions of LFA-1 starting from 1 μM in buffer containing either MnCl2 or CaCl2 and MgCl2. Competition experiments were performed by addition of serial dilutions of antagonists to 2 nM compound 1 and either 3 nM LFA-1 (in MnCl2) or 40 nM LFA-1 (in CaCl2 and MgCl2). In the ICAM-1-Ig competition experiments, the LFA-1 concentrations were reduced to 2 nM and 20 nM LFA-1 in the two divalent cation buffer conditions to maximize inhibition by ICAM-1-Ig. The different LFA-1 concentrations used in the experiments were taken into account in the affinity calculations (see below). The solutions were incubated in 96-well black HE96 plates (Molecular Devices) for 2 h at 37°C. FP measurements were performed on an Analyst platereader (Molecular Devices) using 485 nm excitation, 530 nm emission, and 505 nm dichroic filters. All raw intensity data were corrected for background emissions by subtraction of the intensities measured from the appropriate samples without compound 1. The LFA-1 binding and antagonist competition data were analyzed using a nonlinear least-squares fit of a four-parameter equation with KaleidaGraph software (Synergy Software) to obtain the EC50 values for the LFA-1 titration and the IC50 values of the antagonists. The equation used to fit the data is Y = ((A − D)/(1 + (X/C) exp(B))) + D, where Y is the assay response, A is Y-value at the upper asymptote, B is the slope factor, C is the IC50 or EC50, and D is the Y-value at the lower asymptote. In general, the data measured in both the homogeneous FP and heterogeneous ELISA formats described below, contain relatively large signal-to-background ratios and the error estimates in the fits are typically <10% of the final value of the fitted parameter. The equilibrium dissociation constants (Kd) of LFA-1 for compound 1 with and without A-286982 were calculated using Klotz and Hill analyses (Panvera 1995). The affinities (Ki) of the antagonists for LFA-1 were calculated using the IC50 values, the Kd of compound 1 /LFA-1, and the concentrations of compound 1 and LFA-1 in the competition experiments (Jacobs et al. 1975; Keating et al. 2000).
LFA-1/ICAM-1 and LFA-1/small-molecule enzyme-linked immunosorbent assays (ELISAs)
Antagonist competition
Small molecules and sICAM-1 were assayed for the ability to disrupt binding of ICAM-1-Ig or a fluorescein-labeled small-molecule antagonist, compound 2B, to LFA-1 in a competitive format (Quan et al. 1998; Burdick 1999; Gadek et al. 2002). Compound 2B is similar to compound 1, but with a longer linker between the small molecule and fluorescein to maximize the binding of the anti-fluorescein detection antibody. Ninety-six-well plates were coated with 5 μg/mL (33.3 nM) mouse anti-human β2 integrin (a nonfunction blocking antibody) in phosphate-buffered saline (PBS) overnight at 4°C. The plates were blocked with assay buffer (20 mM HEPES at pH 7.2, 140 mM NaCl, 1 mM MnCl2, 0.5% bovine serum albumin (BSA) and 0.05% Tween-20) for 1 h at room temperature. After washing in buffer (50 mM Tris-Hcl at pH 7.5, 100 mM NaCl, 1 mM MnCl2, and 0.05% Tween-20), 8 nM LFA-1 (LFA-1/ICAM-1 ELISA) or 2 nM LFA-1 (LFA-1/small-molecule ELISA) were added, followed by incubation for 1 h at 37°C. The plates were washed, and for the LFA-1/ICAM-1 ELISA, serial dilutions of the small-molecule antagonists or sICAM-1 were added to the plates for 30 min, followed by addition of 0.89 nM ICAM-1-Ig (final concentration) for 2 h at 37°C. After an additional wash, goat anti-huIgG (Fc specific)-HRP was added and incubated for 1 h at 37°C. In the LFA-1/small-molecule ELISA, the diluted antagonists and 25 nM compound 2B were added concurrently to the plates, followed by a 2-h incubation at 37°C. Sheep anti-fluorescein-HRP was added after a wash and incubated for 1 h at 37°C. For both assays, after washing, the bound HRP-conjugated antibodies were detected by addition of tetramethylbenzidine (TMB) followed by measurement of the absorbance of the product at 450 nm after the addition of 1 M H3PO4 to stop the reaction. The IC50 values for each curve were determined by fitting to the four-parameter equation described above using KaleidaGraph software. The format and the results from this form of the LFA-1/ICAM-1 assay are similar to those previously reported (Burdick 1999; Gadek et al. 2002); however, this format is more robust due to antibody capture of the LFA-1 rather than direct coating onto the ELISA plate.
