

Abstract
The nitric oxide molecule (NO) is involved in many important physiological processes and seems to be stabilized by reduced thiol species, such as S-nitrosoglutathione (GSNO). GSNO binds strongly to glutathione transferases, a major superfamily of detoxifying enzymes. We have determined the crystal structure of GSNO bound to dimeric human glutathione transferase P1-1 (hGSTP1-1) at 1.4 Å resolution. The GSNO ligand binds in the active site with the nitrosyl moiety involved in multiple interactions with the protein. Isothermal titration calorimetry and differential scanning calorimetry (DSC) have been used to characterize the interaction of GSNO with the enzyme. The binding of GSNO to wild-type hGSTP1-1 induces a negative cooperativity with a kinetic process concomitant to the binding process occurring at more physiological temperatures. GSNO inhibits wild-type enzyme competitively at lower temperatures but covalently at higher temperatures, presumably by S-nitrosylation of a sulfhydryl group. The C47S mutation removes the covalent modification potential of the enzyme by GSNO. These results are consistent with a model in which the flexible helix α2 of hGST P1-1 must move sufficiently to allow chemical modification of Cys47. In contrast to wild-type enzyme, the C47S mutation induces a positive cooperativity toward GSNO binding. The DSC results show that the thermal stability of the mutant is slightly higher than wild type, consistent with helix α2 forming new interactions with the other subunit. All these results suggest that Cys47 plays a key role in intersubunit cooperativity and that under certain pathological conditions S-nitrosylation of Cys47 by GSNO is a likely physiological scenario.
Keywords: calorimetry, glutathione S-transferase, nitric oxide, nitrosoglutathione, X-ray crystallography
Glutathione transferases (EC 2.5.1.18; GSTs) are a superfamily of enzymes involved in the biotransformation of numerous carcinogenic, mutagenic, toxic, and pharmacologically active compounds (Jakoby and Habig 1980). The human cytosolic GSTs are dimeric proteins grouped into at least eight species-independent classes (α, κ, μ, π, ω, σ, θ, and ζ) on the basis of their amino acid sequence, substrate specificity, and immunological properties (Mannervik et al. 1985; Meyer et al. 1991; Buetler and Eaton 1992; Ji et al. 1995; Board et al. 2000). Human glutathione transferase P1-1 (hGSTP1-1) (Mannervik et al. 2005), a homodimeric protein of ∼46 kDa, has been extensively studied for its potential use as a marker during chemical carcinogenesis (Kano et al. 1987; Tsuchida et al. 1989) and its possible role in the mechanism of cellular multidrug resistance against a number of anti-neoplastic agents (Black et al. 1990).
The main structural differences among GST classes are located in the GSH binding site (G-site) and, in particular, the helix α2 region, and in the hydrophobic substrate binding site (H-site). Kinetic studies have revealed the importance of helix α2 to the function of hGSTP1-1 (Caccuri et al. 1996; Ricci et al. 1996). The crystal structures suggest that helix α2 and its flanking regions (residues 35–51) are the prime candidates for the flexible regions that are known to influence catalysis. A number of residues in this region (Trp38, Lys44, and Gln51) participate in the binding of GSH. Moreover, some residues, such as Cys47 and Tyr49, appear to participate in intersubunit communication (Reinemer et al. 1992; Oakley et al. 1998). We have initiated a program of structural and thermodynamic studies to evaluate the relative importance of residues in helix α2 and its flanking regions in the binding of ligands of physiological importance to hGSTP1-1 (Ortiz-Salmerón et al. 2003).
Although many of the physiological roles and functions of GSTs are well known, there are certain aspects still unclear, such as their involvement in the complex mechanisms of nitric oxide (NO) detoxification or storage. NO is an important signaling molecule in both physiological and pathological processes. In the central nervous system, NO is a neuronal messenger and is possibly responsible for the development of various diseases (Moncada 1994; Rubanyi 1998). Frequently, NO is transported through the cell attached to low molecular weight compounds possessing thiol groups such as GSH (Hogg et al. 1996; Kluge et al. 1997). S-nitrosothiols, such as S-nitrosoglutathione (GSNO), may function as storage forms of NO in vivo, and may participate in transnitrosylation reactions. It is becoming increasingly evident that S-nitrosylation can modify protein function through alterations in the behavior of sulfhydryl groups.
The cysteine residues of hGSTP1-1 (four per subunit) are not a structural part of the active site, and their nonessential character has also been demonstrated by site-directed mutagenesis of the rat and human class π isoenzymes and the human class μ enzyme (Kong et al. 1991; Tamai et al. 1991; Widersten et al. 1991). However, there is a highly reactive and conserved cysteine residue (Cys47) in the π class isoenzymes, situated close to the G-site (10.7 Å from the sulfur atom of GSH), whose structural integrity seems to be crucial in maintaining an active conformation of the enzyme (Reinemer et al. 1992). Cys47 is located at the C-terminal end of helix α2 and sits in a hydrophobic pocket defined by residues Trp38, Leu43, Leu52, and Tyr63, and the aliphatic portion of Lys54. Although Cys47 is not involved directly in either substrate binding or catalysis (Reinemer et al. 1991), chemical modification of its side chain inactivates the enzyme reversibly, possibly by blocking a structural transition during catalysis (Widersten et al. 1991) or by inducing an unfavorable conformational change for GSH binding (Ricci et al. 1991, 1995; Tamai et al. 1991; Lo Bello et al. 1995; Ji et al. 2002). S-nitrosylation of human GSTP1-1 at Cys47 by GSNO has been demonstrated (Lo Bello et al. 2001), suggesting an important physiological role for GSNO regulation of GST activity.
Here we report a structural and thermodynamic study of the effect of GSNO binding on wild-type enzyme (wt-hGSTP1-1) and a C47S mutant. The crystal structure of GSNO bound to hGSTP1-1 shows that the GSNO ligand binds in the active site with the nitrosyl moiety involved in multiple interactions with the enzyme. The isothermal calorimetry studies demonstrate that either positive or negative cooperativity regulate GSNO binding to mutant and wild-type enzyme, respectively. Differential scanning calorimetry (DSC) demonstrates that thermal denaturation of both enzymes is irreversible, with the thermal stability for the mutant being slightly higher that that for wt-hGSTP1-1. These results highlight the importance of helix α2 and, in particular, Cys47 in intersubunit communications. Furthermore, GSNO is shown to be capable of regulating GST activity in various ways.
Results
Crystal structures of the complex GSNO–hGSTP1-1
Normally hGSTP1-1 only crystallizes in the presence of significant amounts of DTT. However, we were concerned that the presence of the reducing agent would inhibit any covalent modification of protein cysteinyl residues with GSNO. We were fortunate to obtain crystals of the enzyme GSNO complex in the presence and absence of DTT. However, there was no evidence in the electron density maps of either structure for covalent modification of any of the cysteine residues. Indeed, superposition of the α-carbon atoms of the GSNO complex structures, DTT and non-DDT, demonstrates they are essentially identical with a root mean square deviation (RMSD) of 0.2 Å for the 416 residues included in the calculation. The GSNO structures also superimpose closely with the previously published hGSTP1-1–GSH complex (5GSS) yielding a RMSD of ∼0.3 Å, indicating the structures are virtually identical. The electron density clearly showed that the cystienyl moiety of GSH was nitrosylated in both GSNO structures (Fig. 1). The density for the DTT structure revealed three possible conformations of the nitroso group, which could be modeled with ∼33% occupancy for each. In one conformation the NO moiety is stabilized by water-mediated interactions with Arg13 and Tyr108. In the second conformation the NO moiety is stabilized by direct interactions with Arg13 (side chain) and Tyr7. In the third conformation the NO moiety is stabilized by direct interactions with Arg13 (main chain) and Tyr7.
Figure 1.

Stereo diagram of the 2Fo – Fc electron density map of the hGSTP1-1–GSNO complex at 1.4 Å resolution (contoured at the 1σ level). Only one conformer of the nitroso moiety is shown for clarity.
