

Abstract
Androgens exert their effects by binding to the highly specific androgen receptor (AR). In addition to natural potent androgens, AR binds a variety of synthetic agonist or antagonist molecules with different affinities. To identify molecular determinants responsible for this selectivity, we have determined the crystal structure of the human androgen receptor ligand-binding domain (hARLBD) in complex with two natural androgens, testosterone (Testo) and dihydrotestosterone (DHT), and with an androgenic steroid used in sport doping, tetrahydrogestrinone (THG), at 1.64, 1.90, and 1.75 Å resolution, respectively. Comparison of these structures first highlights the flexibility of several residues buried in the ligand-binding pocket that can accommodate a variety of ligand structures. As expected, the ligand structure itself (dimension, presence, and position of unsaturated bonds that influence the geometry of the steroidal nucleus or the electronic properties of the neighboring atoms, etc.) determines the number of interactions it can make with the hARLBD. Indeed, THG—which possesses the highest affinity—establishes more van der Waals contacts with the receptor than the other steroids, whereas the geometry of the atoms forming electrostatic interactions at both extremities of the steroid nucleus seems mainly responsible for the higher affinity measured experimentally for DHT over Testo. Moreover, estimation of the ligand–receptor interaction energy through modeling confirms that even minor modifications in ligand structure have a great impact on the strength of these interactions. Our crystallographic data combined with those obtained by modeling will be helpful in the design of novel molecules with stronger affinity for the AR.
Keywords: human androgen receptor, crystal structure, ligand binding pocket, agonists, tetrahydrogestrinone
The androgen receptor (AR) is a member of the nuclear receptor (NR) superfamily (Mangelsdorf et al. 1995). Like the other NRs, it is constituted by three main functional domains: a variable N-terminal domain (NTD), a highly conserved DNA-binding domain (DBD), and a conserved ligand-binding domain (LBD) (Jenster et al. 1991). After binding of an androgen to its LBD, AR rapidly translocates to the nucleus, where it directly interacts with DNA as a homodimer, at androgen response elements (ARE) found in the regulatory regions of target genes. This complex can thenceforth recruit coactivators (Jenster 1998) through the ligand-dependent transactivation function (AF-2) located in the LBD and hence control transcription of specific genes. Through this mechanism, androgens such as testosterone (Testo) and 5α-dihydrotestosterone (DHT) regulate a wide range of physiological responses, most notably male sexual differentiation and maturation including the development, growth, and maintenance of the normal prostate (Mooradian et al. 1987; Keller et al. 1996; Roy et al. 1999). Defects in AR function are involved in health disorders including prostate cancer's resistance to androgen ablation therapy (Quigley et al. 1995; Heinlein and Chang 2004).
Because of their anabolic characteristics, androgens have been used by athletes for a long time (Evans 2004). It is thus not surprising that chemically modified androgens, often synthesized for pharmacological purposes, have rapidly given rise to interest in elite sports. Indeed, athletes have been using modified steroids with a higher anabolic:androgenic ratio to enhance their performances. Recently, a novel chemically modified steroid, tetrahydrogestrinone (THG), has appeared as a doping agent. A potent androgen and progestin (Death et al. 2004), THG is produced by the hydrogenation of gestrinone, a progestin used to treat endometriosis (Dawood et al. 1997), and has been identified as the first true “designer androgen,” being custom produced to evade detection (Catlin et al. 2004). Indeed, it was undetectable in urine by standard antidoping tests until Catlin et al. (2004) developed a specific test. Using a pangenomic assay, THG has been shown to modulate hundreds of genes in a time-dependent fashion almost superimposable to DHT (Labrie et al. 2005).
All androgens, natural or chemically designed, exert their action via the AR by binding its unique LBD. However, these various ligands bind AR with very different affinities, their Ki values ranging from low nanomolar concentrations for the most potent androgens to micromolar concentrations for the weaker ones. Interestingly, it is almost impossible to predict the strength of the interaction between a ligand and a receptor only on the basis of its structure, since steroids with very similar structures can possess markedly different affinities for a given receptor while structurally different ligands could have similar high affinities. The ligand-binding pocket (LBP) of the NRs is composed of a large number of residues making up the binding interface and involved in ligand–receptor complex formation. It is not clear, however, if all these residues or only a small subset contribute to the binding energy. A complete characterization of the factors contributing to the ligand–AR interaction would thus greatly help us to understand the basis of the ligand specificity and selectivity.
To date, several crystal structures of the human AR ligand-binding domain (hARLBD) have been solved in complex with the natural androgen DHT (Sack et al. 2001) and with chemically modified steroids such as the agonist metribolone (R1881) (Matias et al. 2000) or the corticosteroid agonist 9α-fluorocortisol (Matias et al. 2002). In addition, structures of the liganded hARLDB have been determined in complex with a peptide derived from physiological coactivators (He et al. 2004; Hur et al. 2004; Estebanez-Perpina et al. 2005). In all these complexes, the LBD adopts the same fold, mainly composed of α-helices arranged as a three-layered antiparallel α-helical sandwich, a fold common to all the NRs (Wurtz et al. 1996). These structures show that the LBP is mainly composed of hydrophobic residues, the side chains of which can easily adopt variable positions in order to better fit the hydrophobic core of the steroid and stabilize it.
LBP is also composed of polar amino acids able to establish hydrogen bonds at both extremities of the steroid nucleus of all potent androgens. If several studies have well demonstrated the importance of these polar residues in ligand binding and shown the effect of their substitution, which is associated with disorders that impair androgen-dependent male sexual differentiation or affect the development and/or progression of cancers such as prostate cancer in the human (De Bellis et al. 1992; Pinsky et al. 1992; Gaddipati et al. 1994; Taplin et al. 1995; Poujol et al. 2000; Sakai et al. 2000; Chavez et al. 2001; Labrie et al. 2002, 2005), little is known about the role and the importance of the hydrophobic residues that form the major part of the LBP. Because almost all potent androgen steroids known to bind hAR with high affinity possess the same polar groups at their nucleus extremities, it is more than likely that the hydrophobic residues are of paramount importance, not only in the stabilization of the steroid in its pocket but also in the high selectivity and specificity observed for all members of the NR superfamily.
To better understand the role of the hydrophobic residues found in the LBP, we have determined and compared the crystal structures of the hARLBD in complex with three agonist ligands of similar structure but possessing different levels of affinity for the receptor. Here we report the crystallographic structure of the human ARLBD in complex with natural androgens Testo and DHT and with the designer steroid THG (Fig. 1). Close inspection and comparison of these high-resolution complex structures have allowed identifying in both molecules, ligand and receptor, molecular determinants that appear to be important for high-affinity binding of androgens. To confirm the relative importance of each of these determinants identified by crystallography, ligand–receptor interaction energy has been estimated by modeling and energy minimization. Our results, which led us to propose possible roles for these determinants, will be used in drug design strategies, especially for the conception of new AR antagonists with higher affinity for the human androgen receptor.
Figure 1.

