

Abstract
Molecular chaperones of the Hsp70 family (bacterial DnaK, DnaJ, and GrpE) were shown to be strictly required for refolding of firefly luciferase from a denatured state and thus for effective restoration of its activity. At the same time the luciferase was found to be synthesized in an Escherichia coli cell-free translation system in a highly active state in the extract with no chaperone activity. The addition of the chaperones to the extract during translation did not raise the activity of the enzyme. The abrupt arrest of translation by the addition of a translational inhibitor led to immediate cessation of the enzyme activity accumulation, indicating the cotranslational character of luciferase folding. The results presented suggest that the chaperones of the Hsp70 family are not required for effective cotranslational folding of firefly luciferase.
Keywords: cotranslational folding, specific activity, Hsp70 chaperones, trigger factor
The ribosomal synthesis of polypeptide chains is often accompanied by their folding into functionally active proteins. The cotranslational folding of proteins was demonstrated both in eukaryotic (Frydman et al. 1994; Kolb et al. 1994; Komar et al. 1997) and prokaryotic (Nicola et al. 1999; Kolb et al. 2000) cytosols. The question arises about the participation of molecular chaperones in cotranslational protein folding (Frydman 2001). In this study the effect of Escherichia coli molecular chaperones of the Hsp70 family (DnaK, DnaJ, and GrpE) on the folding of firefly luciferase synthesized in a bacterial cell-free system was studied.
Firefly (Photinus pyralis) luciferase is a monomeric multidomain protein (Conti et al. 1996) that acquires its native structure during translation (Frydman et al. 1994; Kolb et al. 1994, 2000). On the other hand, the refolding of firefly luciferase from a denatured state in the absence of chaperones proceeds inefficiently and very slowly (Herbst et al. 1997; Zako et al. 2000), and requires Hsp70 chaperones to be refolded with a reasonable rate (Schröder et al. 1993; Szabo et al. 1994; Buchberger et al. 1996). The productive assistance of chaperones in refolding of luciferase might suggest their role in cotranslational folding of the protein as well. The interaction of Hsp70 chaperones with newly synthesized proteins, including ribosome-associated nascent chains of luciferase, was reported (Hendrick et al. 1993; Frydman et al. 1994; Teter et al. 1999).
Here we compared the effects of bacterial Hsp70 family chaperones on cotranslational folding of luciferase in a bacterial cell-free translation system and on its refolding from a denatured state in the same bacterial extract.
Results
Refolding of firefly luciferase in E. coli S30 extract
To estimate endogenous chaperone activity in the bacterial extract used for translation, the experiments on refolding of denatured luciferase were carried out in the translation mixture. A sample of commercial firefly luciferase was treated with 7.4 M urea, and the refolding was initiated by 50-fold dilution into the translation mixture without luciferase mRNA (in some refolding experiments GFP mRNA was present instead of luciferase mRNA). Thus, the refolding was performed in the presence of the same components as the folding during translation. The concentration of luciferase in the refolding experiments was 8.4 nM, that is, within the same range as in the cell-free translation system. The course of refolding was recorded in a luminometer cell by monitoring the luciferase activity as a function of time. Samples of nondenatured luciferase diluted with the same translation mixture were taken as controls. As seen in Figure 1, the refolding of luciferase in the E. coli translation mixture was inefficient; only 8% activity was recovered when the plateau level was reached after 90 min incubation (data not shown). In contrast, the refolding became highly efficient when the mixture used for dilution of the denaturant was supplemented with exogenous DnaK, DnaJ, and GrpE: Luciferase restored about 80% of its activity within 60 min (Fig. 1).
Figure 1.

Refolding of urea-denatured firefly luciferase in cell-free translation systems. Refolding of luciferase denatured with buffered 7.4 M urea was initiated by dilution of a 0.5-μL aliquot containing 1.54 ng of the protein with 25 μL of translation system supplemented with luciferin and GFP mRNA. The time course of refolding was recorded at 25°C by recovery of luciferase luminescence. The translation system supplemented with the same amount of native enzyme was used as a control. The addition of Hsp70 chaperones is indicated above the curves; the addition of trigger factor, below the curve.
Thus, the refolding test showed a scarcity of active Hsp70 chaperones in the bacterial S30 extract used. This made it possible to evaluate the contribution of chaperones of the Hsp70 family to cotranslational folding in the cell-free translation system.
