

Abstract
3-Hydroxyanthranilic acid 3,4-dioxygenase (3HAO) is a non-heme ferrous extradiol dioxygenase in the kynurenine pathway from tryptophan. It catalyzes the conversion of 3-hydroxyanthranilate (HAA) to quinolinic acid (QUIN), an endogenous neurotoxin, via the activation of N-methyl-D-aspartate (NMDA) receptors and the precursor of NAD+ biosynthesis. The crystal structure of 3HAO from S. cerevisiae at 2.4 Å resolution shows it to be a member of the functionally diverse cupin superfamily. The structure represents the first eukaryotic 3HAO to be resolved. The enzyme forms homodimers, with two nickel binding sites per molecule. One of the bound nickel atoms occupies the proposed ferrous-coordinated active site, which is located in a conserved double-strand β-helix domain. Examination of the structure reveals the participation of a series of residues in catalysis different from other extradiol dioxygenases. Together with two iron-binding residues (His49 and Glu55), Asp120, Asn51, Glu111, and Arg114 form a hydrogen-bonding network; this hydrogen-bond network is key to the catalysis of 3HAO. Residues Arg101, Gln59, and the substrate-binding hydrophobic pocket are crucial for substrate specificity. Structure comparison with 3HAO from Ralstonia metallidurans reveals similarities at the active site and suggests the same catalytic mechanism in prokaryotic and eukaryotic 3HAO. Based on sequence comparison, we suggest that bicupin of human 3HAO is the first example of evolution from a monocupin dimer to bicupin monomer in the diverse cupin superfamilies. Based on the model of the substrate HAA at the active site of Y3HAO, we propose a mechanism of catalysis for 3HAO.
Keywords: X-ray crystallography; kynurenine pathway; extradiol dioxygenase; cupin superfamily; 2-His-1-carboxylate facial triad; MAD; 3-hydroxyanthranilic acid 3,4-dioxygenase
In humans, the major route of tryptophan catabolism is through the kynurenine pathway, with some of the key metabolic intermediates being kynurenine, 3-hydro- xyanthranilate (HAA), and quinolinic acid (QUIN) (Schwarcz and Pellicciari 2002; Stone and Darlington 2002). 3-Hydroxyanthranilate 3,4-dioxygenase (3HAO, EC 1.13.11.6) catalyzes the final aromatic ring opening reaction, utilizing non-heme Fe2+ to incorporate both atoms of molecular oxygen into 3-hydroxyanthranilate (Scheme 1). The enzymatic product, α-amino-β-carboxymuconic ɛ-semialdehyde is then nonenzymatically transformed to QUIN, which is the source of much of the nicotinic acid required physiologically for the synthesis of nicotinamide and the essential enzymatic cofactors such as nicotinamide adenine dinucleotide (NAD+) (Schwarcz and Pellicciari 2002; Nandi et al. 2003). Interest in the importance of this pathway was aroused with the discovery that it includes several compounds relevant to neurodegeneration and inflammatory neurological diseases (Heyes et al. 1996); 3-hydroxykynurenine and HAA can produce neuronal damage, primarily by the induction of free radical formation (Schwarcz and Pellicciari 2002; Stone and Darlington 2002). Kynurenic acid was shown to be an antagonist at NMDA, kainite, and α-amino-3-hydroxy-5-methyl-isoxazole (AMPA) receptors, whereas QUIN proved to excite neurons via the activation of NMDA receptors, and consequently produced neuronal damage (Schwarcz et al. 1983). Hyperactivity of 3HAO has been implicated as a damaging defect in Huntington's disease (Schwarcz et al. 1988; Guidetti et al. 2004), and the neurotoxic capacity of QUIN in vivo has also implicated it in a variety of other neurodegenerative disorders, including epilepsy, Alzheimer's disease, and AIDS dementia complex (Heyes et al. 1992). Manipulation of the kynurenine pathway of tryptophan metabolism, such as inhibition of the activity of the enzymes responsible for synthesizing QUIN, has yielded a plethora of agents that are now being developed as neuroprotectants and anticonvulsants (Schwarcz and Pellicciari 2002; Stone and Darlington 2002).
Scheme 1.

The reaction catayzed by 3HAO
Most of the enzymes in the kynurenine pathway have been identified in humans and in yeast. Enzymatic properties of 3HAO purified from many mammalian tissues, such as beef liver, beef kidney, rat liver, and rat brain, have been extensively studied since 1962 (Nandi et al. 2003). Recently the similar tryptophan-to-QUIN pathway has been discovered in several bacterial species (Kurnasov et al. 2003), and a novel 2-nitrobenzoate degradation pathway by the formation of HAA as an intermediate has also been revealed in Pseudomonas fluorescens strain KU-7A and Arthrobacter protophormiae RKJ100 (Hasegawa et al. 2000; Pandey et al. 2003). The prokaryotic 3HAO enzymes show apparent homology with the eukaryotic ones (Muraki et al. 2003).
3HAO belongs to the type III non-heme ferrous extradiol dioxygenases. Extradiol dioxygenases are divided into at least three evolutionary independent families: Type I extradiol dioxygenases belong to the vicinal oxygen chelate superfamily. These include one-domain enzymes, such as the 2,3-dihydroxybiphenyl 1,2-dioxygenases II and III from Rhodococcus globerulus P6, and two-do-main enzymes, such as the 2,3-dihydroxybiphenyl 1,2-dioxygenase from Pseudomonas sp. strain KKS102, which correspond to Class I and Class II dioxygenases according to Spence et al. (1996). Type II extradiol dioxygenases are multimers, such as Protocatechuate 4,5-dioxygenase(LigAB) from Sphingomonas paucimobilis SYK-6. Type III extradiol dioxygenases belong to the cupin super-family and include enzymes such as the gentisate, homogentisate dioxygenases, 1-hydroxy-2-naphthoate dioxygenase, and 3HAO (Vetting et al. 2004; Nogales et al. 2005).
The typical catechol extradiol dioxygenases play important environmental roles by facilitating the aerobic degradation of catechol derivatives by soil bacteria (Solomon et al. 2000). Although catecholic extradiol dioxygenases utilize Fe2+ to cleave a bond located adjacent to only one of the ortho-hydroxyl groups, and 3HAO cleaves a bond located adjacent to the hydroxyl group of 3-hydroxyanthranilate, the region believed to be important for iron binding is highly conserved (Bugg et al. 1997). All of these enzymes contain one non-heme Fe2+ and cleave aromatic substrates using dioxygen with no other cosubstrates and cofactors. The mechanism of 3HAO was deduced to be similar to the proposed mechanism of catechol extradiol dioxygenases (Solomon et al. 2000). Here we report the crystal structure of a eukaryotic 3HAO from Saccharomyces cerevisiae (Y3HAO) determined at 2.4 Å resolution. The Y3HAO structure and active site identified here are of critical importance to further understand the catalytic mechanism of 3HAO and the substrate specificity, and also to help in the design of 3HAO inhibitors to efficiently reduce the QUIN level and attenuate neurological deficits. On the other hand, as an extradiol dioxygenase, 3HAO may be utilized in degrading aromatic compounds.