Molecular structures of the ligands used. DHT, dihydrotestosterone; Testo, testosterone; THG, tetrahydrogestrinone; R1881, methyltrienolone. Carbon and oxygen atoms of the steroids are numbered according to the standard steroid nomenclature and the cycles designated by letters. For THG, extra carbon atoms are numbered 20 and 21 for the 17α-ethyl group and 22 for the C18-methyl group.
Results and Discussion
General structure of hARLBD complexed with agonist ligands
We crystallized the human androgen receptor ligand-binding domain in the presence of the natural and synthetic ligands Testo, DHT, and THG. All these ligands have a steroid-based structure (Fig. 1) and are highly potent androgens. These steroids, however, possess different affinities for the hAR. Indeed, when their capacity to displace [3H]R1881 from the human AR has been measured, Labrie et al. (2005) have found that R1881, DHT, and testosterone have relative potencies of 72%, 58%, and 7% compared with the 100% value set for THG, the compound showing the highest affinity for AR. Having obtained the three crystal structures, we performed precise structural analysis and comparison in order to determine the way by which the hAR can adapt the structure of its steroid-binding site to accommodate these structurally different ligands. Our objective was to identify, on the ligand structure and on the protein structure, the molecular determinants responsible for the variability of their binding affinity.
All crystals obtained belonged to the P212121 space group and contained one molecule per asymmetric unit. The hARLBD complex crystal structures presented here were refined to crystallographic R-factors of 21.4% (Rfree = 24.1%), 19.1% (Rfree = 23.0%), and 18.6% (Rfree = 20.9%), at 1.90 Å, 1.64 Å, and 1.75 Å of resolution for DHT, Testo, and THG, respectively (see crystallographic statistics in Table 1). Even if the electronic density maps were very well defined for the three complexes, a complete model, from residues 671 to 919, was finally constructed only for the Testo complex. For the DHT and THG complexes, a part of the loop between helices 9 and 10 (residues 844–849) seemed to be disordered since no electron density was observed. Consequently, no model was built for these six residues. For the Testo complex, we were able to construct the loop since the six Cα atoms had unambiguous matching electron density, but their side chains were built into poorly defined electron density.
Table 1.
Data collection and refinement statistics (data of the last shell of resolution)

a Rsym is a measure of the internal consistency of the data defined as ∑|Ii−<I>|/∑Ii, where Ii is the intensity of the ith observation and <I>, the mean intensity of the reflection.
b Rcryst = ∑|Fo − Fc|/∑FO, where FO and FC are the observed and calculated amplitudes of structure factors.
Analysis of our crystallographic structures with PROCHECK (Laskowski et al. 1993) showed that they are all composed, as expected, of 11 α-helices and four β-strands arranged in two β-hairpin motifs (Fig. 2). The helices are packed together in the typical “helical sandwich” fold seen in all NRs crystallized to date (for review and references, see Gronemeyer et al. 2004). A pairwise comparison revealed that the overall structures of the Testo, DHT, and THG complexes are very similar. Root mean square deviation (RMSD) values calculated with LSQKAB (CCP4 1994) for all their common Cαs are 0.238 Å, 0.330 Å, and 0.229 Å for the Testo/DHT, DHT/THG, and Testo/THG complexes, respectively. Except for the side chain of some residues, especially in the vicinity of the ligand, they are also very similar to the other structures of agonist–hARLBD complexes published so far (Matias et al. 2000; Sack et al. 2001; He et al. 2004; Hur et al. 2004; Bohl et al. 2005a; Estebanez-Perpina et al. 2005).
Figure 2.

Overall view of the structure of hARLBD in complex with Testo.
Structure of the ligand-binding pockets
The electron density maps for the three ligands were clearly visible in difference Fourier maps during model building, and there was no doubt about their identification in the ligand-binding site. For each complex, the ligand was fitted into the Fo − Fc electron density map and then refined with the rest of the model (Fig. 3). The LBP is composed of residues belonging to four helices (H3, H4, H5, and H11) and a β-strand located between H5 and H6. It consists of a large nonspecific apolar cavity where many hydrophobic amino acid residues interact with the steroid nucleus through van der Waals contacts. The binding site is completed by a few polar residues that firmly tether the steroid molecule via hydrogen-bond networks formed with polar atoms found at both extremities of the ligand structures. The inherent nonspecificity of the hydrophobic interactions that loosely maintain the steroid in the steroid-binding cavity combined with the fact that the side chains of these residues are quite mobile and can adopt various conformations may explain how a steroid receptor of the NR family can bind several structurally different ligands. Table 2 presents all the interactions (hydrogen bonds and hydrophobic contacts) observed between the hARLBD and each ligand studied.
Figure 3.
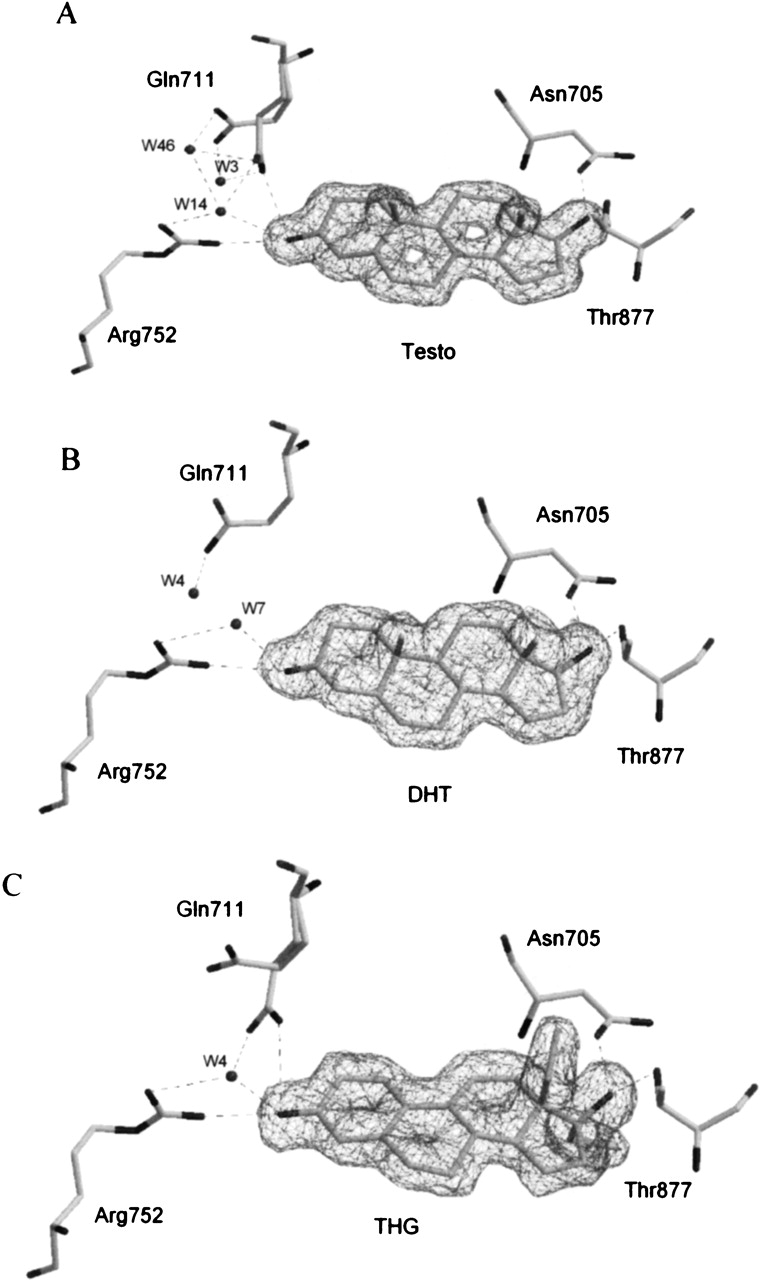
Ligands in androgen receptor ligand-binding pocket (LBP) and residues of interest. The electron density maps, contoured at 1σ, show unambiguously the identity of each ligand in the LBP: Testo (A), DHT (B), and THG (C). Putative H-bonds between ligand, water molecules, and LBP residues are shown as dashed lines (all possible H-bonds are determined from geometric parameters).