To test the ability of trigger factor to catalyze refolding of denatured luciferase, the same refolding experiment was carried out in the translation mixture supplemented with 5 μM trigger factor. No promotion of luciferase refolding by the trigger factor was detected (Fig. 1, lower curve).
Cotranslational folding of firefly luciferase
The efficiency of cotranslational folding at different concentrations of added chaperones and in their absence was judged from specific activity of luciferase synthesized under these different conditions. Results are shown in Figure 2. As seen from the time course of full-length polypeptide accumulation (Fig. 2A), the addition of chaperones to the translation system had little effect on protein synthesis. Similarly, an excess of chaperones in the translation reaction did not lead to a significant increase in luciferase activity accumulation (Fig. 2B). Moreover, the specific activity of luciferase also remained the same, irrespective of the presence or the absence of added chaperones or their concentration (Fig. 2C). The slight increase in specific activity during incubation after reaching the translational plateau seen in Figure 2C (both in the presence and the absence of chaperones) can be attributed to a delayed release of some ribosome-bound luciferase chains from the ribosome.
Figure 2.
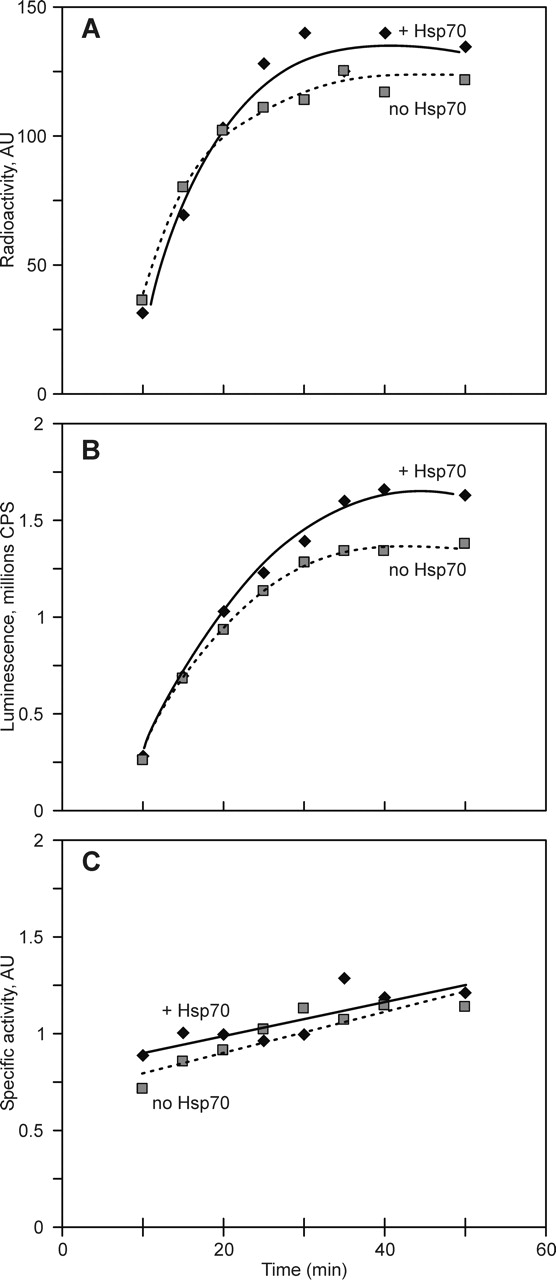
The time course of luciferase mRNA translation in cell-free systems. Translation was performed in an E. coli S30 translation system at 25°C in the presence of 0.1 mM luciferin and [14C]phenylalanine. Black symbols and solid lines correspond to the reaction with addition of Hsp70 chaperones; gray symbols and dashed lines, to the reaction without added chaperones. (A) Full-length luciferase polypeptide accumulation as revealed by 10% SDS-PAGE and phosphor-imaging. (B) Kinetics of luciferase activity accumulation in the same translation mixture. (C) Specific activity of luciferase calculated as the ratio of the luminescence (data from B) to the radioactivity (data from A).
The results shown in Figure 2 demonstrated that the Hsp 70 family chaperones had an effect neither on the productivity of luciferase synthesis nor on the efficiency of its folding into active enzyme during translation.
