

Abstract
The biological activity of DnaK, the bacterial representative of the Hsp70 protein family, is regulated by the allosteric interaction between its nucleotide and peptide substrate binding domains. Despite the importance of the nucleotide-induced cycling of DnaK between substrate-accepting and releasing states, the heterotropic allosteric mechanism remains as yet undefined. To further characterize this mechanism, the nucleotide-induced absorbance changes in the vibrational spectrum of wild-type DnaK was characterized. To assign the conformation sensitive absorption bands, two deletion mutants (one lacking the C-terminal α-helical subdomain and another comprising only the N-terminal ATPase domain), and a single-point DnaK mutant (T199A) with strongly reduced ATPase activity, were investigated by time-resolved infrared difference spectroscopy combined with the use of caged-nucleotides. The results indicate that (1) ATP, but not ADP, binding promotes a conformational change in both subdomains of the peptide binding domain that can be individually resolved; (2) these conformational changes are kinetically coupled, most likely to ensure a decrease in the affinity of DnaK for peptide substrates and a concomitant displacement of the lid away from the peptide binding site that would promote efficient diffusion of the released peptide to the medium; and (3) the α-helical subdomain contributes to stabilize the interdomain interface against the thermal challenge and allows bidirectional transmission of the allosteric signal between the ATPase and substrate binding domains at stress temperatures (42°C).
Keywords: DnaK, Hsp70, chaperones, allosterism, infrared, caged-nucleotides
The Hsp70 (70-kDa heat-shock proteins) family of protein chaperones are involved in a variety of essential cellular functions such as folding, assembly, and transport of proteins (Hartl 1996; Bukau and Horwich 1998). DnaK is a well-characterized member of this protein family in Escherichia coli (Mayer et al. 2000; Slepenkov and Witt 2002), and it consists of two major structurally and functionally different domains: an N-terminal 45-kDa ATPase domain connected by a short and conserved linker to a 25-kDa substrate binding domain (SBD). The latter contains two subdomains, a β-sandwich that holds the peptide binding site and a helical subdomain that folds over the β-sandwich and controls the accessibility of the peptide binding site, acting as a lid.
Different cochaperones regulate DnaK activity through substrate binding and nucleotide hydrolysis (DnaJ), and through nucleotide exchange (GrpE). The activity of DnaK, and of all known Hsp70s, is modulated by an allosteric mechanism where the bound nucleotide at the ATPase domain determines the affinity of the SBD for protein substrates. Since allostery is bidirectional, substrate binding enhances the ATPase activity of the protein. Several biochemical—partial proteolysis (Buchberger et al. 1995) and cross-linking (Farr et al. 1998)—and biophysical—fluorescence spectroscopy (Moro et al. 2003; Slepenkov and Witt 2003) and small-angle X-ray scattering (Wilbanks et al. 1995; Shi et al. 1996)—techniques have shown that ATP induces a global conformational change in the SBD of Hsp70 proteins that involves rearrangements of both β- and α-subdomains. These results demonstrate that ligand binding to one of these domains has an effect on the structural and functional properties of the other domain, although structural details of the intramolecular allosteric regulation are scarce. While the use of lidless mutants demonstrate a direct communication between the ATPase and the β-sandwich subdomain (Pellechia et al. 2000; Buczynski et al. 2001), it is not clear how the allosteric signal is transmited to the α-helical subdomain. High-resolution three-dimensional structures of isolated structural domains in various ligand states have been reported (Zhu et al. 1996; Harrison et al. 1997; Pellechia et al. 2000), but they do not provide information on either the interdomain interface or the residues involved in allosteric signaling. A recent NMR study of a 54-kDa two-domain construct of Hsp70 from Thermus thermophilus, containing the ATPase domain and a truncated SBD that includes the β-strand sandwich and lacks the lid subdomain, has allowed investigators to estimate the relative positioning of both protein domains and therefore to locate at least part of the interdomain interface (Revington et al. 2005).
In this context, infrared difference spectroscopy combined with the use of caged ligands opens the possibility to follow in real time the ATP-induced allosteric conformational change, as already demonstrated for other biological systems, including GroEL (von Germar et al. 1999). The results presented here show that (1) the allosteric transition induced in DnaK by ATP binding simultaneously affects both subdomains of the SBD; (2) ATP hydrolysis relaxes the protein conformation to an ADP-like state, more similar to the apo-protein; and (3) the stabilizing subdomain interactions at the SBD are essential to maintain a functional interdomain interface that allows allosteric signaling at stress temperatures (e.g., 42°C).
Results
TR-DIR (time-resolved difference infrared) spectroscopy is very useful in detecting subtle protein conformational changes associated with ligand binding (Mantele 1993; Barth and Zscherp 2002). Moreover, in combination with the use of caged ligands the interaction of the ligand with the protein can be easily controlled, since illumination of the sample hydrolyzes the cage and leaves the molecule free to interact with the protein. Difference spectra are generated by subtracting spectra recorded at different times after ligand release from the spectrum measured just before sample illumination. In this way, IR bands arising from secondary structures or specific residues that do not take part in the interaction are cancelled out, while those that change their hydrogen bonding or chemical environment upon interaction give off differential spectroscopic signals.
The difference IR spectra contain contributions from different molecular events, such as nucleotide binding and hydrolysis, and the photolysis reaction. ATP binding can only be monitored if the caged-nucleotide is unable to bind DnaK. Filtration experiments were performed to test whether binding of caged-ATP to nucleotide-free DnaK was taking place (Fig. 1A). They demonstrated that this was not the case since unlike free nucleotides which bound tightly DnaK (Kd = 1–10 nM) (Theyssen et al. 1996; Russell et al. 1998), caged-ATP did not bind the protein and all the initially added caged ligand appeared in the filtrate. Therefore, the IR difference spectra also contain information of the binding process.
Figure 1.
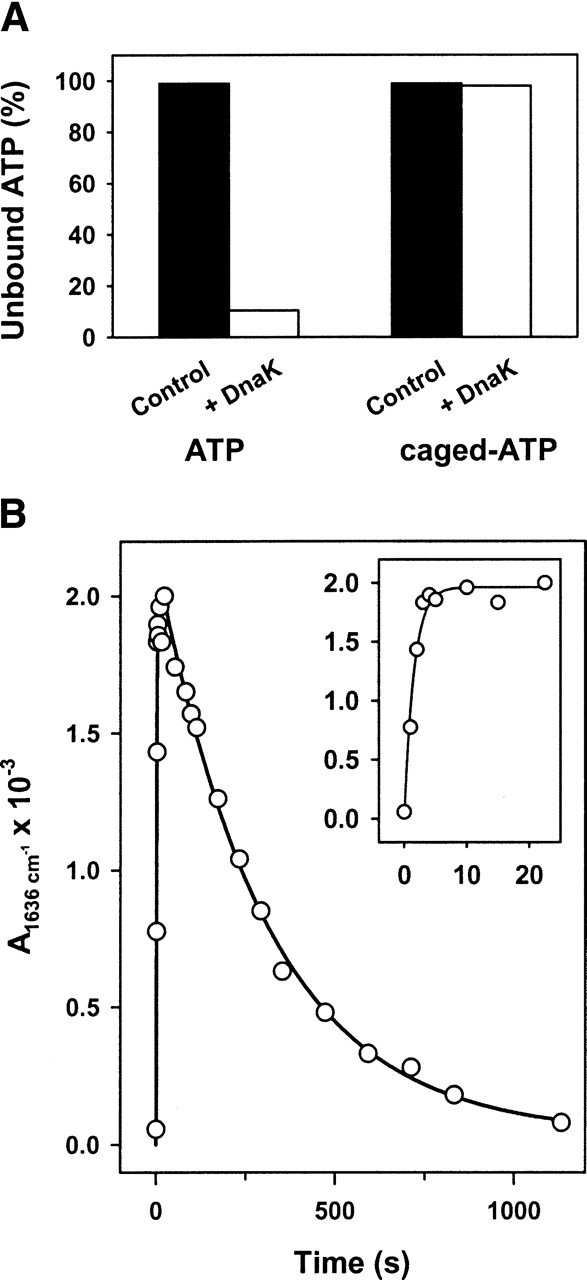
(A) Interaction of caged-ATP with DnaK. Equimolar amounts of DnaK and ATP or caged-ATP were mixed and their free concentration was estimated after centrifugation of the mixture in microconcentration filters. (B) Kinetics of ATP-induced conformational change in DnaK. Time dependence of the absorbance change at 1636 cm−1. Protein concentration was ~1.4 mM and the nucleotide:protein molar ratio was 1. Experiments were performed in H2O-based buffer at 25°C. (Inset) Absorbance changes during the first 25 sec after ATP release.