Table 2.
Ligand-receptor interactions analysis by LIGPLOT

For hydrophobic contacts, only receptor atoms located <4.0 Å from a ligand atom were considered. Interactions with values close to this cutoff are considered as weak interactions. (—) Weak or nonexistent interaction; (X) atom absent in the ligand structure. Carbon and oxygen atoms for the different ligands are numbered as shown in Figure 1.
a Residue in a double conformation.
Among differences in the structure of residues that form the LBP of hAR and that are directly in contact with the three ligands, the most noteworthy is the smaller distance between the side chain of residue Met745 and the steroid skeleton we observed in the THG complex (Fig. 4). This structural difference is probably due to the absence of a C19-methyl group in the THG structure (Fig. 1) and has previously been reported for hAR structures in complex with R1881 (Matias et al. 2000; He et al. 2004), a synthetic agonist lacking the C19-methyl group as well, and with bicalutamide, a nonsteroidal molecule also without atoms in the corresponding area (Bohl et al. 2005a). The movement of the Met745 side chain appears to be sufficient to create room for the second major change, the rotation of the side chain of residue Trp741 around its Cβ. Interestingly, the rotation of this particular residue upon the binding of THG was predicted by molecular modelization in a model with this steroid very recently published (Jasuja et al. 2005). Our crystallographic structure confirms that the Trp741 conformational change that takes the indole cycle of the tryptophan residue far away from its position in the Testo and DHT complexes (Fig. 4) largely contributes to the pocket ability to accommodate the extra C18-methyl group borne by THG. In the THG–hARLBD structure presented here, Trp741 adopts a conformation similar to the one observed in the R1881–hARLBD structure described by He et al. (2004), but different from that reported by Matias et al. (2000) for the same complex. Our high-resolution structure confirms that the actual conformation of Trp741 for steroids lacking a C19 methyl group such as R1881 or THG is more than likely the one seen by He et al. (2004). In addition to Trp741, the extra 18-methyl group of the THG compels Met895 to adopt a new conformation with its Cα at 1.2 Å farther from the ligand nucleus. The consequence of this last movement is a slight deformation of the N-terminal extremity of helix 12, which is, however, well positioned over the LBP in a position similar to that observed in the complexes with Testo or DHT. Finally, the presence of an ethyl group at position 17α in the THG structure also has a visible impact on the structure of residues forming the LBP. Indeed, these extra carbon atoms slightly push out the side chain of residue Leu701, which rotates its Cβ and Cγ to create the necessary space to accommodate this chain (Fig. 4).
Figure 4.

Superposition of the moving residues of the LBP of hARLBD. Stereo view showing conformation of Leu701, Trp741, Met745, and Met895 in the various hARLBD complexes. Testo is depicted in yellow; DHT, in blue; and THG, in gray.
All these observations highlight the flexibility of the residues forming the steroid-binding pocket of the androgen receptor, a structural characteristic that could be exploited in drug design strategies, especially for designing AR antagonists. The capacity of the androgen receptor to modify the shape of its ligand-binding site in order to accommodate ligands with different structures is well demonstrated by the variation of the LBP volumes in our different complexes. Indeed, considering only the non-hydrogen atoms, the size of the ligand-binding pocket measured for each complex (Testo = 584 Å3, DHT = 582 Å3, THG = 605 Å3) is proportional to the volume of the ligand itself (Testo = 252 Å3, DHT = 249 Å3, THG = 275 Å3), each ligand occupying <50% of the available space. The rest of the space is occupied by the hydrogen atoms belonging to the amino acids forming the cavity and to the ligand. A similar observation was done with the human enzyme 17β-hydroxysteroid dehydrogenase type I, known to be specific to estrogens but which is able to bind other steroids by rearranging some side chains of its steroid-binding site (see Blanchet et al. 2005 and references therein).
Hydrogen bonding
As a rule, all potent androgens that bind hAR with high affinity possess a ketone group at position C3 and a hydroxyl group at position 17β. The oxygen atom (O3) of the ketone group has a lone pair of electrons and thus could act as a hydrogen bond acceptor able to establish a strong interaction with polar or charged amino acids. Likewise, the hydrogen atom of the 17β-hydroxyl group bears a partial positive charge that allows its interaction with a highly electronegative atom borne by an adjacent amino acid residue. Because hydrogen bonds (electrostatic interactions) are much stronger (∼2 orders of magnitude) than van der Waals forces (electrodynamic interactions), it is likely that they constitute the main element explaining why hAR binds androgens so strongly with affinities in the low nanomolar range (Mowszowicz et al. 1981). On the other hand, electrostatic interactions are also likely necessary for promoting ligand binding by the free receptor, whereas electrodynamic interactions, because of their larger number, would ensure the high selectivity of the receptor and help stabilize the ligand bound deeply within the receptor pocket.
In the hAR structure, two residues (Arg752 and Gln711) located in the immediate vicinity of cycle A of the steroid ligand are the most likely to create an interaction with the O3 atom (Fig. 3). However, because of its strong positive charge, Arg752 is likely to be the one able to establish the strongest interaction with the ketone group. The position of its side chain is perfectly conserved in the hAR structure in complex with the three ligands studied, thus suggesting that it is particularly important for the binding of androgens by this receptor. This is in agreement with the finding that a mutation at this position (Arg752Gln) affects the AR functional activity. Males bearing this mutation suffer from a genetic disorder called Androgen Insensitivity Syndrome (AIS), the most common form of male pseudohermaphroditism, and present a completely female phenotype (Sakai et al. 2000). On the other hand, as seen in two of our hAR complex structures, the side chain of Gln711 can adopt different conformations according to the ligand. Indeed, Gln711 is observed in a double conformation for Testo and THG complexes, whereas it is far from the ligand in our DHT complex (Figs. 3, 5), thus suggesting that the putative hydrogen contact between this residue and the steroid nucleus is not essential for ligand binding. A point mutation in the androgen receptor gene resulting in a glutamine-to-glutamate replacement (Gln711Glu) has also been found in patients with AIS (Chavez et al. 2001). However, and in agreement with its role in steroid binding deduced from our crystal structures, replacement of residue Gln711 does not apparently affect hAR binding activity. Finally, in addition to the Arg752 and Gln711 residues, a water molecule observed at the same position in all structures could also interact with the O3 atom of the ligands. This water molecule is well stabilized and could form hydrogen bonds with the amido oxygen of Gln711 and the guanidinium group of Arg752 (Fig. 3). This arrangement is quite similar to that reported for other ligand–receptor complexes. For example, in the progesterone receptor, a water molecule seen in the vicinity of the progesterone 3-keto function participates in a hydrogen bond network with Gln725 and Arg766 (Williams and Sigler 1998).