The absence of a post-translational folding phase during cell-free translation
In the next series of experiments, the time course of luciferase synthesis and folding in the presence of luciferase substrates was recorded continuously in a luminometer, as described earlier (Kolb et al. 1994). At some point during the linear phase of active enzyme accumulation the translation was arrested by addition of an inhibitor into the incubation mixture. As the instant block of translation prevented further synthesis of luciferase, a further increase in enzymatic activity would reflect the folding of the polypeptide chains that had been already released from the ribosome (posttranslational phase of folding) (Kolb et al. 1994, 2000; Agashe et al. 2004). Figure 3A shows that the addition of 100 μM minocycline (tetracycline antibiotic) to the translation mixtures resulted in abrupt cessation of the increase in luciferase activity (see also Kolb et al. 1994 see also Kolb et al. 2000). The same effect was observed with other translation inhibitors, such as 20 μM thiostrepton or RNAase A (not shown). The addition of a buffer instead of a translation inhibitor gave just a slight decrease of the slope of the activity accumulation curve due to dilution effect (Fig. 3C). The immediate halt of the activity accumulation upon addition of the translation inhibitors indicates that all luciferase molecules released from the ribosome had already acquired the active conformation.
Figure 3.
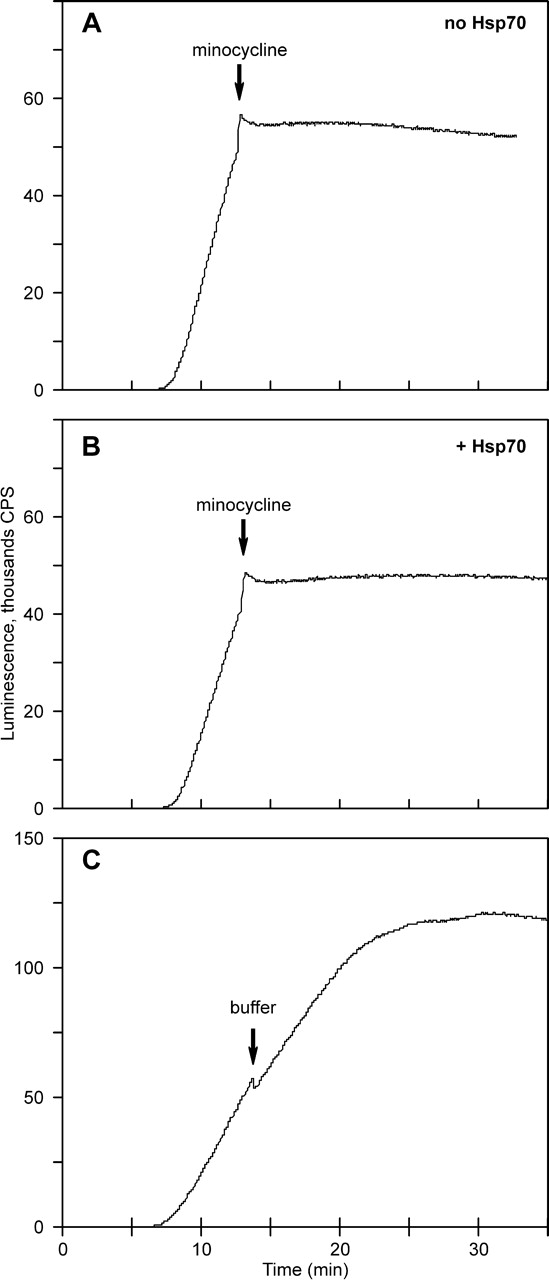
Cessation of luciferase activity accumulation upon arrest of translation. (A) Blocking of luciferase synthesis by injection of minocycline into the translation system to a final concentration of 0.1 mM, no chaperones added. (B) The same as in A with addition of Hsp70 chaperones to the translation system. (C) Control with addition of buffer instead of an inhibitor.
Similar experiments on translation inhibition were carried out with translation systems containing chaperones. The concentration of newly synthesized full-length luciferase in these experiments was estimated to be about 8 nM, and DnaK, DnaJ, and GrpE were added in concentrations of 1.3 μM, 200 nM, and 650 nM, respectively. Figure 3B shows that the abrupt cessation of active luciferase accumulation was observed also in the presence of the chaperones, as in the case when they were absent (Fig. 3, cf. A and B). Hence, the presence of Hsp70 chaperones did not result in the appearance of a detectable posttranslational folding phase during luciferase synthesis.