Isothermal Titration Calorimetry (ITC)
Thermodynamics of GSNO binding to wild-type enzyme
Figure 2 shows a typical ITC profile for the binding of GSNO to dimeric wt-hGSTP1-1 in phosphate buffer (Buffer A) at pH 7.0 and 25.1°C. Identical experiments were carried out at 16.3°C and 20.4°C. Control experiments that involve the same type of injections of GSNO solution into the same buffer were also carried out in order to measure the heat of dilution. A noncooperative model is unable to fit the calorimetric data appropriately. However, a model of two equal and interacting sites is able to fit all the calorimetric data. The binding equilibrium constant value for the first site, K1, is ∼2 orders of magnitude higher than that for the second site, K2 (Table 1). Notwithstanding, it is interesting to observe the calorimetric traces obtained at temperatures >25°C (for example, at 35.1°C) (Fig. 3). Closer examination of the calorimetric traces at this temperature revealed some important features. Firstly, the calorimetric response after each injection of GSNO shows an initial exothermic phase followed by an endothermic phase. Moreover, it is also worth noting that each injection includes ligand dilution, which always occurs in any binding experiment and is evaluated in a separate experiment (Fig. 3a). Secondly, the response time for the exothermic initial phase is smaller than that obtained for the endothermic phase and comparable to the response time in the dilution experiment. Therefore, the presence of these two phases might reveal the occurrence of two separable steps in the binding reaction. The first process (fast) could be the binding of GSNO to the protein and the second one could correspond to a chemical reaction (slow) that occurs concomitant to the binding process. As a result of these observations, Scheme 1 is suggested.
Figure 2.
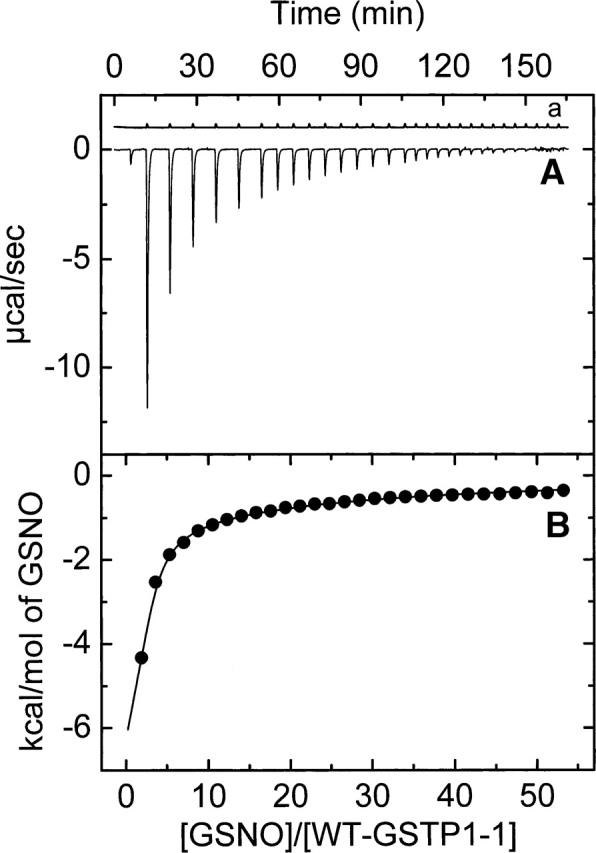
Representative isothermal titration calorimetry measurements for the binding of GSNO to wt-hGSTP1-1 at 25.1°C. Titrations were performed in 20 mM sodium phosphate (pH 7.0), 5 mM NaCl, and 0.1 mM EDTA buffer. A shows the raw data for the titration of enzyme (43.66 μM) with 29 8-μL injections of GSNO (12.7 mM). A preinjection of 1 μL was performed at the beginning. (a) GSNO dilution experiment. The area under each peak was integrated and plotted in B against the molar ratio GSNO/enzyme inside the cell. The solid smooth line represents the best fit of the experimental data to a model of two equal and interacting sites (negative cooperativity).
Table 1.
Thermodynamic parameters for the GSNO binding to wt-hGST P1-1 at pH 7.0 in sodium phosphate buffer (Buffer A)

Figure 3.

ITC data for the binding of GSNO to wt-hGSTP1-1 at 35.1°C. Titrations were performed in 20 mM sodium phosphate (pH 7.0), 5 mM NaCl, and 0.1 mM EDTA buffer. Raw data for the titration of enzyme (39.46 μM) with 29 8-μL injections of GSNO (13.95 mM). A preinjection of 1 μL was performed at the beginning. (a) GSNO dilution experiment.
Scheme 1.

Thus, in Step 1 (exothermic), the protein binds reversibly GSNO and occludes the G-site in a negative cooperative manner. This step is common for all temperatures. In Step 2, the initial complex rearranges in such a way that the Cys47 gets nitrosylated. This step is irreversible, presumably due to the covalent attachment of the NO moiety of GSNO to Cys47. In order to examine if the endothermic phase was a chemical reaction or a random artifact, we performed a series of ITC experiments varying either the GSNO concentration or the protein concentration (i.e., 16.89 μM, 39.46 μM, and 73.87 μM). An increase in the protein concentration increases the extension of this additional chemical process. These results rule out both the existence of a random artifact and the existence of a chemical process associated with the GSNO binding. Moreover, an exhaustive dialysis in phosphate buffer of the GSNO titrated wt-hGSTP1-1 in a calorimetric experiment at 35°C (T > 25°C) was unable to restore the ligand-free enzyme. However, if DTT or 2-mercaptoethanol (2 mM) is included in the dialysis buffer solution, the protein releases the inhibitor and is able to bind GSNO again. These results demonstrate the existence of a covalent and reversible chemical modification of the wild-type enzyme by GSNO. However, when these experiments are carried out with either the wild-type enzyme titrated in a calorimetric experiment at 20°C (T < 25°C) or the C47S mutant titrated at 35°C, the protein releases the inhibitor by dialysis in phosphate buffer without the need of a reducing agent.
We have performed a deconvolution procedure for the calorimetric traces obtained at temperatures >25°C in order to split the two processes and to obtain the kinetic and thermodynamic parameters for this association, according to the model described in Scheme 1. For this purpose, the calorimetric peaks for each injection were isolated and analyzed as described in the Supplemental Material.
After the deconvolution, the resulting binding thermograms were analyzed the same way as those obtained at less than 25°C. The binding thermodynamic parameters at all studied temperatures are shown in Table 1. The observed enthalpy changes for both the first (ΔH1) and the second (ΔH2) sites are negative in every case. An enthalpic–entropic compensation for the GSNO binding to wild-type enzyme was found for both sites in the studied temperature range. The main contribution to the deduced negative cooperativity for binding of GSNO is entropic in origin. A practically linear dependence was deduced for the global thermodynamic parameters [i.e., ΔHg = ΔH1 + ΔH2; ΔG°g = ΔG°1 + ΔG°2; TΔS°g = T(ΔS°1 + ΔS°2)] (Fig. 4). A heat capacity change (ΔCp,g° = −584.7 ± 181.6 cal·mol−1·K−1) was obtained from the slope of ΔHg versus temperature.
Figure 4.

Temperature dependence of the global thermodynamic parameters (ΔHg circles; −TΔS°g, squares; and ΔG°g, triangles) for the binding of GSNO to the C47S mutant and wild-type enzymes. The parameters are shown by hollowed and filled symbols for the C47S and the wild-type enzyme, respectively. The binding heat capacity changes were determined by linear repression analysis as the slopes of the plots of ΔHg vs. temperature.
Thermodynamics of GSNO binding to C47S-hGSTP1-1
A positive cooperativity between the two GSNO binding sites was deduced for the C47S mutant. A typical calorimetric thermogram for this mutant in Buffer A and 29.8°C is illustrated in Figure 5. This cooperativity was corroborated by examining the Hill coefficients, nH, which ranged from 1.31 to 1.37. Experiments identical to that shown in Figure 5 were performed in the temperature range of 13°–35°C. The results are shown in Table 2. A positive cooperativity was also found for the binding of substrate to the mutants C47S and C47A of hGSTP1-1 (Lo Bello et al. 1995; Ricci et al. 1995). These results are consistent with the binding of GSNO in the first subunit inducing a favorable conformational change on the second subunit, which then displays an increased affinity for GSNO. The binding to the first site is enthalpically favorable and entropically unfavorable over the temperature range studied. On the contrary, the binding to the second site is entropically favorable and enthapically unfavorable (Table 2). Global enthalpy change, ΔHg, is practically linear with the temperature, resulting in a constant and negative heat capacity change (ΔCp,g° = −561.5 ± 98.7 cal·mol−1·K−1) (Fig. 4).
