

Abstract
We have determined the crystal structure of the DUF16 domain of unknown function encoded by the gene MPN010 of Mycoplasma pneumoniae at 1.8 Å resolution. The crystal structure revealed that this domain is composed of two separated homotrimeric coiled-coils. The shorter one consists of 11 highly conserved residues. The sequence comprises noncanonical heptad repeats that induce a right-handed coiled-coil structure. The longer one is composed of approximately nine heptad repeats. In this coiled-coil structure, there are three distinguishable regions that confer unique structural properties compared with other known homotrimeric coiled-coils. The first part, containing one stutter, is an unusual phenylalanine-rich region that is not found in any other coiled-coil structures. The second part is a highly conserved glutamine-rich region, frequently found in other trimeric coiled-coil structures. The last part is composed of prototype heptad repeats. The phylogenetic analysis of the DUF16 family together with a secondary structure prediction shows that the DUF16 family can be classified into five subclasses according to N-terminal sequences. Based on the structural comparison with other coiled-coil structures, a probable molecular function of the DUF16 family is discussed.
Keywords: DUF16, MPN010, hypothetical protein, coiled-coil, stutter
Mycoplasma pneumoniae is one of the smallest microbes with ∼677 genes (Himmelreich et al. 1996). The cellular functions found in this organism are quite representative of other organisms, except for the lack of amino acid biosynthetic pathways. Mycoplasmas are primarily mucosal pathogens, living a parasitic existence in close association with epithelial cells of their host, usually in the respiratory or urogenital tracts (Waites and Talkington 2004). M. pneumoniae exclusively parasitizes humans, whereas some of the other human mycoplasmas have also been recovered from nonhuman primates (Waites and Talkington 2004). M. pneumoniae is involved in many human diseases such as asthma, pneumonia, Stevens-Johnson syndrome, Guillain-Barré Syndrome, autoimmune disease, central nerve system disease, and so forth (Waites and Talkington 2004).The Berkeley Structural Genomics Center (BSGC) has a mission to obtain a near-complete structural complement of two closely related pathogens, M. genitalium and M. pneumoniae (Kim et al. 2003). To achieve this goal, the full-size protein targets and domain targets have been selected from those predicted to be most tractable experimentally and likely to yield new structural and functional information. The DUF16 domain of MPN010 is one of the structural genomic domain targets of BSGC that has been selected based on the domain prediction (http://www.strgen. org/status/mptargets.html). MPN010 is a hypothetical protein with a molecular mass of 15.2 kDa. In the Pfam databases (Bateman et al. 2004), MPN010 belongs to the DUF16 family (Pfam01519.11) due to the C-terminal DUF16 domain for which function is not known. Interestingly, out of 33 members of the DUF16 family, 26 appear to occur in M. pneumoniae. Out of 88 hypothetical proteins of M. pneumoniae, 26 proteins are DUF16 family members (Fig. 1). In fact, the DUF16 family is the largest protein family among all M. pneumoniae protein families. All DUF16 family members in M. pneumoniae are essential genes as assayed by global transposon mutagenesis (Hutchison et al. 1999). Therefore, the elucidation of a molecular role of the DUF16 domain may contribute to a better understanding of M. pneumoniae. To gain insight into the molecular function of MPN010, the crystal structure of the DUF16 domain of MPN010 has been solved to 1.8 Å resolution determined by a single wavelength anomalous diffraction (SAD) method.
Figure 1.

Sequence comparisons of the DUF16 family in M. pneumoniae. The yellow region indicates the short helical bundle of the DUF16 domain based on the MPNOIO structure. The front of this region is the approximate N-terminal domain of the DUF 16 family and the back is the long helical bundle region of the DUF 16 domain based on the MPNOIO structure. Except for the secondary structure of MPNOIO, the other secondary structures are predictions obtained with PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred). Blue, α-helix; red, β-strand; minus symbol (−), a gap; period (.), less than mean value plus 1 SD; plus symbol (+), less than mean value plus 2 SD; and asterisk (*), less than mean value plus 3 SD. “TWISTER” represents each position of the heptad repeats of the DUF16 domain of MPN010 obtained using the program TWISTER. In here, “z” indicates a potential location of stutters. The percent sequence identities against MPN010 are also shown. Members of each subclass are as follows: (1) subfamily I: MPN104, MPN675, MPN038, MPN287, MPN484, and MPN151; (2) subfamily II: MPN010, MPN137, and MPN145; (3) subfamily III: MPN283, MPN504, and MPN524; (4) subfamily IV: MPN501, MPN100, MPN204, MPN410, MPN094, MPN130, MPN368, MPN138, and MPN139; and (5) subfamily V: MPN655, MPN344, MPN013, and MPN127. The insertions are abbreviated with the number of amino acids. The proline-rich regions are in these abbreviations.