Discussion
Spontaneous refolding of firefly luciferase from a denatured state in buffer solutions was shown to be slow and inefficient, and Hsp70 family chaperones were strictly required for fast and productive refolding of the enzyme (Schröder et al. 1993; Szabo et al. 1994; Buchberger et al. 1996). At the same time, the cotranslational folding of this protein in cell-free systems was found to be fast and productive (Kolb et al. 2000). The involvement of the Hsp70 chaperones in cotranslational folding could not be excluded, more so as the physical interaction of Hsp70 chaperones with ribosome-associated nascent chains of this protein was reported (Hendrick et al. 1993; Frydman et al. 1994).
The results presented here demonstrate that chaperones of the Hsp70 family are not required for cotranslational folding of firefly luciferase: Their presence in a large excess in translation systems during synthesis of the protein affected neither the specific activity of the enzyme synthesized nor the time course of its cotranslational folding.
Several experimental findings are consistent with the idea of chaperone-independent cotranslational folding of firefly luciferase. First, it was found that specific activity of luciferase synthesized under nonstress conditions in E. coli ΔdnaK52 cells (containing no DnaK and reduced concentration of DnaJ chaperones) is equal to that of the enzyme produced in dnaK+ cells of wild type (Hesterkamp and Bukau 1998). Second, the efficient folding of luciferase was demonstrated upon transport of the growing chains into proteoliposomes, i.e., in the milieu without luminal microsomal chaperones and other folding catalysts (Tyedmers et al. 1996). Furthermore, the folding of luciferase in proteoliposomes was found to be more efficient than that assisted by soluble luminal chaperones in microsomes (Tyedmers et al. 1996). These results call the role of molecular chaperones in cotranslational protein folding into question.
The cooperation of the bacterial Hsp70 system with trigger factor in chaperoning nascent multidomain proteins was recently claimed; in particular, it was reported that the simultaneous addition of Hsp70 with trigger factor to a bacterial translation system resulted in an almost fourfold increase in the specific activity of synthesized luciferase (Agashe et al. 2004). This result is in contradiction with the earlier observation on the parallel synthesis of active firefly luciferase in bacterial (E. coli), plant (wheat germ), and animal (rabbit reticulocyte) cell-free translation systems in which the same high level of specific activity was attained without added chaperones (Kolb et al. 2000). Our results show that trigger factor does not promote refolding of denatured luciferase in the translation mixture (Fig. 1); the effect was absent also in buffer solutions (data not shown). If the participation of trigger factor in folding of nascent luciferase (Agashe et al. 2004) is the case, it shows a fundamental difference in folding pathways between cotranslational folding and refolding from a denatured state: Refolding catalysts are not involved in cotranslational folding and vice versa.
Chaperone-independent folding of newly synthesized proteins was shown also in a number of other cases, including the reovirus cell attachment protein (Gilmore et al. 1996), alphavirus capsid protein (Nicola et al. 1999), bacterial dihydrofolate reductase (DHFR), green fluorescent protein (GFP), and λ-lysozyme (Shimizu et al. 2001). The three latter examples are of special interest because chaperones were absent “by default” in the cell-free system used for the syntheses: This system was reconstituted completely from individual purified or synthetic components without any molecular chaperones. Translation in the system resulted in the synthesis of active proteins; the specific activity of the synthesized DHFR was similar to that of the protein, isolated and purified from overproducing cells (Shimizu et al. 2001).
It is likely that the ribosomal machinery by itself is principally capable of maintaining the correct and fast folding of a nascent polypeptide chain during translation. At least those chaperones that catalyze refolding of denatured luciferase seem to be not required for the cotranslational folding.
Materials and methods
Materials
Mg(OAc)2, KOAc, and MgCl2 were purchased from Fluka; urea was from ICN, [14C]phenylalanine (19 GBq/mmol) was from Amersham Pharmacia Biotech; restriction endonuclease XhoI was from Fermentas; creatine phosphate, creatine kinase, nucleoside triphosphates, luciferin, Photinus pyralis luciferase, T7 RNA polymerase, endonuclease BglII, and RNasin were purchased from Roche. DnaK, DnaJ, and GrpE (dimer) were supplied by Stressgen. Succinyl-Ala-Phe-Pro-Phe-4-nitro-anilide was purchased from Bachem. Minocycline was a gift from Lederle Laboratories. Other chemicals were from Sigma.