Results and Discussion
Overall structure of the Y3HAO homodimer
The structure of Y3HAO was determined by the multiple-wavelength anomalous dispersion method (MAD) using data sets at three wavelengths. A significant part of the anomalous signals was attributed to the four exogenous Ni ions, consistent with the relatively poor dispersive signals, and that most of the side chains of selenomethionines were very flexible (Met1, Met140) or showed apparent dual conformations (Met37). The Ni-binding form of Y3HAO crystallizes in space group P212121 as a dimer. Residues 176–177(SN) in subunit A and 172–177(ARPQSN) in subunit B are missing due to the lack of electron density. The protein appears to correspond to the sequence encoded by the gene YJR025c (Kucharczyk et al. 1998), although Gly47 reported for this gene sequence does not fit to Asp47 of the crystal structure (Fig. 1). However, our sequence is deduced from direct cloning of yeast genomic DNA, rather than from an artificial mutation. In fact, we think the sequence in the databases could be wrong, as the Asp47 residue is critical for the catalytic activity and is present in all homologs (see below). The main chain of the protein folds into a conserved jelly roll with flanking helices.
Figure 1.
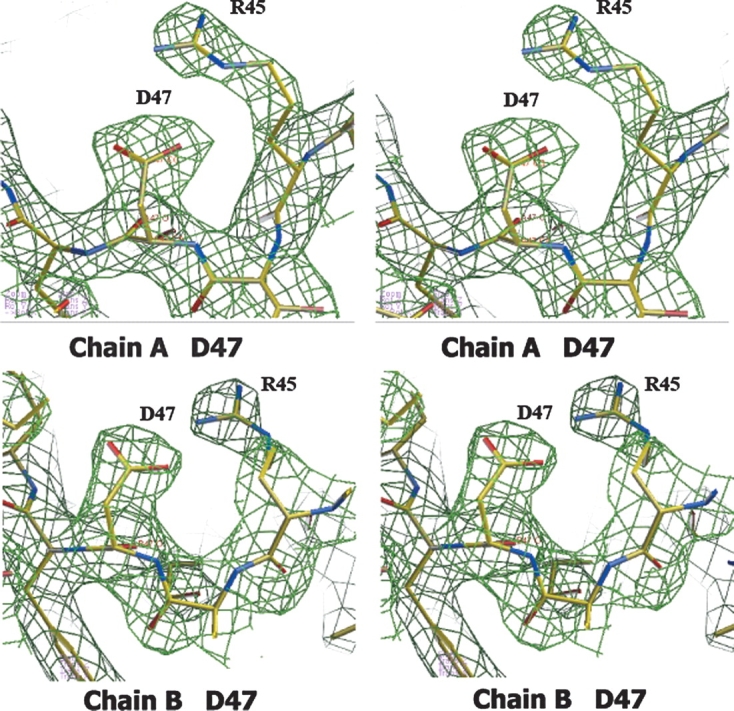
According to the gene sequence, residue 47 is reported to be a glycine. The stereo view of the 2Fo-Fc electron density map contoured at 1σ clearly indicates an aspartate in this position. The map shown in green was superimposed on the refined 2.4 Å resolution coordinates of Y3HAO. Carbon (yellow), oxygen (red), and nitrogen (cyan) atoms are shown.
The asymmetric unit of the Y3HAO crystal contains two molecules related by noncrystallographic twofold symmetry axes (Fig. 2B). Residues 26–137 fold into two antiparallel β-sheets 1↓2↑9↓4↑7↓ and 3↓8↑5↓6↑ (strands labeled in Fig. 2A), with nine strands forming a central β-jelly roll barrel. The N-terminal residues 1–20 comprise an α1 helix and a short 310 helix at the C-terminal end, an additional β-strand (β0) at the N-terminal end that is antiparallel with the β-sheet 7↓4↑9↓2↑1↓ of the other dimer-related molecule and participates in the barrel formation. The jelly roll forms a cupin cavity corresponding to the catalytic site and containing a Ni2+ in the crystal structure. After the core β-sheets are two additional anti- parallel β-strands (10↑11↓), which form an extension to one face of the jelly roll. In a parallel arrangement with them, the longest α helix (α2) forms a lid over the active site. The loop between β10↑and β11↓ and the loop between α3 and C-terminal residues are linked with a coordinated Ni2+. This C-terminal segment is at the far end of the dimer (Fig. 2). The region acting as a lid over the active site appears to be very flexible, as evinced by the markedly high B factors comprising residues at loop β11–α2 and N-terminal of α2, and functions as a gate controlling access to the active site. The dimer interface is formed principally by β-sheet 0↑1↓2↑9↓4↑7↓ (Fig. 2), and the total area buried at the interface is ∼2700 Å2 per subunit, corresponding to 30% of the subunit solvent-accessible surface area and resulting in the high degree of burial of hydrophobic residues (Trp12, Phe34, Val36, Trp56, Tyr58, Leu90, Ile108, Val110) at the interface.
Figure 2.
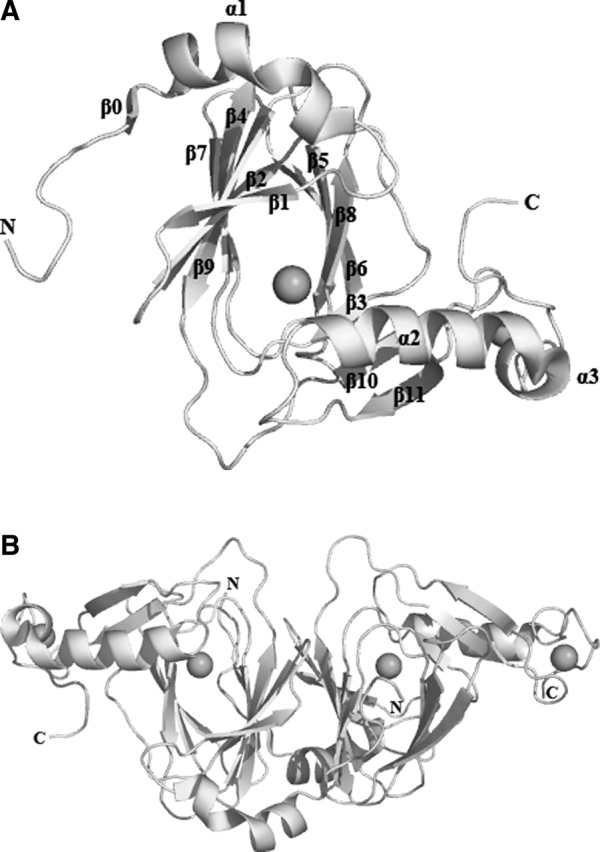
Molecular structure. (A) Ribbon diagram of the Y3HAO subunit. The metal atoms are shown as black spheres. (B) A top view down the axis of the β-barrel of the Y3HAO dimer. The N-terminal strand can be seen to interact with the β-sheet of the other dimer-related subunit. Images here and in the following figures are generated using PYMOL (http://www.pymol.org).