Figure 5.

Double or unique Gln711 conformation in (A) Testo–, (B) DHT–, and (C) THG–hARLBD complexes. Electron density maps are contoured at 1σ.
At the other extremity of the steroid nucleus, Asn705 and Thr877 are well positioned for contacting the 17β-hydroxyl group and for maintaining the steroid firmly inside the LBP (Fig. 3). The importance of these two residues in the hAR steroid binding is well documented since the change of Asn705 for a serine residue results in a complete absence of androgen binding by the mutated hAR (De Bellis et al. 1992; Pinsky et al. 1992), while another mutant receptor (Asn705Ala) shows a decreased specificity toward androgens (Poujol et al. 2000). Replacement of Thr877 has been found many times in specimens obtained from men with metastatic carcinoma of the prostate (Gaddipati et al. 1994). Moreover, the threonine-to-alanine substitution at position 877 is known to alter the selectivity toward androgens with the result that the mutated AR can bind androgens, hydroxyflutamide, progesterone, and estrogen, and be transcriptionally activated in the presence of any of these steroids (Harris et al. 1990, 1991; Veldscholte et al. 1990; Ris-Stalpers et al. 1993; Taplin et al. 1995). The finding that this mutated AR with altered ligand specificity is frequently found in advanced prostatic carcinomas suggests a possible role in tumor progression. Bohl et al. (2005b) have very recently determined the crystallographic structure of the T877A-hAR-LBD mutant in complex with hydroxyflutamide, showing that the T877A mutation results in the presence of an additional water molecule into the LBP. Moreover, this water molecule mediates a hydrogen bond between the carbonyl oxygen of the ligand and the backbone oxygen of residue Leu873 from helix 11. It thus appears that this interaction, present only in AR bearing the T877A mutation, confers to hydroxyflutamide the capacity to stimulate AR-mediated transcription.
Even if the hydrogen bonds made by Asn705 and Thr877 probably constitute the other most important contact between the receptor and its steroidal ligand after the Gln711 and Arg752 interactions, it is very unlikely that they could contribute to the difference of affinity experimentally measured for the three ligands studied. Indeed, the structures of all complexes (Testo, DHT, and THG) are identical at this position and the conformation of the side chain of Asn705 and Thr877 residues is perfectly conserved in the three hAR complex structures. Nonetheless, precise comparison of the angles between the atoms involved in the formation of these hydrogen bonds reveals subtle differences that must be considered to explain the slight affinity variations observed between these highly structurally related ligands (see below).
Hydrophobic interactions
We first observed that the hydrophobic contacts between the receptor and the ligands were very similar, especially for the Testo and DHT complexes. Indeed, considering only atoms of the receptor located <4.0 Å from an atom of the ligand, we counted 20 hydrophobic interactions for Testo, compared to 18 for DHT. The interatomic distances for each of them were also very similar (Table 2). Consequently, the 10-fold higher affinity shown by the hAR for DHT compared to Testo cannot be explained on the basis of the hydrophobic contacts established by the androgen receptor and these androgens. On the other hand, analysis of the THG–hARLBD complex structure using LIGPLOT (Wallace et al. 1995) revealed that the three additional carbon atoms found on the trienolone steroid nucleus of the THG, the 17α-ethyl group and the 18-methyl group, contribute to raising to 26 the number of hydrophobic contacts and largely compensate for the absence of the C19 atom found in the structure of the two other ligands (Fig. 1). It thus appears that these additional atoms allow THG to form more numerous contacts with the receptor via hydrophobic interactions, a difference that could, at least in part, explain why THG possesses an approximately twofold higher affinity than DHT for the hAR. A similar conclusion can be drawn when THG is compared with the highly potent androgen metribolone (R1881), the affinity of which is about twofold lower than that of THG (EC50 values of 2.8 nM for R1881 and 1.86 nM for THG). The only difference between these two synthetic methyltrienolone-based androgens is the presence, on the THG structure, of two extra methyl groups (Fig. 1), which could create additional hydrophobic interactions with the receptor.
Structural elements determining ligand affinity
As mentioned above, hydrogen bonds probably constitute one of the main elements contributing to the binding of ligands by hAR. Consequently, a small variation in these interactions could greatly influence the affinity of the receptor for its ligands. In hydrogen bonding, alignment is critical, with significant weakening if donor D, the H atom, and acceptor A are not collinear. Small deviations from linearity in the bond angle (up to 20°) have a relatively minor effect (Rao 1972), but the strength of an H-bond rapidly decreases from 160° to no interaction energy around 90°. Moreover, the distance separating A and D atoms is also indicative of the bond strength. Indeed, the dependency on bond length is very important, and strength has been shown to exponentially decay with distance (Espinosa et al. 1998). We thus closely examined geometrical characteristics of our hAR complex structures to find differences that could explain the variability in the binding affinity measured for the three ligands studied. To determine these H-bonding geometrical parameters, we used the ZMM molecular modeling program (Zhorov 1983) to add H atoms to the protein and ligands and to refine their position by energy minimization, while the position of non-H atoms was constrained to their crystallographic coordinates (see Materials and Methods).
Structures of the Testo and DHT are very similar (Fig. 1). However, the presence of an unsaturated bond (C4–C5) in the Testo molecule changes the geometry of the A ring and the orientation of the ketone group at position C3, which could modify the strength of the possible H-bond made with residue Arg752. Indeed, the angle (121.1°) measured between donor, hydrogen, and acceptor atoms (here CζArg752, HNη2Arg752, and O3ligand atoms) as well as the distance (2.36 Å) separating the hydrogen and the acceptor atoms clearly favor the DHT–hARLBD interaction over Testo (104.6° and 2.84–2.92 Å). This constitutes the most important difference observed in the binding mode of these two steroids and could mainly explain the difference in their affinity for the androgen receptor. The only other striking difference in this region is the double conformation adopted by the side chain of residue Gln711 in the Testo complex and the possible additional hydrogen bond formed by this residue (HNɛ2 atom) with the O3ligand atom of the Testo observed when the side chain of Gln711 is in its proximal conformation (Fig. 3). However, even if the geometrical characteristics of this H-bond seem close to optimal values (distance of 2.56 Å and angle of 144.3°), this interaction does not appear to compensate sufficiently for the weaker interaction between Arg752 and Testo. Interestingly, Gln711 adopts the same double conformation when THG, which has a 20-fold higher affinity for hAR than Testo, is bound into the ligand-binding pocket (Fig. 3). This observation strongly supports our hypothesis, according to which this hydrogen contact between Gln711 and the steroid does not contribute importantly to ligand binding. It should also be noted that Gln711 is seen in a double conformation, with its side chain near the ligand or in an intermediate conformation, in the structure of all the other DHT–hARLBD complexes published (Sack et al. 2001; Hur et al. 2004; Estebanez-Perpina et al. 2005). However, contrarily to our Testo–hARLBD complex structure, the orientation of the Nɛ2 group of Gln711 in those structures does not allow H-bonding with the ketone group of DHT.