Results
Quality of the model and overall structure
There are three molecules in an asymmetric unit of the unit cell. Most of the residues are well defined by the electron density for the refined models of the DUF16 domain of MPN010 (Fig. 2A). Two models include residues from Gly50 to Ser130, and the third model includes residues Thr51 to Ser130. The final model has been refined at 1.8 Å resolution to a crystallographic R-factor and R-free factor of 22.9% and 28.1%, respectively. The relatively high R-factor and R-free factor may be due to the disorder of the N- and C-terminal residues: The first five residues and the last residue of the DUF16 domain were invisible, and the residues near both termini show higher B-factor than other regions. This may also be the reason for the poor initial electron density map as discussed in Materials and Methods. The averaged B-factors for main-chain atoms and side-chain atoms are 22.3 Å2 and 34.2 Å2, respectively. Table 1B summarizes the refinement statistics as well as model quality parameters. The mean positional error in atomic coordinates for the refined models is estimated to be within 0.23 Å by the Luzzati plot (Luzzati 1952). All residues lie in the allowed region of the ϕ-ψ plot produced with PROCHECK (Laskowski et al. 1993).
Figure 2.
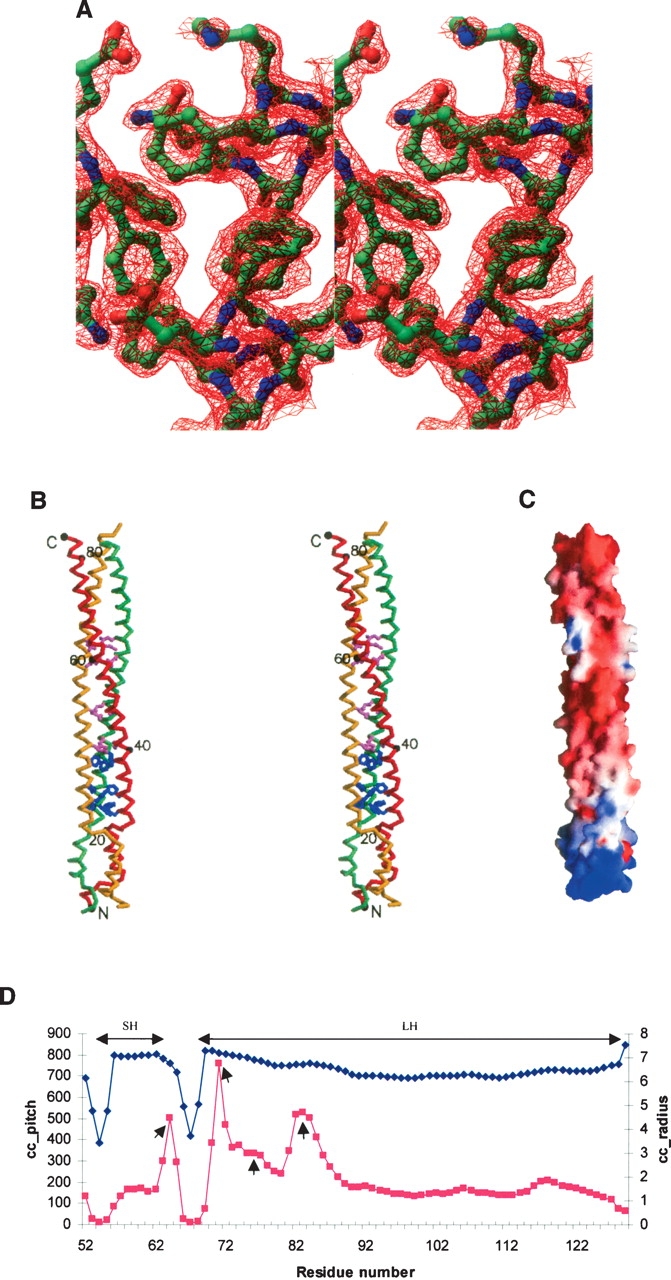
Diagrams of the DUF16 domain of MPN010. (A) A stereo view of a refined electron-density map around a phenyalanine-rich region countered at 1.5σ. The 2Fo-Fc map from finally refined phases was calculated using all reflection data between 20 Å and 1.8 Å. The figure was generated using the program RIBBONS (Carson 1991). The residues around the phenylalanine-rich region are represented by ball-and-stick models (blue, nitrogen atoms; red, oxygen; green, carbon). The front phenylalanines are Phe75 and the ones below are Phe72. (B) A stereo drawing of a Cα atom trace of the DUF16 domain of MPN010. Each model was colored differently. Every 20th residue is numbered and represented by a dot. The phenylalanine (blue) and glutamine (pink) residues that occupy trimer interfaces are represented by a ball-and-stick model. The figure was generated by MOLSCRIPT (Kraulis 1991). (C) The electrostatic surface potential of the DUF16 domain of MPN010. A molecular surface is created by the program GRASP (red, negative; blue, positive; white, uncharged) (Nicholls et al. 1991). (D) Local coiled-coil parameters plotted against the residue numbers. The gray squares represent a coiled-coil radius and the black diamonds indicate coiled-coil pitch. “SH” and “LH” represent the range of the short and the long helical bundles, respectively. The small arrows indicate the positions showing properties of stutters.
