

Abstract
C-terminal binding proteins (CtBPs) are moonlighting proteins involved in nuclear transcriptional corepression and in Golgi membrane tubule fission. Structural information on CtBPs is available for their substrate-binding domain, responsible for transcriptional repressor recognition/binding, and for the nucleotide-binding domain, involved in NAD(H)-binding and dimerization. On the contrary, little is known about the structure of CtBP C-terminal region (∼90 residues), hosting sites for post-translational modifications. In the present communication we apply a combined approach based on bioinformatics, nuclear magnetic resonance, circular dichroism spectroscopy, and small-angle X-ray scattering, and we show that the CtBP C-terminal region is intrinsically unstructured in the full-length CtBP and in constructs lacking the substrate- and/or the nucleotide-binding domains. The flexible nature of this protein region, and its structural transitions, may be instrumental for CtBP recognition and binding to diverse molecular partners.
Keywords: CtBP, circular dichroism, SAXS, protein-NMR, intrinsically disordered proteins, transcription corepressor
C-terminal binding proteins (CtBPs) belong to a recently discovered protein family, hosting isoforms CtBP1, 2, and 3, that show distinct functions according to their cellular localization. In the nucleus, CtBPs act as transcriptional corepressors, targeting DNA-binding proteins. Two-hybrid screens have identified in Drosophila and mammals about three dozen CtBP-interacting transcription factors (Turner and Crossley 2001; Chinnadurai 2002, 2003). CtBP corepressor activity has been proposed to rely on its ability to oligomerize, thus acting as a scaffold in the formation of a multiprotein complex (∼106 Da) that hosts the essential components of both gene targeting and coordinated histone modifications, thus allowing repression of the targeted genes (Shi et al. 2003).
In isolated Golgi apparatus the CtBP3 isoform (previously known as BARS [Brefeldin A-ADP Ribosylated Substrate] and recently renamed as short-CtBP1 or CtBP1-S) has been shown to be a key component of the machinery controlling Golgi tubule fission (Weigert et al. 1999). In recent studies on intact cells, the CtBP fission-inducing activity was shown to participate in the fragmentation of the Golgi complex during mitosis (Hidalgo Carcedo et al. 2004) and in intracellular membrane traffic (Bonazzi et al. 2005). Thus, based on its ability to fulfill different functions depending on cellular localization, CtBP has been described as a moonlighting protein (Turner and Crossley 2001).
From the structural viewpoint, CtBPs are composed of three distinct domains (Nardini et al. 2003). The substrate-binding domain (∼155 residues, mainly N-terminal) is involved in the binding of transcription repressors, relying on the specific recognition of a consensus sequence, -Pro-X-Asp-Leu-Ser-(PXDLS), present in various transcription factors (Turner and Crossley 2001). The CtBP nucleotide-binding domain (or central domain; ∼195 residues), binds a NAD(H) molecule responsible for modulation of corepression activity (Zhang et al. 2002; Fjeld et al. 2003). The C-terminal domain (∼90 residues) is thought to play regulatory functions, being the site of post-translational modifications. Phosphorylation of the C-terminal domain residue Ser422 by UV activated homeodomain interacting protein kinase 2 (HIPK2) targets CtBP1 to the proteasomal degradation pathway. The resultant decrease in CtBP levels, similar to the cellular response to p53 activation by HIPK2, addresses cells to apoptosis (Zhang et al. 2003, 2005). SUMOylation at the C-terminal Lys428 of CtBP1 via the action of PC2, a polycomb group protein, promotes CtBP nuclear localization (Kagey et al. 2003); SUMOylation of CtBP is inhibited by binding of nNOS PDZ domain, in keeping with the known inhibitory effects of nNOS on the nuclear accumulation of CtBP (Lin et al. 2003).
The three-dimensional structures of a truncated form of human CtBP1 (residues 28–353) (PDB code 1MX3; Kumar et al. 2002) and rat CtBP3 (residues 15–345) (PDB codes 1HKU and 1HL3; Nardini et al. 2003), comprising the substrate- and the nucleotide-binding domains (t-CtBP), have been solved by means of X-ray crystallography. On the contrary, the structure of the full-length protein, or of the CtBP C-terminal region (CtBP1 residues 354–440 and CtBP3 residues 346–430), has remained elusive. Here, we investigate the structural features of full-length CtBPs using complementary biophysical approaches (bioinformatics, NMR, CD, and small-angle X-ray scattering [SAXS]) applied to rat CtBP3. We focus specifically on the CtBP C-terminal domain, both in the full-length protein and in two designed protein constructs. The C-terminal region of CtBP3 displays sequence and structural features typical of intrinsically unstructured proteins, a property that may be functional to the assembly of CtBP in the core nuclear complex and/or for the recognition of diverse molecular partners.