At the other extremity of the steroid nucleus, no major difference was apparent between Testo and DHT concerning the geometrical characteristics of the hydrogen-bonding network provided by the side chains of residues Asn705 and Thr877. Indeed, all distances and bond angles were almost the same for these two complexes (Table 3). However, in the THG structure, where the presence of three conjugated double bonds makes the steroid nucleus much more planar and changes the orientation of the hydroxyl group at position 17, the angle between the atoms forming the H-bond (the O atom of the carbonyl group [Asn705] and the H and the O atoms of the hydroxyl group of the ligand) is almost planar while the acceptor and the hydrogen atoms are very close (∼1.9 Å), two characteristics indicating the higher strength of this bond. This information, considered together with the additional hydrophobic interactions found between THG and the hARLBD, could then explain the higher affinity of this ligand over that of the most potent natural androgen, DHT.
Table 3.
H-bonding angles (°) and distances (Å) calculated from modeling results

(1) Complex with Gln 711 in the vicinity of the ligand; (2) complex with Gln 711 farther from the ligand.
Observed hAR ligand preferences confirmed by modeling
Modeling was first used to add hydrogen atoms and to optimize their coordinates. It also allowed estimating the ligand–receptor interaction energy. The modeling procedure was carried out using the crystallographic coordinates determined for each hARLDB–ligand complex and allowing only minimal changes to the crystal structures because of the quality and the precision of these experimentally determined data. As mentioned above, the affinity of the hAR is higher for THG than for DHT and Testo (Labrie et al. 2005), and energy minimization analysis highlighted some distinctive features in their binding mode that could explain these differences. During the molecular modeling of the Testo and THG complex structures, we treated separately the two possible conformations of Gln711 observed in our crystallographic models. For each complex, our calculations predict a final receptor conformation very similar to the X-ray structures and a final global energy value of ∼−1000 kcal/mol. Generally, the ligand–receptor interaction energy was slightly better for DHT (−33.6 kcal/mol) than for Testo (−31.0 kcal/mol) (Table 4). Moreover, in agreement with the affinity comparisons (Labrie et al. 2005), the strongest interaction energy values were calculated for THG (−41.4 kcal/mol) (Table 4). Modeling predicted that the interactions provided by van der Waals contacts are the most important and count for more than two-thirds of all forces between the hAR and the ligand. Moreover, the energy values calculated for the van der Waals interactions are greater for THG than for the two other ligands (Table 4) and certainly contribute to the best affinity of hAR for this ligand. As for the two natural androgens, we surprisingly calculated higher van der Waals interaction energy for Testo, notably because of additional interactions between cycle A of the steroid and the side chain of Gln711 when this residue is closer to the steroid nucleus.
Table 4.
Mean ligand-receptor interaction energy values for DHT–, Testo–, and THG–hARLBD complexes calculated by modeling

For the Testo– and THG–hARLBD complexes, the energy value indicated is an average of values calculated for each of the two conformations of residue Gln711.
Electrostatic energy, however—the second most important contribution to ligand–receptor interaction energy—largely favors DHT over Testo and explains why DHT possesses a higher affinity for the receptor. Finally, whatever the ligand, the global H-bonding energies predicted are very similar for all complexes (Table 4), even if some differences could be highlighted. For each ligand, the simulation program detected an H-bond involving the ligand O3 atom. This H-bonding probably involves the HNη2 atom of Arg752 in the DHT complex and for complexes in which Gln711 conformation is in its distal position. In this case, the best energy value was calculated for DHT (Table 5), in agreement with the H-bonding geometrical parameters calculated from our crystallographic structures (Table 3). The stronger H-bond energy values predicted between Arg752 and the O3 atom of DHT over Testo (Table 5) can be explained by the difference in the electronegativity of the O3 atom, which is significantly lower for the Testo molecule because of the presence of a double bond (C4=C5) in its structure. As explained above, this structural particularity also induces different O3 atom geometry, which leads to better H-bond parameters between Arg752 and DHT compared to Testo (Table 3). Concerning Testo and THG complexes for which Gln711 side chain points toward the ligand, the H-bonding energy value decreases (is more favorable), suggesting a possible additional H-bond with the Gln711 HNɛ2 atom. This additional interaction could thus play a role in Testo binding, maybe compensating for the poor interaction with Arg752 by using both van der Waals and H-bond energies. For THG, the H-bond energy value variation is too small to conclude a real Gln711 contribution. For each complex, stronger and equivalent H-bonds were also predicted with the Asn705 Oɛ2 atom and the ligand HO17 atom, and between the Hγ1 Thr877 and O17 ligand atoms, thus confirming crystallographic results (Tables 2, 3).
Table 5.
Energy values (kcal/mol) for H-bonds predicted by modeling between receptor amino acids and ligand atoms for DHT–, Testo–, and THG–hARLBD complexes

As a whole, energies calculated through molecular modeling are in perfect agreement with our crystal structures and help us to understand, at a molecular level, the difference of affinity for the hAR between the three ligands studied. Above all, however, this approach allows better characterization of the interactions established between the receptor and its ligands and permits us to quantify the relative energy associated with each of them. Molecular determinants responsible for receptor–ligand recognition and binding can thus be identified and, by comparing their position in the different complexes, it becomes possible to determine their flexibility.
Because of its anabolic characteristics, THG (Catlin et al. 2004; Labrie et al. 2005) is one example of the many synthetic anabolic steroids used as doping agents to increase sport performances. THG exerts its activity by binding the androgen receptor with a higher affinity than the natural potent androgens, Testo and DHT (Labrie et al. 2005). Determination and comparison of the crystallographic structures of the human androgen receptor ligand-binding domain in complex with THG, Testo, and DHT have permitted us to find many differences in the binding mode of these molecules. Along with modeling experiments, these crystal structures have allowed the identification of important features implicated in androgen recognition and high-affinity binding. Elucidation of the molecular mechanisms responsible for the ligand–receptor interactions should guide us in the process of developing new molecules able to bind with high affinity and specificity the human androgen receptor.
Materials and methods
Materials
Chemical products were purchased from Sigma-Aldrich Canada while steroids were purchased from Steraloid. Expression vectors and protein chromatography products were obtained from Amersham Pharmacia Biotech. Protein concentration was determined with the Bio-Rad Protein Assay (Bio-Rad Laboratories). Tetrahydrogestrinone was synthesized in our laboratory following the protocol described elsewhere (Labrie et al. 2005).