ATP binding
As mentioned above, the interaction between the nucleotide and DnaK is accomplished by releasing the ligand from an inactive (caged) photolabile analog with a UV flash that has no effect on the protein. Interaction of ATP with DnaK induces absorbance changes in the difference spectra that reflect a nucleotide-induced transition between distinct protein conformations. Previous studies have established that bands assigned to the photolytic release of nucleotides dominate the difference IR spectra <1300 cm−1 (Barth et al. 1996). Above this frequency, i.e., in the 1750–1300 cm−1 spectral region, the experimentally detected absorbance changes contain contributions from molecular events related to the photolysis reaction and to protein conformational changes induced by nucleotide binding and/or hydrolysis.
The time evolution of a representative differential signal at 1636 cm−1 shows two different kinetic events: a first one evidenced by a fast increase in the amplitude of the absorbance change (apparent rate constant of 0.65 sec−1), followed by a second event characterized by a slow decrease with an apparent rate constant of 0.004 sec−1 (Fig. 1B). When the binding event is studied by intrinsic fluorescence, two transitions are observed instead of one (Slepenkov and Witt 1998). The apparent rate constant of the fast event is ATP-concentration dependent with a maximum rate of 20 sec−1 at 25°C, its detection being beyond our experimental time resolution (0.5 sec), while the slow transition shows an apparent rate constant (k = 0.67 sec−1) similar to that described in this study. To further distinguish binding from hydrolysis, caged-ATP was released into ADP-bound DnaK samples to slow down the binding event (Slepenkov and Witt 1998; Grimshaw et al. 2001) and to better mimic the physiological situation that includes nucleotide exchange. Under these experimental conditions, the first kinetic event experimentally detected after ATP release to the medium is characterized by the appearance of the following differential signals: 1693(−)/1685(+); 1674(−)/1660(+); 1650(−)/1636(+); 1567(+); 1543(−), and 1391(+) (Fig. 2A). The 1526(−) cm−1 signal together with a broad band centered at ~1640 cm−1 are due to the photolysis reaction (Fig. 2A). Both the intensity and the position of the absorbance changes were essentially the same regardless of the presence of ADP in the sample before ATP release; thus, nucleotide exchange did not induce significant spectroscopic modifications (cf. top traces, Figs. 2 and 3). It also follows from this comparison that a larger similarity exists between nucleotide-free and ADP-bound DnaK than between any of these states and the ATP-bound form of the protein (see below). Analysis of the differential signals at 1636 cm−1 and 1650 cm−1 shows the expected reduction in the apparent rate constant from 0.65 sec−1 to 0.05 sec−1 (Fig. 2B). The similarity of this rate constant with the value found for ADP dissociation from ADP-DnaK complexes (0.035 sec−1) (Theyssen et al. 1996) suggests, as expected, that ADP-dissociation precedes the ATP binding step that triggers protein conformational changes. Therefore, the fast kinetic event observed in this study represents the conformational change previously assigned to a global structural transition that occurs after ATP binding to the protein (Ha and McKay, 1995; Slepenkov and Witt 1998). The binding reaction is too fast to be detected in this study and was related with an alteration of the local environment around W102 (Slepenkov and Witt 1998).
Figure 2.
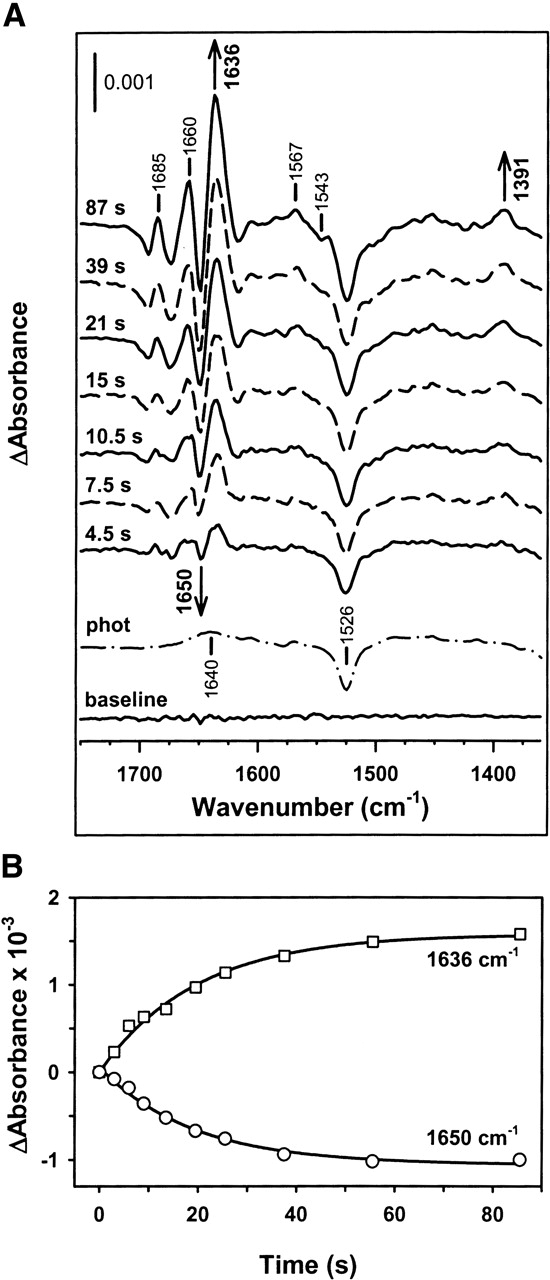
ATP binding to DnaK induces absorbance changes in the protein IR spectrum. (A) Time dependence of the IR difference spectrum of DnaK during the first 2 min after ATP release to an ADP-saturated DnaK sample. The final ATP/protein molar ratio was ~1. Protein concentration was 1.4 mM, the buffer was prepared in H2O, and measurements were carried out at 25°C. For the purpose of comparison, a baseline spectrum recorded with the protein sample and without flash (baseline) and the photolysis spectrum (phot) measured without protein are also shown. Average of 13 independent experiments. (B) Kinetics of the absorbance changes at 1650 cm−1 (circles) and 1636 cm−1 (squares), corresponding to the spectra depicted in A. The best fits obtained with single exponentials are shown in solid lines.
Figure 3.