Figure 5.
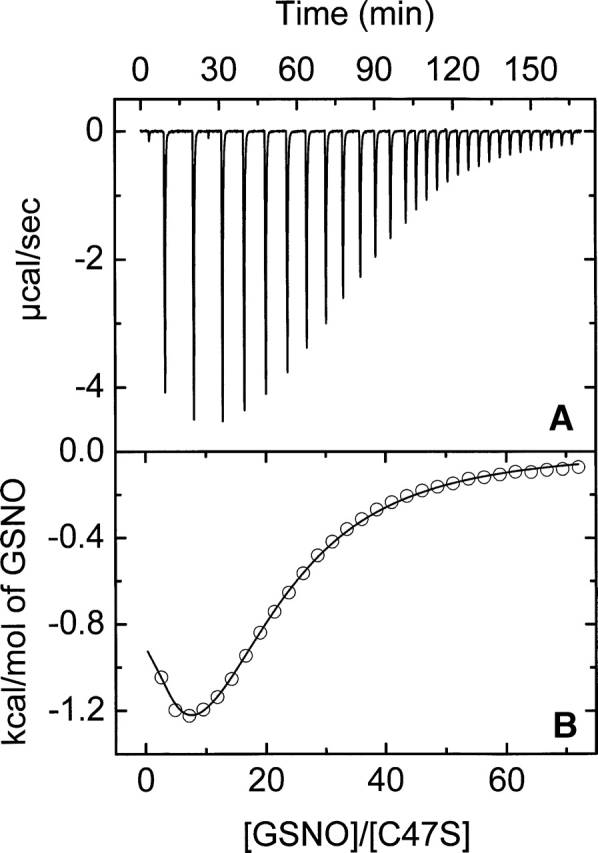
Representative isothermal titration calorimetry measurements for the binding of GSNO to C47S-hGSTP1-1 at 29.8°C. Titrations were performed in 20 mM sodium phosphate (pH 7.0), 5 mM NaCl, and 0.1 mM EDTA buffer. A shows raw data for the titration of 35.56 μM of mutant with 29 8-μL injections of GSNO (14.1 mM). A preinjection of 1 μL was performed at the beginning. The area under each peak in A was integrated and plotted in B against the molar ratio GSNO/mutant enzyme inside the cell. The solid smooth line represents the best fit of the experimental data to a model of two equal and interacting sites (positive cooperativity).
Table 2.
Thermodynamic parameters for the GSNO binding to C47S-hGST P1-1 at pH 7.0 in sodium phosphate buffer (Buffer A)

Differential scanning calorimetry
The thermal denaturation of wt-hGSTP1-1 and the C47S mutant was always irreversible (in the absence and the presence of GSNO ligand), as no transition could be detected in reheating runs (not even when the first run had been stopped immediately after the end of the second transition). No aggregation was observed in the samples extracted from the calorimetric cell.
Profiles of excess heat capacity versus T were obtained for scanning rates of 0.2–1.5 K·min−1. The position of the maximum on the profiles is shifted to higher temperatures as the scanning rate increases (Fig. 6). Such an effect verifies the irreversible character of thermal denaturation of the protein (Sánchez-Ruiz and Mateo 1987; Freire et al. 1990) and therefore a kinetic control for this process. Due to the irreversibility of the thermal denaturation of hGST P1-1, thermodynamic parameters cannot be obtained. Nevertheless, it is possible to obtain some information related to the irreversible process. Therefore, to analyze the profiles obtained by DSC we used a model of irreversible denaturation for oligomeric proteins (Sánchez-Ruiz 1992; Lyubarev and Kurganov 1998). The averaged Ea values over all scan rates examined (0.2, 0.5, 0.83, 1.16, 1.33, 1.50 K/min) were 159.7 kcal/mol and 160.2 kcal/mol for the C47S mutant and wild-type enzymes, respectively. Moreover, the activation energy obtained by Methods II (Fig. 7) and III were 161.3 and 170.4 kcal/mol (Method II) and 150.2 and 167.3 (Method III) for the C47S mutant and wild-type enzymes, respectively. Consequently, the activation energy values averaged over the results obtained from the three methods are 157.1 kcal/mol and 166 kcal/mol for the C47S mutant and the wild-type enzyme, respectively. These values are the same within experimental error. Thus, if it is assumed that a similar thermal denaturation mechanism occurs in both enzymes, the energetic barrier will be also the same (∼160 kcal/mol).
Figure 6.

Differential scanning calorimetry transitions for the C47S mutant thermal denaturation in Buffer B, at six different scan rates: 0.2 (a), 0.5 (b), 0.83 (c), 1.16 (d), 1.33 (e), and 1.5 K/min (f). In every case, the total protein concentration is 1.12 mg/mL. DSC transitions were corrected for the instrumental and chemical baselines.
Figure 7.

Plot of ln(v/T2m,i) vs. 1/Tm,i (i = 1, 2) (Method II). wt- and C47S-hGSTP1-1 data are shown as circles and squares, respectively. Filled and open forms represent the first (Tm,1) and the second (Tm,2) transitions, respectively (Tm,1 < Tm,2). (For details, see Materials and Methods.)
Thermal denaturation pathway for hGSTP1-1
The thermal unfolding for the wt-hGSTP1-1 and the C47S mutant has turned out to be completely irreversible under the examined conditions. Two partially overlapping transitions (centered at ∼55°C and 57°C) are apparent in the thermogram for dimeric wt-hGSTP1-1 (Fig. 8). As mentioned above, both transitions are irreversible. The overall unfolding pathway for a homodimeric protein must begin with a folded dimer (N2) and end with two denatured monomers (2D). This can be achieved in several ways, three of which are described below as examples:
Figure 8.

Differential scanning calorimetry transitions for the thermal denaturation of wt-hGSTP1-1 (filled symbols) and C47S mutant (open symbols) in Buffer B (10 mM HEPES-NaOH at pH 7.5) at 1.5 K/min. DSC transitions were corrected for the instrumental and chemical baselines. The solid lines represent the best two-transition three-state irreversible model according to Method I. (For details, see Materials and Methods.)
Model 1: N2 → 2D
Model 2: N2 → 2I → 2D
Model 3: N2 → I2 → 2D
The presence of two transitions (biphasic unfolding) for wt-hGSTP1-1 indicates that a simple two-state unfolding process (Model 1) involving only native dimer and unfolded monomer was not applicable. However, Models 2 and 3 could both be valid (Sánchez-Ruiz 1992; Lyubarev and Kurganov 1998). The Tm dependence in Cp curve with protein concentration can be used as a definitive diagnostic tool to distinguish between Models 2 and 3. That is, only the bimolecular dissociation steps (the first and second steps in Models 2 and 3, respectively) should be protein concentration–dependent. The first unfolding transition from the native dimer to a monomeric intermediate was confirmed by its protein concentration dependence, indicating the presence of a dissociation process (Takahashi and Sturtevant 1981; Sánchez-Ruiz 1992). The second unfolding transition is concentration-independent. Thus, our results suggest that Model 2 is suitable to describe the wt-hGSTP1-1 thermal unfolding mechanism.
In the case of C47S–hGSTP1-1, a small second transition centered at 58°C was observed in the thermograms obtained at low rate scanning (Fig. 6). This second transition is also scan-rate-dependent but virtually unobservable as the scan rate is increased. By comparing the behavior of C47S–hGSTP1-1 to that obtained for wt-hGSTP1-1 (Fig. 8) we can deduce the following: (1) The C47S mutant is more stable than the wild-type enzyme (Tm1 and Tm2 are ∼2° higher for C47S-hGSTP1-1); (2) a model with two irreversible steps is able to fit satisfactorily the DSC thermograms for the wt-hGSTP1-1 and C47S mutant enzymes, indicating a similar thermal unfolding pathway (Fig. 8).
Influence of GSNO in DSC transitions
The presence of GSNO in the DSC profiles for the C47S mutant and wild-type enzymes has been examined using a scan rate of 1.5 K/min. The C47S mutant DSC thermogram in the presence of 2.08 mM GSNO is similar to that obtained in its absence. However, a very surprising result from the present studies is the wt-hGSTP1-1 thermogram shape change in the presence of GSNO (2.13 mM). In this case, only one transition is obtained, instead of the two transitions arising in its absence (Fig. 9). As illustrated in Figure 9, the Tm is the same as that obtained for the first transition in the absence of GSNO. Thus, the presence of GSNO alters the hGSTP1-1 thermal denaturation pathway, changing it to a high cooperative two-state model where the folded dimer and the denatured monomer are highly populated (Fig. 9).