Preparation of E. coli S30 extract
S30 extract was prepared according to Zubay’s (1973) procedure with minor modifications. E. coli strain A19 cells were grown in LB medium at 37°C with intensive aeration up to OD 0.8 at 590 nm, then chilled to 10°C and collected by centrifugation at 4°C. The cells were washed twice with an equal volume of a buffer containing 10 mM Tris-OAc (pH20 8.2), 14 mM Mg(OAc)2, 60 mM KCl, and 6 mM β-mercaptoethanol, and once in a 10-fold volume of the same buffer. Then the cells were resuspended in an equal volume of the same buffer containing 1 mM DTT instead of β-mercaptoethanol. After addition of 0.1 mM phenylmethyl sulfonyl fluoride to the suspension, the cells were disintegrated in a cooled French press. The homogenate was centrifuged at 30,000g for 30 min and the upper two-thirds of the supernatant was collected and recentrifuged under the same conditions. Again, the upper two-thirds of the supernatant was collected. Then every 10 mL of supernatant received 0.7 mL of a mixture containing 0.75 M Tris-OAc (pH20 8.2), 7.5 mM DTT, 21 mM Mg(OAc)2, 0.5 mM each of the 20 amino acids, 6 mM ATP, and 0.5 M acetylphosphate. After 80 min incubation at 37°C, the homogenate was dialyzed against a buffer containing 10 mM Tris-OAc (pH20 8.2), 14 mM Mg(OAc)2, 60 mM KOAc, and 0.5 mM DTT. The buffer was changed after 2 h; dialysis continued overnight. Finally the S30 extract was clarified by centrifugation at 10,000g, frozen in portions in liquid nitrogen, and stored at −80°C.
Isolation and purification of trigger factor (TF)
This was performed according to Stoller et al. (1995) from the E. coli strain BL21. The factor specific peptidylprolyl cis/trans isomerase activity was tested in a reaction with chromopeptide succinyl-Ala-Phe-Pro-Phe-4-nitroanilide as in Scholz et al. (1997) and found to be similar to that described there.
Preparation of mRNA
In vitro transcription with T7 RNA polymerase was used to prepare mRNA. Luciferase mRNA was transcribed from pT7 luc (Kolb et al. 2000) linearized with XhoI, and mRNA encoding for GFP “cycle3/red shift mutant” was prepared by transcription of pGFPC3M1 plasmid (Chekulaeva et al. 2001) linearized with BglII. The transcription reaction was carried out in 100 μL of 200 mM HEPES-KOH (pH20 7.5), containing 30 mM MgCl2, 2 mM spermidine, 20 mM dithiotreitol, 7 mM ATP, GTP, CTP, and UTP each, 40 units of RNasin, 3 μg of linearized DNA template, and 500 units of T7 RNA polymerase. The reaction was carried out at 37°C for 2 h and stopped by phenolchloroform extraction. Transcript was further purified by precipitation with 3.5 M LiCl. Purity and size of mRNAs synthesized were controlled by 5% PAGE in 7 M urea. A 1-mg/ml solution of the transcript in H2O was used for translation experiments.
In vitro translation
Translation in 30% E. coli S30 extract was carried out at 25°C in the presence of 1.2 mM ATP, 0.8 mM GTP, CTP, and UTP each, 0.03 mg/mL folinic acid, 80 mM creatine phosphate, 0.25 mg/mL creatine kinase, 0.64 mM cAMP, 4% PEG 8000, 1 mM amino acids each except glutamic acid, and 0.175 mg/mL total E. coli tRNA in a buffer containing 14 mM Mg(OAc)2, 26 mM HEPES-KOH (pH20 7.5), 27 mM KOAc, 210 mM potassium glutamate, and 1.7 mM dithiotreitol. The reaction mixture also contained 0.1 mM luciferin. [14C]phenylalanine was added to the translation system when indicated instead of an unlabeled one to the concentration of 1.9 μM. The reaction mixture was preincubated at 4°C for 10 min, and then translation was initiated by addition of mRNA to the concentration of 20 μg/mL. When translation was performed in the presence of chaperones, individual DnaK, DnaJ, and GrpE (dimer) were used in concentrations up to 1.3 μM, 200 nM, and 650 nM, respectively.
