

Abstract
In the last decade enormous advances in life sciences have been possible due to the information obtained from DNA sequencing projects. The optimal interpretation and analysis of genome sequence data requires the precise annotation and classification of proteins deduced from open reading frames, which is usually done with the help of family-specific signatures. Here we report a novel profile for the IclR type of transcriptional activators and repressors. In contrast to profiles for other families of transcriptional regulators, the new IclR profile is located outside the helix-turn-helix DNA-binding motif. We provide evidence that the new profile is more specific than any of the existing signatures for this family of regulators. More than 500 representatives of this family were identified with this profile. A database on bacterial regulators (http://www.bactregulators.org) was built to compile and regroup the sequences with the aid of the new profile.
Keywords: IclR, transcriptional regulator, family profile
Recent developments in functional genomics and the availability of bacterial DNA chips have revealed that microorganisms are able to alter its transcriptome pattern in response to changing environmental conditions. This involves a series of adaptive responses that are mainly triggered by regulatory proteins (Ramos et al. 2001).
The most recurrent DNA-binding motif for the binding of regulators to their corresponding promoters is a conserved DNA recognition motif that consists of an α-helix, a turn, and a second α-helix (referred to as HTH). The latter helix, termed the “recognition helix,” was shown to fit into the DNA major groove (Pabo and Sauer 1992). Among HTH transcriptional regulators, families have been proposed based on common 3D structural motifs, conserved domains, and primary sequences (Nguyen and Saier 1995; Gallegos et al. 1997; Rigali et al. 2002; Ramos et al. 2005). Comparative studies have led to the determination of a specific signature for some families of bacterial regulators, and these signatures have made it possible to detect and classify new family members (Schell 1993; Gallegos et al. 1997; Rigali et al. 2002; Busenlehner et al. 2003).
One of the families of bacterial transcriptional regulators is termed IclR, which has been named after the Escherichia coli IclR protein. This protein controls the glyoxylate shunt and represents the best-characterized member of the family (Nègre et al. 1992; Yamamoto and Ishihama 2003). The specific functions regulated by members of the IclR family are diverse and include, for example, carbon metabolism in enterobacteriaceae (Yamamoto and Ishihama 2003), degradation of aromatic compounds by soil bacteria (Gerischer et al. 1998), solvent tolerance in Pseudomonas (Guazzaroni et al. 2004), inactivation of quorum sensing signals in Agrobacterium (Zhang et al. 2004), plant virulence by certain enterobacteriaceae (Reverchon et al. 1991), and sporulation in Streptomyces (Jiang and Kendrick 2000).
Interpro (Mulder et al. 2003) assigns proteins to the IclR family according to the PROSITE profiles PS51077 and PS51078 (Hofmann et al. 1999), the SMART domain SM00346 (Schultz et al. 2000), or the Pfam Hidden Markov Model (HMM) PF01614 (Bateman et al. 2002). The signatures used by PROSITE, SMART, and Pfam to identify IclR differ significantly and are located in different parts of the protein sequence. Pfam PF01614 HMM and PROSITE PS51078 are based on a very large segment of the proteins, the former comprising residues 82–269 and the latter comprising residues 87–272 in the IclR primary sequence, and do not consider the HTH motif. This contrasts with the SMART domain SM00346 and PROSITE profile PS51077. The former uses the HTH region of the protein and a large adjacent fragment up to residue 114 in the E. coli IclR primary sequence, whereas the latter is located between amino acids 24 and 86 in the primary IclR sequence. These differences between signatures do not guarantee an unequivocal identification of family members. Therefore, efforts were made to define a precise profile for the recognition of members of the IclR family of transcriptional regulators, which is reported here. This profile has allowed the identification of >500 members of the IclR family of transcriptional regulators (as of August 2005), which were found to be widely distributed in bacteria. In addition, data on IclR proteins were collected and deposited in our database of bacterial regulator proteins (http://www.bactregulators.org).