Ligand binding
The LFA-1/ICAM-1 and LFA-1/small-molecule ELISAs were performed as described above except that serial dilutions of either ICAM-1-Ig or compound 2B were added to plates either in the presence or the absence of antagonist. In all cases the ligand was added concurrently with the antagonist. The plates were incubated for 6 h at 37°C to approach equilibrium conditions after antagonist and ligand addition, before wash and addition of the detection antibody. The EC50 values for each curve were determined by fitting with a four-parameter model as described above. The EC50 values generated in the presence and the absence of antagonist were analyzed by Schild regression (Arunlakshana and Schild 1959; Pratt and Taylor 1990; Matthews 1993; Kenakin 1997; Lutz and Kenakin 1999). The Schild plots of Log (Conc. ratio −1) versus antagonist concentration are calculated from (Conc. ratio −1) = ((ligand EC50 with antagonist)/(ligand EC50 without antagonist)) − 1. The slopes of the plots of the Log (Conc. ratio −1) versus Antagonist concentration are calculated by fitting the line to the linear equation, Y = A + BX.
Cross-linking of a radiolabeled, photoactivatable analog of compound 3 to LFA-1
Full-length human membrane-associated LFA-1 or BSA (0.35 mg/mL [1.4 and 5.3 μM, respectively] in 20 mM HEPES, 150 mM NaCl, 5 mM CaCl2, 5 mM MgCl2, 1 mM MnCl2, and 1% n-octylglucoside at pH 7.2) was incubated overnight at 37°C with 4.1 μM compound 5, a tritium-labeled photoactivatable analog of compound 3 (Kauer et al. 1986), in either the presence or the absence of 290 μM compound 3. The molar ratio of compound 5 to LFA-1 was 3:1. A 96-well plate precoated with 1% BSA was used for the incubation. Just prior to cross-linking, excess compound 5 was rapidly removed by gel filtration with a G-25 microspin column in a 96-well format equilibrated with the same buffer. The LFA-1/compound 5 complex was cross-linked by exposure to a high-pressure mercury-vapor lamp (450 watts, Ace Glass). During irradiation, samples were cooled on ice and protected by a 5 mm-thick plate of borosilicate glass to minimize protein degradation. Residual unlinked compound 5 was removed by gel filtration (G-25) as above. The cross-linked complex was then denatured in 8 M guanidine hydrochloride (GuHCl) and reduced and alkylated. The treated proteins were subjected to SDS-PAGE followed by Coomassie blue staining. Radiolabeled proteins were visualized by audioradiography.
To identify compound 5 binding sites, the treated αL and β2 subunits were separated by size-exclusion chromatography in the presence of 6 M GuHCl, 20 mM HEPES, 10 mM EDTA (pH 6.8), and then chemically cleaved with 2.6 M hydroxylamine in 10% acetic acid with 7 M GuHCl for 4 h at 75°C. The radiolabeled protein fragments were separated by SDS-PAGE and either visualized by autoradiography or transferred onto a polyvinylidene fluoride membrane, stained with Coomassie blue, and then identified by N-terminal protein sequencing.