Protein purification
Human ARLBD was expressed and purified as described by Matias et al. (2000). The hARLBD cDNA (residues 654–919) was cloned in the pGEX 5X-2 vector and expressed as a glutathione S-transferase (GST) fusion protein in the Escherichia coli strain BL21 (DE3) pLysS cells. Expression was carried out at room temperature for 15–18 h in LB broth supplemented with the ligand of interest (50 μM for DHT or 400 μM for Testo and THG) after induction with 100 μM isopropyl-β-D-thiogalactoside (IPTG). Harvested cells were lysed with several freeze/thaw cycles and sonication in a buffer containing 50 mM Tris (pH 7.3), 150 mM NaCl, 5 mM EDTA, 10% glycerol, 0.5% CHAPS, 10 mM DTT, 200 μg/mL lysozyme, 1 mM PMSF, and 50 μM of the ligand of interest. The soluble proteins were loaded onto a glutathione Sepharose column, washed, and eluted with 15 mM reduced glutathione. The GST affinity ligand was cleaved with FXa and the protein mixture was loaded onto a DE52 anion exchange column. The eluted hARLBD without GST was concentrated and further purified on a Superdex 75 size exclusion column using 20 mM HEPES (pH 7.5), 150 mM LiSO4, 10% glycerol, 0.1% n-octyl-β-glucoside, and 1 mM DTT. The purified protein was concentrated up to 5 mg/mL, 7 mg/mL, and 12.5 mg/mL, with Testo, DHT, and THG, respectively, as ligand. Approximately 3.5 mg of protein were obtained per liter of cell culture.
Crystallization and data collection
Protein crystallization was achieved using the hanging drop vapor diffusion method at room temperature. Crystals appeared within 2 d in drops of a 1:1 (v/v) ratio of purified protein and well solution. The crystals of Testo–, DHT– and THG–hARLBD complexes appeared in two different conditions: (1) 0.1 M PIPES, bicine, or HEPES buffers with pH ranging from 6.5 to 8.5 and 1.3–1.5 M MgSO4 or (2) the same buffers with 0.45–0.55 M MgSO4, 0.4–0.9 M Na/K tartrate, and 0.3–0.5 M NDSB-195. Crystals grew for 2 wk to a size of ∼400 × 200 × 100 μm3. Crystals used for X-ray diffraction experiments were soaked in paratone oil and flash-cooled in a stream of nitrogen gas at 100 K. Data for the DHT–hARLDB complex were collected on beamline BM30 at the European Radiation Facility (Grenoble, France) using a MarCCD detector. For the Testo–hARLBD and the THG–hARLBD complexes, diffraction experiments were recorded on an R-AXIS IIc image-plate detector mounted on a Rigaku RU-200 rotating-anode generator equipped with focusing mirrors (Rigaku MSC). The observed reflections were integrated and reduced using the XDS package (Kabsch 1993). Details on data collection are presented in Table 1.
Structure determination and refinement
The DHT–hARLBD complex structure was solved by molecular replacement using the AMoRe program (Navaza 1994) and the hARLBD bound to R1881 as the search model (Protein Data Bank accession code 1E3G; Matias et al. 2000). The initial model obtained was first submitted to a simulated annealing cycle at 3000K and then refined using several rounds of conjugate gradient minimization and individual B-factor refinement in CNS (Brünger et al. 1998). Five percent of the data was randomly selected and excluded from the refinement procedure (Brünger 1992). After each refinement cycle, the model was manually corrected using O (Jones et al. 1991) with the 2Fo − Fc and Fo − Fc calculated maps. Ligand and water molecules were progressively added to the model. Since Testo– and THG–hARLBD complexes crystallized in the same P212121 space group and had similar unit cell parameters as the DHT–hARLBD complex, the initial model for Testo– and THG–hARLBD was generated with a rigid body protocol from REFMAC (Murshudov et al. 1997) using the DHT-complex structure. The refinement procedure for these ligands was the same used for DHT. Before the addition of the ligand in the model, the density of Testo, DHT, and THG was clearly visible in all maps. Sulfate ions were added to the three complex models; DTT and glycerol molecules were observed in the Testo– and THG–hARLBD structures and a HEPES molecule was only added to the THG complex model. The quality of all models was monitored with PROCHECK (Laskowski et al. 1993), and the final refinement results and statistics are shown in Table 1. The volume of the cavity occupied by the ligands was calculated with SPDBV (Kaplan and Littlejohn 2001), and all figures were generated with Molscript (Kraulis 1991) and SPDBV. Protein Data Bank accession codes are 2AM9 for Testo–hARLBD, 2AMA for DHT–hARLBD, and 2AMB for THG–hARLBD.
Molecular modeling
In order to find structural elements explaining the variation in androgen receptor affinity of the three ligands, we calculated the interaction energies of the complexes using ZMM (http://www.zmmsoft.com; Zhorov 1983; Zhorov and Bregestovski 2000). The receptor was represented by a double-shell model, as previously described in Zhorov and Lin (2000), based on our three crystallographic structures. The inner flexible shell was composed of hARLDB amino acids having at least one atom within 8 Å of the ligand, thus selecting 56 residues. Residue internal coordinates of the flexible shell were allowed to move during minimization steps. The other amino acids of the model were included in the outer rigid shell, in which they were not allowed to vary during energy minimization. Water molecules were removed from the models, but the hydration effects were simulated by an implicit method (Lazaridis and Karplus 1999). Partial charges of the ligands were determined with the AM1 method of the MOPAC software (Dewar et al. 1985). The models were then energetically minimized using the Monte Carlo minimization (MCM) protocol (Li and Scheraga 1987) with the AMBER force field (Weiner et al. 1984).
The goal of this modeling was not to find the global energy minimum of the system but to estimate the ligand–receptor interaction energy with minimal changes of the crystal structures. For this purpose, we used a multistep relaxation method, which only affects the flexible shell, with very short MCM trajectories (which were terminated after 10 consecutive energy minimizations). In the first relaxation step, only hydrogen atoms were allowed to move. In the second and third steps, side chains and backbone torsion angles were respectively allowed to vary. Finally, in the fourth step, the constraints were removed for all variables (torsion angles, bond angles, and free particle movement). In all steps however, non-hydrogen atoms were not allowed to move by >1 Å from their crystallographic coordinates, a constraint achieved through a flat-bottom energy function.
Acknowledgments
This work was supported by Endorecherche Inc. P.-L.C. is the recipient of a Master's scholarship provided by the Natural Sciences and Engineering Research Council of Canada. We thank Dr. Sylvain Gauthier and his research group for THG synthesis; ZMMSoft for providing us with the ZMM molecular modeling program; and Ms. Sylvie Méthot for careful reading of the manuscript.
Footnotes
Reprint requests to: Rock Breton, Centre de Recherche en Endocrinologie Moléculaire et Oncologique, Centre Hospitalier de l'Université Laval (CHUL), 2705, boul. Laurier, Ste-Foy, QC G1V 4G2, Canada; e-mail: rock.breton@crchul.ulaval.ca; fax: (418) 654-2761.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051905906.
Abbreviations:hAR, human androgen receptor; NR, nuclear receptor; NTD, N-terminal domain; DBD, DNA-binding domain; LBD, ligand-binding domain; ARE, androgen response element; LBP, ligand-binding pocket; DHT, 5α-androstan-3-one, 17β-ol; Testo, testosterone; THG, tetrahydrogestrinone.