Figure 9.
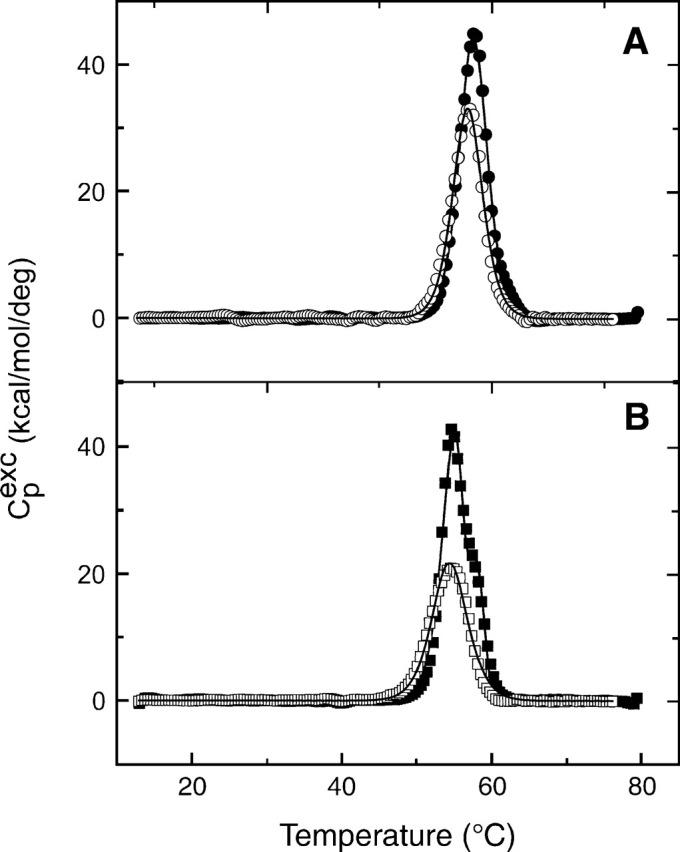
Excess heat capacity vs. temperature for the thermal denaturation of 1.1 mg/mL C47S-hGSTP1-1 (A) and 1.1 mg/mL wt-hGSTP1-1 (B) at 1.5 K/min. Filled and open symbols represent the experimental data in the absence and the presence of 2.1 mM GSNO, respectively. DSC transitions were corrected for the instrumental and chemical baselines. The solid lines represent the best one-transition non-two-state model. The cooperativity ratio ΔHcal/ΔHVH was ∼1 in all cases.
Discussion
ITC and crystallographic experiments
Structural and thermodynamic data reported in this article indicate that GSNO acts as a competitive inhibitor for hGSTP1-1, in concordance with the previous studies (Clark and Debnam 1988; Keese et al. 1997). However, Clark and Debnam (1988) reported that the inhibition mode of rat liver cytosolic GST by GSNO was competitive with respect to GSH, and that there was no evidence for a covalent modification of the enzyme. From our calorimetric thermograms obtained for the interaction of GSNO to wt-hGSTP1-1 at temperatures >25°C, two separable steps are visualized: a reversible binding of GSNO (exothermic phase, fast) and an irreversible reaction, possibly a covalent modification of the enzyme, which is corroborated by dialysis of titrated samples (see Results).
The function and chemical reactivity of cysteinyl residues in cytosolic GST has been extensively investigated (Kong et al. 1991; Tamai et al. 1991; Park et al. 2001). Thus, chemical modification and site-directed mutagenesis studies on hGSTP1-1 suggested that Cys47 and Cys101, the most reactive among the four cysteines in each subunit (Ricci et al. 1991; Lo Bello et al. 1995), are the likely targets of thiol reagents such as GSNO (Lo Bello et al. 1995, 2001; Ricci et al. 1995). Perhaps one possible reason for this high reactivity of Cys47 may rely on the particular position of this residue, situated at the end of the flexible helix α2, close to the G-site. Crystallographic analysis reveals that Cys47 is located on the surface, with its thiol group pointing into a small hydrophobic pocket formed by main chain atoms of Gln51 and Lys44 and the side chain atoms of Trp38 and Leu52. Previous studies suggest that residues such as Cys47 and Tyr49 (which interacts with helices α4 and α5 of the adjacent subunit) participate in intersubunit communication between active sites of the dimer (Reinemer et al. 1992; Oakley et al. 1998).
The results reported here indicate that GSNO inhibits hGSTP1-1 both competitively and through a covalent modification, presumably by S-nitrosylation of a sulfhydryl group in the enzyme. Since the thermogram peaks for the binding of this ligand to the C47S mutant did not show the biphasic effect observable for wt-hGSTP1-1, probably only Cys47 acts as target for GSNO. On the basis of these observations Scheme 1 was postulated. This S-nitrosylation reaction is only observable at temperatures >25°C. The kinetic constant corresponding to this process increases with increasing temperature, resulting in an activation energy of 11.64 ± 1.23 kcal/mol (Supplemental Material). Thus, only when the molecules have sufficient energy to overtake this energetic barrier does S-nitrosylation take place.
However, no kinetic effect can be seen in the calorimetric thermograms at temperatures <25°C. As a result of these and other observations the following can be inferred: (1) GSNO binds competitively to wt-hGSTP1-1 at temperatures <25°C, without appreciable covalent modification of the enzyme; (2) a covalent binding of GSNO to wt-hGSTP1-1 takes place at temperatures >25°C, presumably by S-nitrosylation of the highly reactive Cys47 sulfhydryl group. Thus, the reason that Cys47 did not appear covalently modified in the crystals of the non-DTT structure may be the temperature at which these crystals were obtained.
Moreover, the competitive binding to the G-site or the covalent modification of a cysteinyl residue located nearby (probably Cys47) by GSNO reported here can be explained on the basis of previous studies on GSTP1-1 modification by alkylating reagents. The enzyme adopts at least two different conformations, depending on the presence or the absence of GSH, because of the mobile α2 loop lining the G-site. In the first case, the Cys47 side chain is buried into a hydrophobic pocket and not accessible to the solvent and GSNO can act as a competitive inhibitor of the natural substrate. In the absence of GSH, the mobile α2 loop is more flexible and Cys47 becomes more accessible to the solvent and acts as a preferential target of S-nitrosylation by GSNO. The temperatures used in this work (13°–41°C) have enabled both scenarios to be observed. The effects observed at 37°C are likely to be the more physiological relevant scenario where the GSNO molecule acts as a nitrosant agent toward GSTP1-1 inside the cell but could only be relevant in certain pathological conditions where intracellular GSH depletion occurs. Under healthy conditions the enzyme would have its active sites fully occupied by GSH, and thus Cys47 would be not accessible to the solvent. However, in the presence of large amounts of GSNO and small quantities of iron, a different type of nitrosylation can occur, as suggested previously by Lo Bello et al. (2001) and confirmed now by recent studies in vitro and in vivo (Cesareo et al. 2005).
From the binding process of GSNO to wt-hGSTP1-1 at all studied temperatures, a negative cooperativity is deduced. However, a positive cooperativity was found for the binding of this inhibitor to the C47S mutant. Some studies reported in literature (Lo Bello et al. 1993; Ricci et al. 1995) indicated the importance of an electrostatic interaction (Cys47− Lys54+) for anchoring the flexible helix α2 and shaping the correct special arrangement for the binding of substrate in the active site. This ion pair might act as a “hinge” that limits the number of potential conformations. When this ion pair is disrupted by mutation of either residue the flexibility of this region could be greatly increased, causing helix α2 to contact the other subunit to generate a structural communication, which could be the basis for the observed positive cooperativity in the mutant C47S. A similar behavior was observed in the binding of GSH to this mutant (Lo Bello et al. 1995). Moreover, the GSNO complex structure reported here shows that GSNO binds to the G-site in a fashion identical to the way GSH binds to the enzyme (Oakley et al. 1997). The nitroso moiety adopts three alternative conformations in the DTT structure, each of them stabilized, in part, by interactions with Arg13 and/or one of the active site tyrosines. These results reinforce the role of GSNO as a competitive inhibitor.