Generation of the αL construct lacking the I domain
The construct used, pLFA.huID.Δp, contains the sequence of the αL gene from the Nar1 restriction site 5′ of the I domain to the second PflM1 restriction site 3′ of the I domain in which the first PflM1 restriction site 3′ of the I domain was abolished (Edwards et al. 1995). To generate the mutant lacking the I domain, the following primers were made: the forward primer CACTGTGGCGCCCTGGTTTTCAGGAAGGTAGTGGA TCAGGCACAAGCAAACAGGACCTGACTTC, containing the sequence from the Nar1 site to the start of the I domain, a sequence of DNA encoding GSGSG and the 23 bp of the αL sequence after the end of the I domain, and the reverse primer TCTGAGCCATGTGCTGGTATCGAGGG GC, which primes at the second PflM1 restriction site after the I domain. PCR was performed using these primers and the pLFA.huID.Δp linearized with Bgl II, which cut at a site within the I domain. A DNA fragment was amplified that contained the sequence from the Nar1 site to the second PflM1 site, in which the entire I domain, from C125 through G311, was replaced with a DNA sequence encoding GSGSG. This piece of DNA was purified, digested with Nar1 and PflM1, and inserted into the human αL plasmid (pRKLFAαm) at the corresponding Nar1 and PflM1 sites. Correct insertion of the DNA sequence encoding GSGSG was confirmed by sequence analysis.
Binding of LFA-1 lacking the I domain to ICAM-1 or compound 2B
Human 293 cells were transfected with the β2 construct alone (mock) or with either the wild-type αL construct or the αL construct lacking the I domain (I-less) and allowed to recover for 3 d. The cells were detached and resuspended in adhesion buffer (0.02 M HEPES at pH 7.2, 0.14 M NaCl, 0.2% glucose). Binding to plate bound ICAM-1-Ig was performed as described (Edwards et al. 1998). For binding of compound 2B, 2 × 105 cells were added per well in a round-bottom 96-well plate in adhesion buffer containing 0.5% BGG, 0.1 mM MnCl2, 1 μg/mL anti-β2 activating antibody MEM-48, and 1 μM compound 2B. The cells were incubated for 1 h at 37°C, washed with cold PBS, and fixed with 1% formaldehyde/PBS. The cells were then incubated with a 1:500 dilution of sheep anti-fluorescein-HRP for 1 h at room temperature, washed with PBS, and incubated with TMB for 15 min. The reaction was stopped with 1 M H3PO4 and read at 450 nm. In parallel, the transfectants were tested for the structural integrity of the surface-expressed αL/β2 complexes and for the presence or the absence of the I domain by FACS analysis using a panel of antibodies with known binding epitopes (Edwards et al. 1998).
Acknowledgments
We thank Dan Burdick for synthesis of A-286982; Yu-Ying Liu and Lucy Chen at Hoffman-La Roche for the synthesis of compound 5; and Drs. M. Shimaoka, T. Springer, and coauthors (Shimaoka et al. 2003a,b) for sharing a preprint of their manuscripts describing the crystal structure of the I domain/ICAM-1 complex and the LFA-1 SDS-PAGE stabilization studies.
Abbreviations
LFA-1, lymphocyte function-associated antigen-1
ICAM-1, intercellular adhesion molecule-1
I domain, inserted domain
MIDAS, metal ion-dependent adhesion site
IDAS, I domain allosteric site
ICAM-1-Ig, ICAM-1-Immunoglobulin G fusion
sICAM-1, soluble ICAM-1
HRP, horseradish peroxidase
FITC, fluorescein isothiocyante
FP, fluorescence polarization
BGG, bovine γglobulins
PBS, phosphate-buffered saline
BSA, bovine serum albumin
TMB, tetramethylbenzidine
GuHCl, guanidine hydrochloride
SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051583406.
References
- Arunlakshana, O. and Schild, H.O. 1959. Some quantitative uses of drug antagonists. Br. J. Pharmacol. 14: 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein, P. 1969. The nature of a hydroxylamine-sensitive bond in collagen. Biochem. Biophys. Res. Commun. 36: 957–964. [DOI] [PubMed] [Google Scholar]
- Burdick, D.J. 1999. LFA-1 antagonist compounds. PCT Int. Appl. (Genentech, USA) WO9949856.
