

Abstract
Allosteric interactions between residues that are spatially apart and well separated in sequence are important in the function of multimeric proteins as well as single-domain proteins. This observation suggests that, among the residues that are involved in long-range communications, mutation at one site should affect interactions at a distant site. By adopting a sequence-based approach, we present an automated approach that uses a generalization of the familiar sequence entropy in conjunction with a coupled two-way clustering algorithm, to predict the network of interactions that trigger allosteric interactions in proteins. We use the method to identify the subset of dynamically important residues in three families, namely, the small PDZ family, G protein–coupled receptors (GPCR), and the Lectins, which are cell-adhesion receptors that mediate the tethering and rolling of leukocytes on inflamed endothelium. For the PDZ and GPCR families, our procedure predicts, in agreement with previous studies, a network containing a small number of residues that are involved in their function. Application to the Lectin family reveals a network of residues interspersed throughout the C-terminal end of the structure that are responsible for binding to ligands. Based on our results and previous studies, we propose that functional robustness requires that only a small subset of distantly connected residues be involved in transmitting allosteric signals in proteins.
Keywords: structure, protein families, evolutionary relationships, proteomics
Long-range communications among a network of residues that are far apart in sequence and in structure are crucial for biological functions. The classic example is the allosteric communication in which binding of a ligand to a specific region of a protein often triggers large conformational changes in a distant part (Monod et al. 1965). Starting from the well-studied case of oxygen binding to hemoglobin (Perutz et al. 1998), large-scale domain movements in response to ligand binding have been noted in other systems. For example, binding of ATP and the co-chaperonin GroES to the oligomeric Escherichia coli chaperonin triggers dramatic rigid body motions in different subdomains of GroEL (Xu and Sigler 1998; Horovitz et al. 2001). Similarly, examination of the structures of DNA polymerases and the dNTP–polymerase–DNA complexes shows evidence of such large movements (Steitz 1999). In the case of polymerases, whose structure is analyzed using the right-hand metaphor, the initial step involving binding of the unliganded polymerase to DNA triggers the thumb to close around the DNA. Subsequent binding of dNTP to the binary complex results in the rotation of the fingers from the open conformation to a closed catalysis-ready state. These and other examples show that the set of residues that induce the long-range allosteric communications in oligomeric biological nanomachines may well be encoded in the structures and hence, in the sequence itself.
More recently, it has been argued that allosteric communication may well be a part of the observed dynamical fluctuations in small single-domain proteins (Kern and Zuiderweg 2003). Many NMR experiments have shown that even after reaching the native state, proteins undergo conformational fluctuations with time scales from several nanoseconds to milliseconds. Surprisingly, it has been suggested that such functionally important fluctuations are triggered by long-range interactions among a network of residues. Just as with multimeric proteins, these functionally important sets of residues are also encoded in the structures.
The above-mentioned, rather ubiquitous examples, naturally raise an important question, namely, how can we identify the network of residues that mediate allosteric communication? There have been several distinct approaches to answer this question, each with a varying degree of success. Although they differ significantly in detail, the methods are either based on probing changes in the thermodynamics and dynamics of proteins with known structures or using probabilistic methods to analyze evolutionarily related sequences. The double mutant cycles, which probe the responses of specific sequence pairs to mutations (Schreiber and Fersht 1995), can be used to obtain the thermodynamics of interactions between the mutated residues. It is also possible to probe interactions among residues by examining the dynamics of specific residues using NMR (Kern and Zuiderweg 2003). Recently, we introduced a method that monitors the response of a region of a folded protein, represented using the Elastic Network Model, in response to a perturbation at another site (Zheng et al. 2005). The application of the method to the polymerase family successfully identified a network of dynamically relevant residues that are involved in the open/closed transition.
In contrast to the relatively few structure-based methods, numerous techniques that exploit the properties of families of sequences have been developed to infer correlations between amino acids in protein families. A sequence-based method (Lesk and Chothia 1980; Altschuh et al. 1987; Neher 1994; Taylor and Hatrick 1994; Lichtarge et al. 1996; Pazos et al. 1997; Pollock and Taylor 1997; Olmea et al. 1999; Sowa et al. 2001; Kass and Horovitz 2002; Valencia and Pazos 2002) is desirable because a larger database of evolved sequences with related functions can be studied. It is logical to postulate that the distribution of amino acid types at a given position in a multiple sequence alignment (MSA) is the manifestation of evolutionary changes under constraints imposed by function. In addition, it is likely that for functional reasons coevolution of a network of residues in a sequence also occurs. If so, such correlations should appear as statistically significant signals when analyzing a MSA. Using sequence correlation entropy (SCE) (Dima and Thirumalai 2004), which does not involve the standard preaveraging of site-dependent probabilities in the MSA, we showed that statistically significant correlations between charged residues exist in a number of protein families. Interestingly, most of these proteins are associated with various diseases. Thus, functionally important signals can be obtained using a large data set of sequences alone.
In a series of insightful papers, Ranganathan and coworkers (Lockless and Ranganathan 1999; Hatley et al. 2003; Suel et al. 2003; Shulman et al. 2004) have introduced a method based on statistical coupling analysis (SCA) to identify the relevant energetically coupled residues. The basic premise of the SCA method is that, from the sequence family, the coevolution of positions, either for structural or functional reasons, can be captured by comparing the statistical properties of amino acids in the full MSA and its statistically significant subsets. In the applications so far, the SCA method has revealed coevolution between distant sites with functional roles. The good agreement between the SCA predictions and limited experimental data (Lockless and Ranganathan 1999; Hatley et al. 2003; Suel et al. 2003; Shulman et al. 2004) lends credence to this approach.
In this paper we introduce a variant of the SCA method that builds on the hypothesis that signals for coevolving residues are encrypted in the database of sequences, provided the number of sequences in the MSA is large enough. By insisting that the central limit theorem be obeyed, namely, the statistical properties of a large enough subset of the MSA be the same as in the MSA, we present an automated method for identifying allosteric sites using a family of sequences. After establishing that our method provides consistent results for the PDZ and GPCR families, we describe the mapping of interacting residues for the selectin family, which are cell adhesion proteins. Our results for all three families show that the predictions of the automated sequence-based approach can be used to target the functionally or dynamically relevant residues in double mutant cycle experiments (Schreiber and Fersht 1995) or NMR.
Results
Size of subalignments
The choice of appropriate size for subalignments is critical in obtaining statistically significant correlations between residues in a protein family. The smallest value of f should be chosen to satisfy the central limit theorem. We applied our criteria to the families included in the present study (GPCR and Lectin C) and also to the globin family analyzed by Lockless and Ranganathan, in Suel et al. 2003.