References
- Blanchet J., Lin S.-X., Zhorov B.S. 2005. Mapping of steroids binding to 17β-hydroxysteroid dehydrogenase type 1 using Monte Carlo energy minimization reveals alternative binding modes Biochemistry 44 7218–7227. [DOI] [PubMed] [Google Scholar]
- Bohl C.E., Gao W., Miller D.D., Bell C.E., Dalton J.T. 2005a. Structural basis for antagonism and resistance of bicalutamide in prostate cancer Proc. Natl. Acad. Sci. 102 6201–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohl C.E., Miller D.D., Chen J., Bell C.E., Dalton J.T. 2005b. Structural basis for accommodation of nonsteroidal ligands in the androgen receptor J. Biol. Chem. 280 37747–37754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T. 1992. The free R value: A novel statistical quantity for assessing the accuracy of crystal structures Nature 355 472–474. [DOI] [PubMed] [Google Scholar]
- Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S.et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Catlin D.H., Sekera M.H., Ahrens B.D., Starcevic B., Chang Y.C., Hatton C.K. 2004. Tetrahydrogestrinone: Discovery, synthesis, and detection in urine Rapid Commun. Mass Spectrom. 18 1245–1249. [DOI] [PubMed] [Google Scholar]
- Chavez B., Mendez J.P., Ulloa-Aguirre A., Larrea F., Vilchis F. 2001. Eight novel mutations of the androgen receptor gene in patients with androgen insensitivity syndrome J. Hum. Genet. 46 560–565. [DOI] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project, Number 4). 1994. The CCP4 suite: Programs for protein crystallography Acta Crystallogr. D Biol. Crystallogr. 50 760–763. [DOI] [PubMed] [Google Scholar]
- Dawood M.Y., Obasiolu C.W., Ramos J., Khan-Dawood F.S. 1997. Clinical, endocrine, and metabolic effects of two doses of gestrinone in treatment of pelvic endometriosis Am. J. Obstet. Gynecol. 176 387–394. [DOI] [PubMed] [Google Scholar]
- Death A.K., McGrath K.C., Kazlauskas R., Handelsman D.J. 2004. Tetrahydrogestrinone is a potent androgen and progestin J. Clin. Endocrinol. Metab. 89 2498–2500. [DOI] [PubMed] [Google Scholar]
- De Bellis A., Quigley C.A., Cariello N.F., El-Awady M.K., Sar M., Lane M.V., Wilson E.M., French F.S. 1992. Single base mutations in the androgen receptor gene causing complete androgen insensitivity: Rapid detection by a modified denaturing gradient gel electrophoresis technique Mol. Endocrinol. 6 1909–1920. [DOI] [PubMed] [Google Scholar]
- Dewar M.J.S., Zoebisch E.G., Healy E.F., Stewart J.J.P. 1985. AM1: A new general purpose quantum mechanical molecular model J. Am. Chem. Soc. 107 3902–3909. [Google Scholar]
- Espinosa E., Molins E., Lecomte C. 1998. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities Chem. Phys. Lett. 285 170–173. [Google Scholar]
- Estebanez-Perpina E., Moore J.M., Mar E., Delgado-Rodrigues E., Nguyen P., Baxter J.D., Buehrer B.M., Webb P., Fletterick R.J., Guy R.K. 2005. The molecular mechanisms of coactivator utilization in ligand-dependent transactivation by the androgen receptor J. Biol. Chem. 280 8060–8068. [DOI] [PubMed] [Google Scholar]
- Evans N.A. 2004. Current concepts in anabolic-androgenic steroids Am. J. Sports Med. 32 534–542. [DOI] [PubMed] [Google Scholar]
- Gaddipati J.P., McLeod D.G., Heidenberg H.B., Sesterhenn I.A., Finger M.J., Moul J.W., Srivastava S. 1994. Frequent detection of codon 877 mutation in the androgen receptor gene in advanced prostate cancers Cancer Res. 54 2861–2864. [PubMed] [Google Scholar]
- Gronemeyer H., Gustafsson J.A., Laudet V. 2004. Principles for modulation of the nuclear receptor superfamily Nat. Rev. Drug Discov. 3 950–964. [DOI] [PubMed] [Google Scholar]
- Harris S.E., Rong Z., Harris M.A., Lubahn D.B. 1990. Androgen receptor in human prostate carcinoma LNCaP/ADEP cells contains a mutation which alters the specificity of the steroid-dependent transcriptional activation region (Abstract 275) Program of the 72nd Annual Meeting of The Endocrine SocietyAtlanta, GA.
- Harris S.E., Harris M.A., Rong Z., Hall J., Judge S., French F.S., Joseph D.R., Lubahn D.B., Simental J.A., Wilson E.M. 1991. Androgen regulation of HBGF-1 (aFGF) mRNA and characterization of the androgen-receptor mRNA in the human prostate carcinoma cell line -LNCaP/A-DEP In Molecular and cellular biology of prostate cancer (eds. Karr J.P.et al.) . pp. 315–330. Plenum Press, New York.