- Burdick, D.J., Paris, K., Weese, K., Stanley, M., Beresini, M., Clark, K., McDowell, R.S., Marsters Jr., J.C., and Gadek, T.R. 2003. N-Benzoyl amino acids as LFA-1/ICAM inhibitors 1: Amino acid structure–activity relationship. Bioorg. Med. Chem. Lett. 13: 1015–1018. [DOI] [PubMed] [Google Scholar]
- Burdick, D.J., Marsters Jr., J.C., Aliagas-Martin, I., Stanley, M., Beresini, M., Clark, K., McDowell, R.S., and Gadek, T.R. 2004. N-Benzoyl amino acids as ICAM/LFA-1 inhibitors. Part 2: Structure–activity relationship of the benzoyl moiety. Bioorg. Med. Chem. Lett. 14: 2055–2059. [DOI] [PubMed] [Google Scholar]
- Coultrap, S.J., Sun, H., Tenner Jr., T.E., and Machu, T.K. 1999. Competitive antagonism of the mouse 5-hydroxytryptamine3 receptor by bisindolylmaleimide I, a “selective” protein kinase C inhibitor. J. Pharmacol. Exp. Ther. 290: 76–82. [PubMed] [Google Scholar]
- Dennis, M.S., Henzel, W.J., Pitti, R.M., Lipari, M.T., Napier, M.A., Deisher, T.A., Bunting, S., and Lazarus, R.A. 1990. Platelet glycoprotein IIb–IIIa protein antagonists from snake venoms: Evidence for a family of platelet-aggregation inhibitors. Proc. Natl. Acad. Sci. 87: 2471–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dransfield, I., Cabanas, C., Craig, A., and Hogg, N. 1992. Divalent cation regulation of the function of the leukocyte integrin LFA-1. J. Cell Biol. 116: 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, C.P., Champe, M., Gonzalez, T., Wessinger, M.E., Spencer, S.A., Presta, L.G., Berman, P.W., and Bodary, S.C. 1995. Identification of amino acids in the CD11a I-domain important for binding of the leukocyte function-associated antigen-1 (LFA-1) to intercellular adhesion molecule-1 (ICAM-1). J. Biol. Chem. 270: 12635–12640. [DOI] [PubMed] [Google Scholar]
- Edwards, C.P., Fisher, K.L., Presta, L.G., and Bodary, S.C. 1998. Mapping the intercellular adhesion molecule-1 and -2 binding site on the inserted domain of leukocyte function-associated antigen-1. J. Biol. Chem. 273: 28937–28944. [DOI] [PubMed] [Google Scholar]
- Fisher, K.L., Lu, J., Riddle, L., Kim, K.J., Presta, L.G., and Bodary, S.C. 1997. Identification of the binding site in intercellular adhesion molecule 1 for its receptor, leukocyte function-associated antigen 1. Mol. Biol. Cell 8: 501–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenette, P.S. 2001. Locking a leukocyte integrin with statins. N. Engl. J. Med. 345: 1419–1421. [DOI] [PubMed] [Google Scholar]
- Gaddum, J.H., Hameed, K.A., Hathway, D.E., and Stephens, F.F. 1955. Quantitative studies of antagonists for 5-hydroxytryptamine. Q. J. Exp. Physiol. 40: 49–74. [DOI] [PubMed] [Google Scholar]
- Gadek, T.R., Burdick, D.J., McDowell, R.S., Stanley, M.S., Marsters Jr., J.C., Paris, K.J., Oare, D.A., Reynolds, M.E., Ladner, C., Zioncheck, K.A., et al. 2002. Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science 295: 1086–1089 (and online supplementary material). [DOI] [PubMed] [Google Scholar]
- Gahmberg, C.G. 1997. Leukocyte adhesion: CD11/CD18 integrins and intercellular adhesion molecules. Curr. Opin. Cell Biol. 9: 643–650. [DOI] [PubMed] [Google Scholar]