GPCR family
The full MSA for the GPCR family (reported in the supplementary material of Suel et al. 2003) contains 940 sequences. From the MSA, we build subalignments with different f (<1) values by randomly choosing fNMSA sequences. To perform the averages in Equations 6 and 7, we generated 1000 subalignments for a given f and we computed 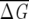 λ using Equation 6. The distribution of these values for increasing f from Figure 1A shows that for
λ using Equation 6. The distribution of these values for increasing f from Figure 1A shows that for 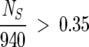 the subalignments satisfy the conditions from Equations 6 and 7 and are therefore statistically significant. This value of f is virtually identical to the f = 0.33 value chosen by Lockless and Ranganathan (Suel et al. 2003) for the minimal size of a subalignment.
the subalignments satisfy the conditions from Equations 6 and 7 and are therefore statistically significant. This value of f is virtually identical to the f = 0.33 value chosen by Lockless and Ranganathan (Suel et al. 2003) for the minimal size of a subalignment.
Figure 1.

Distribution of 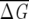 λ for various sizes (
λ for various sizes (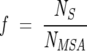 ) of subalignments for the GPCR, globin, and Lectin families. The index λ refers to the one of the subsets with NS sequences. Insets show the dependence of 〈
) of subalignments for the GPCR, globin, and Lectin families. The index λ refers to the one of the subsets with NS sequences. Insets show the dependence of 〈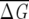 〉f and σf for various f. As expected from Equation 6, 〈
〉f and σf for various f. As expected from Equation 6, 〈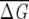 〉f shows a plateau starting at a given value of f (indicated by the arrow), while σf for f larger than this value decreases according to Equation 7. The minimum size of f should correspond to the value indicated by the arrow. (A) GPCR family (940 sequences in full MSA). (B) Globin family (880 sequences in full MSA). (C) Lectin C family (1126 sequences in full MSA).
〉f shows a plateau starting at a given value of f (indicated by the arrow), while σf for f larger than this value decreases according to Equation 7. The minimum size of f should correspond to the value indicated by the arrow. (A) GPCR family (940 sequences in full MSA). (B) Globin family (880 sequences in full MSA). (C) Lectin C family (1126 sequences in full MSA).
Globin family
The MSA for the globin family contains 880 sequences (Suel et al. 2003). The distribution of 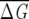 λ for the increasing fraction of sequences in a subalignment shows (Fig. 1B) that subalignments containing more than 55% of the original MSA are statistically significant. Lockless and Ranganathan chose, based on their ad hoc criterion, f = 0.68. In globins,
λ for the increasing fraction of sequences in a subalignment shows (Fig. 1B) that subalignments containing more than 55% of the original MSA are statistically significant. Lockless and Ranganathan chose, based on their ad hoc criterion, f = 0.68. In globins, 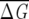 MAS ~ 0.65 in globins, while in GPCR,
MAS ~ 0.65 in globins, while in GPCR, 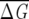 MAS ~ 0.17. There is also a greater variation in GPCR between distributions corresponding to different sizes. These results reflect the degree of variation among the sequences in a family, with the sequence similarity in GPCR being smaller than in globins.
MAS ~ 0.17. There is also a greater variation in GPCR between distributions corresponding to different sizes. These results reflect the degree of variation among the sequences in a family, with the sequence similarity in GPCR being smaller than in globins.
Lectin C
The Lectin C family, obtained starting from the Pfam (Bateman et al. 2002) entry and the CLUSTALW software (Higgins et al. 1994) for realignment of the sequences that remain after weeding out all sequence fragments, contains 1126 sequences. The distribution of 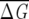 λ for increasing f shows (Fig. 1C) that subalignments containing more than 15% of the MSA are statistically significant. In our application, we chose a cut-off of 20%. This example illustrates that for f > 15%, the average of the distribution no longer changes, while the variance decreases as 1\Ns and the distribution becomes more Gaussian-like. We also note that the average sequence similarity in the Lectin C family is smaller than in globins, but still bigger than in GPCR. These examples clearly show that the choice of f, based on Equations 6 and 7, depends on the family.
λ for increasing f shows (Fig. 1C) that subalignments containing more than 15% of the MSA are statistically significant. In our application, we chose a cut-off of 20%. This example illustrates that for f > 15%, the average of the distribution no longer changes, while the variance decreases as 1\Ns and the distribution becomes more Gaussian-like. We also note that the average sequence similarity in the Lectin C family is smaller than in globins, but still bigger than in GPCR. These examples clearly show that the choice of f, based on Equations 6 and 7, depends on the family.
Correlation between residues in PDZ and GPCR families
To test the efficacy of our procedure, we applied our version of the SCA to identify a set of correlated residues in the PDZ and GPCR families. These two cases allow us to compare our results with those of Lockless and Ranganathan. After demonstrating the equivalence between the two procedures, we apply our formulation to identify the network of correlated residues in the family of cell adhesion molecules.
We calculated ΔΔGij for all positions in the MSA using Equations 4 and 5 for the PDZ domains that represent protein-binding motifs. Following Lockless and Ranganathan, we chose the subset of sequences in the MSA in which His at position 76 is perfectly conserved and calculated the response to this perturbation at all other positions. Comparison of the Lockless and Ranganathan results and our calculations for ΔΔGi,76 shows excellent agreement with a better than 0.95 correlation (Fig. 2A). The set of identified residues that are coupled, a few long-range pairs, may be relevant in the dynamics of the PDZ domains.
Figure 2.

Comparison between ΔΔGij values obtained as described in Lockless and Ranganathan (1999) and using our procedure. The panel on the left is for the GPCR family, while the one on the right corresponds to the PDZ family. For each family, we selected a perturbation at the most functionally important position. The data for the two families show a very large degree of correlation (≥ 0.95), indicating that our procedure captures all the crucial details of the previous implementation of the statistical coupling analysis (SCA) method.
The application of the present simplified procedure to the transmembrane G protein–coupled receptor (GPCR) family also yields results in near quantitative agreement with the Lockless and Ranganathan algorithm. Using the moderately conserved position Tyr296, which is involved in the ligand interactions in GPCR family, as a perturbation we calculated ΔΔGi,296. There is a small subset of residues that are uniformly spread throughout the sequence and are coupled to Tyr296. The magnitudes of ΔΔGi,296 for all i calculated using the two procedures are nearly identical (Fig. 2B). We conclude that the present version of the statistical coupling analysis can identify the network of interacting residues provided the data set of sequences is large enough that meaningful statistical mechanics can be used.