The global binding enthalpy change values for both enzymes are negative at all studied temperatures and are very similar to each other (Fig. 4). Therefore, although the GSNO binding process to both enzymes is different, the global thermodynamic parameters are analogous.
The global thermodynamic parameters reported here indicate that the mutation leads to increased changes in both negative enthalpy and negative entropy (Table 3; Fig. 4). The binding Gibbs energy increases as a consequence of the mutation, and so the affinity of GSNO for C47S is lower than that for the wild-type enzyme. These results suggest that although the interaction between the C47S mutant and the GSNO is enthalpically more favorable (albeit slightly) than that for the wild-type enzyme, the entropic loss due to binding is also increased, indicating the mutation is enthalpically favorable but entropically unfavorable (Fig. 4). The unfavorable entropy change outweighs the slight enthalpic advantage, resulting in an affinity lower for the binding of GSNO to the C47S mutant (Table 3). The temperature dependence of global ΔH and −TΔS° for the GSNO–wt-hGSTP1-1 binding exhibits an enthalpic–entropic compensation, and as a consequence, ΔG° remains relatively constant with temperature. A similar behavior was observed in the binding of S-hexylglutathione to the Y49F mutant (Ortiz-Salmerón et al. 2003). Moreover, the global ΔCp° values for wild-type and C47S mutant are similar. Therefore, even though the GSNO binding processes for both enzymes are different, as demonstrated by the thermodynamic properties for each site, their global values are almost the same, suggesting that the final and initial states are very similar in both cases. The very close similarity between the C47S and wt-hGSTP1-1 crystal structures further supports this idea (A.J. Oakley, M. Lo Bello, and M.W. Parker, unpubl.).
Table 3.
Global thermodynamic parameters for the GSNO binding to the hGST P1-1 C47S mutant and wild-type enzymes, at 29.8°C and pH 7.0

Experimental conditions are given in Materials and Methods. ΔG°, ΔH, and TΔS° are change in Gibbs energy, binding enthalpy, and entropy, respectively. ΔΔG°, ΔΔH and Δ(TΔS°) are the differences in each of the values from those of wt-hGST P1-1.
DSC experiments
We have shown that the DSC thermograms for the thermal denaturation of wt-hGSTP1-1 and C47S mutant can be interpreted in terms of a kinetic process with two steps (three states; Scheme 2, below). This model predicts that (1) the calorimetric traces should be highly dependent on the scan rate, and (2) two transitions should be apparent in the dimeric hGSTP1-1 thermogram, as is the case for hGSTP1-1 (wild-type and C47S mutant). A study varying the protein concentration has allowed us to demonstrate that the first step in Scheme 2 corresponds to dimer dissociation into a monomeric intermediate. However, since this concentration dependence is very slight in the protein concentration range 0.4–3.5 mg/mL, we may conclude that association of monomers does not contribute significantly to the kinetic behavior of the hGSTP1-1 thermal denaturation. Therefore, our results indicate that the homodimeric hGSTP1-1 denaturation pathway is a multistep process. A two-step mechanism was also deduced for chemical unfolding of wt-hGSTP1-1 (Aceto et al. 1992). However, no information is available about the thermal unfolding pathway for this enzyme. Assuming a high homology in the sequence and structure of all GSTs within the same class (π for h-GSTP1-1), a similar unfolding pathway should be expected for class π porcine lung GST. However, in this last case, a simple reversible two-state mechanism (N2 ↔ 2D) was obtained using urea and guanidinium chloride as chemical denaturants (Dirr and Reinemer 1991; Erhardt and Dirr 1995), in contrast with our own and previous results (Aceto et al. 1992). Equally, highly cooperative and concerted two-state models have been proposed for class α and Sj26 GSTs (Kaplan et al. 1997; Wallace et al. 1998). However, a mechanism with two steps (three states), similar to that observed for hGSTP1-1, was demonstrated for a class μ-GSTs (GSTM1-1 and GSTM2-2) (Hornby et al. 2000). Different multistep pathways have been reported in the literature for the unfolding/refolding of μ- and σ-GST using chemical denaturants (Stevens et al. 1998; Hornby et al. 2000). Therefore, an unfolding/refolding common pathway for all GST classes is not possible. What's more, even for GSTs within the same class, different mechanisms are possible, as happens to be the case for some π-class GSTs. Finally, it is also worth noting that the chemical unfolding mechanism for a protein can be different from the thermal unfolding mechanism (Dirr and Reinemer 1991; Erhardt and Dirr 1995; this work).
Scheme 2.

By comparing the Tm values for the two transitions (Fig. 8) we conclude that the thermal stability of the C47S mutant is slightly higher than that for the wild-type enzyme. Thus this mutation generates an enzyme with an increased stability. A reason for this could be higher flexibility in helix α2 facilitating a structural communication between the subunits as a consequence of this mutation.
The presence of GSNO hardly changes the temperature values corresponding to the maximum of the heat capacity curve, Tm, for both enzymes. However, its presence seems to change the shape, the height, and the area (enthalpy change) of the thermogram, mainly for the wild-type enzyme (Fig. 9). Therefore, the thermal unfolding mechanism for the wild-type enzyme in the presence of GSNO may be different from that in the absence of this inhibitor. A possible explanation for this different behavior could be the existence of S-nitrosylation of Cys47. Since a simple transition that is nearly symmetric and highly cooperative is observed (ΔHcal/ΔHVH ≈ 1), it is expected that the binding of GSNO renders a more flexible complex than the free wild-type enzyme. Assuming the existence of an electrostatic interaction between Cys47 and Lys54 that decreases the flexibility of helix α2 and therefore the communication between the subunits (Oakley et al. 1998), the binding and nitrosylation of Cys47 removes this electrostatic restriction, favoring mobility and flexibility and, consequently, communication between the subunits.
Concluding remarks
The crystallographic results provide a molecular basis for understanding the competitive inhibitory behavior of GSNO toward hGST P1-1. The calorimetry results show that Cys47, a residue that forms part of the highly flexible helix α2 region, plays an important role in intersubunit communications. At low temperatures helix α2 is not sufficiently mobile to allow S-nitrosylation of Cys47, and thus GSNO acts as a competitive inhibitor. At more physiological temperatures helix α2 is sufficiently mobile to allow chemical modification of Cys47, and such modification can inactivate the enzyme by producing an altered G-site since helix α2 forms one of the walls. Thus GSNO is capable of regulating GST activity in a number of ways.
Materials and methods
GSNO synthesis
GSNO was prepared by a modification of the original published procedure (Hart 1985; Cavero et al. 2000; Lo Bello et al. 2001). A solution with 3 mL of equimolar GSH and NaNO2 (33 mM) was vortexed and placed on ice for 1 h. To this solution 45 μL of 11 M HCl was added and vortexed, and the solution again was placed on ice for a further 2 h. A 40-μL portion of 10 M NaOH was then added to neutralize the solution. After, 4 mL of acetone were added and the GSNO sank to the bottom of the tube as a pink oil. The upper layer of acetone/H2O was aspirated off. GSNO was resuspended in 500 μL H2O and again 4 mL of acetone was added. The solution was mixed and then left to sit for 1 h until the GSNO oil sank to the bottom. The upper solution was again aspirated away. The resulting pink oil was frozen to −196°C and placed on a freeze dryer for 4 h. The resulting pink solid (yield of ∼300 mg) had a UV spectrum consistent with published reports (Hart 1985; Hogg et al. 1996). The compound was wrapped in foil to exclude light and stored at 4°C. The final concentration was calculated from absorbance at 332 nm using the extinction coefficient 750 M−1·cm−1.
Expression plasmids and site-directed mutagenesis
The plasmid pGST-1, producing large amounts of recombinant wild-type GST P1-1 in the cytoplasm of Escherichia coli, has been described previously (Battistoni et al. 1995). The expression plasmid p18Seq-1, reported previously (Lo Bello et al. 1995), was used to generate the single-stranded DNA template to be used for site-directed mutagenesis of Cys47, according to the method described by Kunkel and coworkers (Kunkel et al. 1991) with minor modifications.