Results and Discussion
The first step in the development of the new signature for IclR family members was the selection of a seed containing 53 sequences based on the following two criteria: (1) InterPro entry IPR005471 identifies the protein unequivocally as an IclR family member; (2) the proteins were similar in size, i.e., 240–280 amino acids. BLASTCLUST analysis showed that each of the 53 proteins could be clearly distinguished from each other. The sequences were subsequently aligned with CLUSTAL (http://www.clustalw.genome.jp), which revealed three regions that were particularly well conserved in the multialignments (Fig. 1). One of the conserved regions comprises the HTH DNA binding motif located at the N terminus, a second region covers part of the N-terminal portion of the proteins toward the central region, and the third one corresponds to a segment from the central region of the protein toward the C terminus (see Fig. 1). The conserved regions were progressively extended in both directions until the global score of the multialignment diminished. The resulting alignments of these three regions were used as a seed to construct different conventional profiles, each covering a conserved region (available at http://www.bactregulators.org/docs.php). The profiles were built with the “pfmake” program available at the Swiss Institute of Bioinformatics (http://www.isrec.isb-sib.ch/ftp-server/pftools) (Bucher et al. 1996). The different profiles were confronted against all entries in the SWISS-PROT and TREMBL databases (released July 2005). We found that the profile covering the central region toward the C-terminal end (amino acids 151–229 in E. coli IclR) identified all IclR members recognized as such by PROSITE PS51078 and Pfam PF01614, whereas the profiles based on other segments of the protein had a reduced discriminatory capacity, and identified not only IclR family members but also regulators unequivocally ascribed to other families. A profile based on the combination of any of the conserved regions was found to be less precise than the profile that was based solely on the central region toward the C-terminal end of the multialignment. We thus considered that members of the IclR family are best identified by a profile that does not include the HTH domain of this set of proteins, and that covers a significant portion of the C terminus of the proteins. This contrasts with findings for the AraC/XylS (Gallegos et al. 1997), TetR (Orth et al. 2000; Schumacher et al. 2002; Ramos et al. 2005), and GntR (Rigali et al. 2002) families, which are best defined by a specific profile that includes the HTH DNA binding domain.
Figure 1.

(Continued on next page)
Figure 1.

Multialignment of the 53 sequences used as the seed to construct the IclR family profile. Shaded in green is the conserved segment that best defined the IclR family. Bars in blue above the sequence indicate the HTH binding motif. Highlighted in light brown are the residues that are conserved in ≥60% of the aligned sequences, and in purple are shown the amino acids with ≥80% conservation.
The IclR profile, available at the BacTregulators database (http://www.bactregulators.org/docs.php), was confronted against all prokaryotic proteins in the SWISS-PROT and TrEMBL (SPTR) databases (release 13-8-05) using the “pfsearch” program available at http://www.isrec.isb-sib.ch/ftp-server/pftools (Bucher et al. 1996). The program, which proposes a tentative threshold N-score of 8.5 to consider a protein as member of the IclR family, selected 546 proteins as putative members of the IclR family, of which 34 were encoded by plasmids.
To evaluate the specificity (false positives) and sensitivity (false negatives) of the new IclR profile, we used an in-house developed tool termed “Provalidator.” Provalidator is a PHP-based tool that assists in the automation of profile construction and validation, and will be available free of charge at http://www.bactregulators.org. Our analysis revealed no apparent false positive proteins. A search in Interpro (Zdobnov and Apweiler 2001), a database containing all currently available classification methods for IclR proteins, assigned 587 proteins to the IclR family. The 41 proteins assigned to the IclR family by Interpro, although not identified with the new profile constructed in this study, were considered as incorrectly assigned to the family. In fact, among these 41 proteins there were three truncated polypeptides (Table 1, proteins 30, 31, and 35) and six polypeptides of reduced size (71–137 amino acids, namely proteins 23, 26, 30, 32, 34, 36, and 37 in Table 1), which made it unlikely for the latter being part of the IclR family, since our analysis revealed that these polypeptides do not possess an HTH DNA binding domain. The remaining 32 proteins assigned by Interpro (listed in Table 1) were divided into two groups according to their score with the new profile developed here. A group of 25 proteins (Table 1, proteins 8–39, not considering the above-mentioned small or truncated proteins) yielded N-score values between 2.1 and 6.4. Alignment of these 25 proteins with IclR family members revealed substantial sequence conservation at the HTH DNA binding domain, but less sequence conservation in the C-terminal where the new profile is located (not shown). The reason why Interpro assigns these proteins to the IclR family is because PROSITE PS51077 and SMART SM00346 include the nondiscriminatory HTH region. In agreement with this observation is that, with the exception of protein Q57K18 that is exclusively recognized by SM00346, all proteins listed in Table 1 are recognized by PS51077, which is the other profile including the HTH sequence. Therefore, these proteins should be considered as incorrectly assigned to the IclR family, since a profile based on the HTH as PROSITE PS51077 lacks the necessary discriminatory potential.