Correlation between residues in the GPCR family
In order to identify the network of residues that are correlated, we performed the CTWC analysis using the ΔΔGij values. We used the Euclidean distance (see Eq. 10) as a similarity metric for comparison of the ΔΔGij values at two sites k and j (Eq. 9). We used K = 20 order nearest neighbors and q = 20 in clustering the positions and K = 10 in clustering of perturbations. We performed two rounds of coupled SPC clustering: (1) the clustering of positions in the presence of all the perturbations and the clustering of perturbations in the presence of all the positions. The size of the clusters is determined as described in Materials and Methods at each temperature. (2) In the second round, we cluster the positions using the already clustered perturbations in the previous step. In addition, we cluster the perturbations in the presence of the positions clustered at step 1. At each step we selected the cluster corresponding to the largest ΔΔGij values. The results of clustering the G matrix led to 55 clustered positions and 18 clustered perturbations (see Tables 1, 2). Out of the 18 clustered perturbations, 17 correspond to clustered positions, which shows that the statistical procedure leads to self-consistent results. Moreover, all 10 perturbations that were reported to be clustered in Suel et al. (2003) are among the 18 clustered perturbations and 41 (including two positions found at −1 or +1 from an actually clustered position) of the 47 clustered positions in Suel et al. (2003) are among our 55 clustered positions. Therefore, we can recognize 100% of the perturbations and 87% of the positions identified by Suel et al. (2003). In addition, we also identify three (121, 294, and 313) new positions functionally important for GPCRs. These were not found using the Lockless and Ranganathan procedure (Suel et al. 2003).
Table 1.
List of 55 positions clustered
| 44 | 51 | 58 | 59 | 61 | 66 | 67 | 75 | 78 | 90 |
| 91 | 92 | 113 | 115 | 117 | 120 | 121 | 122 | 123 | |
| 124 | 125 | 126 | 129 | 131 | 134 | 136 | 140 | 141 | 149 |
| 152 | 157 | 164 | 168 | 213 (212) | 219 | 222 | 230 | 249 | 253 |
| 254 | 257 | 258 | 259 | 265 | 268 | 269 | 293 | 294 | 295 |
| 296 | 298 | 299 | 300 | 312 (313) | 317 (316) |
The list of the 55 positions clustered in GPCR. The notation is according to the PDB file 1l9h. Bold entries correspond to results from Suel et al. (2003); italic entries are positions identified in Suel et al. (2003) as functionally important. A bold entry in brackets is the position clustered in Suel et al. (2003), which in our case is situated at (−1) from a clustered position.
GPCR, notation from PDB file 1L9H.
Table 2.
List of 18 perturbations (position and amino acid type selected) clustered
| 51G | 61V | 66K | 69R | 78S | 92P | 123A | 144Y | 157I |
| 164A | 203Y | 219M | 222C | 254L | 268F | 268Y | 296F | 317Y |
The list of the 18 perturbations (position and amino acid type selected) clustered in GPCR. The notation is according to the PDB file 1l9h. Italic entries correspond to perturbations not clustered in Suel et al. (2003) but occurring at clustered positions. Bold entries correspond to results from Suel et al. (2003).
GPCR, notation from PDB file 1L9H.
Network of functionally important residues in the Lectin C family
Background
The selectin family contains proteins that are involved in the cell adhesion process. These proteins and their glycoconjugate ligands are implicated in the tethering and rolling of circulating leukocytes on blood vessels, endothelial cells, and platelets. The first step in a multistage dynamics process involves binding of proteins in the selectin family, which are expressed in leukocytes, to ligands in the endothelial cells. The recognition of the ligands involves a coordinated interaction between L-selectin and the various glycoproteins. The crystal structures of the complex between selectin constructs and different ligands have provided insights into the set of residues in selectins that mediate the initial steps in the tethering and rolling process. The sequence-based approach allows us to map the network of residues that signal the tethering process. The success of our predictions is validated by explicit comparison of the predicted binding sites to those identified in the crystal structures.
The structures of the complexes between P-selectin and E-selectin and a weakly bound ligand and the stereospecifically bound P-selectin glycoprotein ligand PSGL-1 (Somers et al. 2000) identify several sites that not only bind to the ligand, but also respond dynamically to the ligand binding process. Comparison of the liganded and nonliganded structures also reveals large-scale movements in the loops connecting Asn83–Asn89 (we use the numbering in PDB entry 1g1s for LEM3_HUMAN positions 42–188). The crystal structures clearly identify specific, discrete binding sites. There are three classes of sites that are coordinated to different ligands: (1) The metal (Ca2+) ion-dependent weak binding of certain ligands occurs by coordination of Ca2+ to side chains of Gln80, Asn82, Asn105, and Asp106. (2) The interaction of glycans with P- or E-selectin occurs by coordination to residues Tyr48, Gln92, Tyr94, Ser97, Pro98, and Ser99. (3) It is well known that selectins bind strongly to glycoproteins like PSGL-1. Several residues have been identified in the crystal structure of PSGL-1 in complex with P- and E-selectins. These include Ser47, Arg85, His108, Lys111, Lys112, and Lys113. Several of these residues form a network of hydrogen bonds upon ligand binding.
Besides the three classes of residues, regions of the P-and E-selectins apparently undergo large conformational changes upon complexation (Somers et al. 2000). Comparison of the structures of unliganded and liganded complexes shows that upon binding, the loop Asn83–Asn89 moves from the periphery to the sugar-binding sites. In addition, the group of residues Arg54–Glu74 also undergoes large-scale displacement into the region occupied by the Asn83–Asn89 loop in the unliganded state. From the perspective of allosteric signaling, it is unclear which of the 21 residues in the Arg54–Glu74 loop are directly linked in the signaling pathway. It is logical to suggest that only a subset of these residues is coupled directly to other parts of the structure. The movement of Arg54–Gln74 as a whole is likely to be a consequence of chain connectivity and stereochemistry.
Sequence entropy is not an indicator of sequence correlation
Sequence entropy for the Lectin C family shows that not many residues are strongly conserved [S(i) < 0.5] (Fig. 3). With this as the cut-off, we find that only residues Cys19, Leu26, Trp50, Gly52, Pro81, Cys90, Cys109, and Cys117 exhibit strong conservation. A key characteristic of cell-adhesion proteins is the preponderance of a large number of Cys residues that form disulfide bonds. From this perspective, strong conservation of the four Cys residues is not a surprise. The other six strongly conserved residues do not seem to be associated with identified functions. In contrast, the crystal structures identify many nonconserved long-range interacting residues to be relevant for some aspects of function. It is obvious from S(i) alone that it is impossible to decipher the set of coevolving or interacting residues.