- Gottlieb, A., Krueger, J.G., Bright, R., Ling, M., Lebwohl, M., Kang, S., Feldman, S., Spellman, M., Wittkowski K., Ochs, H.D., et al. 2000. Effects of administration of a single dose of a humanized monoclonal antibody to CD11a on the immunobiology and clinical activity of psoriasis. J. Am. Acad. Dermatol. 42: 428–435. [DOI] [PubMed] [Google Scholar]
- Huang, C. and Springer, T.A. 1995. A binding interface on the I domain of lymphocyte function-associated antigen-1 (LFA-1) required for specific interaction with intercellular adhesion molecule 1 (ICAM-1). J. Biol. Chem. 270: 19008–19016. [DOI] [PubMed] [Google Scholar]
- Humphries, M.J. 1996. Integrin activation: The link between ligand binding and signal transduction. Curr. Opin. Cell Biol. 8: 632–640. [DOI] [PubMed] [Google Scholar]
- Huth, J.R., Olejniczak, E.T., Mendoza, R., Liang, H., Harris, E.A.S., Lupher Jr., M.L., Wilson, A.E., Fesik, S.W., and Staunton, D.E. 2000. NMR and mutagenesis evidence for an I domain allosteric site that regulates lymphocyte function-associated antigen 1 ligand binding. Proc. Natl. Acad. Sci. 97: 5231–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes, R.O. 1992. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell 69: 11–25. [DOI] [PubMed] [Google Scholar]
- Jacobs, S., Chang, K.-J., and Cuatrecasas, P. 1975. Estimation of hormone receptor affinity by competitive displacement of labeled ligand: Effect of concentration of receptor and of labeled ligand. Biochem. Biophys. Res. Commun. 66: 687–692. [DOI] [PubMed] [Google Scholar]
- Kage, R., Leeman, S.E., Krause, J.E., Costello, C.E., and Boyd, N.D. 1996. Identification of methionine as the site of covalent attachment of a p-benzoyl-phenylalanine-containing analogue of substance P on the substance P (NK-1) receptor. J. Biol. Chem. 271: 25797–25800. [DOI] [PubMed] [Google Scholar]
- Kallen, J., Welzenbach, K., Ramage, P., Geyl, D., Kriwacki, R., Legge, G., Cottens, S., Weitz-Schmidt, G., and Hommel, U. 1999. Structural basis for LFA-1 inhibition upon lovastatin binding to the CD11a I-domain. J. Mol. Biol. 292: 1–9. [DOI] [PubMed] [Google Scholar]
- Kauer, J.C., Erickson-Viitanen, S., Wolfe Jr., H.R., and DeGrado, W.F. 1986. p-Benzoyl-L-phenylalanine, a new photoreactive amino acid. Photolabeling of calmodulin with a synthetic calmodulin-binding peptide. J. Biol. Chem. 261: 10695–10700. [PubMed] [Google Scholar]
- Keating, S., Marsters, J., Beresini, M., Ladner, C., Zioncheck, K., Clark, K., Arellano, F., and Bodary, S. 2000. Putting the pieces together: Contribution of fluorescence polarization assays to small molecule lead optimization. SPIE Proc. 3913: 128–137. [Google Scholar]
- Kelly, T.A., Jeanfavre, D.D., McNeil, D.W., Woska Jr., J.R., Reilly, P.L., Mainolfi, E.A., Kishimoto, K.M., Nabozny, G.H., Zinter, R., Bormann, B.-J., et al. 1999. Cutting edge: A small molecule antagonist of LFA-1-mediated cell adhesion. J. Immunol. 163: 5173–5177. [PubMed] [Google Scholar]
- Kenakin, T. 1997. Pharmacologic analysis of drug-receptor interaction. Lippincott-Raven, Philadelphia.