Results
Signals from sequence analysis
The C-terminal region of CtBP (CtBP C-term) displays an amino acid composition different from that of any protein of known three-dimensional structure, being rich in Gly and Pro residues (Fig. 1). Bioinformatic analysis of CtBP C-term indicates very little predicted secondary structure, essentially consisting of few very short beta segments (Fig. 1), as typical of unstructured protein regions (Liu et al. 2002). Furthermore, only 23% of the CtBP3 C-term sequence (residues 345–430) is composed of “order promoting” residues (such as Asn, Cys, Ile, Phe, Trp, and Tyr), while almost 56% is composed of “disorder promoting” residues (especially Ala, Gly, Pro, and Ser), as described for several intrisically disordered proteins (Dunker et al. 2001). These values should be compared to an average of 36% and 47% for the two residue classes, respectively, reported for regularly folded globular proteins.
Figure 1.

CtBP sequence alignments. Rat CtBP3 (CtBP3_r, SWISS-PROT:09Z2F5) is aligned with human CtBP1 (CtBP1_h, SWISS-PROT:Q13363), mouse CtBP1 (CtBP1_m, SWISS-PROT:088712), human CtBP2 (CtBP2_h, SWISS-PROT:P56545), mouse CtBP2 (CtBP2_m, SWISS-PROT:P56546), and Xenopus laevis CtBP (isoforms CtBP_xa, SWISS-PROT:Q9YHU; CtBP_xb, SWISS-PROT:Q9W758). The sequence alignment was performed using the CLUSTALW program (http://www.ebi.ac.uk/clustalw). Residues identical in all sequences are highlighted in yellow. Residues identical to CtBP3 in >50% of the aligned sequences are highlighted in gray. Pro and Gly residues in the C-terminal region are shown in blue and in black, respectively. Secondary structure of t-CtBP3 (residues 15–345) and secondary structure prediction for the C-terminal region are shown in black and in orange, respectively, with a = helix and b = strand. The C-term(1) and C-term(2) constructs are indicated by green and by red horizontal bars, respectively.
Using both the GlobPlot approach (Linding et al. 2003) and the VL-XT PONDR predictor of naturally disordered regions (Li et al. 1999), CtBP3 C-term is predicted to be natively unfolded. GlobPlot does not recognize any potential globular domain on CtBP3 C-term, indicating two main regions (residues 366–380 and 396–427) as disordered (Fig. 2A). Accordingly, VL-XT PONDR predicts most of CtBP3 C-term as intrinsically disordered (PONDR score >0.5; residues underlined in Fig. 2B). Interestingly, as positive control, both GlobPlot and PONDR clearly identify the globular and ordered fold of the substrate- and nucleotide-binding domains in the full-length CtBP3 sequence (Fig. 2). Conversely, disordered regions are predicted at the N terminus of the protein (disordered in the crystal structure of t-CtBP3) and mainly in loops connecting secondary structure elements (Nardini et al. 2003).
Figure 2.
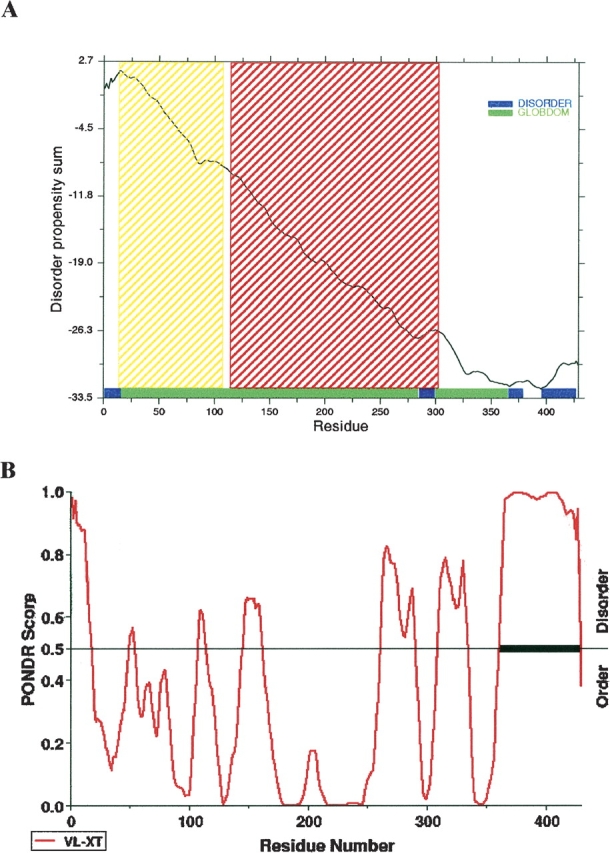
Sequence prediction of unstructured regions. (A) GlobPlot: The disorder propensity score is plotted against residue number. Residue ranges predicted for globular and disordered segments are indicated by green and by blue bars, respectively. Shaded regions (yellow and red) correspond to the substrate- and nucleotide-binding domains of CtBP3, predicted on the basis of its sequence similarity to D-2-hydroxyacid-specific NAD-dependent dehydrogenases (SMART/Pfam prediction: http://smart.embl.de). (B) PONDR: The prediction score is plotted against residue number. Residues with a score above a threshold of 0.5 are considered disordered. The disordered C-terminal region is marked by a thick bar.