Table 1.
Statistics of the peak wavelength SAD data set and crystal parameters and refinement statistics
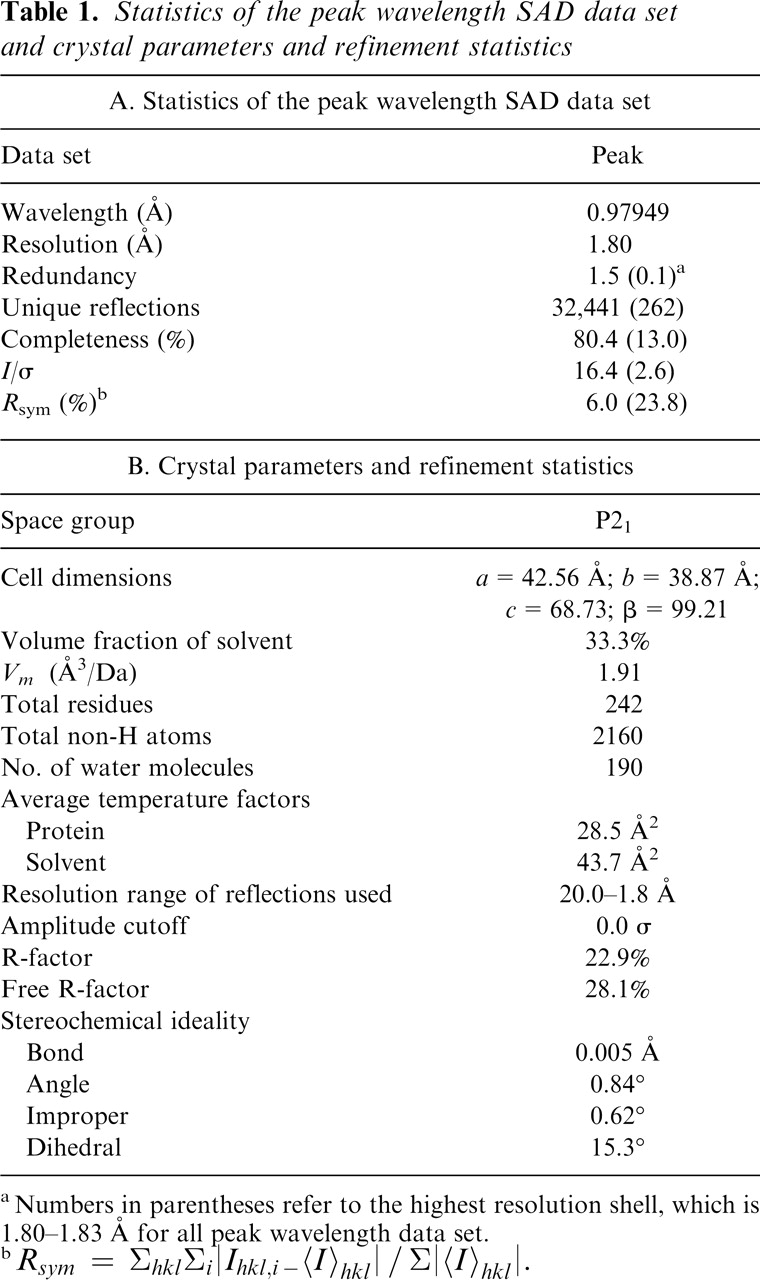
aNumbers in parentheses refer to the higest resolution shell, which is 1.80–1.83 Å for all peak wavelength data set.