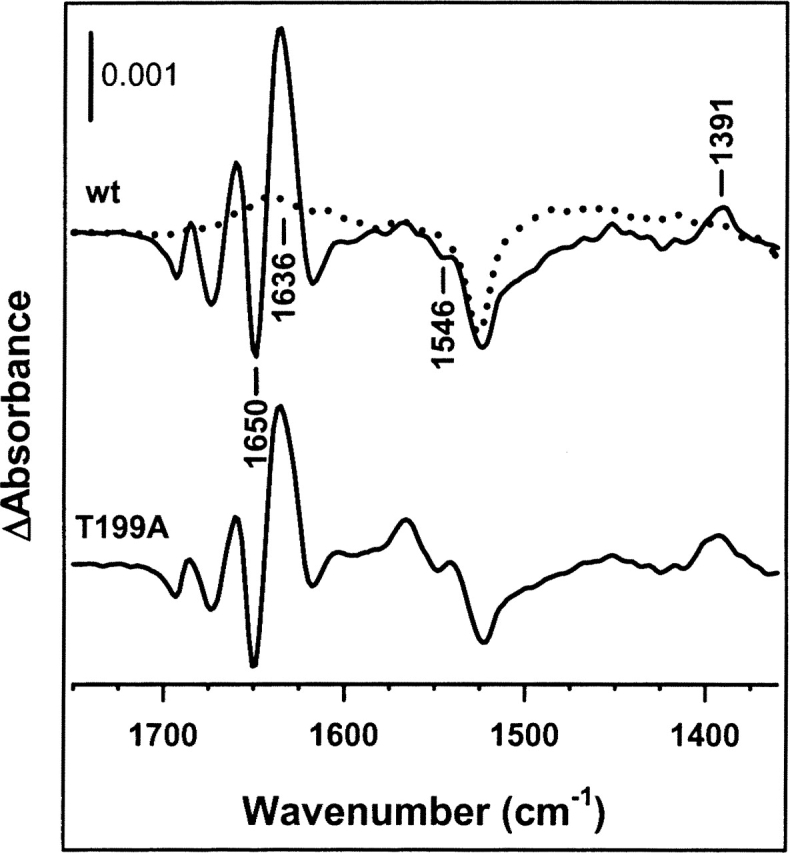
Assignment of the ATP-binding event. Comparison of the IR difference spectrum of wild-type apo-DnaK obtained during the first 10 sec after ATP release to the medium (upper trace) with the spectrum corresponding to an ATPase defective protein mutant (T199A) (lower trace). Experiments were performed in H2O-based buffer, at a protein concentration of 1.4 mM and 25°C. The spectrum corresponding to the photolysis reaction is shown in dotted line. The nucleotide:protein molar ratio was 1. Average of 10 (wild-type DnaK) and 6 (T199A DnaK) experiments.
When the same kinetic analysis is carried out in D2O-based buffer, similar apparent rate constants are obtained for absorbance changes that might be attributed to different structural elements (0.02 sec−1 and 0.019 sec−1 for the bands at 1645 cm−1 and 1631 cm−1, respectively) and to acidic residues (0.022 sec−1 for the signal at 1578 cm−1) (data not shown; see below). The twofold higher values of the time constant obtained in H2O medium might be explained by the higher protein concentration used in this buffer compared with D2O-based samples (see Materials and Methods). The assignment of these absorbance changes to a conformational change associated with nucleotide binding to DnaK was further tested by analyzing the ATP-induced difference spectrum of DnaK T199A. This single-point mutant, although unable to hydrolyze ATP under our experimental conditions (25°C), undergoes a nucleotide-induced allosteric transition similar to that of wild-type DnaK (Buchberger et al. 1995; Barthel et al. 2001; data not shown). As seen in Figure 3, the spectra recorded during the first 10 sec after ATP release to samples containing nucleotide-free wild type or DnaK T199A look alike, indicating that the above-described difference spectra of wild-type DnaK are indeed reflecting a conformational transition induced immediately after ATP binding to the protein.
Allosteric communication
An important question that would be of interest to address is whether these absorbance changes are due to interdomain allosteric communication or to a more direct nucleotide-induced conformational change located at the ATPase domain. To answer this question the ATP-induced IR differential signals of wild-type and two DnaK deletion mutants that lack the helical lid subdomain (DnaK1–507) or the entire SBD (ATPase domain) were compared (Fig. 4A). The results obtained in H2O buffer indicate that progressive deletion of the SBD results in a significant reduction of the intensity of some absorbance changes. Removal of the helical subdomain of the SBD reduces the amplitude of the differential signal at 1650(−)/1636(+) cm−1, as well as that of the positive absorbance change at 1391 cm−1. Since the positive component of the first differential feature would overlap with that of the β-structure (see below), the conformational change at the lid subdomain can be experimentally characterized by a reduction of the intensity at 1650 cm−1. As expected, deletion of the β-sandwich subdomain of the SBD results in a decrease in the intensity of the differential signal at 1636 cm−1, characteristic of extended structures (Susi and Byler 1986), and a further decrease in the amplitude of the 1395 cm−1 absorbance change that becomes negative. To prove that these absorbance changes were due to the allosteric transition that immediately follows ATP binding, the difference IR spectra of these proteins were also analyzed in the presence of ADP (Fig. 4A, lower panel). In contrast to what was found for ATP, the absorbance changes induced by ADP binding to the three proteins were similar and include only weak differential signals in the amide I region (1700–1610 cm−1). Thus, compared to ATP, ADP induces small conformational rearrangements that mostly affect to the ATPase domain of the protein. Taken together, these results demonstrate that ATP binding to the ATPase domain of DnaK induces conformational changes in both subdomains of the SBD of the protein that can be independently followed by difference IR spectroscopy.
Figure 4.

Assignment of absorbance changes due to interdomain interaction. (A) (Upper panel) ATP-binding spectrum of wild-type DnaK, DnaK(1–507), and the ATPase domain of the protein, recorded in H2O medium at 25°C. Average of 10 (wild-type DnaK), 7 (DnaK1–507), and 13 (ATPase) independent experiments. Protein concentration was 1.4 mM and the nucleotide:protein molar ratio was 1. Spectra were recorded during the first 10 sec after nucleotide release. (Lower panel) ADP-binding spectra of the same samples recorded in H2O-based buffer under the same experimental conditions. Average of seven (wild-type DnaK), six (Dnak1–507), and eight (ATPase) different experiments. (B) (Upper panel) ATP-induced absorbance changes in the IR difference spectrum of wild-type DnaK, DnaK1–507, and the ATPase domain of the protein. Protein concentration was 1 mM and equimolecular amounts of nucleotide were released to the medium. Experiments were performed in D2O-based buffer at 25°C and are the average of five (wild-type DnaK), four (DnaK1–507), and seven (ATPase) different experiments. (Lower panel) ADP-binding spectra of the same samples using the same experimental conditions. Average of 12, 5, and 4 independent experiments for wild-type DnaK, DnaK1–507, and ATPase domain, respectively. The photolysis spectra are shown in dotted lines. Other details are as in Figure 2.
A comparison between the difference spectra of wild-type DnaK in H2O (Fig. 4A) and D2O (Fig. 4B) recorded during the first 10 sec after nucleotide release to the medium, helps to assign the spectral features to secondary structure elements or to side chains of specific residues. Deuteration shifts the amide I modes (1700–1610 cm−1), mainly due to peptide C=O groups stretching, by up to 10 cm−1, and the amide II band, mainly due to amide-backbone NH groups, from ≈1550 cm−1 to 1460 cm−1 (Byler and Susi 1986; Arrondo et al. 1993; Jackson and Mantsch 1995). In contrast, larger downshifts (≥30 cm−1) are characteristic for the side-chain absorptions of solvent-accessible Asn, Gln, Lys, and Arg, as are small upshifts for COO− bands (Chirgadze et al. 1975; Venyaminov and Kalnin 1990; Barth 2000). The comparison shows the following differences upon deuteration: (1) Differential features between 1630–1700 cm−1 are down-shifted by 4–7 cm−1; (2) the negative signal at 1618 cm−1 becomes positive; and (3) a differential signal at 1581(−)/1560(+) cm−1 is clearly detected only in D2O. Therefore, the components between 1700 and 1630 cm−1 can be assigned to secondary structure elements while the one at 1560 cm−1 might arise from side chains of acidic amino acids. The reason why this signal is not well defined in H2O could be related to the possible overlapping with other deuteration-sensitive vibrations.