Figure 3.

Sequence entropy for the 117 positions in 1g1s using the multiple sequence alignment from PROSITE (with 287 sequences). The circles correspond to S(i) for the 20 types of amino acids obtained according to Equation 3, while the diamonds represent the chemical sequence entropy obtained by classifying the amino acids into four classes, namely, hydrophobic, polar, and positively and negatively charged. The hydrophobic residues are Ala, Leu, Ile, Val, Trp, Tyr, Cys, Met, and Phe. The positively charged residues are Arg and Lys. The negatively charged residues are Glu and Asp. The polar residues are Thr, Gly, Ser, His, Gln, Asn, and Pro. The reduction in the number of the classes of amino acids leads to conservation at a larger number of positions. By using only four types of amino acids and a cut-off for strong conservation at S(i) < 0.25, we find that positions Thr2, Tyr3, Tyr5, Trp12, Ser15, Val27, Ile29, Tyr37, Leu38, Tyr49, Ile51, Gly52, Ile53, Trp60, Trp62, Trp76, Val91, Ile93, Trp104, Ala115, and Leu116 are also conserved. Among these 21 sites, only Trp60 and Trp62 are crystallographically identified to be functionally relevant (they belong to the Arg54–Glu74 loop). Therefore, sequence entropy alone cannot lead to the network of amino acids identified by our procedure.
Signaling involves a sparse network
We have obtained the network of coupled residues that may be involved in the allosteric signaling in the selectin family upon binding to glycoproteins and sugars. We used the Lectin_C Prosite (PS50041) (Hulo et al. 2004) entry, which contains 287 sequences. The sequences are aligned against the sequence of LEM3_HUMAN from positions 39–159 using the CLUSTALW package (Higgins et al. 1994). The total length of the alignment (including gaps) is 214 residues. With f = 0.2, 98 perturbations are allowed at the various positions in the Lectin_C family. We applied successive rounds of SPC clustering for the 214 positions and the 98 perturbations. For all steps, we used K = 10, q = 20 and the standard rescaling of input matrix values (described in the Materials and Methods section). The measure of similarity used to cluster both the positions and the perturbations is SEik from Equation 10. After three rounds of SPC, we obtained a cluster of 28 positions (see Table 3) and a cluster of 24 perturbations (see Table 4). Nineteen of these 24 perturbations occur at positions that cluster as well.
Table 3.
List of 28 clustered positions in Lectin C
| 4 | 5 | 6 | 15 | 27 | 28 | 29 | 38 | 49 | 51 |
| 55 | 57 | 58 | 63 | 73 | 76 | 83 | 89 | 90 | 92 |
| 93 | 94 | 104 | 105 | 106 | 113 | 115 | 116 |
The list of the 28 clustered positions in Lectin C (notation from PDB file 1g1s). Bold entries correspond to positions that are determined to be important for function based on analysis of crystal structures. Notation from PDB file 1g1s.
Table 4.
List of 24 perturbations (position and amino acid type selected) clustered in Lectin C
| 5A | 6H | 28S | 35Q | 38L | 38V | 54S | 55D | 57E | 58G |
| 63V | 63S | 80E | 82N | 83N | 87G | 89E | 90D | 92A | 93E |
| 105N | 106D | 115F | 116V |
The list of the 24 perturbations (position and amino acid type selected) clustered in Lectin C (notation from PDB file 1g1s). Italic entries denote perturbations at clustered positions; bold entries denote perturbations at positions that are structurally important.
Notation from PDB file 1g1s.
It is logical to suggest that the list of residues whose interactions are coupled and form a network for functional reasons are the union of positions and perturbations that are coupled. Based on this supposition, we find that all of the residues involving binding to Ca2+ are identified (Table 3). Among the six residues that bind directly to sugar, our method is able to predict only three. We do find that residues that are close to the crystallographically identified binding sites are found in the largest cluster. Similarly, nearly all the residues in the neighborhood of these that interact directly with PSGL-1 are correctly identified by our method (Table 3). In addition to these discrete binding sites, we successfully identify the beginning and end of the Asn83–Asn89 loop. Several of the residues in the Arg54–Glu74 loop are also predicted to be significant in the response to ligand binding. Taken together, the comparison between the prediction of the sequence-based approach and the crystallographic method that provides a direct glimpse of the network of residues playing a key role in response to ligand binding is good.
Allosteric network involves predominantly long-range contacts
The mapping of the 28 clustered positions on the structure of the complex between P-selectin and PSGL-1 (1g1s) from Figure 4 reveals the extent to which they form a spatially correlated network. We find, from the contact map of the complex, that the identified positions are connected either by covalent bonds or by nonbonded contacts. The various views of the cluster of residues and the set of experimentally proposed positions with functional roles (Fig. 4) show a large degree of spatial overlap between the two sets. The nature of interactions among the residues in the network may be classified in terms of the contact map of 1g1s. If the distance between a pair of heavy atoms in two residues is within 5.2 Å, we assume that they are in contact. There are 244 nonbonded contacts among the 117 residues of chain A in 1g1s.
Figure 4.

The network of 28 clustered residues in 1g1s (two different orientations: A, top view; B, side view). The yellow surface represents the 28 clustered positions, while the orange surface represents the 42 positions that have been identified using crystal structures of liganded and unliganded P- and E-selectins as functionally important. Half of the 42 residues are in the Arg54–Glu74 loop. It is unlikely that all of these residues are equally important for function. Their coordinated motion upon glycoprotein binding is, in all likelihood, due to chain connectivity. There is a large degree of overlap between our predictions and experiments. The clustered positions also comprise a physically connected network either by bonded or nonbonded contacts. The mapping to the structure shows that, except for residues in the N-terminus end, they are interspersed throughout the structure. The figures were produced using the packages VMD (Humphrey et al. 1996) and Povray (http://www.povray.org/). Cylinders represent α-helices and arrows represent β-strands.
We denote all contacts between amino acids separated by 11 or more positions as long-range and all other contacts as short-range. In 1g1s there are 146 long-range contacts and 98 short-range contacts. In the contact map of 1g1s, we find 64 long-range and 18 short-range contacts between the 33 clustered positions, while the crystallographically identified 43 functional positions, which include all the 21 residues in the Arg54–Glu74 loop, have 53 long-range and 63 short-range contacts. Thus, the set of 33 clustered positions is composed of very well interconnected residues that are located far away along the sequence. Therefore, in view of their large structural overlap, we propose that the correlated residues can act as connectors between various functional regions. This finding resembles the idea that allosteric communications are transmitted throughout a structure by means of a sparse but well connected network of interacting residues. In the lectin family, the signaling occurs predominantly through long-range contacts.