The RMS distance between the 170 Cα atoms of subunit A and subunit B is 1.2 Å. The principal differences between the subunits occur in three surface loops, loop α1–β1 (residues 21–25), loop β9–β10 (residues 113–120), loop β11–α2 (residues 138–144), and N-terminal of α2. These segments are flexible and consistent with their high temperature factors and may be involved in substrate entry and product release. In subunit B, loop α1– β1, β1, and α2 are closer than in subunit A. In the present Y3HAO structure, one of the bound nickel atoms occupies the proposed Fe2+-coordinated active site. It is coordinated by the Nδ1 atom of His49, two Oɛ atoms of Glu55, and the Nɛ2 atom of His97, with metal–ligand distances of 2.1–2.2 Å except for the Glu55 Oɛ1 atom where the distance is 2.5 Å, shorter than the distance of 2.7 Å from the Glu57 Oɛ1 to Fe3+ ion in the recently solved 3HAO from Ralstonia metallidurans (RM3HAO) (Zhang et al. 2005). The octahedral coordination environment is completed by two water molecules positioned further away from the metal center. In subunit A, the distance of the two water molecules to Ni2+ is 3.0 and 3.4 Å, respectively (Fig. 3). In subunit B of Y3HAO, the coordinate bond distances are similar to that of subunit A except that the coordinate bond distance of one of the water molecules to Ni2+ is 2.1 Å, and another water molecule to Ni2+ is 2.9 Å. In RM3HAO, the distance of the two water molecules to Fe3+ is the same and is 2.4 Å (Fig. 3). In solution, Y3HAO also appears as an intimate dimer identified by gel-filtration chromatography (data not shown). However, all of the mammalian 3HAOs characterized so far were found to be present and active as a monomer (∼33 kDa) (Calderone et al. 2002). Further sequence analysis suggests that the additional C-terminal segment present in metazoan enzymes may contribute to this dissimilarity (see below).
Figure 3.

Metal binding site (Ni2+ in the crystal but Fe2+ in vivo) of subunit A of Y3HAO and Fe3+ binding site of RM3HAO. The Ni2+ ion is coordinated by the Nδ1 atom of His49, two Oɛ atoms of Glu55, the Nɛ2 atom of His97 (dashed lines), and two water molecules (W, dashed lines). The Fe3+ ion is coordinated by the Nδ1 atom of His51, two Oɛ atoms of Glu57, the Nɛ2 atom of His95, and two water molecules. The distances between atoms in Å are also shown. The coordinating residues are represented in ball-and-stick models.
Structural comparison of Y3HAO and RM3HAO
The secondary structure and overall fold of the Y3HAO homodimer is very similar to that of RM3HAO. The crystal structure of RM3HAO reveals a monomer in the asymmetric unit; the monomers in adjacent asymmetric units interact, forming a dimer related by twofold symmetry. The sequence identity between Y3HAO and RM3HAO is 38%. Upon superposition of subunit A to that of RM3HAO, the RMS distance between the 169 Cα atoms is 1.5 Å. Figure 4 shows the superposition of the Cα backbones of the two structures, as well as structure-based sequence alignment of the corresponding sequences. 3HAO contains the two characteristic sequences of the cupin superfamily, namely G-(X)5-HXH-(X)3,4-E-(X)6-G and G-(X)5-PXG-(X)2-H-(X)3-N, each of which corresponds to two β strands, separated by a less conserved region of 15–50 amino acids composed of another two β strands with an intervening loop of variable length (Dunwell et al. 2001). In Y3HAO, these signature sequences are separated by 23 amino acids. The intervening loop, β5–β6, has six residues (71–76), and three of them (Glu71, Asp73, and Glu75) are acidic . Glu71, with Arg123 and Tyr125 (two residues in β10), and Glu135 (in β11) form a hydrogen-bonding network and make the overall structure compact. Asp73, together with Asp70, Thr72, and Lys77, forms another hydrogen-bonding network, whereas the intervening loop in RM3HAO is shorter and has only two residues, Asp and Gly. In RM3HAO, Glu122 and Tyr124 (two residues in β10) and Arg134 and Glu136 (two residues in β11) form a hydrogen-bonding network with Arg94, a residue in loop β7–β8. Another difference occurs in β1, which is two residues longer, and loop β1– β2 (residues 31–33), which is three residues shorter in Y3HAO. Differences occur also in loop β11–α2, which is three residues longer than in RM3HAO, and α2 (residues 145–155), which is two residues shorter. These segments act as a lid region over the active site of RM3HAO and are more compact than that of Y3HAO.
Figure 4.
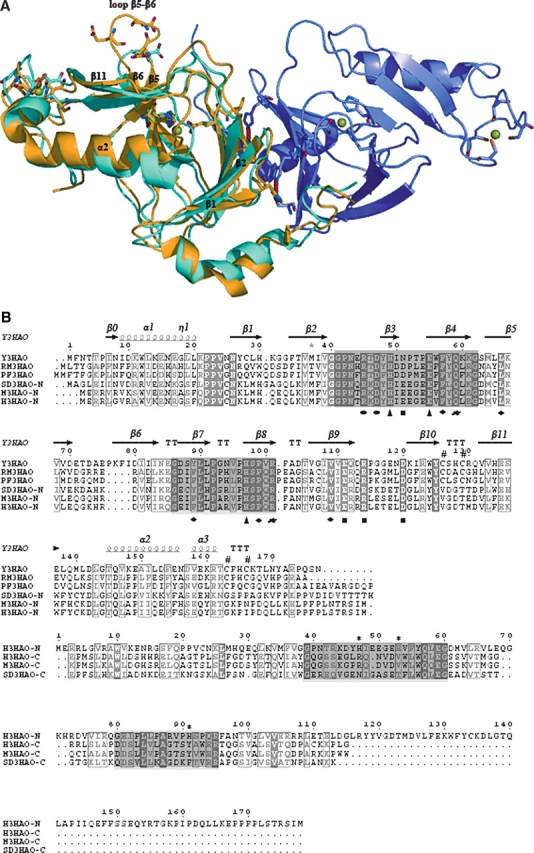
(A) Superimpositioning of the Cα backbones of subunit A of Y3HAO (yellow) and RM3HAO (cyan) (PDB code 1YFU; Zhang et al. 2005). Subunit B of Y3HAO is shown in violet. The view is related by ∼25° rotation about the horizontal axis of Figure 2B. (Green spheres) The Ni2+ ions in Y3HAO; (red spheres) iron atoms in RM3HAO. The residues coordinating metal ions, three acid residues in loop β5–β6 of Y3HAO, and one acid residue in the same loop of RM3HAO are shown as ball-and-stick. (Red dashed lines) Hydrogen bonds between Ser88 and His30 and Trp56 of the other dimer-related subunit in Y3HAO. (B) Structure-based sequence alignment of Y3HAO with its homologs. Y3HAO from Saccharomyces cerevisiae; RM3HAO from Ralstonia metallidurans; PF3HAO from Pseudomonas fluorescens; SD3HAO-N, N-terminal domain from Suberites domuncula; SD3HAO-C, C-terminal domain from Suberites domuncula; M3HAO-N, N-terminal domain from Mus musculus; M3HAO-C, C-terminal domain from Mus musculus; H3HAO-N, N-terminal domain from Homo sapiens; H3HAOC, C-terminal domain from Homo sapiens. The secondary structure and residue numbering for Y3HAO are shown above its sequence. (Arrows) β-strands; (large coils) α-helices; (η1) 310 helices. Fully conserved residues (white letters on black background) and conservatively substituted residues (black letters in black boxes) across this group of 3HAOs are indicated. (Black star) Residue in alternate conformations. The image was generated using the program ESPript (Gouet et al. 1999), with secondary structure elements assigned based on 1ZVF for Y3HAO. (Black triangle) Residues binding active-site iron ion; (black rectangles) residues forming the hydrogen-bonding network; (black circle) residues binding two oxygens; (black blob) residues involved in binding the substrate; (black diamond) residues forming hydrophobic pocket; (number sign) conserved cysteines in the monocupin 3HAOs; β0, β1, β2, β4, β7, β9 involved in dimerization are shown. The two characteristic conserved cupin sequences are shaded. Proposed metal coordinating residues of N-terminal domain of H3HAO are indicated with *.