Protein expression and purification
Native and mutant GST P1-1 enzymes were produced as described previously (Battistoni et al. 1995; Lo Bello et al. 1995). Briefly, TOP 10 E. coli cells, harboring plasmid pGST-1 or plasmid expressing Cys47 mutant enzyme (pGST-A47), were grown in Luria Bertani medium containing 100 μg/mL ampicillin and 50 μg/mL streptomycin. The synthesis of GST was induced by the addition of 0.2 mM isopropyl-1-thio-β-galactopyranoside when the absorbance at 600 nm was 0.5. Eighteen hours after induction, cells were harvested by centrifugation and lysed as previously described (Battistoni et al. 1995). Native and GST mutant enzymes were purified by affinity chromatography on immobilized glutathione (Simons and Van der Jagt 1977). After affinity purification, the native and the mutant enzymes (C47S) were homogeneous as judged by SDS-PAGE (Laemmli 1970). Protein concentration was determined by the method of Lowry et al. (1951).
Solutions of wild-type and C47S mutant were prepared by dialysis of the enzyme against several changes of buffer solution at 4°C. Protein concentrations were determined from absorbance measurements at 280 nm using an extinction coefficient of 5.44 × 104 M−1·cm−1 for the dimer.
Crystallization
Wild-type hGSTP1-1 was crystallized using the hanging drop vapor diffusion method as described elsewhere (Oakley et al. 1997). Briefly, a 2-μL drop of protein (concentration of 6.7 mg/mL in 0.1 mM EDTA and 10 mM phosphate buffer at pH 7.0) was mixed with the same volume of reservoir buffer comprising 100 mM MES buffer (pH 5.5 or 6.0), 22% (w/v) polyethylene glycol (PEG) 8000, 20 mM CaCl2, in the presence of 10 mM DTT. Crystals grew at 22°C and reached a suitable size in ∼1 wk. The crystals were then transferred into 4-μL drops of a new reservoir solution containing 10 mM GSNO, 100 mM MES buffer (pH 6), 22% (w/v) PEG 8000, and 20 mM CaCl2. These crystals were left to soak for ∼1 wk. Alternatively, the above procedure was repeated in the absence of DTT with crystals grown in the presence of 10 mM GSNO (i.e., cocrystallized in the presence of GSNO).
Data collection and processing
The X-ray diffraction data were collected at the Advanced Photon Source (Chicago, Illinois), beam line 14-ID-B using a MAR165 CCD MARResearch detector. The wavelength was set to 0.99 Å. For cryoprotection the crystals were soaked for 2 min in the well solution containing 5% (v/v) methyl-2, 4-pentanediol (MPD), then dipped briefly in a solution of the well solution containing 10% (v/v) MPD. The crystals were then snap-frozen at 100 K in the cryostream. Diffraction data were processed and scaled with HKL (Otwinowski and Minor 1997). Data collection statistics are presented in Table 4. The crystals were shown to belong to a monoclinic lattice, with the space group C2, as seen previously for the wt-hGSTP1-1 (Oakley et al. 1997).
Table 4.
Data collection and processing for the GSNO complex

The values in parentheses are for the highest resolution bin.
aRmerge = ∑ hkl ∑i|Ii − <I>|/|<I>|, where Ii is the intensity for the ith measurement of an equivalent reflection with indices h,k,l.
Structure determination
Refinement of both GSNO structures (in the presence or the absence of DTT) began with the π class GST in the C2 space group (5GSS; Oakley et al. 1997) that had GSH and water molecules removed. Rigid body refinement in CNS (Brünger et al. 1998) was used to compensate for any possible changes in crystal packing. The models were then refined by a round of simulated annealing using CNS. As the asymmetric unit of the crystal contained two GST monomers, use was made of the noncrystallographic symmetry restraints on all nonhydrogen atoms in the initial rounds of refinement but were released in the later stages. The models were rebuilt with TURBO (A. Roussel and C. Cambillau, Silicon Graphics Inc.), and GSNO, MES, carbonate, calcium, and water molecules were added. The models were further refined with cycles of positional and isotropically restrained B-factor refinement. In the final rounds of refinement three different conformers of the NO moiety of GSNO and two alternative conformers of Cys101 were observed in the DTT data set. In the non-DTT data set two of three conformers of the GSNO ligand were observed. After multiple rounds of refinement and rebuilding the final R-factor was 19.7% (Rfree = 21.3%) for all data to 1.4 Å resolution for the DTT data set and 17.8% (Rfree = 20.8%) for all data to 1.7 Å resolution for the non-DTT data set. The stereochemistry was analyzed with the program PROCHECK (Laskowski et al. 1993) and gave values either similar or better than expected for structures refined at similar resolutions. A summary of the refinement statistics is given in Table 5.
Table 5.
Refinement statistics for the GSNO complex

Isothermal titration calorimetry
ITC experiments were carried out using an MCS titration microcalorimeter (Wiseman et al. 1989) (Microcal, Inc.). Ligand solutions were prepared in the buffer from the last dialysis change. ITC measurements were routinely performed in 20 mM sodium phosphate, 5 mM NaCl, 0.1 mM EDTA (pH 7) (Buffer A). In control experiments, under identical experimental conditions, the GSNO was injected into buffer. Raw data were collected, corrected for the ligand heats of dilution, and integrated using the Microcal Origin software supplied with the instrument. A two equal and interacting sites model was used to fit the data for both wt-hGSTP1-1 and the C47S mutant. The experimental data were fitted using Scientist software (Micromath Scientific Software) to the model algorithm implemented by us and using this model equation.
Differential scanning calorimetry
Measurements were performed on a MicroCal VP-DSC with cell volumes of 0.5 mL at the indicated scan rates. Prior to the DSC experiments, all samples were exhaustively dialyzed against 10 mM HEPES-NaOH (pH 7.5) (Buffer B). Calorimetric cells were kept under an excess pressure of 30 psi to prevent degassing during the scan. Protein concentrations were ∼12 μM per dimer GSTP1-1 in all experiments, except as otherwise indicated. The DSC curves were obtained by scanning the solutions of wt-hGSTP1-1 or C47S mutant in Buffer B at temperatures from 10°C to 80°C. Reversibility of the thermal transitions was checked by reheating the samples after fast cooling from the previous protein solution scan. Since thermal transitions were always found to be completely irreversible, the calorimetric traces were baseline-corrected by subtracting the sample reheating scans. The chemical baseline was subtracted according to the procedure of Takahashi and Sturtevant (1981). After baseline subtraction the excess molar heat capacity and the temperature were included as initial data in software Scientist (MicroMath Scientific Software). The DSC data were analyzed to a model including two consecutive irreversible steps (Lyubarev and Kurganov 1998) (Method I),
where k1 and k2 are the first-order kinetic constants of the corresponding reactions, the temperature dependence of which follows the Arrhenius equation
where Ai is the Arrhenius constant, Ea,i is the activation energy for each step, R is the universal gas constant, and T is the temperature in Kelvin.
The kinetic behavior of a system described by Scheme 2 is determined by the differential equations
 |
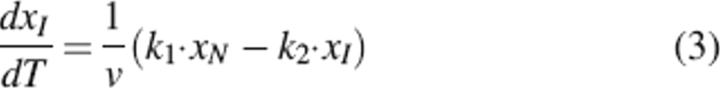 |
where xN and xI are the mole fractions of native and partially unfolded protein, respectively; v is the scanning rate; and T is the absolute temperature. Besides, xN + xI + xD = 1.
Conversely, the excess heat capacity is expressed by the equation
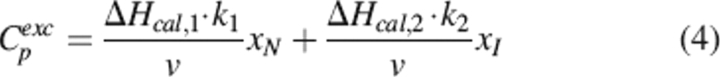 |
In the case when k1 > k2 (T2* > T1*, Ea,1 = Ea,2 = Ea), significant amounts of intermediate I are accumulated, making the profile of Cp versus T acquire a two-humped form (Lyubarev and Kurganov 1998). Using software Scientist and a mathematical algorithm implemented by us with equations 1–4, we were able to determine ΔHcal,1, ΔHcal,2, and Ea.
From the relationship v/T2m,i = (Ai·R/Ea,i)·e−Ea,i/RTm,i (Method II), one can obtain Ea,i from the linear regression of ln(v/Tm,i2) versus 1/Tm,i, where v is the scan rate and Tm,i is the transition temperature for each transition. Ea,i was also calculated from the equation Ea,i = eRT2m,iCmp,i/ΔHcal,i (Method III), where e = 2.7183 and Cp,im is the experimental heat capacity obtained at the maximum of the calorimetric curve for each transition.