Discussion
Discovering residues involved in long-range communications is important for understanding the molecular basis of allostery (Perutz et al. 1998; Lockless and Ranganathan 1999; Kern and Zuiderweg 2003; Dima and Thirumalai 2004), and in several contexts including proteins that are implicated in diseases (Dima and Thirumalai 2004). It is logical to suggest, as proposed in several previous studies (Lesk and Chothia 1980; Altschuh et al. 1987; Neher 1994; Taylor and Hatrick 1994; Pazos et al. 1997; Pollock and Taylor 1997; Olmea et al. 1999; Valencia and Pazos 2002; Dima and Thirumalai 2004), that the interaction between residues must be encrypted as a statistically significant signature in the evolutionary catalog of sequences. Here we have proposed a variant of the sequence entropy, which embodies the principles of the SCA, to infer the network of interacting residues in three protein families. For the PDZ and the GPCR families, our predictions coincide with the ones reported elsewhere. Moreover, for the GPCR family, we not only reproduce known functionally relevant residues that are involved in signaling and binding, but also predict previously unidentified residues that could play a relevant role in the interhelical packing of the rhodopsin family. The application of our procedure to the lectin family leads to the correct prediction of all the important sets of residues that could play a key role in the tethering and rolling processes. Just as in the previous applications (Suel et al. 2003), our results also show that the network of correlated residues is sparse. This finding may be fairly general and is consistent with the notion that proteins can tolerate a substantial number of mutations at many positions without sacrificing functional efficiency.
In two recent papers (Fodor and Aldrich 2004a,b), the efficacy of using the SCA in predicting covariation of residues based on evolutionary information has been questioned. It appears that there are major differences in the way the SCA algorithm was used in these studies and the basis on which it was formulated. The specific differences are: (1) As shown here, the subset of sequences retained must be chosen to satisfy the demands of the central limit theorem (see Materials and Methods). If these conditions are violated, then one can get spurious results. The inclusion of poorly conserved columns can compromise the quality of a subset of alignments, thus leading to spurious results. (2) Only elements of the perturbation matrix ΔΔGi,j with j > i were retained and not the entire matrix. While this may have been deemed necessary to compare with other methods, it is a violation of the basis on which SCA is proposed. Retaining only elements in the upper half of G tacitly assumes that G is symmetric. This is not necessarily the case because the number or nature of contacts residue i makes can be very different from the ones j makes. Since these are context-dependent, we expect G to be asymmetric in general. Thus, all the elements (ΔΔGi,j for all i and j) have to be analyzed to obtain the network of residues implicated in allostery. (3) The goal of allostery is to identify spatially long-range signals between residues that are not in contact. Thus, identifying residues that are in contact does not constitute an appropriate measure of success of covariance algorithms. For example, methods that identify neighbors of atoms in a liquid cannot predict the long-range response to perturbations that reflect their underlying elasticity. Such long-range propagation of perturbations, which occurs in solids but not in liquids both of which share similar short-range order, is at the heart of allostery. Because of these fundamental differences, it is difficult to assess if the proposed invalidity of SCA has been correctly established (Fodor and Aldrich 2004a,b).
In the current formulation of the SCA, only pairwise interactions between residues are probed. It is likely that variations among more than two residues are possible because of simultaneous interactions among three or more residues. In order to decipher correlations between three residues, it is necessary to perturb sites j and k and probe the response at site i. Because this will require obtaining subsets of the MSA in which sites j and k are conserved, the statistics might not be as good as for probing pairwise interactions. Nevertheless, by using our procedure, the coupling ΔΔGi,{jk} can be computed to test which of the perturbations are pairwise additive. This valuable information is extremely difficult to obtain from experiments.
All sequence-based approaches are “thermodynamic” in nature and only consider evolutionary sequence changes. From the perspective of function, it is necessary to consider dynamic changes to perturbations that can be induced either by mutations or by changes in external conditions (pH, temperature, denaturant, mechanical force, etc.). Such changes require structural probes. Our previous work using the elastic network model (Zheng et al. 2005) was an attempt to integrate sequence- and structure-based methods to identify the sparse network of correlated residues that dynamically trigger allosteric transitions in polymerases. It is desirable to develop a theoretically based method, along the lines developed here, that focuses on residue-dependent structural perturbations for probing dynamical responses.
Materials and methods
Response of a position in the MSA to perturbations
Following Lockless and Ranganathan (1999), we defined, for each position i in the MSA, a statistical “free energy”:
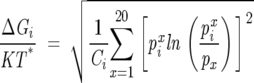 |
(1) |
where kT* is an arbitrary energy scale (which we set to 10 in our calculations), LMSA is the length of the sequences in the MSA including the gaps, and i = 1, 2, 3,…, LMSA. The number of types of amino acids that appear at least once at i is Ci (i.e., Ci ≤ 20), and px is the mean frequency of amino acid type x in the MSA. The statistical free energy is the excess value of ΔGi when pix deviates from px. We computed pix using
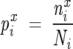 |
(2) |
where nix is the number of sequences in the MSA with amino acid x at position i, and Ni is the total number of sequences in the MSA that have one of the 20 types of amino acids at position i, so that Ni = ∑20x =1nxi.
Our procedure differs from the one used by Lockless and Ranganathan in two important aspects: (1) Instead of the binomial density function (Lockless and Ranganathan 1999) for the distribution of amino acid type x at a random position in the MSA, we use the typical frequency of x in the MSA. (2) We use the frequency of each of the 20 types of amino acids in the given MSA rather than in the entire SWISS-PROT database (Appel et al. 1994). With our procedure, the free energy ΔGi in Equation 1 is a straightforward extension of the familiar sequence entropy
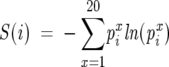 |
(3) |
at i in the MSA.
It is preferable to use, for each protein family, its typical amino acid distribution, rather than a general and maybe sometimes even incorrect set of frequencies. For example, the native state of BPTI has three disulfide bonds formed between six cysteine residues (out of the total of 56 positions in the chain). Therefore, for the BPTI family the frequency of finding CYS is ~ 12%, which is considerably bigger than the 1% frequency (Creighton 1993) found in a random protein sequence. Thus, context dependence is automatically accounted for in our procedure.