Essentially the same results are obtained when the spectroscopic effect of ATP binding to the three proteins is analyzed in D2O, apart from the isotopic downshifts mentioned above for wild-type DnaK (Fig. 4). The only significant difference is that in deuterated medium the 1600–1540 cm−1 spectral region seems to be more sensitive to the presence of different subdomains of the SBD. The positive signal observed for wild-type DnaK at 1560 cm−1 evolves to a broad one that might be composed of two components at 1572 and 1560 cm−1 upon lid removal, and to one major absorbance change at 1572 cm−1 for ATP-induced conformational change for the ATPase domain.
In light of the recent finding that interactions between helix B at the lid and loops at the β-sandwich of the SBD are essential to maintain the stability of the substrate binding site that allows formation of peptide–DnaK complexes at stress temperatures (e.g., 42°C) (Moro et al. 2004), we also tested a putative role of these interactions in the thermal stabilization of the interdomain interface. If this were the case, only the IR difference spectrum of DnaK(1–507) should be temperature sensitive while that of wild-type DnaK should remain unchanged. Figure 5A shows that this is indeed the case, since absorbance changes at the same positions and with similar amplitudes are observed for wild-type DnaK within the temperature range 25°–42°C. In contrast, the fine structure of the difference spectrum of the deletion mutant observed at 25°C is lost at 42°C, the spectrum resembling that of the ADP-bound state of the protein (see Fig. 4). To rule out the possibility that the nucleotide did not bind to the deletion mutant at stress temperatures we also analyzed the ATPase activity of wild-type and DnaK(1–507) at both permissive and stress temperatures. The activity values for wild-type and DnaK(1–507) at 42°C were 6.4 times and 7.5 times higher, respectively (data not shown), than those measured at 25°C (Moro et al. 2004), indicating that at 42°C the deletion mutant binds ATP.
Figure 5.
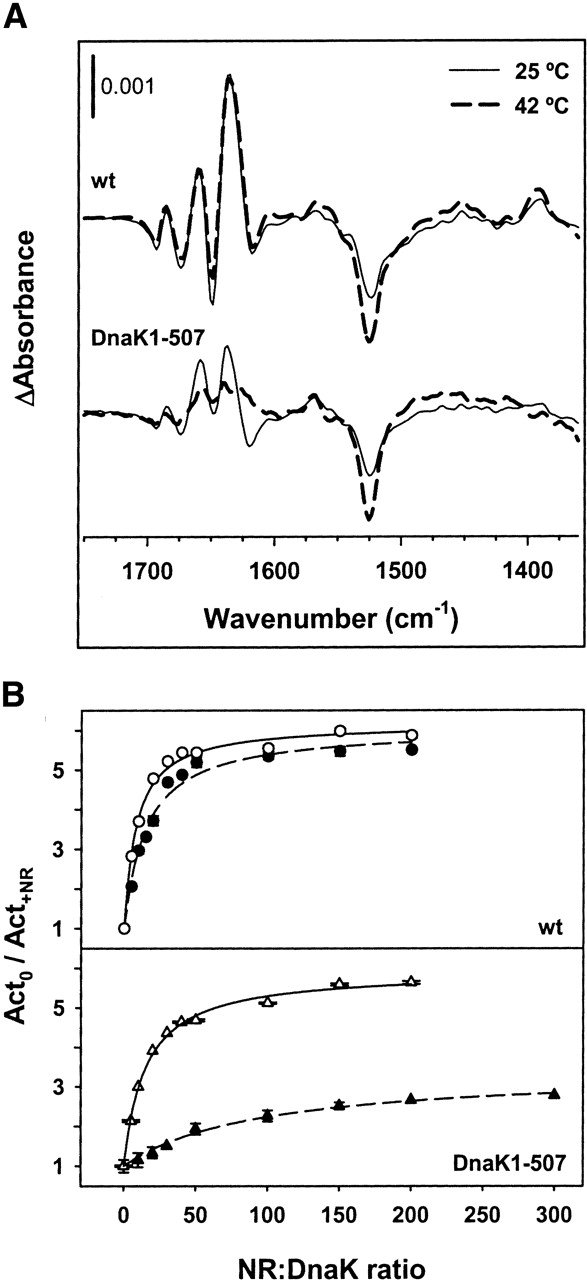
Thermal stability of the interdomain interface. (A) ATP-binding IR difference spectra of wild-type DnaK (upper traces) and DnaK1–507 (lower traces) recorded at 25°C (solid line) and 42°C (broken line) in H2O medium. Spectra were recorded during the first 10 sec after ATP release into samples containing ~1.4 mM DnaK or DnaK1–507 saturated with equimolecular amounts of ADP. ADP was added to stabilize the ATPase domain of the protein so that it can stand stress temperatures (e.g., 42°C). The concentration of ATP released from the cage was 1.4 mM and 4.2 mM at 25°C and 42°C, respectively, to account for the temperature-induced activation of the protein. Spectra at 25°C are taken from Figure 4. Average of seven (wild-type) and seven (DnaK1–507) independent experiments at 42°C. (B) Peptide-induced stimulation of the ATPase activity of wild-type DnaK (upper panel) and DnaK1–507 (lower panel) measured at 25°C (empty symbols) and 42°C (filled symbols). Relative values are the average of at least two independent measurements of the ATPase activity of the proteins in the absence (Act0) and in the presence of different concentrations of NR peptide (Act+NR). The best fits obtained, as described in Materials and Methods, are shown in solid (25°C) and broken (42°C) lines.
The effect of temperature on the communication in the opposite direction, e.g., from the SBD to the NBD domain, is usually studied by following the peptide-induced stimulation of DnaK ATPase activity (Jordan and McMacken 1995; Montgomery et al. 1999). Using this method, our data clearly show that at saturating peptide concentrations the stimulation is similar for wild-type and lidless DnaK at 25°C (~5.5-fold), while at 42°C this value remains unchanged for wild-type DnaK and is reduced to 2.7 for the deletion mutant (Fig. 5B). These data also show that the relative affinity of these proteins for NR peptide varies differently with increasing temperatures. The peptide:protein molar ratio at which half maximum activation is achieved (apparent Kd) changes slightly with temperature for wild-type DnaK (8 and 15 μM at 25° and 42°C, respectively), while for the deletion mutant the apparent Kd value increases from 15 to 95 μM when the temperature is raised from 25° to 42°C. Therefore, these results point out that both sub-domains of the SBD are important not only to maintain the conformation of the binding site but also to stabilize the interdomain interface through which the bidirectional allosteric signal is transmitted.
ATP hydrolysis
The second kinetic event shown in Figure 1, characterized by an exponential decrease of absorbance changes, reflects nucleotide hydrolysis. The time evolution of the difference spectra measured in H2O medium is shown in Figure 6, and indicates that during hydrolysis a slow conformational change drives DnaK to a state similar, but not identical, to ADP-bound DnaK (Fig. 6, top trace). Spectroscopically, this conformational change is characterized by (1) a decrease in the amplitude of differential signals appearing >1630 cm−1 (1693/1685, 1674/1660, and 1650/1636 cm−1), (2) the appearance of negative signals at 1559 cm−1, and (3) the positive absorbance change at 1391 turns into a negative signal at 1396 cm−1. When the same experiment is performed in D2O, a similar change in the difference spectrum is observed as a function of time, considering the effect of deuteration on the location of the differential signals (see Fig 4; data not shown). The differences observed between wild-type DnaK and the two deletion mutants in the spectral regions 1560–1590 cm−1 and 1390–1400 cm−1, and the development of a negative signal at ~1560 cm−1 during ATP hydrolysis indicate that the chemical environment of —COOH group(s) from acidic residue(s) is modified. The development of this negative signal does not arise from a change in the protonation state of acidic residues since there is not experimental evidence of differential signals at 1700–1750 cm−1, where the —COOH groups absorb (Barth 2000). The alternative explanation, as found for GroEL, would be a modification of specific salt bridges (von Germar et al. 1999). If this were the case, the assignment of counterions for these —COO−group(s) is not straightforward due to the low extinction coefficient of absorbance bands of Arg and Lys residues, and the overlap with stronger signals coming from the polypeptide backbone in this spectral region, and therefore it would require the characterization of selected mutants.