The atomic coordinates for the GSNO (DDT) complex (code 2A2R) and GSNO (no DTT) complex (code 2A2S) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics (http://www.rscb.org/) at Rutgers University (New Brunswick, NJ).
Electronic supplemental material
Deconvolution and analysis procedure of the calorimetric traces can be found.
Acknowledgments
This work was supported in part by grant BIO2004-02112 from DGICYT, Ministerio de Ciencia y Tecnología (Spain), the Plan Andaluz de Investigación (CVI-290). M.L.B. was partially supported by MURST-Italy (COFIN 2004). This work was also supported by a grant from the Australian Research Council to M.W.P. L.J.P. is supported by a National Health and Medical Research Council of Australia (NHMRC) Dora Lush Scholarship, and M.W.P. is a NHMRC Senior Principal Research Fellow. We thank Harry Tong and other BioCARS staff for their help at the Advanced Photon Source. This work was supported by the Australian Synchrotron Research Program, which is funded by the Commonwealth of Australia under the Major National Research Facilities Program. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Energy Research.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Luis García-Fuentes, Department of Physical Chemistry, Biochemistry and Inorganic Chemistry, Faculty of Experimental Sciences, University of Almería, La Cañada de San Urbano, 04120 Almería, Spain; e-mail: lgarcia@ual.es; fax: +34-950-015008.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.052055206.
Abbreviations: DSC, differential scanning calorimetry; DTT, dithiothreitol; GSNO, S-nitrosoglutathione; GST, glutathione S-transferase; hGSTP1-1, human glutathione transferase P1-1; HEPES, N-(2-hydroxyethyl)-piperazine-N′-2-ethanesulfonic acid; ITC, isothermal titration calorimetry; MES, 2-morpholinoethanesulfonic acid; PEG, polyethylene glycol
References
- Aceto A., Caccuri A.M., Sacchetta P., Bucciarelli T., Dragani B., Rosato N., Federici G., Di Ilio C. 1992. Dissociation and unfolding of Pi-class glutathione transferase. Evidence for a monomeric inactive intermediate J 285 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistoni A., Mazzetti A.P., Petruzzelli R., Muramatsu M., Federici G., Ricci G., Lo Bello M. 1995. Cytoplasmic and periplasmic production of human placental glutathione transferase in Escherichia coli Protein Expr. Purif. 6 579–587. [DOI] [PubMed] [Google Scholar]
- Black S.M., Beggs J.D., Hayes J.D., Bartoszek A., Muramatsu M., Sakai M., Wolf C.R. 1990. Expression of human glutathione S-transferases in Saccharomyces cerevisiae confers resistance to the anticancer drugs adriamycin and chlorambucil Biochem. J. 268 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Board P.J., Coggan M., Chelvanayagam G., Easteal S., Jermiin L.S., Schulte G.K., Danley D.E., Hoth L.R., Griffor M.C., Kamath A.V.et al. 2000. Identification, characterization, and crystal structure of the Omega class glutathione transferases J. Biol. Chem. 275 24798–24806. [DOI] [PubMed] [Google Scholar]
- Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.-S., Kuszewski J., Nilges M., Pannu N.S.et al. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Buetler T.M. and Eaton D.L. 1992. Complementary DNA cloning, messenger RNA expression, and induction of alpha-class glutathione S-transferases in mouse tissues Cancer Res. 52 314–318. [PubMed] [Google Scholar]
- Caccuri A.M., Ascenzi P., Antonini G., Parker M.W., Oakley A.J., Chiessi E., Nuccetelli M., Battistoni A., Bellizia A., Ricci G. 1996. Structural flexibility modulates the activity of human glutathione transferase P1-1. Influence of a poor co-substrate on dynamics and kinetics of human glutathione transferase J. Biol. Chem. 271 16193–16198. [DOI] [PubMed] [Google Scholar]
- Cavero M., Hobbs A., Madge D., Motherwell W.B., Selwood D., Potier P. 2000. Synthesis and biological evaluation of enantiopure thionitrites: The solid-phase synthesis and nitrosation of D-glutathione as a molecular probe Bioorg. Med. Chem. Lett. 10 641–644. [DOI] [PubMed] [Google Scholar]
- Cesareo E., Parker L.J., Pedersen J.Z., Nuccetelli M., Mazzetti A.P., Pastore A., Federici G., Caccuri A.M., Ricci G., Adams J.J.et al. 2005. Nitrosylation of human glutathione transferase P1-1 with dinitrosyl diglutathionyl iron complex in vitro and in vivo. J. Biol. Chem. 280 42172–42180. [DOI] [PubMed] [Google Scholar]
- Clark A.G. and Debnam P. 1988. Inhibition of glutathione S-transferases from rat liver by S-nitroso-L-glutathione Biochem. Pharmacol. 37 3199–3201. [DOI] [PubMed] [Google Scholar]
- Dirr H.W. and Reinemer P. 1991. Equilibrium unfolding of class pi glutathione S-transferase Biochem. Biophys. Res. Commun. 180 294–300. [DOI] [PubMed] [Google Scholar]
- Erhardt J. and Dirr H. 1995. Native dimer stabilizes the subunit tertiary structure of porcine class pi glutathione S-transferase Eur. J. Biochem. 230 614–620. [PubMed] [Google Scholar]
- Freire E., van Osdol W.W., Mayorga O.L., Sánchez-Ruiz J.M. 1990. Calorimetrically determined dynamics of complex unfolding transitions in proteins Annu. Rev. Biophys. Biophys. Chem. 19 159–188. [DOI] [PubMed] [Google Scholar]
- Hart T.W. 1985. Some observations concerning the S-nitroso and S-phenylsulphonyl derivates of L-cysteine and glutathione Tetrahedron Lett. 26 2013–2016. [Google Scholar]
- Hogg N., Singh R.J., Kalyanaraman B. 1996. The role of glutathione in the transport and catabolism of nitric oxide FEBS Lett. 382 223–228. [DOI] [PubMed] [Google Scholar]
- Hornby J.A., Luo J.K., Stevens J.M., Wallace L.A., Kaplan W., Armstrong R.N., Dirr H.W. 2000. Equilibrium folding of dimeric class μ glutathione transferases involves a stable monomeric intermediate Biochemistry 39 12336–12344. [DOI] [PubMed] [Google Scholar]
- Jakoby W.B. and Habig W.H. 1980. Glutahione transferases In Enzymatic basis of detoxification (ed. Jacoby W.B.) . pp. 63–94. Academic Press, New York Vol. 2. [Google Scholar]
- Ji X., von Rosenvinge E.C., Johnson W.W., Tomarev S.L., Piatigorsky J., Armstrong R.N., Gilliland G.L. 1995. Three-dimensional structure, catalytic properties, and evolution of a sigma class glutathione transferase from squid, a progenitor of the lens S-crystallins of cephalopods Biochemistry 34 5317–5328. [DOI] [PubMed] [Google Scholar]
- Ji Y., Toader V., Bennett B.M. 2002. Regulation of microsomal and cytosolic glutathione S-transferase activities by S-nitrosylation Biochem. Pharmacol. 63 1397–1404. [DOI] [PubMed] [Google Scholar]
- Kano T., Sakai M., Muramatsu M. 1987. Structure and expression of a human class pi glutathione S-transferase messenger RNA Cancer Res. 47 5626–5630. [PubMed] [Google Scholar]
- Kaplan W., Husler P., Klump H., Erhardt J., Sluis-Cremer N., Dirr H. 1997. Conformational stability of pGEX-expressed Schistosoma japonicum glutathione S-transferase: A detoxification enzyme and fusion-protein affinity tag Protein Sci. 6 399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keese M.A., Bose M., Mulsch A., Schirmer R.H., Becker K. 1997. Dinitrosyl-dithiol-iron complexes, nitric oxide (NO) carriers in vivo, as potent inhibitors of human glutathione reductase and glutathione-S-transferase Biochem. Pharmacol. 54 1307–1313. [DOI] [PubMed] [Google Scholar]
- Kluge I., Gutteck-Amsler U., Zollinger M., Do K.Q. 1997. S-nitrosoglutathione in rat cerebellum: Identification and quantification by liquid chromatography-mass spectrometry J. Neurochem. 69 2599–2607. [DOI] [PubMed] [Google Scholar]