For a given MSA, we build subsets of the whole alignment in which we retain only sequences that have only one type of amino acid at a position j, that is, in the subset pix = 1 for x. Let us assume that for functional and structural reasons, positions i and j are coupled, i.e., substitutions in i would affect j. If this is the case, we expect that evolutionary pressure, under functional constraints, would lead to a correlated change in the amino acid type at i in the restricted (the subset of the original MSA) alignment. In other words, the residues at i and j might “communicate” in the course of function or upon binding to ligands. A measure of the correlation between two positions in the MSA is the statistical free energy change at i as a result of a perturbation at j:
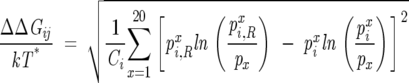 |
(4) |
with
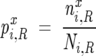 |
(5) |
where nxi,R is the number of sequences in the restricted MSA that have amino acid x at i, Ni,R is the total number of sequences in the restricted MSA that have a valid type of amino acid at position i, and Ni,R = ∑x = 120 nxi,R. It follows that ΔΔGij = 0 for both a perfectly conserved position (pix = 1) and for a position where all amino acids are found at their mean frequencies in the MSA (pix = px).
Below we give examples to show that there is an almost perfect correlation between the ΔΔGij obtained with our method and the Lockless and Ranganathan method. As stated above, our method is an extension of the sequence entropy method that is used to infer conservation of amino acids in a MSA. Our formulation of SCA lends support to the finding of Fodor and Aldrich (2004a) that the predictions of the SCA method are similar to those obtained from the sequence entropy alone. In contrast with the SCE method, the SCA approach involves averaging of the probabilities pi,Rx and pix over sequences. Such an averaging can sometimes obscure real correlations.
Size of subalignments must obey central limit theorem
In the implementation of the procedure, it is crucial to choose an optimal size of the subalignment, while previously (Suel et al. 2003) the size of the subalignments was chosen arbitrarily based only on intuitive arguments. Several subsets each containing a fraction of the total number of sequences in the MSA were chosen. For each set, ΔΔGij values for a few (usually about five) least conserved positions are computed. The size of the subalignment was chosen so that 〈ΔΔGi〉 = ∑Nsj = 1 ΔΔGij ~ 0, where the angle brackets indicate an average over the NS sequences in the subalignment.
We appeal to statistical mechanics in choosing the size of the subalignments that contain P = fNMSA sequences. The number of alignments for a fixed f is 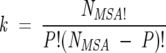 . To obtain statistically meaningful results, the general properties of the subalignments must be similar to the original MSA. In analogy with statistical mechanics, we suggest that the smallest value of f be chosen so that the law of large numbers is obeyed. In particular, we choose f so that the following criteria are satisfied:
. To obtain statistically meaningful results, the general properties of the subalignments must be similar to the original MSA. In analogy with statistical mechanics, we suggest that the smallest value of f be chosen so that the law of large numbers is obeyed. In particular, we choose f so that the following criteria are satisfied:
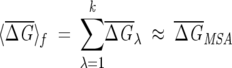 |
(6) |
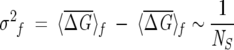 |
(7) |
where 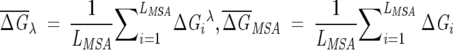 , δf is the width of the distribution of
, δf is the width of the distribution of 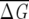 λ, and NS is the number of sequences in the subalignment. Operationally, the second criterion (Eq. 7) is valid, provided that the variance in the subalignments satisfies
λ, and NS is the number of sequences in the subalignment. Operationally, the second criterion (Eq. 7) is valid, provided that the variance in the subalignments satisfies
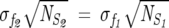 |
(8) |
where NS1 and NS2 are the number of sequences in subalignments with f1 and f2, respectively. The advantage of using our criteria (Eqs. 6, 7) is that f is automatically chosen from the MSA alone without having to compute ΔΔGij. Failure to satisfy these criteria can give spurious results in the application of SCA.
Clustering procedure and similarity measures
The matrix G, whose elements are the ΔΔGij values for a protein family, represents the response of positions i in the MSA to all allowed perturbations at site j provided the perturbations satisfy the acceptance criteria stated above. The rows of the matrix correspond to positions in the MSA and the columns to perturbations. Our objective is to reliably determine the network(s) of positions that change in a correlated manner starting from this matrix. To this end, we used the coupled two-way clustering (CTWC) that was developed to analyze DNA-microarray data (Getz et al. 2000). The basic idea is to carry out successive elementary rounds of Superparamagnetic clustering (SPC) (Blatt et al. 1996). At each step, the submatrix that contains positions and perturbations that cluster together in the previous iteration with large signals is extracted.
An important ingredient in the SPC technique is the choice of a similarity measure between a pair of entries that are to be clustered. In the context of clustering of positions in an MSA, there are at least two natural choices for similarity measures, (1) the Euclidean distance and (2) the Pearson correlation coefficient. In what follows, we give the rationale and the details for using these measures. The collection of the ΔΔGij values (with j varying from 1 to LMSA with LMSA being the total number of positions, including gaps, of the alignment) for a given position i in the MSA can be thought of as a vector with LMSA components, v→i = {ΔΔGi1,…, ΔΔGiLMSA}. Therefore, the degree of similarity between two positions i and k can be represented by the Euclidean distance between the two corresponding vectors, i.e.,
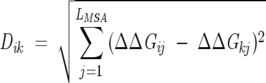 |
(9) |
For each MSA there is a spread in the magnitudes of the ΔΔGij values (e.g., from ~ 0.01 to ~ 10). Thus, for a pair of small matrix elements, Dik will be small even if the two vectors are not similar. On the other hand, for two related positions i and k with large ΔΔGij values, a difference in any of their components could lead to a large Dik value that would not reflect their true similarity. Positions with small ΔΔGij values are of little interest because they show basically no response to changes in other positions in the MSA.
To correct for the potentially spurious results indicated above, we use the following protocol: (1) We eliminate entries (positions) that show virtually no response to the overwhelming majority of perturbations. (2) We scale the ΔΔGij values so that only a few categories of the matrix elements are included in the analysis. To a large extent, the results do not depend on the precise boundaries used in the classification of ΔΔGij. (3) The Dik values are suitably normalized. If all or all but one of the corresponding ΔΔGij values are <1.0, then the row corresponding to position i is deleted from the input data matrix. The scaling of the ΔΔGij values is achieved by assigning them to two or three characteristic entries. For example, all the small ΔΔGij values (i.e., ΔΔGij < 1.0) are kept unchanged, while the intermediate ΔΔGij values (i.e., 1.0 ≤ ΔΔGij < 2.0) are assigned a value αi and the remaining (large) ΔΔGij values are assigned a value α2 such that α1 ~ 10 × maxij{min(ΔΔGij)} and α1 < α2. We normalize Dik using
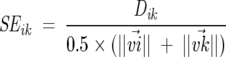 |
(10) |
where ||v→i|| is the norm of the vector v→i. The SEik is small for pairs of vectors that have components of similar values, and it is independent of the actual magnitude of the individual components. In addition, because positions that show reduced or no response to the majority of the perturbations are eliminated, a small SEik value indicates that the two positions show large responses to the same set of perturbations.