![]()
A Cα atom trace of a monomer is shown in Figure 2B. Each monomer consists of a long α-helix with length of 120 Å. The three monomers form continuous two separated helical bundles. The short helical bundle starts from residue Val54 to Lys64, and the other long one starts from residue Thr68 to Glu129. The surface area buried by each monomer is ∼1860 Å2.
Properties of the short helical bundle
Eleven residues form the short helical bundle. These residues comprise noncanonical heptad repeats that induce a right-handed coiled-coil. Generally, canonical heptad repeats, (abcdefg)n with the occurrence of apolar residues preferentially in the first a and fourth d positions of the repeat, result in a left-handed coiled-coil (Gruber and Lupas 2003). The program TWISTER (Strelkov and Burkhard 2002), which analyzes coiled-coil properties based on tertiary structure, confirms that the small helical bundle is a right-handed coiled-coil (Fig. 2D). However, these noncanonical heptad sequences are not predicted to form a coiled-coil structure based on the primary sequence based coiled-coil prediction program Multicoil (Wolf et al. 1997).
The core of the short helix bundle is stabilized by four hydrophobic residues: Val54, Leu59, Leu63, and Phe66. Interestingly, Val54 is located on the most highly conserved 53YVT55 motif that is located between the N-terminal domain and the short helical bundle (Fig. 1). The outside surface of the short helical bundle is positively charged. The whole electrostatic surface potential of the DUF16 domain shows an uneven distribution of charges (Fig. 2C). Since the sequences of short helical bundle are relatively well conserved among the whole DUF16 family, these noncanonical heptad repeats may perform a structurally and biologically important and unique role in this family.
Properties of the long helical bundle
Approximately nine heptad repeats are found in this coiled-coil structure. This length is longer than those of several common three-stranded α-fibrous proteins (Conway and Parry 1991). However, the prediction programs based on primary sequence did not detect the first three heptad repeats, probably due to the presence of one stutter and several phenylalanines in the beginning of the long helical bundle. A stutter is interpreted as an addition of four residues in the heptad repeat and is known to play many roles in coiled-coil structures including underwinding of coiled-coils. The program TWISTER confirmed the presence of a stutter between Phe75 and Glu76. It is known that stutters result in a local increase of the coiled-coil pitch value and the coiled-coil radius (Strelkov and Burkhard 2002). According to these properties, there are three more positions for putative stutters at residues around Lys64, Glu71, and Phe83, as shown in Figure 2D. However, the first two putative stutters cannot be interpreted as canonical stutters, because they are located on the edge of the helical bundles, which results in locally irregular repeats. Interestingly, there is no stutter around Phe83. Therefore, this can be an example that bulky phenylalanines at the core may induce effects similar to those of stutters.
Detailed analyses show that the long helical bundle can be divided into three sections according to the residues in the core region. The beginning portion can be called a phenylalanine-rich region with Phe72, Phe75, and Phe83 occupying a core at the beginning (Fig. 2B). Since the large aromatic rings induce a steric hindrance at the trimer interfaces, the presence of several phenylalanine residues at the core is quite surprising. The phenylalanine residue is very rare in two or three helical bundle structures (Woolfson and Alber 1995).
The middle portion of the long helical bundle is a highly conserved glutamine-rich region (Fig. 2B). Glutamine is known to be enriched at positions of trimers because of its particular modulation property (Gonzalez et al. 1996). Out of seven glutamine residues in this region, four glutamine residues—Gln86, Gln93, Gln104, and Gln 107—are located in the core of this region. The last portion is a prototype coiled-coil region of which the core is composed of Leu 112, Ile115, Leu119, Ile122, Leu126, and Leu 129. Being at the end of the long helical bundle, this region may contribute stability to the whole helical bundle.
The major side-chain interaction of the long helical bundle is a type IV “knobs-into-holes” interaction as found in GCN4 leucine zipper, influenza virus hemagglutinin, and SNARE complex (Walshaw and Woolfson 2001). Stability of coiled-coils is mainly achieved by a distinctive knobs-into-holes packing of the apolar side chains into a hydrophobic core.
In summary, the beginning of the long helical bundle has a strong possibility for underwinding and opening due to the one stutter and phenylalanine residues. But this tendency is gradually decreased in the middle region and vanishes at the last prototype coiled-coil region. Interestingly, the two connecting residues, one between the N-terminal domain and the short helical bundle (residues 50GTR52) and the other between helical bundles (residues 65NFV67) are wrapped in a left-handed fashion (Fig. 2B). These help to stabilize the whole helical bundle structures by increasing the tendency for overwinding.