The orientations and positions of the active-site residues —Asn43, Arg45, Asp47, His49, Glu55, Gln59, His97, Arg101, Glu111, Arg114, and Asp120—are especially well conserved, with only one conservative substitution at position 51, an Asn residue in yeast and an Asp residue in Ralstonia metallidurans (Fig. 4B), which suggests the same catalytic mechanism in prokaryotic and eukaryotic 3HAO. The km value of HAA of 19.2 μM for wild-type Y3HAO is similar to those obtained for RM3HAO (22.4 μM) and for the cloned H3HAO (2.3 μM) (Kucharczyk et al. 1998; Zhang et al. 2005). Loop α1–β1 in RM3HAO is aligned well with subunit A of Y3HAO; furthermore, this loop in the complex structure of RM3HAO with substrate reveals a closed conformation, suggesting that substrate binding would induce a closed conformation. Buried hydrophobic residues (Phe34, Val36, Tyr58, Leu90) at the interface are conserved in the yeast enzyme (177 residues) and its prokaryotic homologs which are all of a similar length, indicative of a biological dimer, except that the Oγ atom of Ser88 of subunit A forms a bifurcated hydrogen bond with the Nɛ2 atom of His30 and the Nɛ1 atom of Trp56 of the other dimer-related subunit in yeast (Fig. 4A).
Alignment of 3HAO sequences
Using the program Clustal W (Thompson et al. 1994), we aligned the 30 most homologous 3HAO sequences and show six representative homologous 3HAO sequences (Fig. 4B). Based on the structure of Y3HAO, and the complex structures of RM3HAO with the inhibitor 4-chloro-3-hydroxyanthranilate (ClHAA) and either molecular oxygen or NO (nitric oxide), active site residues are strictly conserved among these 30 orthologous sequences. Iron-binding residues, including two histidine residues (His49 and His97) and a glutamic acid (Glu55), not only are conserved, but also are located in similar spatial arrangements (Fig. 5). Together with His49 and Glu55, four hydrophilic residues near the active iron site, Asp120 (or E), Asn51 (or D, E), Glu111, and Arg114 form a hydrogen-bonding network (Fig. 5); this hydrogen-bond network may participate in a charge-relay system and seems to be important in the catalysis of 3HAO. Two oxygen-binding residues, Arg45 and Asp47, help O2 bind to ferrous ion; upon the substrate binding, Arg45 moves toward the bound O2 molecule to form a hydrogen bond with the noncovalently bound oxygen atom, and at the same time Asp47 moves to Arg45 to keep two hydrogen bonds with Arg45 (Fig. 5) (Zhang et al. 2005). Residues Arg101, Gln59, and the substrate-binding hydrophobic pocket are also highly conserved; upon substrate binding, Arg101 forms two hydrogen bonds to the carboxylate group of the substrate, Gln59 forms a hydrogen bond with Arg101 (Zhang et al. 2005; Fig. 5), and the hydrophobic face of the substrate is located in a hydrophobic pocket, primarily formed by Phe57, Leu66 (or V), Tyr89 (or F), Pro99, and Val109. Asn43 may form a hydrogen-bond network with Arg45 and Arg101 by solvent molecules, and also presents in all homologs. Other conserved residues—Pro22, Pro23, Gly40, Gly62, and Pro92—are located at the loops, which are likely for maintaining structural integrity (Fig. 4B).
Figure 5.

Stereo view of superposition of the active sites of subunit A of Y3HAO (yellow), RM3HAO (PDB code 1YFU, cyan), and the complex structures of the RM3HAO with the inhibitor ClHAA (magenta) and molecular oxygen (red) (PDB code 1YFW, magenta). The metal ions are shown in Y3HAO (green) and RM3HAO (red). The water molecules are shown in Y3HAO (gray) and RM3HAO (cyan). The residue numbering is that of Y3HAO except that E110 is the residue in RM3HAO. Conservative substitution residue in RM3HAO is given in parentheses. D120, N51, E111, and R114, together with H49 and E55, form a hydrogen-bonding network. D47 keeps two hydrogen bonds with Arg45, and Q59 forms a hydrogen bond with R101. Hydrogen bonds are shown as red broken lines.
As has been predicted on the basis of primary sequence, the β-jelly roll barrel subunit structure of 3HAO indicates that it is a member of a class of proteins known as cupins, with a double-stranded β-helix fold in SCOP (Murzin et al. 1995) structural classification characterized by a conserved β-barrel fold from which the name of the superfamily is derived. In spite of the low sequence identity among the superfamily (9%–20%), cupin is possibly the most functionally diverse protein superfamily described to date, comprising both enzymatic and nonenzymatic members, with either one or two cupin domains (Dunwell et al. 2004). With respect to the metazoan 3HAOs, the conservation of an extra C-terminal segment (∼100 residues) absent in the lower species is also obvious. Secondary structure of the C-terminal domain is predicted to be mainly eight β-strands, much like the composition of the core N-terminal fold, and consistent with a circular dichroism analysis of the secondary structure of human 3HAO, which is mainly of β strands and a minority of α (Calderone et al. 2002). It tempted us to speculate that the C-terminal domain may also adopt a cupin fold to make the enzyme a bicupin monomer with a similar overall geometry as that of the Y3HAO dimer.
It is the first example of evolution from a monocupin dimer to bicupin monomer in the diverse cupin super-families. It is also a little reminiscent of the evolution of class I to class II of the type I extradiol dioxygenases (Spence et al. 1996), while the mutation and loss of function happened in the N-terminal domain to give class II. Alignments showed that these C-terminal domains of the metazoan 3HAOs share a low homology with their N-terminal portions as well as other known cupin sequences, reflecting a rather unconstrained evolution (Fig. 4B). The two conserved cupin sequence motifs are best conserved in the N-terminal domain, where the conserved histidine and glutamic acid residues correspond to the metal-coordinating residues. The C-terminal domain motifs lack the metal-binding residues normally associated with the cupin fold, which indicates an abolishment of metal binding and activity of the C-terminal domain. The C-terminal region is unrelated to the enzymatic reaction, against the proposed deprotonation function of His190 in human 3HAO (Mendel et al. 2004). Identification of the metazoan bicupin 3HAOs is of significance in that the higher species enzymes are experimentally intractable due to instability and inclusion body formation. An exception is human homo-gentisate dioxygenase (Titus et al. 2000), a hexamer arranged as a dimer of trimers, which also belongs to the type III non-heme ferrous extradiol dioxygenases; but it is an unusual type of cupin as it has the active site metal ion coordinated near the interface between subunits within the partially formed cupin domain, and it contains the metal binding site in its C-terminal domain. However, 3HAO shares a characteristic metal coordination site within the jellyroll in cupins as a monocupin dimer or bicupin monomer; therefore, 3HAO is a special subgroup of the type III extradiol dioxygenases.