A second similarity measure that can be used is the Pearson correlation coefficient
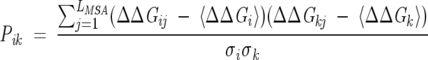 |
(11) |
where 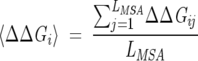 is the average ΔΔGij value for position i and
is the average ΔΔGij value for position i and
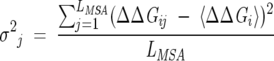 |
is the variance. Just as with the Euclidean distance similarity measure, Pik is small for two positions with little or no responses to the majority of perturbations. Prior to calculating Pik, we eliminate all such positions. The procedure used is the same as described above. For two perfectly correlated (anticorrelated) positions, Pik = 1 (−1), while for uncorrelated positions, Pik = 0. Because the Euclidean distance measure (SEik) has small values for two correlated positions and we want to be able to use the two similarity measures interchangeably, we replaced Pik with
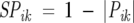 |
(12) |
where |Pik| is the absolute value of Pik. Therefore, both SEik and SPik are zero when the two positions are perfectly correlated.
The Euclidean similarity measure SEik is best suited when the individual ΔΔGij values are not broadly distributed, that is, when the largest ΔΔGij value is ~ 3.0. In such a case, the responses of each position in the alignment to the various perturbations are similar in magnitude. Therefore, the use of the Pearson correlation coefficient SPik would lead to the majority of positions being clustered. On the other hand, using the SEik and the associated rescaling of entries allows us to distinguish between positions, and therefore only a handful of positions turn out to be clustered after the application of the CTWC procedure. It follows then that the Pearson similarity measure is best suited for MSA for which there is a broad distribution in the magnitude of the responses of positions to perturbations.
Clustering algorithm
The clustering algorithm consists of several steps. We first calculate the similarity measure between all the pairs of data points. Based on these values, we select, for each input data point, its K nearest neighbors (n.n.) (K varies between 10 and 20). The next step consists in retaining, for each input data point, only its K-order neighbors, that is, i is considered a K-order neighbor of j if and only if i is one of the K n.n. of j and j is one of the K n.n. of i. Using only the pairs of data points in this K-order neighbors list, we calculate the parameters in the q-state Potts spin representation of the data points (Blatt et al. 1996). The Swendsen-Wang algorithm (Wang and Swendsen 1990), which we use to determine the conformations of the spin system in the SPC step (Blatt et al. 1996), starts with all the data points being assigned to the same value of the spin (i.e., with the spin system in the ferromagnetic phase) and unconnected with each other and the temperature T ~ 0. To map out the conformational space of the spin system, we increase the temperature linearly in small steps (such that at step t the temperature is Tt = Tt − 1 + δT with δT ~ 0.001) from T ~ 0 to T = Tmax (usually Tmax ~ 1.0). At each temperature we go through each site i that has at least one K-order neighbor, and we randomly connect it with its n.n. with the same spin value using the probability P(ij) from Wang and Swendsen (1990). We pick a random number r1 between 0 and 1, and we connect i with j (with si = sj) if and only if r1 ≤ P(ij). Then we identify the clusters in the system of spins as the collections of connected points with the same value of the spin. In the next step, we reassign the value of the spin in each of these clusters to a randomly picked value (picked with equal probability among the q available values). Finally, we loop through all the data points and their K-order n.n. and determine all the points that belong to the same cluster. Two positions with the same spin value are considered part of the same cluster. As in a typical Monte-Carlo simulation, this procedure is repeated for Nsteps (usually 10,000) number of steps at each temperature and, to allow for the equilibration of the system at the given temperature, in the calculation of the averages that enter in the average correlation between spins and the susceptibility, we disregard the data from the first Neq steps (usually 3000).
The spin conformations are used to calculate the average spin–spin correlation (〈δsi,Sj〉) for each data point and its K-order n.n. at each temperature. From the distribution of spin–spin correlations we choose the clustering temperature (Tc) as the temperature where this distribution has equal height peaks at values 1 (more precisely, 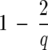 ) and 0 (more precisely,
) and 0 (more precisely, 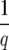 ) and is small in between. At Tc any two points for which the corresponding 〈δsi, sj〉 are assigned to a cluster. This assigns the core region for each of the clusters of data points. Because at nonzero temperatures some data points might not have any pair that satisfies the threshold for Φ, following Domany (1999), we capture the points lying at the periphery of the clusters by linking each data point i with its K-order n.n. j of maximum correlation 〈δsi, sj〉.
) and is small in between. At Tc any two points for which the corresponding 〈δsi, sj〉 are assigned to a cluster. This assigns the core region for each of the clusters of data points. Because at nonzero temperatures some data points might not have any pair that satisfies the threshold for Φ, following Domany (1999), we capture the points lying at the periphery of the clusters by linking each data point i with its K-order n.n. j of maximum correlation 〈δsi, sj〉.
Acknowledgments
We acknowledge Jie Chen for useful discussions. This work was supported in part by grants from the National Institutes of Health and the National Science Foundation through grant number NSF CHE05-14056.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051767306.
References
- Altschuh, D., Lesk, A.M., Bloomer, A.C., and Klug, A. 1987. Correlation of co-ordinated amino acid substitutions with function in viruses related to tobacco mosaic virus. J. Mol. Biol. 193: 693–707. [DOI] [PubMed] [Google Scholar]
- Appel, R.D., Bairoch, A., and Hochstrasser, D.F. 1994. A new generation of information retrieval tools for biologists: The example of the ExPASy WWW server. Trends Biochem. Sci. 19: 258–260. [DOI] [PubMed] [Google Scholar]
- Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S.R., Griffith-Jones, S., Howe, K.L., Marshall, M., and Sonnhammer, E.L. 2002. The Pfam protein families database. Nucleic Acids Res. 30: 276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatt, M., Wiseman, S., and Domany, E. 1996. Superparamagnetic clustering of data. Phys. Rev. Lett. 76: 3251–3254. [DOI] [PubMed] [Google Scholar]
- Creighton, T.E. 1993. Proteins: Structures and molecular properties. W.H. Freeman and Company, New York.