Comparison with other coiled-coil proteins
Coiled-coils consist of two to five amphipathic α-helices that twist around one another to form a supercoil. This structural motif, which is found in up to 10% of all protein sequences (Walshaw and Woolfson 2001), mediates sub-unit oligomerization of a large number of proteins. Their functional roles are categorized as skeletal protein, motor protein, pH-dependent coiled-coil switches, and molecular recognition (Burkhard et al. 2001). Structural comparison of the DUF16 domain of MPN010 with the above protein families reveals some similarities and dissimilarities: (1) Usually skeletal proteins form a dimeric domain (Herrmann and Aebi 2004) instead of a trimeric coiled-coil structure as shown in the DUF 16 domain. (2) Motor proteins have an ATPase domain connected to a dimeric coiled-coil structure (Kozielski et al. 1997). However, MPN010 does not contain any ATPase homologous sequences. (3) pH-dependent coiled-coil switches form homotrimeric coiled-coils as shown in influenza virus hemagglutinin (Swalley et al. 2004). From a structural point of view, the DUF 16 domain is most similar to these proteins. However, the rest of the MPN010 sequence is totally different from these proteins. Sequences in viral fusion peptides or other fusogenic peptides such as lysine-or arginine-rich peptides (Lochmann et al. 2004), which are involved in membrane fusion, are not found in MP010 either. (4) Molecular recognition proteins form a heterotetrameric coiled-coil as shown in the SNARE structure (Sutton et al. 1998) instead of a homotrimeric coiled-coil found in the DUF16 domain. In summary, the DUF16 domain has some structural similarities with other coiled-coil-containing proteins but still retains unique structural features.
From a structural point of view, the DUF16 domain shows the most homology with homotrimeric coiled-coil structures, often seen in viral membrane fusion proteins, a coiled-coil cartilage matrix protein (Dames et al. 1998), lung surfactant protein D (Kovacs et al. 2002; Hakansson et al. 1999), and bacteriophage T4 fibritin (Tao et al. 1997). However, some features found in the DUF 16 domain are not detected in other homotrimeric coiled-coil structures.
Phylogenetic and sequence analyses
There are two major domains in this family. The N-terminal domains are quite diverse (Fig. 1), unlike the C-terminal domains. A phylogenetic analysis of the DUF 16 and a primary sequence analysis of the N-terminal sequences (Fig. 1) suggest that the DUF 16 family can be classified into at least five sequence subfamilies. A secondary structure prediction with PSIPRED shows that most of the N-terminal domain structures may belong to the α + β class. MPN104, MPN675, MPN038, MPN287, MPN-484, and MPN151 belong to the first subfamily. This subfamily has the smallest N-terminal domain with a highly conserved motif “FxxNLNHMEKxKSG.” MPN010, MPN137, and MPN145 comprise the second subfamily. They have various N-terminal sequences except one highly conserved motif, “FExxxKxP.” The third and the fourth subfamilies are quite similar to each other. However, the N-terminal domain of the third family may form an all-α structure. The fourth, the largest subfamily, is composed of about one-third of all DUF 16 family members. The fifth subfamily has the longest N-terminal length without a common motif, and the high content of prolines is one of the features of this subfamily.
The program Multicoil (Wolf et al. 1997) predicted most of the C-terminal DUF 16 domains to form helical bundles. However, it failed to predict a helical bundle structure for the C-terminal domain of MPN010, probably because of the relatively high content of phenylalanine in the DUF16 domain in MPN010. The amino acid is usually not detected in a coiled-coil structure (Woolfson and Alber 1995). We predict the C-terminal domains of MPN151 and MPN675 to be coiled-coil since they also have relatively high phenylalanine content like MPN010 (Fig. 1). Therefore, we predict that all the C-terminal DUF 16 domains form a homotrimeric coiled-coil structure and play a similar molecular role for the members of this sequence family.
Biological implication
Certain bacteria move many cell lengths over surfaces in the direction of their long axis, unaided by flagella, in a process called gliding. Gliding motility is observed in a large variety of phylogenetically unrelated bacteria including Mycoplasma (McBride 2001). Transformants of M. pneumoniae with transposon insertions within MPN104 and MPN524 exhibit an altered satellite growth phenotype perhaps involving gliding motility (B. Hasselbring and D. Krause, pers. comm.). Interestingly, MPN104 and MPN524 contain a DUF16 domain (Fig. 1) and are a homolog of MPN010.