Figure 6.
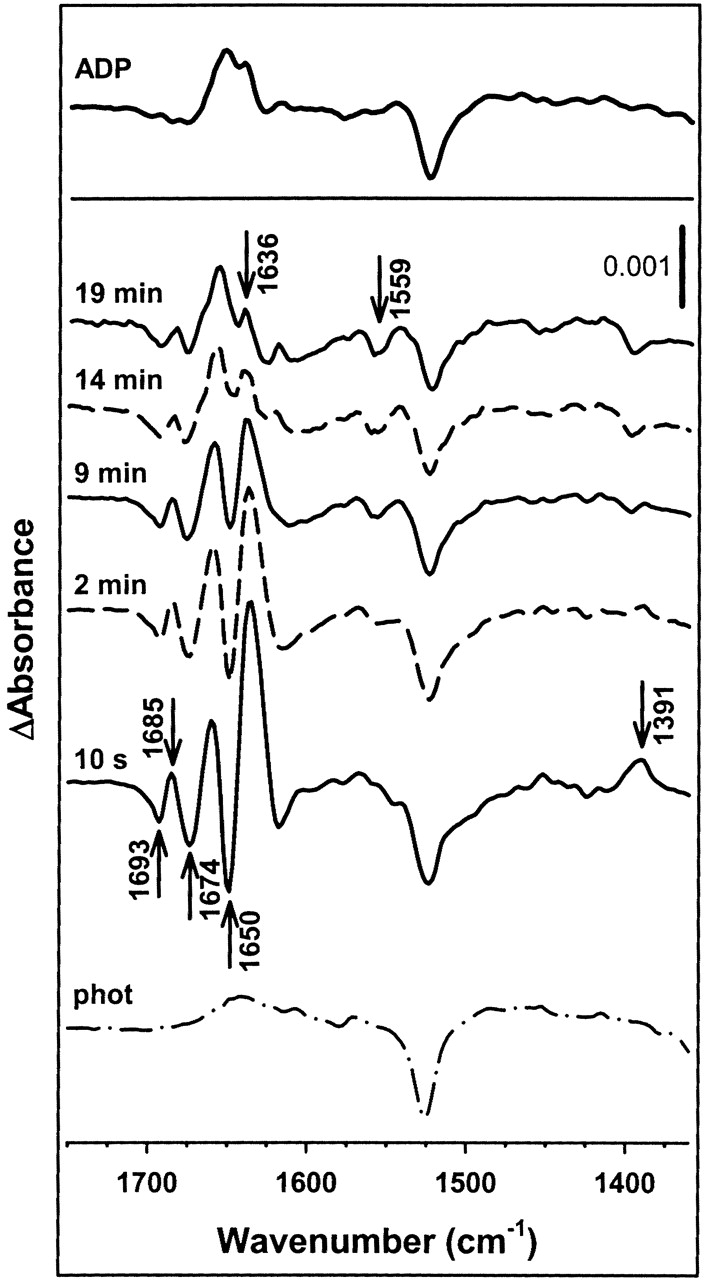
ATP hydrolysis relaxes DnaK to an ADP-like conformation. Time evolution of the IR difference spectrum of DnaK at times >10 sec after ATP release. DnaK concentration was 1.4 mM. Measurements were performed at a nucleotide:protein molar ratio of 1 at 25°C in H2O-based buffer. The lower spectrum corresponds to the photolysis reaction in the absence of protein. Spectra were recorded at the indicated times after the photolysis flash. Average of 10 independent experiments. For the sake of comparison, the difference spectrum corresponding to the ADP-bound state is shown in the upper panel.
The course of several absorbance changes analyzed in both solvents is shown in Figure 7. The differential signals chosen for this analysis include vibrations from secondary structure elements (1650 and 1645 cm−1; 1636 and 1631 cm−1); from phosphate groups that are hydrolyzed during the enzymatic reaction (1245 and 1250 cm−1); and from acidic amino acid side chains (1391 and 1389 cm−1). The time dependence of all the absorbance changes can be fitted to single exponential functions that give apparent rate constants ranging from 0.21 to 0.27 min−1 in H2O and from 0.12 to 0.20 min−1 in D2O medium. The almost twofold reduction of the hydrolysis reaction rate in D2O might be explained by the higher protein concentration used in H2O (see Materials and Methods), although a secondary isotope effect cannot be ruled out (Barth et al. 1996). These rate constants are approximately two orders of magnitude slower than those estimated for ATP binding, and 5–10 times faster than those reported for the hydrolysis reaction by fluorescence and electrophoretic methods (Farr et al. 1998; Slepenkov and Witt 1998). This discrepancy might be explained by the significantly different protein concentrations used in these studies (1–10 μM) and the present work (1–1.4 mM). Finally, it is interesting to note that the absorbance changes assigned to the subdomains of the SBD (α-helical lid, 1650 cm−1, and β-sandwich, 1636 cm−1 in H2O) show the same rate constant for the conformational change that follows ATP binding and for that associated to its subsequent hydrolysis, suggesting a concerted allosteric conformational change at the SBD in response to ATP.
Figure 7.
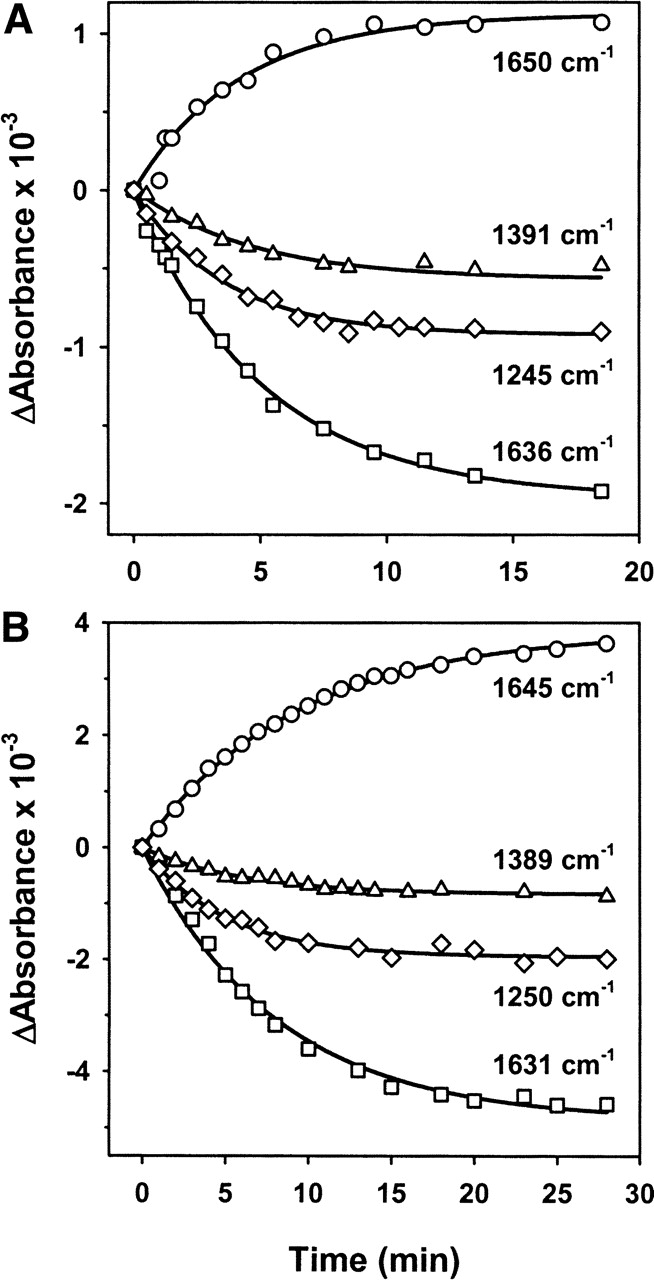
Kinetics of absorbance changes detected during ATP hydrolysis. The samples contained equimolar amounts of ATP and protein, prepared in H2O- (A) and D2O-based (B) buffers, as detailed in Materials and Methods. Absorbance changes in H2O- and D2O-based buffers are respectively assigned to α-helix (1650 and 1645 cm−1), β-structure (1636 and 1631 cm−1), carboxylate groups (1391 and 1389 cm−1), and phosphate vibrations (1245 and 1250 cm−1).