- Knorr, R. and Dustin, M.L. 1997. The lymphocyte function-associated antigen 1 I domain is a transient binding module for intercellular adhesion molecule (ICAM)-1 and ICAM-3 in hydrodynamic flow. J. Exp. Med. 186: 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz, J.R. 1999. Principles of fluorescence spectroscopy, 2nd ed. Plenum Press, New York.
- Larson, R.S., Corbi, A.L., Berman, L., and Springer, T. 1989. Primary structure of the leukocyte function-associated molecule-1 α subunit: An integrin with an embedded domain defining a protein superfamily. J. Cell Biol. 108: 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, G. 2001a. Inhibitors of LFA-1/ICAM-1 interaction: From monoclonal antibodies to small molecules. Drugs Future 26: 767–778. [Google Scholar]
- ———. 2001b. Small molecule antagonists of the LFA-1/ICAM-1 interaction as potential therapeutic agents. Expert Opin. Ther. Patents 11: 1383–1393. [Google Scholar]
- Liu, G., Link, J.T., Pei, Z., Reilly, E.B., Leitza, S., Nguyen, B., Marsh, K.C., Okasinski, G.F., von Geldern, T.W., Ormes, M., et al. 2000. Discovery of novel p-arylthio cinnamides as antagonists of leukocyte function-associated antigen-1/intracellular adhesion molecule-1 interaction. 1. Identification of an additional binding pocket based on an anilino diaryl sulfide lead. J. Med. Chem. 43: 4025–4040. [DOI] [PubMed] [Google Scholar]
- Liu, G., Huth, J.R., Olejniczak, E.T., Mendoza, R., DeVries, P., Leitza, S., Reilly, E.B., Okasinski, G.F., Fesik, S.W., and von Geldern, T.W. 2001. Novel p-arylthio cinnamides as antagonists of leukocyte function- associated antigen-1/intracellular adhesion molecule-1 interaction. 2. Mechanism of inhibition and structure-based improvement of pharmaceutical properties. J. Med. Chem. 44: 1202–1210. [DOI] [PubMed] [Google Scholar]
- Lu, C., Shimaoka, M., Ferzly, M., Oxvig, C., Takagi, J., and Springer, T.A. 2001. An isolated, surface-expressed I domain of the integrin αLβ2 is sufficient for strong adhesive function when locked in the open conformation with a disulfide bond. Proc. Natl. Acad. Sci. 98: 2387–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz, M. and Kenakin, T. 1999. Quantitative molecular pharmacology and informatics in drug discovery. John Wiley & Sons, Ltd., New York.
- Marlin, S.D. and Springer, T.A. 1987. Signalling and adhesive properties of the integrin leucocyte function-associated antigen 1 (LFA-1). Cell 51: 813–819. [DOI] [PubMed] [Google Scholar]
- Matthews, J.C. 1993. Fundamentals of receptor, enzyme, and transport kinetics. CRC Press, Boca Raton, FL.
- Panvera Corp. 1995. Fluorescence polarization applications guide. Panvera Corp., Madison, WI.
- Pratt, W.B. and Taylor, P. 1990. Principles of drug action: The basis of pharmacology. Churchill Livingstone, New York.