Refolding of luciferase
P. pyralis luciferase, 40 μg/mL in 0.5 M HEPES-KOH (pH20 7.5) was denatured by mixing with 12 volumes of a solution containing 8 M urea, 14 mM Mg(OAc)2, 26 mM HEPES-KOH (pH20 7.5), 27 mM KOAc, and 1.7 mM dithiotreitol with subsequent incubation at 20°C for 5 min. Refolding was initiated by dilution of a 0.5-μL aliquot of the denatured enzyme solution with 25 μL of translation mixture containing luciferase substrates (including 0.1 mM luciferin) but programmed with 20 μg/mL GFP mRNA. The time course of refolding was determined at 25°C by continuously recording the light-emitting activity in a luminometer (Kolb et al. 1994). When the refolding was performed in the presence of chaperones, DnaK, DnaJ, and GrpE (dimer) were used in concentrations up to 1.3 μM, 200 nM, and 650 nM, respectively. Trigger factor was added to the refolding mixture to the concentration of 5 μM when indicated.
Specific activity determination
Synthesis of luciferase was performed at 25°C in E. coli S30 extract in the presence of [14C]phenylalanine as described above. To determine enzymatic activity of luciferase, 10-μL aliquots were withdrawn from the translation mixture at the indicated time points and immediately added to 10 μL of a solution containing 40 μM thiostrepton, 1.2 mM ATP, 0.1 mM luciferin, 14 mM Mg(OAc)2, 26 mM HEPES-KOH (pH20 7.5), 27 mM KOAc, and 1.7 mM dithiotreitol; the intensity of emitted light in the aliquots was recorded at 25°C in a luminometer. The same aliquots were then analyzed with 10% SDS-PAGE according to Schägger and von Jagow (1987), and the radioactivity of the full-length luciferase bands was determined with a Cyclone PhosphorImager (Packard Instrument Co.). The specific activity of luciferase was estimated as the ratio of the luminescence to the amount of the synthesized protein calculated from radioactivity.
Acknowledgments
This work was supported by grant no. 02-04-48706 from the Russian Foundation for Basic Research and grant no. 1966.2003.4 from the President of the Russian Federation, and by the Program on Molecular and Cellular Biology of the Russian Academy of Sciences.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051752506.
References
- Agashe, V.R., Guha, S., Chang, H.-C., Genevaux, P., Hayer-Hartl, M., Stemp, M., Georgopoulos, C., Hartl, F.U., and Barral, J.M. 2004. Function of trigger factor and DnaK in multidomain protein folding: Increase in yield at the expense of folding speed. Cell 117: 199–209. [DOI] [PubMed] [Google Scholar]
- Buchberger, A., Schröder, H., Hesterkamp, T., Schönfeld, H.-J., and Bukau, B. 1996. Substrate shuttling between the DnaK and GroEL systems indicates a chaperone network promoting protein folding. J. Mol. Biol. 261: 328–333. [DOI] [PubMed] [Google Scholar]
- Chekulaeva, M.N., Kurnasov, O.V., Shirokov, V.A., and Spirin, A.S. 2001. Continuous-exchange cell-free protein-synthesizing system: Synthesis of HIV-1 antigen Nef. Biochem. Biophys. Res. Commun. 280: 914–917. [DOI] [PubMed] [Google Scholar]
- Conti, E., Francs, N.P., and Brick, P. 1996. Crystal structure of firefly luciferase throws light on a superfamily of adenylate-forming enzymes. Structure 4: 287–298. [DOI] [PubMed] [Google Scholar]
- Frydman, J. 2001. Folding of newly translated proteins in vivo: The role of molecular chaperones. Annu. Rev. Biochem. 70: 603–647. [DOI] [PubMed] [Google Scholar]
- Frydman, J., Nimmesgern, E., Ohtsuka, K., and Hartl, F.U. 1994. Folding of nascent polypeptide chains in a high molecular mass assembly with molecular chaperones. Nature 370: 111–117. [DOI] [PubMed] [Google Scholar]