Two other extradiol dioxygenases also cleave ortho–aminophenol: One of them is 2-aminophenol 1,6-dioxygenase (Lendenmann and Spain1996), which belongs to the type II extradiol dioxygenases; the other is 4-amino-3-hydroxybenzoate 2,3-dioxygenase (4A3HBA23D) from Bordetella sp. 10d (Murakami et al. 2004). 4A3HBA23D from Bordetella sp. 10d (175 residues), also as a biological dimer, has 30% and 24% identities with Y3HAO and 3HAO from Homo sapiens (H3HAO), respectively, and no identities with other extradiol dioxygenases. However, 3HAO and 4A3HBA23D show high substrate specificity only for their natural substrates, respectively, which differ in the position of an amino group. Figure 6 shows a sequence alignment of Y3HAO and 4A3HBA23D. Active-site residues Arg45, His49, Asn51, Glu55, His97, Glu111, Arg114, and Asp120 are identical; Asn43 and Asp47 are conservatively replaced by Glu46 and Glu50 in Bordetella sp. 10d. Two substrate-binding residues, Arg101 and Gln59, are replaced by Phe101 and Ser62, respectively, in Bordetella sp. 10d. The hydrophobic residues lining the substrate are highly conserved, except that Phe57 is replaced by hydrophilic residue Gln60 in Bordetella sp. 10d. Therefore, residues Arg101, Gln59, and Phe57 are crucial for the substrate specificity of 3HAO.
Figure 6.

Sequence alignment between Y3HAO and 4A3HBA23D from Bordetella sp. 10d. The image was generated using the program ESPript (Gouet et al. 1999). Fully conserved residues (white letters on black background) and conservatively substituted residues (black letters in black boxes) are shown. (#) Residues Arg101 and Gln59 are replaced by Phe101 and Ser62 in Bordetella sp. 10d; ($) Phe57, hydrophobic residue lining the substrate, replaced by hydrophilic residue Gln60 in Bordetella sp. 10d.
Metal coordination
The crystal structure of Y3HAO shows that the enzyme contains two nickel centers per monomer, in agreement with results from metal elementary analysis. One metal center is located at the end of the molecule and is solvent-exposed. The nickel ion is coordinated mainly by two typical Cys-XX-Cys motifs (Cys126, Cys129, Cys163 and Cys166) in the C-terminal sequence, with a tetrahedral geometry. This Cys-XX-Cys motif is common to the divalent metals. The nickel ion or the iron ion in the RM3HAO all may be introduced during purification, and the biologically relevant metal ion needs further study. There is strict conservation of this motif in the monocupin 3HAOs, but it is absent in the bicupin homologs. This metal center is apparently unrelated to enzymatic activity, and a structural stabilization role is proposed. Like RM3HAO, the two metal sites in Y3HAO are separated by ∼24 Å.
The experimental electron density map clearly reveals the presence of the other metal ion coordinated by the side chains of His49, Glu55, and His97 located in the β-barrel. In fact, tertiary structure of the cupin domain has the advantage of providing a flexible “active site” within the center of the β-barrel, resulting from the different metals bound by the relatively conserved ligands within the characteristic motifs in the superfamily. A ligand environment with three histidines (via their NE2 atoms) and one glutamate (via the OE2) has been observed in many cupin members such as germin with a Mn2+ center (Woo et al. 2000), phosphomannose isomerase with a Zn2+ center (Cleasby et al. 1996), oxalate decarboxylase with two Mn2+ centers (Anand et al. 2002), copper-or iron-containing quercetin 2,3-dioxygenase (Gopal et al. 2005), zinc ion-containing auxin-binding 1 protein (Woo et al. 2002), and a Ni2+-binding dioxygenase, acireductone dioxygenase (Pochapsky et al. 2002). In some members, just as in 3HAO, one of the three histidines is lost, resulting in a His2 Glu/Asp coordination sphere. This common 2-His-1-car-boxylate facial triad motif is also shared by many mononuclear non-heme iron(II) enzymes from different families that catalyze different reactions and have distinct requirements for catalysis. For example, 4-hydroxyphenylpyruvate dioxygenase (HPPD) (Fritze et al. 2004), one of the α-ketoglutarate iron-dependent dioxygenases, and the predominant subclass of cupins, in spite of divergent structures, all feature an active site in which two histidines and one carboxylate ligand occupy one face of the iron(II) coordination sphere. This 2-His-1-carboxylate facial triad was proposed as a common platform to coordinate an iron(II) center in non-heme iron enzymes and is one of nature's recurring bioinorganic motifs (Koehntop et al. 2005). The three sites at the opposite face allow metal to bind O2 and a variety of substrates,providing the flexibility for the activesite to effect the remarkable range of reactions.
A comparison of the catalytic metal-binding site of 3HAO with the facial triads of known non-heme iron(II) oxygenase reveals an apparent difference: His49 in Y3HAO ligates the metal ion by the Nδ1 atom rather than a Nɛ2 as in most of the oxygenases (Fig. 3), despite the similar overall geometry. The only exception is that in human homogentisate dioxygenase (HGO), a similar Nδ1–Fe2+ ligation is observed on the spacial equivalent His335 (Titus et al. 2000). Both 3HAO and HGO catalyze aromatic ring-cleaving reactions different from the typical catechol extradiol dioxygenases, with one hydroxyl substituted by an ortho-amino or para-hydroxyl group, respectively. However, it was not mentioned by Titus et al. (2000), possibly due to an uncertainty that this kind of ligation would be preserved after substrate binding and during the reaction. In Y3HAO, a strictly conserved Asp120 on loop β9–β10 limits the side chain of His49 by an Oδ1–Nɛ2 hydrogen bond, and the electron density map clearly indicates the nearest atom in His49 to the metal ion should be at the δ position. Rotating the imidazole ring to form a similar Nɛ2–metal bond would need considerable movement of the main chain of β3 and disrupt the hydrogen bond to Asp120. Conformation of the active-site residues is sustained by the hydrogen-bonding network.
Active site and proposed catalytic mechanism
Despite the lack of overall structural similarity, the activesite structures of 3HAO and 2,3-dihydroxybiphenyl 1,2-dioxygenase (BphC) derived from Pseudomonas sp. strain KKS102 (Sato et al. 2002), one of the class II of the type I extradiol dioxygenases, are similar. Like BphC, the substrate HAA binds to the iron of RM3HAO as an asymmetrical bidentate ligand, at sites trans to the two imidazole ligands. Also, the oxygen atom adjacent to the C–C bond that is cleaved is ionized, in which C3-OH is deprotonated (Zhang et al. 2005). An initial deprotonation of the hydroxyl of the substrate is crucial for O2 activation; in RM3HAO, Glu110 forms a hydrogen bond with the C-3 hydroxyl group of HAA and deprotonates C3-OH. The O2 binding site is also opposite to the endogenous iron ligand Glu. These similarities suggest that there may have been a convergent evolution among extradiol dioxygenases and that they probably share similar catalytic mechanisms (Bugg and Lin 2001; Vaillancourt et al. 2002; Siegbahn and Haeffner 2004; Vetting et al. 2004; Viggiani et al. 2004).