Thus, though the specific molecular function is not yet inferable, there is a strong possibility that the coiled-coil structure of the DUF 16 domain of MPN010 is involved in motility of M. pneumoniae.
Materials and methods
Cloning of DUF 16 domain of MPN010
The DUF16 domain (residues Val46 to Lys131) of MPN010 from M. pneumoniae gene (gi no. 13,507,749) was amplified by PCR using genomic DNA template and primers designed for ligation-independent cloning (LIC) (Aslanidis and de Jong 1990). The amplified PCR product was prepared for vector insertion by purification, quantitation, and treatment with T4 DNA polymerase (New England Biolabs) in the presence of 1 mM dTTP. The prepared insert was annealed into the LIC expression vector pB3, a derivative of pET21a (EMD Bioscience Inc.) that expresses the cloned gene with an N-terminal His6 tag with a Tobacco Etch Virus (TEV) cleavage sequence, and transformed into chemical competent DH5α to obtain fusion clones. Clones were screened by plasmid DNA analysis and transformed into BL21(DE3)/pSJS1244 for protein expression (Kim et al. 1998).
Protein expression, purification, and crystallization
Selenomethionine derivative of the protein was expressed in a methionine auxotroph, Escherichia coli strain B834(DE3)/pSJS1244 (Leahy et al. 1992), grown in autoinducing medium (W. Studier, pers. comm.) supplied with selenomethionine. Cells were disrupted by microfluidization (Microfluidics) in 50 mM HEPES (pH 8), 100 mM NaCl, 1 mM PMSF, 10 μg/ mL DNase, and Roche protease inhibitor cocktail (Roche Applied Science) and cell debris was pelleted by centrifugation at 15,000 rpm for 20 min in a Sorvall centrifuge. The supernatant was then spun in a Beckman ultracentrifuge Ti45 rotor at 35,000 rpm for 30 min at 4°C. The fusion protein was affinity purified using 5 mL HiTrap Chelating column on an AKTA Explorer (GE Healthcare). The fusion protein was eluted with a gradient of 250–400 mM imidazole. Fractions were pooled and dialyzed overnight at room temperature against 50 mM HEPES (pH 8.0) and 500 mM NaCl, in the presence of TEV protease. After centrifugation, the supernatant was applied onto a 5-mL HiTrap Chelating column. The cleaved recombinant protein was found in the flow through. Further purification was performed using size-exclusion chromatography (Superdex 75; GE Healthcare) in 20 mM Tris-HCl (pH 8) and 200 mM NaCl. One fraction was chosen based on SDS-PAGE and dynamic light scattering results (DynaPro 99; Wyatt Technology Corp.).
Screening for crystallization conditions was performed using the sparse-matrix method (Jancarik and Kim 1991) with several screens from Hampton Research (Hampton Research) and the Wizard Screen (deCODE genetics). The crystallization robot Hydra Plus-One (Matrix Technologies) was used to set the screens using the sitting drop vapor diffusion method at room temperature. In the optimized crystallization conditions, 1 μL of the protein (18 mg/mL) in 25 mM Tris-HCl (pH 8.5) and 150 mM NaCl was mixed with 1 mL of the mother liquor containing 0.2 M lithium sulfate, 25% PEG3350, and 0.1 M HEPES (pH 7.5) using the hanging drop vapor diffusion method. Thick rod-shaped crystals grew in a few days to approximate dimensions of 0.05×0.05×0.02 mm3.
Data collection and reduction
The crystals were soaked in a drop of mother liquor for ∼1 min before being flashed-cooled in liquid nitrogen and used for X-ray data collection. X-ray diffraction data set to 1.8 Å were collected at a single wavelength (0.97938 Å) at the Macro-molecular Crystallography Facility beamline 8.2.2 at the Advanced Light Source at Lawrence Berkeley National Laboratory using an Area Detector System Co. Quantum 4 CCD detector placed 200 mm from the sample. The oscillation range per image was 1.0° with no overlap between two contiguous images. X-ray diffraction data were processed and scaled using DENZO and SCALEPACK from the HKL program suite (Otwinowski and Minor 1997). Data statistics are summarized in Table 1A.