- Gilmore, R., Coffey, M.C., Leone, G., McLure, K., and Lee, P.W.K. 1996. Cotranslational trimerization of the reovirus cell attachment protein. EMBO J. 15: 2651–2658. [PMC free article] [PubMed] [Google Scholar]
- Hendrick, J.P., Langer, T., Davis, T.A., Hartl, F.U., and Wiedmann, M. 1993. Control of folding and membrane translocation by binding of the chaperone DnaJ to nascent polypeptides. Proc. Natl. Acad. Sci. 90: 10216–10220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst, R., Schafer, U., and Seckler, R. 1997. Equilibrium intermediates in the reversible unfolding of firefly (Photinus pyralis) luciferase. J. Biol. Chem. 272: 7099–7105. [DOI] [PubMed] [Google Scholar]
- Hesterkamp, T. and Bukau, B. 1998. Role of the DnaK and HscA homologs of Hsp70 chaperones in protein folding in E. coli. EMBO J. 17: 4818–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb, V.A., Makeyev, E.V., and Spirin, A.S. 1994. Folding of firefly luciferase during translation in a cell-free system. EMBO J. 13: 3631–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2000. Co-translational folding of an eukaryotic multidomain protein in a prokaryotic translation system. J. Biol. Chem. 275: 16597–16601. [DOI] [PubMed] [Google Scholar]
- Komar, A.A., Kommer, A., Krasheninnikov, I.A., and Spirin, A.S. 1997. Cotranslational folding of globin. J. Biol. Chem. 272: 10646–10651. [DOI] [PubMed] [Google Scholar]
- Nicola, A.V., Chen, W., and Helenius, A. 1999. Co-translational folding of an alphavirus capsid protein in the cytosol of living cells. Nat. Cell Biol. 1:341–345. [DOI] [PubMed] [Google Scholar]
- Schägger, H. and von Jagow, G. 1987. Tricinesodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166: 368–379. [DOI] [PubMed] [Google Scholar]
- Scholz, C., Stoller, G., Zarnt, T., Fischer, G., and Schmid, F.X. 1997. Cooperation of enzymatic and chaperone functions of trigger factor in the catalysis of protein folding. EMBO J. 16: 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder, H., Langer, T., Hartl, F.-U., and Bukau, B. 1993. DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J. 12: 4137–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu, Y., Inoue, A., Tomary, Y., Suzuki, T., Yokogawa, T., Nishikawa, K., and Ueda, T. 2001. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 19: 751–755. [DOI] [PubMed] [Google Scholar]
- Stoller, G., Rücknagel, K.P., Nierhaus, K.H., Schmid, F.X., Fischer, G., and Rahfeld, J.-U. 1995. A ribosome-associated peptidylprolyl cis/trans isomerase identified as the trigger factor. EMBO J. 14: 4939–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo, A., Langer, T., Schröder, H., Flanagan, J., Bukau, B., and Hartl, F.U. 1994. The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system—DnaK, DnaJ, and GrpE. Proc. Natl. Acad. Sci. 91: 10345–10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teter, S.A., Houry, W.A., Ang, D., Tradler, T., Rockabrand, D., Fischer, G., Blum, P., Georgopoulos, C., and Hartl, F.U. 1999. Polypeptide flux through bacterial Hsp70: DnaK cooperates with trigger factor in chaperoning nascent chains. Cell 97: 755–765. [DOI] [PubMed] [Google Scholar]
- Tyedmers, J., Brunke, M., Lechte, M., Sandholzer, U., Dierks, T., Schlotterhose, P., Schmidt, B., and Zimmermann, R. 1996. Efficient folding of firefly luciferase after transport into mammalian microsomes in the absence of luminal chaperones and folding catalysts. J. Biol. Chem. 271: 19509–19513. [DOI] [PubMed] [Google Scholar]
- Zako, T., Deguchi, H., Kitayama, A., Ueda, H., and Nagamune, T. 2000. Refolding of firefly luciferase immobilized on agarose beads. J. Biochem. 127: 351–354. [DOI] [PubMed] [Google Scholar]
- Zubay, G. 1973. In vitro synthesis of protein in microbial systems. Annu. Rev. Genet. 7: 267–287. [DOI] [PubMed] [Google Scholar]