- Quan, C., Skelton, N.J., Clark, K., Jackson, D.Y., Renz, M.E., Chiu, H.H., Keating, S.M., Beresini, M.H., Fong, S., and Artis, D.R. 1998. Transfer of a protein binding epitope to a minimal designed peptide. Biopolymers 47: 265–275. [DOI] [PubMed] [Google Scholar]
- Randi, A.M. and Hogg, N. 1994. I domain of β 2 integrin lymphocyte function-associated antigen-1 contains a binding site for ligand intercellular adhesion molecule-1. J. Biol. Chem. 269: 12395–12398. [PubMed] [Google Scholar]
- Salas, A., Shimaoka, M., Kogan, A.N., Harwood, C., von Andrian, U.H., and Springer, T.A. 2004. Rolling adhesion through an extended conformation of integrin αLβ2 and relation to α I and β I-like domain interaction. Immunity 20: 393–406. [DOI] [PubMed] [Google Scholar]
- Shimaoka, M. and Springer, T.A. 2004. Therapeutic antagonists and the conformational regulation of the β2 integrins. Curr. Topics Med. Chem. 4: 1485–1495. [DOI] [PubMed] [Google Scholar]
- Shimaoka, M., Lu, C., Palframan, R.T., von Andrian, U.H., McCormack, A., Takagi, J., and Springer, T.A. 2001. Reversibly locking a protein fold in an active conformation with a disulfide bond: Integrin αL I domains with high affinity and antagonist activity in vivo. Proc. Natl. Acad. Sci. 98: 6009–6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimaoka, M., Xiao, T., Liu, J.-H., Yang, Y., Dong, Y., Jun, C.-D., McCormack, A., Zhang, R., Joachimiak, A., Takagi, J., et al. 2003a. Structures of the α L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell 112: 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimaoka, M., Salas, A., Yang, W., Weitz-Schmidt, G., and Springer, T. 2003b. Small molecule integrin antagonists that bind to the β2 subunit I-like domain and activate signals in one direction and block them in another. Immunity 19: 391–402. [DOI] [PubMed] [Google Scholar]
- Van Kooyk, Y. and Figdor, C.G. 1997. Signalling and adhesive properties of the integrin leucocyte function-associated antigen 1 (LFA-1). Biochem. Soc. Trans. 25: 515–520. [DOI] [PubMed] [Google Scholar]
- Weitz-Schmidt, G., Welzenbach, K., Brinkmann, V., Kamata, T., Kallen, J., Bruns, C., Cottens, S., Takada, Y., and Hommel, U. 2001. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat. Med. 7: 687–692. [DOI] [PubMed] [Google Scholar]
- Welzenbach, K., Hommel, U., and Weitz-Schmidt, G. 2002. Small molecule inhibitors induce conformational changes in the I domain and the I-like domain of Lymphocyte Function-Associated Antigen-1. J. Biol. Chem. 277: 10590–10598. [DOI] [PubMed] [Google Scholar]
- Wong, A., Hwang, S.M., Johanson, K., Samanen, J., Bennett, D., Landvatter, S.W., Chen, W., Heys, J.R., Ali, F.E., Ku, T.W., et al. 1998. Binding of [3H]-SK&F 107260 and [3H]-SB 214857 to purified integrin αIIbβ3: Evidence for a common binding site for cyclic arginyl-glycinyl-aspartic acid peptides and nonpeptides. J. Pharmacol. Exp. Ther. 285: 228–235. [PubMed] [Google Scholar]
- Woska Jr., J.R., Morelock, M.M., Jeanfavre, D.D., Caviness, G.O., Bormann, B.-J., and Rothlein, R. 1998. Molecular comparison of soluble intercellular adhesion molecule (sICAM)-1 and sICAM-3 binding to lymphocyte function-associated antigen-1. J. Biol. Chem. 273: 4725–4733. [DOI] [PubMed] [Google Scholar]
- Yalamanchili, P., Lu, C., Oxvig, C., and Springer, T.A. 2000. Folding and function of I domain-deleted Mac-1 and lymphocyte function-associated antigen-1. J. Biol. Chem. 275: 21877–21882. [DOI] [PubMed] [Google Scholar]
- Yang, W., Shimaoka, M., Salas, A., Takagi, J., and Springer, T.A. 2004. Intersubunit signal transmission in integrins by a receptor-like interaction with a pull spring. Proc. Natl. Acad. Sci. 101: 2906–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf-Makagiansar, H., Anderson, M.E., Yakovleva, T.V., Murray, J.S., and Siahaan, T.J. 2002. Inhibition of LFA-1/ICAM-1 and VLA-4/ VCAM-1 as a therapeutic approach to inflammation and autoimmune diseases. Med. Res. Rev. 22: 146–167. [DOI] [PubMed] [Google Scholar]