Table 1.
Proteins listed as IclR family members in Interpro (Zdobnov and Apweiler 2001) but detected as non-IclR family members by the new profile
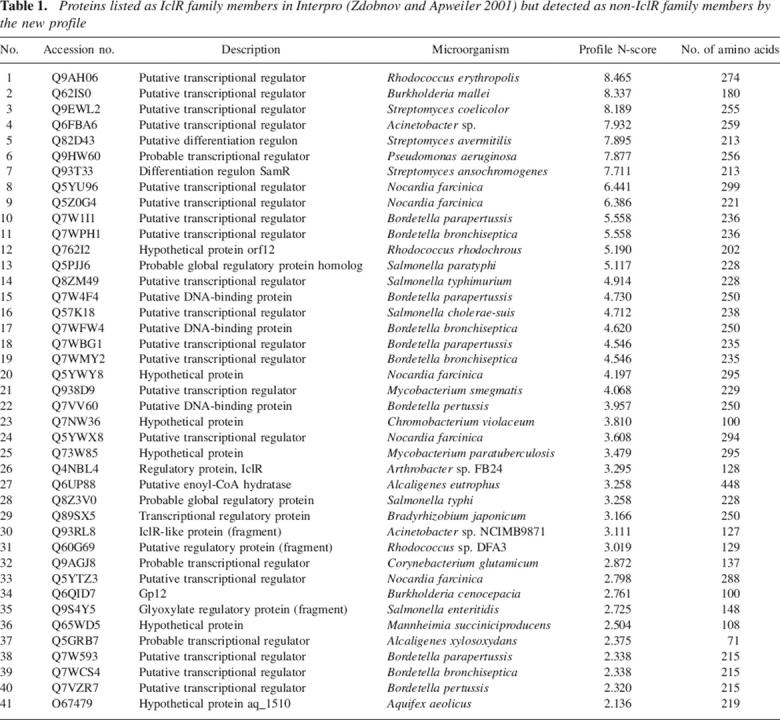
The second group consisted of seven proteins with N-score values between 8.46 and 7.71 (Table 1, proteins 1–7). The alignment of these proteins to IclR family members revealed significant sequence conservation in the fragment spanning the new profile, and thus, it cannot be ruled out that these proteins are IclR family members. However, the N-score threshold of 8.5, as proposed by the “pfsearch” program, cannot be lowered in order to avoid the inclusion of non-IclR proteins. The zone between N-scores of 8.5 and 7.5 is an empirically determined buffer zone where it is recommended to consider the assignment with caution. Sequence annotation is rarely a clear-cut issue, and the purpose of this zone is to prevent the detection of false positives. We consider precision in avoiding false positives more important than the possible exclusion of any family member. Experimental characterization of these proteins will provide support for their identification as members of the Ic1R family, but at present, such information is not available.
The IclR profile with an N-score threshold of 8.5 unequivocally identified proteins as members of the IclR family, and no false positives were found among all prokaryotic proteins that were analyzed. These results indicate that the new profile is highly effective in detecting members of the IclR family.
Using the profile defined above for the IclR family, we searched for members of this family in 228 complete microbial genomes available in NCBI (release 13-8-05). This resulted in the detection of 477 IclR members in 91 microbial genomes belonging to 60 genera of Gram-positive, α, β, and γ-proteobacteria and archaea, indicating a wide taxonomic distribution. This information can be accessed at http://www.bactregulators.org/.
The database of bacterial transcriptional regulators: BacTregulators
The profile that best defines the IclR family members, the sequences of all members of the family, their sequence alignment, as well as the available structural information together with a number of references on IclR proteins have been gathered in the BacTregulators database (http://www.bactregulators.org). This database, which can be searched with a number of different parameters such as organism, name of the regulator, accession code, or simple text information as input information, is, in our view, a convenient tool to identify and study IclR family members.