- He B., Gampe Jr. R.T., Kole A.J., Hnat A.T., Stanley T.B., An G., Stewart E.L., Kalman R.I., Minges J.T., Wilson E.M. 2004. Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance Mol. Cell 16 425–438. [DOI] [PubMed] [Google Scholar]
- Heinlein C.A. and Chang C. 2004. Androgen receptor in prostate cancer Endocr. Rev. 25 276–308. [DOI] [PubMed] [Google Scholar]
- Hur E., Pfaff S.J., Payne E.S., Gron H., Buehrer B.M., Fletterick R.J. 2004. Recognition and accommodation at the androgen receptor coactivator binding interface PLoS Biol. 2 E274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasuja R., Catlin D.H., Miller A., Chang Y.C., Herbst K.L., Starcevic B., Artaza J.N., Singh R., Datta G., Sarkissian A.et al. 2005. Tetrahydrogestrinone is an androgenic steroid that stimulates androgen receptor-mediated, myogenic differentiation in C3H10T1/2 multipotent mesenchymal cells and promotes muscle accretion in orchidectomized, male rats Endocrinology 146 4472–4478. [DOI] [PubMed] [Google Scholar]
- Jenster G. 1998. Coactivators and corepressors as mediators of nuclear receptor function: An update Mol. Cell. Endocrinol. 143 1–7. [DOI] [PubMed] [Google Scholar]
- Jenster G., van der Korput H.A., van Vroonhoven C., van der Kwast T.H., Trapman J., Brinkmann A.O. 1991. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization Mol. Endocrinol. 5 1396–1404. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou J.Y., Cowan S.W., Kjeldgaard M. 1991. Improved methods for the building of protein models in electron density maps and the location of errors in these models Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch W. 1993. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants J. Appl. Crystallogr. 26 795–800. [Google Scholar]
- Kaplan W. and Littlejohn T.G. Swiss-PDB Viewer (Deep View). Brief Bioinform. 2001. 2 195–197. [DOI] [PubMed] [Google Scholar]
- Keller E.T., Ershler W.B., Chang C. 1996. The androgen receptor: A mediator of diverse responses Front. Biosci. 1 d59–d71. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Labrie F., Candas B., Gomez J.L., Cusan L. 2002. Can combined androgen blockade provide long-term control or possible cure of localized prostate cancer? Urology 60 115–119. [DOI] [PubMed] [Google Scholar]
- Labrie F., Luu-The V., Calvo E., Martel C., Cloutier J., Gauthier S., Belleau P., Morissette J., Levesque M.H., Labrie C. 2005. Tetrahydrogestrinone induces a genomic signature typical of a potent anabolic steroid J. Endocrinol. 184 427–433. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures J. Appl. Crystallogr. 26 283. [Google Scholar]
- Lazaridis T. and Karplus M. 1999. Effective energy function for proteins in solution Proteins 35 152–156. [DOI] [PubMed] [Google Scholar]
- Li Z. and Scheraga H.A. 1987. Monte Carlo-minimization approach to the multiple-minima problem in protein folding Proc. Natl. Acad. Sci. 84 6611–6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangelsdorf D.J., Thummel C., Beato M., Herrlich P., Schutz G., Umesono K., Blumberg B., Kastner P., Mark M., Chambon P.et al. 1995. The nuclear receptor superfamily: The second decade Cell 83 835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matias P.M., Donner P., Coelho R., Thomaz M., Peixoto C., Macedo S., Otto N., Joschko S., Scholz P., Wegg A.et al. 2000. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations J. Biol. Chem. 275 26164–26171. [DOI] [PubMed] [Google Scholar]
- Matias P.M., Carrondo M.A., Coelho R., Thomaz M., Zhao X.Y., Wegg A., Crusius K., Egner U., Donner P. 2002. Structural basis for the glucocorticoid response in a mutant human androgen receptor (AR(ccr)) derived from an androgen-independent prostate cancer J. Med. Chem. 45 1439–1446. [DOI] [PubMed] [Google Scholar]
- Mooradian A.D., Morley J.E., Korenman S.G. 1987. Biological actions of androgens Endocr. Rev. 8 1–28. [DOI] [PubMed] [Google Scholar]
- Mowszowicz I., Riahi M., Wright F., Bouchard P., Kuttenn F., Mauvais-Jarvis P. F. 1981. Androgen receptor in human skin cytosol J. Clin. Endocrinol. Metab. 52 338–344. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin A.A., Dodson E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method Acta Crystallogr. D Biol. Crystallogr. 53 240–255. [DOI] [PubMed] [Google Scholar]
- Navaza J. 1994. AMoRe: An automated package for molecular replacement Acta Crystallogr. A 50 157–163. [Google Scholar]
- Pinsky L., Trifiro M., Kaufman M., Beitel L.K., Mhatre A., Kazemi-Esfarjani P., Sabbaghian N., Lumbroso R., Alvarado C., Vasiliou M.et al. 1992. Androgen resistance due to mutation of the androgen receptor Clin. Invest. Med. 15 456–472. [PubMed] [Google Scholar]
- Poujol N., Wurtz J.M., Tahiri B., Lumbroso S., Nicolas J.C., Moras D., Sultan C. 2000. Specific recognition of androgens by their nuclear receptor. A structure-function study J. Biol. Chem. 275 24022–24031. [DOI] [PubMed] [Google Scholar]
- Quigley C.A., De Bellis A., Marschke K.B., el-Awady M.K., Wilson E.M., French F.S. 1995. Androgen receptor defects: Historical, clinical, and molecular perspectives Endocr. Rev. 16 271–321. [DOI] [PubMed] [Google Scholar]
- Rao C.N.R. 1972. Theory of hydrogen bonding in water In Water: A comprehensive treatise (ed. Franks F.) . pp. 93–114. Plenum Press, New York Vol. 1. [Google Scholar]
- Ris-Stalpers C., Verleun-Mooijman M.C., Trapman J., Brinkmann A.O. 1993. Threonine on amino acid position 868 in the human androgen receptor is essential for androgen binding specificity and functional activity Biochem. Biophys. Res. Commun. 196 173–180. [DOI] [PubMed] [Google Scholar]
- Roy A.K., Lavrovsky Y., Song C.S., Chen S., Jung M.H., Velu N.K., Bi B.Y., Chatterjee B. 1999. Regulation of androgen action Vitam. Horm. 55 309–352. [DOI] [PubMed] [Google Scholar]
- Sack J.S., Kish K.F., Wang C., Attar R.M., Kiefer S.E., An Y., Wu G.Y., Scheffler J.E., Salvati M.E., Krystek Jr. S.R.et al. 2001. Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone Proc. Natl. Acad. Sci. 98 4904–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai N., Yamada T., Asao T., Baba M., Yoshida M., Murayama T. 2000. Bilateral testicular tumors in androgen insensitivity syndrome Int. J. Urol. 7 390–392. [DOI] [PubMed] [Google Scholar]
- Taplin M.E., Bubley G.J., Shuster T.D., Frantz M.E., Spooner A.E., Ogata G.K., Keer H.N., Balk S.P. 1995. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer N. Engl. J. Med. 332 1393–1398. [DOI] [PubMed] [Google Scholar]
- Veldscholte J., Ris-Stalpen C., Kuiper G.G., Jenster G., Berrevoets C., Claassen E., van Rooij H.C., Trapman J., Brinkmann A.O., Mulder E. 1990. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens Biochem. Biophys. Res. Commun. 173 534–540. [DOI] [PubMed] [Google Scholar]
- Wallace A.C., Laskowski R.A., Thornton J.M. 1995. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions Protein Eng. 8 127–134. [DOI] [PubMed] [Google Scholar]
- Weiner S.J., Kollman P.A., Case D.A., Snigh U.C., Ghio C., Alagona G., Profeta Jr. S., Weiner P. 1984. A new force field for molecular mechanical simulation of nucleic acids and proteins J. Am. Chem. Soc. 106 765–784. [Google Scholar]
- Williams S.P. and Sigler P.B. 1998. Atomic structure of progesterone complexed with its receptor Nature 393 392–396. [DOI] [PubMed] [Google Scholar]
- Wurtz J.M., Bourguet W., Renaud J.P., Vivat V., Chambon P., Moras D., Gronemeyer H. 1996. A canonical structure for the ligand-binding domain of nuclear receptors Nat. Struct. Biol. 3 87–94. [DOI] [PubMed] [Google Scholar]
- Zhorov B.S. 1983. Vector method for calculating derivatives of the energy deformation of valence angles and torsion energy of complex molecules according to generalized coordinates J. Struct. Chem. 23 649–655. [Google Scholar]
- Zhorov B.S. and Bregestovski P.D. 2000. Chloride channels of glycine and GABA receptors with blockers: Monte Carlo minimization and structure-activity relationships Biophys. J. 78 1786–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhorov B.S. and Lin S.-X. 2000. Monte Carlo-minimized energy profile of estradiol in the ligand-binding tunnel of 17β-hydroxysteroid dehydrogenase: Atomic mechanisms of steroid recognition Proteins 38 414–427. [PubMed] [Google Scholar]