- Kong K.H., Inoue H., Takahashi K. 1991. Non-essentiality of cysteine and histidine residues for the activity of human class Pi glutathione S-transferase Biochem. Biophys. Res. Commun. 181 748–755. [DOI] [PubMed] [Google Scholar]
- Kunkel T.A., Bebenek K., McClary J. 1991. Efficient site-directed mutagenesis using uracil-containing DNA Methods Enzymol. 204 125–139. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4 Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Lo Bello M., Parker M.W., Desideri A., Polticelli F., Falconi M., Del Boccio G., Pennelli A., Federici G., Ricci G. 1993. Peculiar spectroscopic and kinetic properties of Cys-47 in human placental glutathione transferase. Evidence for an atypical thiolate ion pair near the active site J. Biol. Chem. 268 19033–19038. [PubMed] [Google Scholar]
- Lo Bello M., Battistoni A., Mazzetti A.P., Board P.G., Muramatsu M., Federici G., Ricci G. 1995. Site-directed mutagenesis of human glutathione transferase P1-1. Spectral, kinetic, and structural properties of Cys-47 and Lys-54 mutants J. Biol. Chem. 270 249–253. [PubMed] [Google Scholar]
- Lo Bello M., Nuccetelli M., Caccuri A.M., Stella L., Parker M.W., Rossjohn J., McKinstry W.J., Mozzi A.F., Federici G., Polizio F.et al. 2001. Human glutathione transferase P1-1 and nitric oxide carriers: A new role for an old enzyme J. Biol. Chem. 276 42138–42145. [DOI] [PubMed] [Google Scholar]
- Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. 1951. Protein measurement with the Folin phenol reagent J. Biol. Chem. 193 265–275. [PubMed] [Google Scholar]
- Lyubarev A.E. and Kurganov B.I. 1998. Modeling of irreversible thermal protein denaturation at varying temperature. I. The model involving two consecutive irreversible steps Biochemistry (Mosc.) 63 434–440. [PubMed] [Google Scholar]
- Mannervik B., Alin P., Guthenberg C., Jensson H., Tahir M.K., Warholm M., Jornvall H. 1985. Identification of three classes of cytosolic glutathione transferase common to several mammalian species: Correlation between structural data and enzymatic properties Proc. Natl. Acad. Sci. 82 7202–7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannervik B., Board P.G., Hayes J.D., Listowsky I., Pearson W.R. 2005. Nomenclature for mammalian soluble glutathione transferases Methods Enzymol. 401 1–8. [DOI] [PubMed] [Google Scholar]
- Meyer D.J., Coles B., Pemble S.E., Gilmore K.S., Fraser G.M., Ketterer B. 1991. Theta, a new class of glutathione transferases purified from rat and man Biochem. J. 274 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S. 1994. Nitric oxide J. Hypertens. Suppl. 12 35–39. [PubMed] [Google Scholar]
- Oakley A.J., Lo Bello M., Battistoni A., Ricci G., Rossjohn J., Villar H.O., Parker M.W. 1997. The three-dimensional structure of the human Pi class glutathione transferase P1-1 in complex with the inhibitor ethacrynic acid and its glutathione conjugate J. Mol. Biol. 274 84–100. [DOI] [PubMed] [Google Scholar]
- Oakley A.J., Lo Bello M., Ricci G., Federici G., Parker M.W. 1998. Evidence for an induced-fit mechanism operating in Pi class glutathione transferases Biochemistry 37 9912–9917. [DOI] [PubMed] [Google Scholar]
- Ortiz-Salmerón E., Nuccetelli M., Oakley A.J., Parker M.W., Lo Bello M., García-Fuentes L. 2003. Thermodynamic description of the effect of the mutation Y49F on human glutathione transferase P1-1 in binding with glutathione and the inhibitor S-hexylglutathione J. Biol. Chem. 278 46938–46948. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Park H.-J., Lee K.-S., Cho S.-H., Kong K.-H. 2001. Functional studies of cysteine residues in human glutathione S-transferase P1-1 by site-directed mutagenesis Bull. Korean Chem. Soc. 22 77–83. [Google Scholar]
- Reinemer P., Dirr H.W., Ladenstein R., Schaffer J., Gallay O., Huber R. 1991. The three-dimensional structure of class pi glutathione S-transferase in complex with glutathione sulfonate at 2.3 Å resolution EMBO J. 10 1997–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinemer P., Dirr H.W., Ladenstein R., Huber R., Lo Bello M., Federici G., Parker M.W. 1992. Three-dimensional structure of class π glutathione S-transferase from human placenta in complex with S-hexylglutathione at 2.8 Å resolution J. Mol. Biol. 227 214–226. [DOI] [PubMed] [Google Scholar]
- Ricci G., Del Boccio G., Pennelli A., Lo Bello M., Petruzzelli R., Caccuri A.M., Barra D., Federici G. 1991. Redox forms of human placenta glutathione transferase J. Biol. Chem. 266 21409–21415. [PubMed] [Google Scholar]
- Ricci G., Lo Bello M., Caccuri A.M., Pastore A., Nuccetelli M., Parker M.W., Federici G. 1995. Site-directed mutagenesis of human glutathione transferase P1-1. Mutation of Cys-47 induces a positive cooperatively in glutathione transferase P1-1 J. Biol. Chem. 270 1243–1248. [DOI] [PubMed] [Google Scholar]
- Ricci G., Caccuri A.M., Lo Bello M., Rosato N., Mei G., Nicotra M., Chiessi E., Mazzetti A.P., Federici G. 1996. Structural flexibility modulates the activity of human glutathione transferase P1-1. Role of helix 2 flexibility in the catalytic mechanism J. Biol. Chem. 271 16187–16192. [DOI] [PubMed] [Google Scholar]
- Rubanyi G.M. 1998. Nitric oxide and circulatory shock Adv. Exp. Med. Biol. 454 165–172. [DOI] [PubMed] [Google Scholar]
- Sánchez-Ruiz J.M. 1992. Theoretical analysis of Lumry-Eyring models in differential scanning calorimetry Biophys. J. 61 921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Ruiz J.M. and Mateo P.L. 1987. Differential scanning calorimetry of membrane proteins Revis. Biol. Celular 11 15–45. [PubMed] [Google Scholar]
- Simons P.C. and Van der Jagt D.L. 1977. Purification of glutathione S-transferases from human liver by glutathione-affinity chromatography Anal. Biochem. 82 334–341. [DOI] [PubMed] [Google Scholar]
- Stevens J.M., Hornby J.A., Armstrong R.N., Dirr H.W. 1998. Class sigma glutathione transferase unfolds via a dimeric and a monomeric intermediate: Impact of subunit interface on conformational stability in the superfamily Biochemistry 37 15534–15541. [DOI] [PubMed] [Google Scholar]
- Takahashi K. and Sturtevant J.M. 1981. Thermal denaturation of streptomyces subtilisin inhibitor, subtilisin BPN′, and the inhibitor–subtilisin complex Biochemistry 20 6185–6190. [DOI] [PubMed] [Google Scholar]
- Tamai K., Shen H.X., Tsuchida S., Hatayama I., Satoh K., Yasui A., Oikawa A., Sato K. 1991. Role of cysteine residues in the activity of rat glutathione transferase P (7-7): Elucidation by oligonucleotide site-directed mutagenesis Biochem. Biophys. Res. Commun. 179 790–797. [DOI] [PubMed] [Google Scholar]
- Tsuchida S., Sekine Y., Shineha R., Nishihira T., Sato K. 1989. Elevation of the placental glutathione S-transferase form (GST-pi) in tumor tissues and the levels in sera of patients with cancer Cancer Res. 49 5225–5229. [PubMed] [Google Scholar]
- Wallace L.A., Sluis-Cremer N., Dirr H.W. 1998. Equilibrium and kinetic unfolding properties of dimeric human glutathione transferase A1-1 Biochemistry 37 5320–5328. [DOI] [PubMed] [Google Scholar]
- Widersten M., Holmstrom E., Mannervik B. 1991. Cysteine residues are not essential for the catalytic activity of human class Mu glutathione transferase M1-1 FEBS Lett. 293 156–159. [DOI] [PubMed] [Google Scholar]
- Wiseman T., Williston S., Brandts J.F., Lin L.-N. 1989. Rapid measurement of binding constants and heats of binding using a new titration calorimeter Anal. Biochem. 17 131–137. [DOI] [PubMed] [Google Scholar]