On the basis of the coordinates of the RM3HAO–HAA complex structure, a HAA moiety was modeled into the active site of subunit A of Y3HAO, as seen in Figure 7. There is no major difference in the substrate binding pocket between Y3HAO and RM3HAO. The crystal structure of the RM3HAO–HAA complex shows clear differences from that of the native enzyme in that as loop α1–β1, β1, loop β11–α2, and α2 move toward the center of β-barrel, the active site conformation is more closed, but the positions of the residues forming the hydrogen-bond network remain the same. Based on this, we propose a catalytic mechanism for 3HAO (Fig. 8). The first step of the reaction involves the displacement of one solvent ligand and Oɛ1 atom of Glu55 from the enzyme resting state (Fig. 8A) by the substrate to form a five-coordinate iron(II) species with a chelated monoanionic 3-hydroxyanthranilate (Fig. 8B); the substrate HAA binds to the iron as a bidentate ligand in which C3-OH is deprotonated. In the ES complex, the 3-hydroxy group of HAA forms a hydrogen bond to the Oɛ2 atom of Glu111; the hydrogen-bonding network makes it easy to induce deprotonation of C3-OH. Together with the hydrogen-bonding network, the Oɛ1 atom of Glu55 makes a hydrogen bond from the site opposite the C-2 amino group of the substrate HAA (Fig. 7). The carboxylate group of HAA forms two hydrogen bonds to Arg101, and the Nɛ2 atom of Gln59 forms a hydrogen bond with the NH1 group of Arg101; this interaction and hydrophobic interactions stabilize the substrate binding. The next step would then be O2 binding to the vacant site of the ES complex; its binding site is opposite to endogenous iron ligand Glu55. The O2 binding is supposed to induce complete deprotonation of the C2-NH2 group of the substrate (Fig. 8C). At this stage, the bound dioxygen molecule acquires a negative charge due to electron transfer from the metal center, resulting in the formation of an imine–Fe2+-superoxide intermediate. Because the bound dioxygen molecule is located in the cavity that is lined with a mixture of hydrophobic and hydrophilic residues, the negative charge on the superoxide may be stabilized by the NH2 group of Arg45 or the protonated Glu55 (Fig. 8C). Then the activated dioxygen molecule attacks the C3 atom of the substrate to give an iron–alkylperoxo intermediate (Fig. 8D), which undergoes alkenyl migration, Criegee rearrangement, and O–O bond cleavage to give an unsaturated seven-membered lactone intermediate and an iron(II)-bound hydroxide ion (Fig. 8E). The NH2 group of Arg45 or the protonated Glu55 assists cleavage of the O–O bond in the Criegee rearrangement, and iron(II)-bound hydroxide ion then hydrolyzes the lactone to afford product. The precise reaction mechanism will be solved by the complex structures of 3HAO–HAA with active site Fe2+ under anaerobic condition, and mutational studies.
Figure 7.

Model of the substrate HAA at the active site of subunit A of Y3HAO based on the crystal structure of the substrate-bound RM3HAO (PDB code 1YFY) as reported by Zhang et al. (1995). The Oɛ2 atom of E111 forms a hydrogen bond with the C-3 hydroxyl group of HAA, and the Oɛ1 atom of Glu55 forms a hydrogen bond with the C-2 amino group of HAA. These two hydrogen bonds are shown as black broken lines.
Figure 8.

The proposed reaction mechanism catalyzed by 3HAO.
Materials and methods
Cloning, expression, purification, and crystallization
The complete gene fragment encoding Y3HAO protein was subcloned into the pET-28a expression vector from extracted Saccharomyces cerevisiae genomic DNA, and the Y3HAO protein was highly expressed as a soluble protein in Escherichia coli strain BL21(DE3) with a six-residue His tag attached to its N terminus. Purification of the Y3HAO protein was carried out through an affinity chromatography Ni-NTA His bind column (Qiagen) followed by size-exclusion chromatography on a Superdex 75 column (Amersham Biosciences). Crystals of Y3HAO were grown using the hanging drop-vapor diffusion method from a solution containing 1 mg/mL Y3HAO in 30% polyethylene glycol 4000, 0.2 M NaAc, 0.1 M Tris-HCl (pH 8.5), at 20°C. For phase determination, a selenomethionyl derivative was produced and crystallized under similar conditions.
Structure determination
Data sets were collected at three wavelengths from a single selenomethionine derivative crystal at 100 K on Beamline F2 of MacCHESS, using energies corresponding to the peak (λ1 = 0.9798 Å) and the edge (λ2 = 0.9802 Å) of the experimentally determined selenium K-edge. All processing, scaling, and merging of data sets were performed using the HKL2000 package (Otwinowski and Minor 1997). Initial phases were calculated to 3.5 Å resolution with SHARP (de la Fortelle et al. 1997) from seven heavy atom sites. DM (Cowtan 1999) was used for density modification and phase extension to 2.4 Å. A partial Y3HAO model was initially built using ARP/wARP (Perrakis et al. 1999), and then the sequence of Y3HAO was built into the resultant density maps using a threading model as a guide. The electron density allowed very clearly the identification of the two molecules in the asymmetric unit. Refinement was performed with Refmac5.0 (Murshudov et al. 1997) using the peak data, and the noncrystallographic symmetry constraints were released in the latter steps. The metal ions of Y3HAO (four per asymmetric unit) could be unambiguously detected in anomalous difference Fourier maps calculated from both the peak and the edge data. The Ni2+ was refined at full occupancy with a temperature factor of 36.7–42.8 Å2. The final model contains 348 residues, 60 water molecules, and 4 Ni2+ ions. The model was validated using PROCHECK (Laskowski et al. 1993). The Ramachandran plot produced by PROCHECK shows 91.2% of all the residues in the most favored regions, 8.1% in additional allowed regions, and 0.7% generously allowed regions. For details of the R-factor, Rfree, and stereochemistry of the final model, see Table 1.
Table 1.
Data collection and refinement statistics

aRmerge = ∑|Ihkl –<Ihkl>|/∑Ihkl, where <Ihkl> is the average of Ihkl over all symmetry equivalents.
bPhasing power as defined in SHARP.
cFOM (figure of merit) as defined in SHARP.
dRfactor = ∑|Fobs – Fcalc|/∑Fobs. The Rfree factor is calculated using a randomly selected 5% of the reflections set aside throughout the refinement.
Identification of the metal ion
The presence of the metals Mg, Ca, Fe, Co, Ni, and Zn were analyzed by an atomic absorption spectrophotometry experiment using PE 1100B, Atom abs (PE, USA). The results indicated that, after gel-filtration chromatography in 20 mM Tris-HCl (pH 8.5) and 150 mM NaCl, only Ni2+ (the molar ratio of Ni2+ to Y3HAO is 2.2) was concomitant with the protein, apparently introduced during the Ni2+-affinity chromatography step.