- Dima, R.I. and Thirumalai, D. 2004. Proteins associated with diseases show enhanced sequence correlation between charged residues. Bioinformatics 20: 2345–2354. [DOI] [PubMed] [Google Scholar]
- Domany, E. 1999. Superparamagnetic clustering of data—The definitive solution of an ill-poised problem. Physica A 263: 158–169. [Google Scholar]
- Fodor, A. and Aldrich, R.W. 2004a. Influence of conservation on calculations of amino acid covariance in multiple sequence alignments. Proteins 56: 211–221. [DOI] [PubMed] [Google Scholar]
- ———. 2004b. On evolutionary conservation of thermodynamic coupling in proteins. J. Biol. Chem. 279: 19046–19050. [DOI] [PubMed] [Google Scholar]
- Getz, G., Levine, E., and Domany, E. 2000. Coupled two-way clustering analysis of gene microarray data. Proc. Natl. Acad. Sci. 97: 12079–12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatley, M.E., Lockless, S.W., Gibson, S.K., Gilman, A.G., and Ranganathan, R. 2003. Allosteric determinants in guanine nucleotide-binding proteins. Proc. Natl. Acad. Sci. 100: 14445–14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins, D., Thompson, J., Gibson, T., Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horovitz, A., Fridmann, Y., Kafri, G., and Yifrach, O. 2001. Allostery in chaperonins. J. Struct. Biol. 135: 104–114. [DOI] [PubMed] [Google Scholar]
- Hulo, N., Sigrist, C.J.A., Saux, V.L., Langendijk-Genevaux, P., Bordoli, L., Gattiker, A., Castro, E.D., Bucher, P., and Bairoch, A. 2004. Recent improvements to the PROSITE database. Nucleic Acids Res. 32: D134–D137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey, W., Dalke, A., and Schulten, K. 1996. VMD–Visual Molecular Dynamics. J. Mol. Graph. 14: 33–38. [DOI] [PubMed] [Google Scholar]
- Kass, I. and Horovitz, A. 2002. Mapping pathways of allosteric communication in GroEL by analysis of correlated mutations. Proteins 48: 611–617. [DOI] [PubMed] [Google Scholar]
- Kern, D. and Zuiderweg, E.R. 2003. The role of dynamics in allosteric regulation. Curr. Opin. Struct. Biol. 13: 748–757. [DOI] [PubMed] [Google Scholar]
- Lesk, A.M. and Chothia, C. 1980. How different amino acid sequences determine similar protein structures: The structure and evolutionary dynamics of the globins. J. Mol. Biol. 136: 225–270. [DOI] [PubMed] [Google Scholar]
- Lichtarge, O., Bourne, H.R., and Cohen, F.E. 1996. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 257: 342–358. [DOI] [PubMed] [Google Scholar]
- Lockless, S.W. and Ranganathan, R. 1999. Evolutionary conserved pathways of energetic connectivity in protein families. Science 286: 295–299. [DOI] [PubMed] [Google Scholar]
- Monod, J., Wyman, J., and Changeux, J.P. 1965. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 12: 88–118. [DOI] [PubMed] [Google Scholar]
- Neher, E. 1994. How frequent are correlated changes in families of protein sequences? Proc. Natl. Acad. Sci. 91: 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmea, O., Rost, B., and Valencia, A. 1999. Effective use of sequence correlation and conservation in fold recognition. J. Mol. Biol. 295: 1221–1239. [DOI] [PubMed] [Google Scholar]
- Pazos, F., Helmer-Citterich, M., Ausiello, G., and Valencia, A. 1997. Correlated mutations contain information about protein-protein interaction. J. Mol. Biol. 271: 511–523. [DOI] [PubMed] [Google Scholar]
- Perutz, M.F., Wilkinson, A.J., Paoli, M., and Dobson, G.G. 1998. Stereochemistry of cooperative mechanisms in hemoglobin revisited. Annu. Rev. Biophys. Biomol. Struct. 27: 1–34. [DOI] [PubMed] [Google Scholar]
- Pollock, D.D. and Taylor, W.R. 1997. Effectiveness of correlation analysis in identifying protein residues undergoing correlated evolution. Protein Eng. 10: 647–657. [DOI] [PubMed] [Google Scholar]
- Schreiber, G. and Fersht, A.R. 1995. Energetics of protein–protein interactions: Analysis of the barnase–barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 248: 478–486. [DOI] [PubMed] [Google Scholar]
- Shulman, A.J., Larson, C., Mangelsdorf, D.J., and Ranganathan, R. 2004. Structural determinants of allosteric ligand activation in RXR heterodimers. Cell 116: 417–429. [DOI] [PubMed] [Google Scholar]
- Somers, W.S., Tang, J., Shaw, G.D., and Camphausen, R.T. 2000. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-Selectin bound to SLex and PSGL-1. Cell 103: 467–479. [DOI] [PubMed] [Google Scholar]
- Sowa, M.E., Hen, W., Slep, K.C., Kercher, M.A., Lichtarge, O., and Wensel, T.G. 2001. Prediction and confirmation of a site critical for effector regulation of RGS domain activity. Nat. Struct. Biol. 8: 234–237. [DOI] [PubMed] [Google Scholar]
- Steitz, T.A. 1999. DNA polymerases: Structural diversity and common mechanisms. J. Biol. Chem. 274: 17395–17398. [DOI] [PubMed] [Google Scholar]
- Suel, G.M., Lockless, S.W., Wall, M.A., and Ranganathan, R. 2003. Evolutionary conserved networks of residues mediate allosteric communication in proteins. Nat. Struct. Biol. 10: 59–69. [DOI] [PubMed] [Google Scholar]
- Taylor, W.R. and Hatrick, K. 1994. Compensating changes in protein multiple sequence alignments. Protein Eng. 7: 341–348. [DOI] [PubMed] [Google Scholar]
- Valencia, A. and Pazos, F. 2002. Computational methods for prediction of protein interactions. Curr. Opin. Struct. Biol. 12: 368–373. [DOI] [PubMed] [Google Scholar]
- Wang, J.-S. and Swendsen, R.H. 1990. Cluster Monte Carlo algorithms. Physica A 167: 565–579. [Google Scholar]
- Xu, Z. and Sigler, P.B. 1998. GroEL/GroES: Structure and function of a two-stroke folding machine. J. Struct. Biol. 124: 129–141. [DOI] [PubMed] [Google Scholar]
- Zheng, W.J., Brooks, B.R., Doniach, S., and Thirumalai, D. 2005. Network of dynamically important residues in the open/closed transition in polymerases is strongly conserved. Structure 13: 565–577. [DOI] [PubMed] [Google Scholar]