Discussion
Allosteric signaling is known to be a mandatory requisite for proper chaperone function. In the case of DnaK, the correct interplay between the ATPase domain and SBD allows the protein to cycle between substrate releasing and accepting states in response to ATP binding and hydrolysis. To further explore the conformational changes associated with the ATPase cycle of the protein, which in turn governs the access to these conformational states, we have applied infrared difference spectroscopy in combination with the use of caged nucleotides.
The experimental evidence for the existence of an ATP-induced allosteric conformational change, characterized by absorbance changes clearly assigned to structural elements at the SBD, raises two questions. The first one is related to the type of secondary structures involved and their location within the SBD. The assignment of differential absorbance features ~1620 cm−1 to protein backbone conformational changes is based on theoretical and systematic IR studies of model proteins and amino acid side chains (Chirgadze et al. 1975; Byler and Susi 1986; Venyaminov and Kalnin 1990; Arrondo et al. 1993; Jackson and Mantsch 1995; Barth 2000) and on a previous spectroscopic characterization of the same deletion mutants (Moro et al. 2004). Removal of the helical lid results in the disappearance of a differential feature at 1650(−)/1636(+) and 1645(−)/1631(+) in H2O and D2O, respectively. The isotopic shift observed upon sample deuteration (5 cm−1) is compatible with a solvent-exposed helical segment(s) undergoing a conformational change once ATP is bound. The observed negative/positive differential signal might be a consequence of a downshift of the helical component that could be due to hydrogen bond shortening. Regarding the biochemical behavior of the lidless mutant, it is important that it maintains the capacity of peptide release stimulation by ATP (Pellecchia et al. 2000; Buczynski et al. 2001; Moro et al. 2004), and of ATPase activity enhancement by peptides (Fig. 7) at 25°C, indicating that the two domains of the deletion mutant are allosterically communicated. This indicates, as previously suggested (Pellecchia et al. 2000; Moro et al. 2004), that under these experimental conditions at least part of the allosteric transition involves the β-sandwich and does not require the lid to occur. Therefore, the differences observed between the ATP-induced IR spectra of DnaK(1–507) and the ATPase domain are due to the allosteric interaction between the ATPase domain and the β-sandwich subdomain of the SBD that takes places immediately after nucleotide binding. The main difference between these spectra in the amide I region is the drastic reduction of the intensity of the positive differential feature at 1636 cm−1 and 1631 cm−1 in H2O and D2O, respectively. Again, the position in both solvents and the downshift observed upon deuteration is compatible with a solvent-exposed β-structure changing its conformation after ATP binding (Arrondo et al. 1993; Jackson and Matsch 1995). NMR studies of the isolated β-sandwich subdomain have also suggested that the loops forming the peptide binding pocket are highly flexible and that one of the β-strands (B3) is less ordered than in the crystal structure (Pellecchia et al. 2000; Stevens et al. 2003). Furthermore, peptide binding rigidifies the β-subdomain and reduces the mobility of the above-mentioned loops, locking in B3. Conformational changes in the same structural elements of the entire chaperone molecule would be compatible with the assignment and the small amplitude of the differential signals observed in this study.
The second question is related to the quantitative aspect of this conformational change. An attempt to quantify the structural changes of the protein backbone upon ATP binding might be carried out using the COBSI (change of backbone structure and interaction) index. This parameter, defined by Barth et al. (1996), relates the integrated intensity of the absorbance changes to the total protein absorbance in the amide I region (1700–1610 cm−1). The small value of the COBSI index (10−5), four orders of magnitude lower than that of the thermal denaturation of the protein (0.12, obtained from Moro et al. 2004), suggests that the conformational rearrangement is localized, and most likely involves modifications of the hydrogen bonding pattern within specific secondary structures rather than a net change in secondary structure. Regardless of which of the proposed alternatives to open the substrate binding site in response to ATP binding takes place—bending of helix B (Zhu et al. 1996) or displacement of the complete lid (Wang et al. 1998)—our data indicate that neither of them is accompanied by a net change in secondary structure of the full-length protein.
A comparison of ours and NMR data is possible for the ATPase domain of the protein, since the effect of ADP and ATP on the nucleotide binding domain (NBD) of T. thermophilus has been characterized by NMR (Revington et al. 2004). In good agreement with the results presented here, it was shown that the differences between the nucleotide-bound states are less widespread than those observed between the apo and the ADP states. The residues involved in these conformational differences were located around the nucleotide binding site (Revington et al. 2004), and those showing the largest chemical shifts when comparing the nucleotide states were clustered at the interface between subdomains 1A and 1B. This region was also identified as the binding channel for DnaJ and is close to the linker that connects the ATPase and the SBD of DnaK. Therefore, the small absorbance differences between both nucleotide containing protein states are in agreement with NMR data.
While other techniques detect a global conformational change in the SBD of DnaK or in one of its subdomains (Buchberger et al. 1995; Farr et al. 1998; Moro et al. 2003; Slepenkov and Witt 2003), TR-IR can resolve in real time structural rearrangements affecting both the βand the α-subdomains of the wild-type DnaK. An interesting finding of this study is that absorbance changes assigned to the lid and the β-subdomains of the SBD show the same time dependence after ATP binding and its subsequent hydrolysis. If the conformational change at the β-subdomain reduces the affinity of DnaK for peptide substrates, and the conformational change at the lid subdomain is involved in moving the lid away from the peptide binding site, the functional consequence of this kinetic coupling is that ATP binding would efficiently mediate diffusion of the released peptide to the medium. A recent NMR study of a deletion mutant of T. thermophilus Hsp70 lacking the lid subdomain has shown that the ATPase domain and the β-subdomain of this protein behave as a single unit and not as two individual domains connected by a flexible linker, indicating that these protein regions should be closely docked (Revington et al. 2005). The kinetic coupling found in this work for the full-length protein indicates that the lid subdomain also forms part of the structural unit closely connected to the ATPase domain. Furthermore, we have also found an important role for the lid subdomain in stabilizing the binding site to display a convenient affinity for peptide substrates, as well as the interdomain interface at stress temperatures to allow bidirectional transmission of the allosteric signal. The interactions that the lid establishes with the β-subdomain, through helix B and the loops flanking the peptide binding site, have been proposed to stabilize the ability of DnaK to stably bind substrates against the thermal challenge (Moro et al. 2004). Our present observation, regarding defective allosteric signaling at stress temperatures, might be explained considering that these interactions either stabilize the region of the β-subdomain that forms part of the interdomain interface, or that the lid is close enough to the ATPase domain and also contributes to the interdomain interaction and hence stabilizes the allosteric interface. Both observations support the view of SBD and ATPase domain functioning as a single unit, e.g., in a concerted manner, in the full-length DnaK.