Acknowledgments
Financial support for this project to L.N. and M.T. was provided by research grants from the Chinese National Natural Science Foundation (grant nos. 30121001, 30025012, 30130080, 30571066), the “973” and “863” Plans of the Chinese Ministry of Science and Technology (grant nos. G1999075603, 2004CB520801, and 2002BA711A13), and the Chinese Academy of Sciences (grant no. KSCX1-SW-17). This work is also based upon research conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the NSF under award no. DMR 0225180, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, which is supported by award no. RR-01646 from the NIH, through its National Center for Research Resources.
Footnotes
Reprint requests to: Maikun Teng or Liwen Niu, Department of Molecular and Cell Biology, School of Life Sciences, University of Science and Technology of China, 96 Jinzhai Road, Hefei, Anhui, 230026, China; e-mail: mkteng@ustc.edu.cn or lwniu@ustc.edu.cn; fax: 86-551-3603046.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051967906.
Abbreviations: Y3HAO, 3-hydroxyanthranilic acid 3,4-dioxygenase from Saccharomyces cerevisiae; RM3HAO, 3-hydroxyanthranilic acid 3,4-dioxygenase from Ralstonia metallidurans; HAA, 3-hydroxyan-thranilate; QUIN, quinolinic acid; NMDA, N-methyl-D-aspartate; NAD+, nicotinamide adenine dinucleotide; AMPA, α-amino-3- hydroxy-5-methyl-isoxazole; 4A3HBA23D, 4-amino-3-hydroxybenzo- ate 2,3-dioxygenase from Bordetella sp. 10d; HPPD, 4-hydroxyphenyl-pyruvate dioxygenase; HGO, human homogentisate dioxygenase; BphC, 2,3-dihydroxybiphenyl 1,2-dioxygenase from Pseudomonas sp. strain KKS102; MAD, multiple wavelength anomalous dispersion; EC, enzyme classification.
References
- Anand R., Dorrestein P.C., Kinsland C., Begley T.P., Ealick S.E. 2002. Structure of oxalate decarboxylase from Bacillus subtilis at 1.75 Å resolution Biochemistry 41: 7659–7669. [DOI] [PubMed] [Google Scholar]
- Bugg T.D.H. and Lin G. 2001. Solving the riddle of the intradiol and extradiol catechol dioxygenases: How do enzymes control hydroperoxide rearrangements? Chem. Commun. 2001: 941–952. [Google Scholar]
- Bugg T.D., Sanvoisin J., Spence E.L. 1997. Exploring the catalytic mechanism of the extradiol catechol dioxygenases Biochem. Soc. Trans. 25: 81–85. [DOI] [PubMed] [Google Scholar]
- Calderone V., Trabucco M., Menin V., Negro A. 2002. Cloning of human 3-hydroxyanthranilic acid dioxygenase in Escherichia coli: Characterisation of the purified enzyme and its in vitro inhibition by Zn2+ Biochim. Biophys. Acta 1596: 283–292. [DOI] [PubMed] [Google Scholar]
- Cleasby A., Wonacott A., Skarzynski T., Hubbard R.E., Davies G.J., Proudfoot A.E., Bernard A.R., Payton M.A., Wells T.N. 1996. The x-ray crystal structure of phosphomannose isomerase from Candida albicans at 1.7 angstrom resolution Nat. Struct. Biol. 3: 470–479. [DOI] [PubMed] [Google Scholar]
- Cowtan K. 1999. Error estimation and bias correction in phase-improvement calculations Acta Crystallogr. D Biol. Crystallogr. 55: 1555–1567. [DOI] [PubMed] [Google Scholar]
- de la Fortelle E., Irwin J.J., Bricogne G. 1997. SHARP: A smaximum-likelihood heavy-atom parameter refinement and phasing program for the MIR and MAD methods In Crystallographic computing (eds. Bourne P. and Watenpaugh K.) . pp. 250–262. IUCR, Oxford, UK.
- Dunwell J.M., Culham A., Carter C.E., Sosa-Aguirre C.R., Good-enough P.W. 2001. Evolution of functional diversity in the cupin superfamily Trends Biochem. Sci. 26: 740–746. [DOI] [PubMed] [Google Scholar]
- Dunwell J.M., Purvis A., Khuri S. 2004. Cupins: The most functionally diverse protein superfamily? Phytochemistry 65: 7–17. [DOI] [PubMed] [Google Scholar]
- Fritze I.M., Linden L., Freigang J., Auerbach G., Huber R., Steinbacher S. 2004. The crystal structures of Zea mays and Arabidopsis 4-hydroxyphenylpyruvate dioxygenase Plant Physiol. 134: 1388–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal B., Madan L.L., Betz S.F., Kossiakoff A.A. 2005. The crystal structure of a quercetin 2,3-dioxygenase from Bacillus subtilis suggests modulation of enzyme activity by a change in the metal ion at the active site(s) Biochemistry 44: 193–201. [DOI] [PubMed] [Google Scholar]
- Gouet P., Courcelle E., Stuart D.I., Metoz F. 1999. ESPript: Multiple sequence alignments in PostScript Bioinformatics 15: 305–308. [DOI] [PubMed] [Google Scholar]
- Guidetti P., Luthi-Carter R.E., Augood S.J., Schwarcz R. 2004. Neostriatal and cortical quinolinate levels are increased in early grade Huntington's disease Neurobiol. Dis. 17: 455–461. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y., Muraki T., Tokuyama T., Iwaki H., Tatsuno M., Lau P.C.K. 2000. A novel degradative pathway of 2-nitrobenzoate via 3-hydroxyanthranilate in Pseudomonas fuorescens strain KU-7 FEMS Microbiol. Lett. 190: 185–190. [DOI] [PubMed] [Google Scholar]
- Heyes M.P., Saito K., Jacobowitz D., Markey S.P., Takikawa O., Vickers J.H. 1992. Poliovirus induces indoleamine-2,3-dioxygenase and quinolinic acid synthesis in macaque brain FASEB J. 6: 2977–2989. [DOI] [PubMed] [Google Scholar]
- Heyes M.P., Achim C.L., Wiley C.A., Major E.O., Saito K., Markey S.P. 1996. Human microglia convert L-tryptophan into the neurotoxin quinolinic acid Biochem. J. 320: 595–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehntop K.D., Emerson J.P., Que Jr. L. 2005. The 2-His-1-carboxylate facial triad: A versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes J. Biol. Inorg. Chem. 10: 87–93. [DOI] [PubMed] [Google Scholar]
- Kucharczyk R., Zagulskia M., Rytkaa J., Herbertb C.J. 1998. The yeast gene YJR025c encodes a 3-hydroxyanthranilic acid dioxygenase and is involved in nicotinic acid biosynthesis FEBS Lett. 424: 127–130. [DOI] [PubMed] [Google Scholar]
- Kurnasov O., Goral V., Colabroy K., Gerdes S., Anantha S., Osterman A., Begley T.P. 2003. NAD biosynthesis: Identification of the tryptophan to quinolinate pathway in bacteria Chem. Biol. 10: 1195–1204. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Lendenmann U. and Spain J.C. 1996. 2-Aminophenol 1,6-dioxygenase: A novel aromatic ring cleavage enzyme purified from Pseudomonas pseudoalcaligenes JS45 J. Bacteriol. 178: 6227–6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel S., Arndt A., Bugg T.D.H. 2004. Acid–base catalysis in the extradiol catechol dioxygenase reaction mechanism: Site-directed mutagenesis of His-115 and His-179 in Escherichia coli 2,3-dihydroxyphe-nylpropionate 1,2-dioxygenase (MhpB) Biochemistry 43: 13390–13396. [DOI] [PubMed] [Google Scholar]