The structural information available for IclR family proteins supports the profile as a useful tool for assigning proteins to this family
Currently, five PDB entries are available that contain structural information on IclR family members. The only full-length 3D structure of an IclR-family member is that of Thermotoga maritima TM0065 (PDB: 1MKM) (Zhang et al. 2002). The other four structures correspond to the effector binding domains of IclR, the glyoxylate shunt regulatory protein, YaiJ and KdgR from E. coli (PDB: 1TF5, 1TF1, 1YSQ, and 1YSP, respectively). All structures have in common that they were obtained in the absence of target promoter DNA or effector molecules. Structural alignments with the DALI algorithm have shown that these proteins share a similar structure, as witnessed by Z-scores >22 (see http://www.bactregulators.org/structure.php).
The TM0065 IclR protein was shown by X-ray crystallography (Zhang et al. 2002) to consist of two α/β domains: a small N-terminal DNA-binding domain with the HTH motif and a larger C-terminal effector-binding domain (Fig. 2). The latter domain consists of a five-stranded, curved β-sheet, which is flanked on both sides by several α-helices. The 79–amino acid fragment that contains the IclR profile is highlighted in yellow in Figure 2. The profile sequence forms a long loop starting at Gly151, followed by a sequence of three helices (H6–H8, of which H6 is buried and H7 and H8 are surface-exposed), and terminates with strands S5 and S6, which form the flanking part of the sheet (Fig. 2). The amino acids with the highest score (indicating that little variation is tolerated) in this new profile are shown in ball-and-stick mode. Gly151, which is labeled in Figure 2, has been proposed to play a key role in tetramerization of the protein, which is likely to occur when the protein is bound to DNA (Zhang et al. 2002). This role in tetramerization is thus likely to be responsible for the high score of Gly151 in the IclR profile. The remaining high-scoring amino acids are all located on the loop, the short buried helix, and the two strands. None of the important amino acids is located on the two long surface-exposed H7 and H8 helices. All the important amino acids are buried to a large degree and maintain multiple interactions with neighboring residues. These residues thus fulfill an important structural role, which accounts for their weight in the IclR profile.
Figure 2.

Schematic representation of the three-dimensional structure of the IclR dimer of Thermotoga maritima. Secondary structure elements are annotated, and the helix-turn-helix DNA binding domain (HTH) is shown in purple. The 79–amino acid fragment comprising the new IclR profile is highlighted in yellow. The nine amino acids with the highest score in the IclR profile are shown in ball-and stick-mode. Gly151 proposed to be involved in tetramerization is annotated.
Acknowledgments
Work at the authors' laboratory was supported by grants CICYT (2003-0515), Ministerio de Medio Ambiente (059/2004/3), and Junta de Andalucía to group CIV191. We thank M. Mar Fandila and Carmen Lorente for secretarial assistance, and Karen Shashok for improving the use of English in the manuscript.
Footnotes
Reprint requests to: Juan L. Ramos, Estación Experimental del Zaidín, Consejo Superior de Investigaciones Científicas, C/ Prof. Albareda 1, 18008 Granada, Spain; e-mail: jlramos@eez.csic.es; fax: +34-958-135740.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051857206.
References
- Bateman A., Birney E., Cerruti L., Durbin R., Etweiler L., Eddy S.R., Griffiths-Jones S., Howe K.L., Marshall M., Sonnhammer E.L. 2002. The Pfam protein families database Nucleic Acids Res. 30: 276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher P., Karplus K., Moeri N., Hoffman K. 1996. A flexible motif search technique based on generalized profiles Comput. Chem. 20: 3–24. [DOI] [PubMed] [Google Scholar]
- Busenlehner L.S., Pennella M.A., Giedroc D.P. 2003. The SmtB/ArsR family of metalloregulatory transcriptional repressors: Structural insights into prokaryotic metal resistance FEMS Microbiol. Rev. 27: 131–143. [DOI] [PubMed] [Google Scholar]