Structure determination and refinement
The program SOLVE (Terwilliger and Berendzen 1999) was used to locate the selenium sites in the crystal and found a best interpretable map from 20.0 to 2.8 Å resolution data. The initial single-wavelength anomalous dispersion phases were further improved by solvent flattening and histogram matching with the DM program in the CCP4 package (Collaborative Computational Project, Number 4 1994). The map calculated by using the improved phases was not good enough to trace the backbone structure of the protein, though the threefold non-crystallographic symmetry (NCS) was recognized and used. Only 20 residues of each monomer were traceable in the initial map. The rest of the protein was built using the arbitrary models made with the aid of secondary structure prediction using PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred). The three NCS matrices were further refined using a software in the RAVE package of real-space averaging and density-manipulation programs (Kleywegt and Jones 1999). A model containing 81 residues was derived from progressive improvement of the electron density map using DMMULTI (Collaborative Computational Project, Number 4 1994). The three models were built using the O program (Jones et al. 1991).
The preliminary model was then refined using the program CNS (Brünger et al. 1998). The reflections between 20.0 Å and 1.8 Å were included throughout the refinement calculations. Ten percent of the data were randomly chosen for free R-factor cross validation. The refinement statistics are shown in Table 1B. Isotropic B-factors for individual atoms were initially fixed to 20 Å2 and were refined in the last stages. The 2Fo-Fc and Fo-Fc maps were used for the manual rebuilding between refinement cycles and for the location of solvent molecules. Atomic coordinates have been deposited in the Protein Data Bank (PDB) with the access code 2BA2.
Acknowledgments
We thank Barbara Gold for cloning, Marlene Henriquez and Bruno Martinez for expression studies and cell paste preparation, and John-Marc Chandonia for bioinformatics search of the gene. We also thank the staff at the Advanced Light Source, which is supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Sciences Division, of the U.S. Department of Energy under contract no. DE-AC03-76SF00098 at Lawrence Berkeley National Laboratory. The work described here was supported by the Protein Structure Initiative grant from the NIH (GM 62412).
Footnotes
Reprint requests to: Sung-Hou Kim, Berkeley Structural Genomics Center, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA; e-mail: shkim@cchem.berkeley.edu; fax: (510) 486-5272.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051993506.
References
- Aslanidis C. and de Jong P.J. 1990. Ligation-independent cloning of PCR products (LIC-PCR) Nucleic Acids Res. 20: 6069–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A., Coin L., Durbin R., Finn R.D., Hollich V., Griffiths-Jones S., Khanna A., Marshall M., Moxon S., Sonnhammer E.L.L.et al. 2004. The Pfam protein families database Nucleic Acids Res. 32:Database Issue D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S.et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Burkhard P., Stetefeld J., Strelkov S.V. 2001. Coiled coils: A highly versatile protein folding motif Trends Cell Biol. 11: 82–88. [DOI] [PubMed] [Google Scholar]
- Carson M. 1991. RIBBONS 2.0 J. Appl. Crystallogr. 24: 958–961. [Google Scholar]
- Collaborative Computational Project, Number 4. 1994. The CCP4 Suite: Programs for protein crystallography Acta Crystallogr. D 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Conway J.F. and Parry D.A. 1991. Three-stranded α-fibrous proteins: The heptad repeat and its implications for structure Int. J. Biol. Macromol. 13: 14–16. [DOI] [PubMed] [Google Scholar]
- Dames S.A., Kammerer R.A., Wiltscheck R., Engel J., Alexandrescu A.T. 1998. NMR structure of a parallel homotrimeric coiled coil Nat. Struct. Biol. 5: 687–691. [DOI] [PubMed] [Google Scholar]
- Gonzalez L. Jr., Woolfson D.N., Alber T. 1996. Buried polar residues and structural specificity in the GCN4 leucine zipper Nat. Struct. Biol. 3: 1011–1018. [DOI] [PubMed] [Google Scholar]
- Gruber M. and Lupas A.N. 2003. Historical review: Another 50th anniversary—New periodicities in coiled coils Trends Biochem. Sci. 28: 679–685. [DOI] [PubMed] [Google Scholar]
- Hakansson K., Lim N.K., Hoppe H., Reid K.B.M. 1999. Crystal structure of the trimeric α-helical coiled-coil and the three lectin domains of human lung surfactant protein D Structure 7: 255–264. [DOI] [PubMed] [Google Scholar]