A comparison of the results presented here with the proposed mechanisms of DnaK action (Theyssen et al. 1996; Slepenkov and Witt 1998) also indicates:
The fast collisional binding process (k ≈ 20 sec−1) described by fluorescence spectroscopy is not accompanied by a net change in secondary structure, in spite of inducing local alterations in the local environment of W102.
In contrast, the second kinetic event (k ≈ 0.65 sec−1) triggered by ATP binding is associated with a conformational transition that as seen by IR mainly affects the SBD and therefore reflects a concerted allosteric conformational modification of both subdomains of the SBD. A concerted structural transition in the ATPase domain of DnaK in response to peptide binding, e.g., in the opposite direction to that described here, has also been described (Slepenkov and Witt 2003), suggesting that both protein domains are tightly communicated.
Nucleotide hydrolysis resets the system and slowly (k ≈ 0.004 sec−1) relaxes the protein conformation to an ADP-like state.
In summary, these results suggest that the SBD, in spite of having two structurally different subdomains, has been designed to function as a single functional unit in connection with the ATPase domain, in the sense that both subdomains (1) respond concomitantly to the allo-steric signal generated by ATP binding and its subsequent hydrolysis and (2) are required to stabilize the interdomain interface.
Materials and methods
Protein expression and purification
Wild-type DnaK, deletion, and single-point mutants were expressed in the BB1553 strain, purified as described (Moro et al. 2003), and concentrated using concentration filters (Centriprep 30, Millipore-Amicon). The endogenous nucleotide content in DnaK preparations was estimated by the A260:A280 ratio, which was in every case <0.7, indicating that the protein was nucleotide-free (Montgomery et al. 1999).
Sample preparation
Sample buffer was exchanged by extensive dialysis against 100 mM MOPS (pH 7.0), 50 mM KCl, 10 mM MgCl2 in H2O or repeated concentration and dilution steps with the same buffer made in D2O, using Centricon 30 (Millipore-Amicon) filters. IR samples were prepared by drying onto a CaF2 window 1 μL of the following reagents: 9 mM caged nucleotide (Molecular Probes-Invitrogen) and 50 mM DTT. They were rehydrated with 2.5 μL of the desired protein dissolved in the above buffer, and the samples were sealed with a second window. Protein concentration was ≈1 and 1.4 mM in D2O and H2O, respectively, as estimated spectrophotometrically using an extinction coefficient of ɛ280 = 15,800 M−1cm−1 (Montgomery et al. 1999). All experiments were carried out using thermostated cell holders at 25°C unless otherwise stated.
Time-resolved infrared spectroscopy (TR-IR)
Experiments were performed in a Nexus 870 (Thermo Nicolet) IR spectrophotometer equipped with an MCT detector. Photolytic release of nucleotides from their caged derivatives was triggered with a Xenon flash tube. The voltage of the flash power supply was adjusted to release the desired nucleotide concentration that in all cases was the same as the protein concentration in the sample (single-turnover conditions). Data were acquired with double-sided interferograms, at a spectral resolution of 4 cm−1.
To improve the signal-to-noise ratio, signals obtained from several samples were averaged after normalizing the spectra to an identical protein concentration. Normalization prevents the possible predominance of individual samples with the highest protein concentrations in the averaged spectra, and was performed as previously described (vonGermar et al. 2000). Control experiments on samples prepared as described above but without protein were performed in the same time interval to obtain the photolysis spectra. For the kinetic analysis of ATP-induced difference spectra, selected bands were integrated as described previously (Barth et al. 1991; vonGermar et al. 2000). The time slots of spectra recording were represented by their average times. The band intensities were fitted to single exponential equations (Sigmaplot 8.0, SPSS Inc.).
Nucleotide binding assay
Free and caged nucleotide (10 μM) were centrifuged in the absence or the presence of 10 μM DnaK, using microconcentration filters (Microcon 30, Millipore-Amicon). The filtration membrane retained the protein and the bound nucleotide while the unbound nucleotide passed through it. Nucleotide concentration in the filtrate was estimated from the absorbance at 260 nm, using an extinction coefficient of 15,400 M−1 cm−1.
ATPase activity
Steady-state ATPase activity measurements were carried out in 40 mM HEPES (pH 7.5), 50 mM KCl, 10 mM MgCl2 in a thermostated cuvette, using an enzymatic ATP-regenerating system (Moro et al. 2003). Protein and ATP concentrations were 5 μM and 1 mM, respectively. ATP hydrolysis was followed recording the NADH absorbance decrease at 340 nm in a Cary (Varian) spectrophotometer. NRLLLTG (NR) peptide was added at the desired final concentration, and the ATPase activity stimulation curves were fitted to the equation Ai = Amax * [NR]i/(Kd + [NR]i), Kd being an apparent binding constant.
Acknowledgments
We thank B. Bukau and M. Mayer (ZMBH, Heidelberg, Germany) for the DnaK T199A mutant, and Stefka Taneva, Felix Goñi and Jose L.R. Arrondo for critically reading the manuscript. This work was supported by grants from the MEC (BFU2004-03452/BMC) and the University of the Basque Country (13505/2001). F.M. is supported by the I3P program. V.F.-S. is recipient of a predoctoral fellowship from the Basque Government.
Abbreviations
SBD, substrate binding domain
TR-IR, time resolved infrared spectroscopy
IR, infrared spectroscopy
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051732706.
References
- Arrondo, J.L.R., Muga, A., Castresana, J., and Goñi, F.M. 1993. Quantitative studies of the structure of proteins in solution by Fourier-transform infrared spectroscopy. Prog. Biophys. Mol. Biol. 59: 23–56. [DOI] [PubMed] [Google Scholar]
- Barth, A. 2000. The infrared absorption of amino acid side chains. Prog. Biophys. Mol. Biol. 74: 141–173. [DOI] [PubMed] [Google Scholar]
- Barth, A. and Zscherp, C. 2002. What vibrations tell us about proteins. Q. Rev. Biophys. 35: 369–430. [DOI] [PubMed] [Google Scholar]
- Barth, A., Mantele, W., and Kreutz, W. 1991. Infrared spectroscopic signals arising from ligand binding and conformational changes in the catalytic cycle of sarcoplasmic reticulum calcium ATPase. Biochim. Biophys. Acta 1057: 115–123. [DOI] [PubMed] [Google Scholar]
- Barth, A., von Germar, F., Kreutz, W., and Mantele, W. 1996. Time-resolved infrared spectroscopy of the Ca2+-ATPase. The enzyme at work. J. Biol. Chem. 271: 30637–30646. [DOI] [PubMed] [Google Scholar]
- Barthel, T.K., Zhang, J., and Walker, G.C. 2001. ATPase-defective derivatives of Escherichia coli DnaK that behave differently with respect to ATP-induced conformational change and peptide release. J. Bacteriol. 183: 5482–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger, A., Theyssen, H., Schroder, H., McCarty, J.S., Virgallita, G., Milkereit, P., Reinstein, J., and Bukau, B. 1995. Nucleotide-induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication. J. Biol. Chem. 270: 16903–16910. [DOI] [PubMed] [Google Scholar]