- Gallegos M.T., Schleif R., Bairoch A., Hofmann K., Ramos J.L. 1997. The AraC/XylS family of transcriptional regulators Microbiol. Mol. Biol. Rev. 61: 393–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerischer U., Segura A., Ornston N.L. 1998. PcaU, a transcriptional activator of genes for protocatechuate utilization in Acinetobacter. J. Bacteriol. 180: 1512–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guazzaroni M.-E., Terán W., Zhang X., Gallegos M.T., Ramos J.L. 2004. TtgV bound to a complex operator site represses transcription of the promoter for the multidrug and solvent extrusion TtgGHI pump J. Bacteriol. 186: 2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K., Bucher P., Falquet L., Bairoch A. 1999. The Prosite database, its status in 1999 Nucleic Acids Res. 27: 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H. and Kendrick K.E. 2000. Characterization of ssfR and ssgA, two genes involved in sporulation of Streptomyces griseus J. Bacteriol. 182: 5521–5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder N.J., Apweiler R., Attwood T.K., Bairoch A., Barrell D., Bateman A., Binns D., Biswas M., Bradley P., Bork P.et al. 2003. The InterPro Database, 2003 brings increased coverage and new features Nucleic Acids Res. 31: 315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nègre D., Cortay J.C., Galinier A., Sauve P., Cozzone A.J. 1992. Specific interactions between the IclR repressor of the acetate operon of Escherichia coli and its operator J. Mol. Biol. 228: 23–29. [DOI] [PubMed] [Google Scholar]
- Nguyen C.C. and Saier Jr. M.H. Jr. 1995. Phylogenetic, structural and functional analyses of the LacI-GalR family of bacterial transcription factors FEBS Lett. 377: 98–102. [DOI] [PubMed] [Google Scholar]
- Orth P., Schnappinger D., Hillen W., Saenger W., Hinrichs W. 2000. Structural basis of gene regulation by the tetracycline inducible Tet repressor-operator system Nat. Struct. Biol. 7: 215–219. [DOI] [PubMed] [Google Scholar]
- Pabo C.O. and Sauer R.T. 1992. Transcription factors: Structural families and principles of DNA recognition Annu. Rev. Biochem. 61: 1053–1095. [DOI] [PubMed] [Google Scholar]
- Ramos J.L., Gallegos M.T., Marqués S., Ramos-González M.I., Espinosa-Urgel M., Segura A. 2001. Response of gram-negative bacteria to certain environmental stresses Curr. Opin. Microbiol. 4: 166–171. [DOI] [PubMed] [Google Scholar]
- Ramos J.L., Martínez-Bueno M., Molina-Henares A.J., Terán W., Watanabe K., Zhang X., Gallegos M.T., Brennan R., Tobes R. 2005. The TetR family of transcriptional repressors Microbiol. Mol. Biol. Rev. 69: 326–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reverchon S., Nasser W., Robert-Baudouy J. 1991. Characterisation of kdgR, a gene of Erwinia chrysanthemi that regulates pectin degradation Mol. Microbiol. 5: 2203–2216. [DOI] [PubMed] [Google Scholar]
- Rigali S., Derouaux A., Giannotta F., Dusart J. 2002. Subdivision of the helix-turn-helix GntR family of bacterial regulators in the FadR, HutC, MocR, and YtrA subfamilies J. Biol. Chem. 277: 12507–12515. [DOI] [PubMed] [Google Scholar]
- Schell M.A. 1993. Molecular biology of the LysR family of transcriptional regulators Annu. Rev. Microbiol. 47: 597–626. [DOI] [PubMed] [Google Scholar]
- Schultz J., Copley R.R., Doerks T., Ponting C.P., Bork P. 2000. SMART: A Web-based tool for the study of genetically mobile domains Nucleic Acids Res. 28: 231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher M.A., Miller M.C., Grkovic S., Brown M.H., Skurray R.A., Brennan R.G. 2002. Structural basis for cooperative DNA binding by two dimers of the multidrug-binding protein QacR EMBO J. 21: 1210–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K. and Ishihama A. 2003. Two different modes of transcription repression of the Escherichia coli acetate operon by IclR Mol. Microbiol. 47: 183–194. [DOI] [PubMed] [Google Scholar]
- Zdobnov E.M. and Apweiler R. 2001. InterProScan—An integration platform for the signature-recognition methods in InterPro Bioinformatics 17: 847–848. [DOI] [PubMed] [Google Scholar]
- Zhang R.G., Kim Y., Skarina T., Beasley S., Laskowski R., Arrowsmith C., Edwards A., Joachimiak A., Savchenko A. 2002. Crystal structure of Thermotoga maritima 0065, a member of the IclR transcriptional factor family J. Biol. Chem. 277: 19183–19190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.-B., Wang C., Zhang L.H. 2004. The quormone degradation system of Agrobacterium tumefaciens is regulated by starvation signal and stress alarmone (p)ppGpp Mol. Microbiol. 52: 1389–1401. [DOI] [PubMed] [Google Scholar]