- Herrmann H. and Aebi U. 2004. Intermediate filaments: Molecular structure, assembly mechanism, and integration into functionally distinct intracellular scaffolds Annu. Rev. Biochem. 73: 749–789. [DOI] [PubMed] [Google Scholar]
- Himmelreich R., Hilbert H., Plagens H., Pirkl E., Li B.C., Herrmann R. 1996. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae Nucleic Acids Res. 24: 4420–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison C.A., Peterson S.N., Gill S.R., Cline R.T., White O., Fraser C.M., Smith H.O., Venter J.C. 1999. Global transposon mutagenesis and a minimal Mycoplasma genome Science 286: 2165–2169. [DOI] [PubMed] [Google Scholar]
- Jancarik J. and Kim S.-H. 1991. Sparse matrix sampling: A screening method for crystallization of proteins J. Appl. Crystallogr. 24: 409–411. [Google Scholar]
- Jones T.A., Zou J.Y., Cowan S.W., Kjeldgaard M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models Acta Crystallogr. A 47: 110–119. [DOI] [PubMed] [Google Scholar]
- Kim R., Sandler S.J., Goldman S., Yokota H., Clark A.J., Kim S.-H. 1998. Overexpression of archaeal proteins in Escherichia coli Biotechnol. Lett. 20: 207–210. [Google Scholar]
- Kim S.-H., Shin D.H., Choi I.G., Schulze-Gahmen U., Chen S., Kim R. 2003. Structure-based functional inference in structural genomics J. Struct. Funct. Genomics 4: 129–135. [DOI] [PubMed] [Google Scholar]
- Kleywegt G.J. and Jones T.A. 1999. Software for handling macromolecular envelopes Acta Crystallogr. D Biol. Crystallogr. 55: 941–944. [DOI] [PubMed] [Google Scholar]
- Kovacs H., O'Donoghue S.I., Hoppe H.J., Comfort D., Reid K.B., Campbell D., Nilges M. 2002. Solution structure of the coiled-coil trimerization domain from lung surfactant protein D J. Biomol. NMR 24: 89–102. [DOI] [PubMed] [Google Scholar]
- Kozielski F., Sack S., Marx A., Thormählen M., Schönbrunn E., Biou V., Thompson A., Mandelkow E.M., Mandelkow E. 1997. The crystal structure of dimeric kinesin and implications for microtubule-dependent motility Cell 91: 985–994. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures J. Appl. Crystallogr. 24: 946–950. [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Leahy D.J., Hendrickson W.A., Aukhil I., Erickson H.P. 1992. Structure of a fibronectin type III domain from tenascin phased by MAD analysis of the selenomethionyl protein Science 258: 987–991. [DOI] [PubMed] [Google Scholar]
- Lochmann D., Jauk E., Zimmer A. 2004. Drug delivery of oligonucleotides by peptides Eur. J. Pharm. Biopharm. 58: 237–251. [DOI] [PubMed] [Google Scholar]
- Luzzati V. 1952. Traitement statistique des erreurs dans la determinationn des structures cristallines Acta Crystallogr. 5: 802–810. [Google Scholar]
- McBride M.J. 2001. Bacterial gliding motility: Multiple mechanisms for cell movement over surfaces Annu. Rev. Microbiol. 55: 49–75. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp K.A., Honig B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons Proteins 11: 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Strelkov S.V. and Burkhard P. 2002. Analysis of α-helical coiled coils with the program TWISTER reveals a structural mechanism for stutter compensation J. Struct. Biol. 137: 54–64. [DOI] [PubMed] [Google Scholar]
- Sutton R.B., Fasshauer D., Jahn R., Brünger A.T. 1998. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution Nature 395: 347–353. [DOI] [PubMed] [Google Scholar]
- Swalley S.E., Baker B.M., Calder L.J., Harrison S.C., Skehel J.J., Wiley D.C. 2004. Full-length influenza hemagglutinin HA2 refolds into the trimeric low-pH-induced conformation Biochemistry 43: 5902–5911. [DOI] [PubMed] [Google Scholar]
- Tao Y., Strelkov S.V., Mesyanzhinov V.V., Rossmann M.G. 1997. Structure of bacteriophage T4 fibritin: A segmented coiled coil and the role of the C-terminal domain Structure 5: 789–798. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen J. 1999. Automated MAD and MIR structure solution Acta Crystallogr. D Biol. Crystallogr. 55: 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waites K.B. and Talkington D.F. 2004. Mycoplasma pneumoniae and its role as a human pathogen Clin. Microbiol. Rev. 17: 697–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walshaw J. and Woolfson D.N. 2001. Socket: A program for identifying and analysing coiled-coil motifs within protein structures J. Mol. Biol. 307: 1427–1450. [DOI] [PubMed] [Google Scholar]
- Wolf E., Kim P.S., Berger B. 1997. MultiCoil: A program for predicting two- and three-stranded coiled coils Protein Sci. 6: 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfson D.N. and Alber T. 1995. Predicting oligomerization states of coiled coils Protein Sci. 4: 1596–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]