- Murakami S., Sawami Y., Takenaka S., Aoki K. 2004. Cloning of a gene encoding 4-amino-3-hydroxybenzoate 2,3-dioxygenase from Bordetella sp. 10d Biochem. Biophys. Res. Commun. 314: 489–494. [DOI] [PubMed] [Google Scholar]
- Muraki T., Taki M., Hasegawa Y., Iwaki H., Lau P.C.K. 2003. Prokaryotic homologs of the eukaryotic 3-hydroxyanthranilate 3,4-dioxygenase and 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase in the 2-nitrobenzoate degradation pathway of Pseudomonas fluorescens strain KU-7 Appl. Environ. Microbiol. 69: 1564–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin A.A., Dodson E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Murzin A.G., Brenner S.E., Hubbard T., Chothia C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures J. Mol. Biol. 247: 536–540. [DOI] [PubMed] [Google Scholar]
- Nandi D., Lightcap E.S., Koo Y.K., Lu X.L., Quancard J., Silverman R.B. 2003. Purification and inactivation of 3-hydroxyanthranilic acid 3,4-dioxygenase from beef liver Int. J. Biochem. Cell Biol. 35: 1085–1097. [DOI] [PubMed] [Google Scholar]
- Nogales J., CanalesÁ., Jiménez-Barbero J., García J.L., Díaz E. Molecular characterization of the gallate dioxygenase from Pseudomonas putida KT2440: The prototype of a new subgroup of extradiol dioxygenases J. Biol. Chem. 280: 35382–35390. [DOI] [PubMed]
- Otwinowski Z. and Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode Methods Enzymol. 276A: 307–326. [DOI] [PubMed] [Google Scholar]
- Pandey J., Paul D., Jain R.K. 2003. Branching of o-nitrobenzoate degradation pathway in Arthrobacter protophormiae RKJ100: Identification of new intermediates FEMS Microbiol. Lett. 229: 231–236. [DOI] [PubMed] [Google Scholar]
- Perrakis A., Morris R., Lamzin V.S. 1999. Automated protein model building combined with iterative structure refinement Nat. Struct. Biol. 6: 458–463. [DOI] [PubMed] [Google Scholar]
- Pochapsky T.C., Pochapsky S.S., Ju T.T., Mo H.P., Al-Mjeni F., Maroney M.J. 2002. Modeling and experiment yields the structure of acireductone dioxygenase from Klebsiella pneumoniae Nat. Struct. Biol. 9: 966–972. [DOI] [PubMed] [Google Scholar]
- Sato N., Uragami Y., Nishizaki T., Takahashi Y., Sazaki G., Sugimoto K., Nonaka T., Masai E., Fukuda M., Senda T. 2002. Crystal structures of the reaction intermediate and its homologue of an extra-diol-cleaving catecholic dioxygenase J. Mol. Biol. 321: 621–636. [DOI] [PubMed] [Google Scholar]
- Schwarcz R. and Pellicciari R. 2002. Manipulation of brain kynurenines: Glial targets, neuronal effects, and clinical opportunities J. Pharmacol. Exp. Ther. 303: 1–10. [DOI] [PubMed] [Google Scholar]
- Schwarcz R., Whetsell W.O., Mangano R.M. 1983. Quinolinic acid: An endogenous metabolite that produces axon-sparing lesions in rat brain Science 219: 316–318. [DOI] [PubMed] [Google Scholar]
- Schwarcz R., Okuno E., White R.J., Bird E.D., Whetsell Jr. W.O. 1988. 3-Hydroxyanthranilate oxygenase activity is increased in the brains of Huntington disease victims Proc. Nat. Acad. Sci. 85: 4079– 4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegbahn P.E.M. and Haeffner F. 2004. Mechanism for catechol ring-cleavage by non-heme iron extradiol dioxygenases J. Am. Chem. Soc. 126: 8919–8932. [DOI] [PubMed] [Google Scholar]
- Solomon E.I., Brunold T.C., Davis M.I., Kemsley J.N., Lee S.K., Lehnert N., Neese F., Skulan A.J., Yang Y.S., Zhou J. 2000. Geometric and electronic structure/function correlations in non-heme iron enzymes Chem. Rev. 100: 235–349. [DOI] [PubMed] [Google Scholar]
- Spence E.L., Kawamukai M., Sanvoisin J., Braven H., Bugg T.D.H. 1996. Catechol dioxygenases from Escherichia coli (MhpB) and Alcaligenes eutrophus (MpcI): Sequence analysis and biochemical properties of a third family of extradiol dioxygenases J. Bacteriol. 178: 5249–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone T.W. and Darlington L.G. 2002. Endogenous kynurenines as targets for drug discovery and development Nat. Rev. Drug Discov. 1: 609–620. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus G.P., Mueller H.A., Burgner J., Córdoba S.R.D., Peñalva M.A., Timm D.E. 2000. Crystal structure of human homogentisate dioxygenase Nat. Struct. Biol. 7: 542–546. [DOI] [PubMed] [Google Scholar]
- Vaillancourt F.H., Barbosa C.J., Spiro T.G., Bolin J.T., Blades M.W., Turner R.F., Eltis L.D. 2002. Definitive evidence for monoanionic binding of 2,3-dihydroxybiphenyl to 2,3-dihydroxybiphenyl 1,2-dioxygenase from UV resonance Raman spectroscopy, UV/Vis absorption spectroscopy, and crystallography J. Am. Chem. Soc. 124: 2485–2496. [DOI] [PubMed] [Google Scholar]
- Vetting M.W., Wackett Jr. L.P., Que L., Lipscomb J.D., Ohlendorf D.H. 2004. Crystallographic comparison of manganese-and iron-dependent homoprotocatechuate 2,3-dioxygenases J. Bacteriol. 186: 1945–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viggiani A., Siani L., Notomista E., Birolo L., Pucci P., Donato A.D. 2004. The role of the conserved residues His-246, His-199, and Tyr-255 in the catalysis of catechol 2,3-dioxygenase from Pseudomonas stutzeri OX1 J. Biol. Chem. 279: 48630–48639. [DOI] [PubMed] [Google Scholar]
- Woo E.J., Dunwell J.M., Goodenough P.W., Marvier A.C., Pickersgill R.W. 2000. Germin is a manganese containing homohexamer with oxalate oxidase and superoxide dismutase activities Nat. Struct. Biol. 7: 1036–1040. [DOI] [PubMed] [Google Scholar]
- Woo E.J., Marshall J., Bauly J., Chen J.G., Venis M., Napier R.M., Pickersgill R.W. 2002. Crystal structure of auxin-binding protein 1 in complex with auxin EMBO J. 21: 2877–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Colabroy K.L., Begley T.P., Ealick S.E. 2005. Structural studies on 3-hydroxyanthranilate-3,4-dioxygenase: The catalytic mechanism of a complex oxidation involved in NAD biosynthesis Biochemistry 44: 7632–7643. [DOI] [PubMed] [Google Scholar]