- Buczynski, G., Slepenkov, S.V., Sehorn, M.G., and Witt, S.N. 2001. Characterization of a lidless form of the molecular chaperone DnaK: Deletion of the lid increases peptide on- and off-rate constants. J. Biol. Chem. 276: 27231–27236. [DOI] [PubMed] [Google Scholar]
- Bukau, B. and Horwich, A.L. 1998. The Hsp70 and Hsp60 chaperone machines. Cell 92: 351–366. [DOI] [PubMed] [Google Scholar]
- Byler, D.M. and Susi, H. 1986. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers 25: 469–487. [DOI] [PubMed] [Google Scholar]
- Chirgadze, Y.N., Fedorov, O.V., and Trushina, N.P. 1975. Estimation of amino acid residue side-chain absorption in the infrared spectra of protein solutions in heavy water. Biopolymers 14: 679–694. [DOI] [PubMed] [Google Scholar]
- Farr, C.D., Slepenkov, S.V., and Witt, S.N. 1998. Visualization of a slow, ATP-induced structural transition in the bacterial molecular chaperone DnaK. J. Biol. Chem. 273: 9744–9748. [DOI] [PubMed] [Google Scholar]
- Grimshaw, J.P., Jelesarov, I., Schonfeld, H.J., and Christen, P. 2001. Reversible thermal transition in GrpE, the nucleotide exchange factor of the DnaK heat-shock system. J. Biol. Chem. 276: 6098–6104. [DOI] [PubMed] [Google Scholar]
- Ha, J.H. and McKay, D.B. 1995. Kinetics of nucleotide-induced changes in the tryptophan fluorescence of the molecular chaperone Hsc70 and its subfragments suggest the ATP-induced conformational change follows initial ATP binding. Biochemistry 34: 11635–11644. [DOI] [PubMed] [Google Scholar]
- Harrison, C.J., Hayer-Hartl, M., Di Liberto, M., Hartl, F., and Kuriyan, J. 1997. Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science 276: 431–435. [DOI] [PubMed] [Google Scholar]
- Hartl, F.U. 1996. Molecular chaperones in cellular protein folding. Nature 381: 571–579. [DOI] [PubMed] [Google Scholar]
- Jackson, M. and Mantsch, H.H. 1995. The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit. Rev. Biochem. Mol. Biol. 30: 95–120. [DOI] [PubMed] [Google Scholar]
- Jordan, R. and McMacken, R. 1995. Modulation of the ATPase activity of the molecular chaperone DnaK by peptides and the DnaJ and GrpE heat shock proteins. J. Biol. Chem. 270: 4563–4569. [DOI] [PubMed] [Google Scholar]
- Mantele, W. 1993. Reaction-induced infrared difference spectroscopy for the study of protein function and reaction mechanisms. Trends Biochem. Sci. 18: 197–202. [DOI] [PubMed] [Google Scholar]
- Mayer, M.P., Rudiger, S., and Bukau, B. 2000. Molecular basis for interactions of the DnaK chaperone with substrates. Biol. Chem. 381: 877–885. [DOI] [PubMed] [Google Scholar]
- Montgomery, D.L., Morimoto, R.I., and Gierasch, L.M. 1999. Mutations in the substrate binding domain of the Escherichia coli 70 kDa molecular chaperone, DnaK, which alter substrate affinity or interdomain coupling. J. Mol. Biol. 286: 915–932. [DOI] [PubMed] [Google Scholar]
- Moro, F., Fernandez, V., and Muga, A. 2003. Interdomain interaction through helices A and B of DnaK peptide binding domain. FEBS Lett. 533: 119–123. [DOI] [PubMed] [Google Scholar]
- Moro, F., Fernández-Sáiz, V., and Muga, A. 2004. The lid subdomain of DnaK is required for the stabilization of the substrate-binding site. J. Biol. Chem. 279: 19600–19606. [DOI] [PubMed] [Google Scholar]
- Pellecchia, M., Montgomery, D.L., Stevens, S.Y., Vander Kooi, C.W., Feng, H.P., Gierasch, L.M., and Zuiderweg, E.R. 2000. Structural insights into substrate binding by the molecular chaperone DnaK. Nat. Struct. Biol. 7: 298–303. [DOI] [PubMed] [Google Scholar]
- Revington, M., Holder, T.M., and Zuiderweg, E.R. 2004. NMR study of nucleotide-induced changes in the nucleotide binding domain of Thermus thermophilus Hsp70 chaperone DnaK: Implications for the allosteric mechanism. J. Biol. Chem. 279: 33958–33967. [DOI] [PubMed] [Google Scholar]
- Revington, M., Zhang, Y., Yip, G.N., Kurochkin, A.V., and Zuiderweg, E.R. 2005. NMR investigations of allosteric processes in a two-domain Thermus thermophilus Hsp70 molecular chaperone. J. Mol. Biol. 349: 163–183. [DOI] [PubMed] [Google Scholar]
- Russell, R., Jordan, R., and McMacken, R. 1998. Characterization of the ATPase cycle of the DnaK molecular chaperone. Biochemistry 37: 596–607. [DOI] [PubMed] [Google Scholar]
- Shi, L., Kataoka, M., and Fink, A.L. 1996. Conformational characterization of DnaK and its complexes by small-angle X-ray scattering. Biochemistry 35: 3297–3308. [DOI] [PubMed] [Google Scholar]
- Slepenkov, S.V. and Witt, S.N. 1998. Kinetics of the reactions of the Escherichia coli molecular chaperone DnaK with ATP: Evidence that a three-step reaction precedes ATP hydrolysis. Biochemistry 37: 1015–1024. [DOI] [PubMed] [Google Scholar]
- ———. 2002. The unfolding story of the Escherichia coli Hsp70 DnaK: Is DnaK a holdase or an unfoldase? Mol. Microbiol. 45: 1197–1206. [DOI] [PubMed] [Google Scholar]
- ———. 2003. Detection of a concerted conformational change in the ATPase domain of DnaK triggered by peptide binding. FEBS Lett. 539: 100–104. [DOI] [PubMed] [Google Scholar]
- Stevens, S.Y., Cai, S., Pellecchia, M., and Zuiderweg, E.R. 2003. The solution structure of the bacterial HSP70 chaperone protein domain DnaK(393–507) in complex with the peptide NRLLLTG. Protein Sci. 12: 2588–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susi, H. and Byler, D.M. 1986. Resolution-enhanced Fourier transform infrared spectroscopy of enzymes. Methods Enzymol. 130: 290–311. [DOI] [PubMed] [Google Scholar]
- Theyssen, H., Schuster, H.P., Packschies, L., Bukau, B., and Reinstein, J. 1996. The second step of ATP binding to DnaK induces peptide release. J. Mol. Biol. 263: 657–670. [DOI] [PubMed] [Google Scholar]
- Venyaminov, S.Yu. and Kalnin, N.N. 1990. Quantitative IR spectrophotometry of peptide compounds in water (H2O) solutions. I. Spectral parameters of amino acid residue absorption bands. Biopolymers 30: 1243–1257. [DOI] [PubMed] [Google Scholar]
- von Germar, F., Galan, A., Llorca, O., Carrascosa, J.L., Valpuesta, J.M., Mantele, W., and Muga, A. 1999. Conformational changes generated in GroEL during ATP hydrolysis as seen by time-resolved infrared spectroscopy. J. Biol. Chem. 274: 5508–5513. [DOI] [PubMed] [Google Scholar]
- von Germar, F., Barth, A., and Mantele, W. 2000. Structural changes of the sarcoplasmic reticulum Ca(2+)-ATPase upon nucleotide binding studied by Fourier transform infrared spectroscopy. Biophys. J. 78: 1531–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H., Kurochkin, A.V., Pang, Y., Hu, W., Flynn, G.C., and Zuiderweg, E.R. 1998. NMR solution structure of the 21 kDa chaperone protein DnaK substrate binding domain: A preview of chaperone-protein interaction. Biochemistry 37: 7929–7940. [DOI] [PubMed] [Google Scholar]
- Wilbanks, S.M., Chen, L., Tsuruta, H., Hodgson, K.O., and McKay, D.B. 1995. Solution small-angle X-ray scattering study of the molecular chaperone Hsc70 and its subfragments. Biochemistry 34: 12095–12106. [DOI] [PubMed] [Google Scholar]
- Zhu, X., Zhao, X., Burkholder, W.F., Gragerov, A., Ogata, C.M., Gottesman, M.E., and Hendrickson, W.A. 1996. Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272: 1606–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]