A further bioinformatic analysis was carried out to check whether the CtBP C-term sequence shows a combination of low mean hydrophobicity, compared to the boundary hydrophobicity (see Materials and Methods for definition of these parameters), coupled to a relatively high net charge, as typically found in natively unfolded proteins or domains. The CtBP3 C-term mean hydrophobicity H, and the mean net charge R, calculated according to the Uversky method (Uversky et al. 2000) are 0.508 and 0.047, respectively; the CtBP3 C-term boundary hydrophobicity was calculated to be 0.430. Unexpectedly, such figures locate CtBP3 C-term in a region of the (R,H) space assigned to folded proteins. However, it has been suggested that proteins identified as unstructured by disorder predictors, such as PONDR, but predicted as ordered by charge-hydropathy calculations, should have properties consistent with a dynamic, collapsed disordered state (i.e., molten globule) (Oldfield et al. 2005).
It is worth noting that the above considerations on the intrinsically disordered nature of the CtBP3 C-term equally extend to the corresponding regions of CtBP1 and CtBP2 from different species, which display closely related amino acid sequences (Fig. 1).
NMR analysis of the C-terminal region
A second approach to the study of the CtBP C-term structural properties was based on the 1H-NMR investigation of the C-term(1) construct. Figure 3 reports an overlap of TOCSY (total correlation spectroscopy) (blue) and NOESY (nuclear Overhauser effect spectroscopy) (red) cross-peaks in the fingerprint region, corresponding to correlations between amidic (7.0–9.5 ppm) and side chain (0.5–5.5 ppm) proton resonances. These signals appear to be poorly resolved, mainly due to the reduced spread of amide resonances, falling all in the 7.6–8.7 ppm chemical shift range. This observation usually indicates the absence of structural organization of the backbone (Wüthrich 1986).
Figure 3.

NMR spectra of the CtBP3 C-term(1) construct. Expansion of the TOCSY (blue) and NOESY (red) spectra, corresponding to residues Ala306–Leu430, in 10 mM potassium phosphate buffer (10.7 mg/mL, 298 K at pH 6.6) showing the fingerprint region (F2, 9.5–7.0 ppm; F1, 5.5–0.5 ppm).
Characteristic indole spin systems in the TOCSY spectrum were detected, corresponding to residues Trp307 and Trp351; however, their assignment was precluded by the spectral crowding in the NOESY spectrum. For the same reason, it was not possible to detect medium-range NOE peaks characteristic of secondary structures. Although the X-ray structure of t-CtBP3 (Nardini et al. 2003) and the secondary structure prediction of the 130 amino acid C-term(1) construct both indicate occurrence of the α5 helix in the Glu310–Thr329 stretch (Fig. 1), typical contacts between the Cα proton of residue i and the NH proton of residue i + 3 or i + 4 cannot be recognized in the NMR spectra of the C-term(1) construct (Wüthrich 1986).
CD spectroscopy comparison of full-length CtBP3 and t-CtBP3
In order to evaluate the secondary structure composition of the C-term region of CtBP3, CD spectra of both full-length CtBP3 (residue 1–430) and t-CtBP3 (residues 1–350) were collected in the far-UV under the same experimental conditions; the t-CtBP3 spectrum was then subtracted from that of full-length CtBP3 (Fig. 4). Both full-length and t-CtBP3 CD spectra are typical of α/β proteins, in agreement with the crystal structure of t-CtBP3 (Nardini et al. 2003). The difference spectrum shows a slightly positive trend in the 208–240 nm region, indicating no significant increase of α or β secondary structure in the full-length protein relative to t-CtBP3, and a clear minimum around 200 nm, in the region associated to random coil/unfolded conformation (Hollósi et al. 1993).
Figure 4.

Far-UV CD measurements. (Continuous line) CD spectra of full-length CtBP3, (dotted line) t-CtBP3, and (dashed line) their difference.
SAXS analysis of the full-length CtBP3 and of the C-term(2) construct
The experimental SAXS curve for full-length CtBP3 is displayed in Figure 5A. The estimated molecular weight of the solute (170 ± 10 kDa) suggests that the protein is tetrameric in solution. Accordingly, the excluded volume of the particle in solution (Porod volume) is (330 ± 15) × 103 Å3, noting that for globular proteins the hydrated volume in Å3 should numerically be about twice the molecular mass in Da. The experimental radius of gyration Rg and maximum size Dmax (52.3 ± 0.8 Å and 190 ± 10 Å, respectively) point to an elongated particle structure. The low-resolution shape of tetrameric CtBP3, reconstructed ab initio (with 222 symmetry constraint) using DAMMIN (Svergun 1999), has the overall size of ∼80 × 80 × 190 Å3, fitting the experimental data with discrepancy χ = 1.36 (Fig. 5A). Very similar shapes were obtained in the absence of symmetry.
Figure 5.
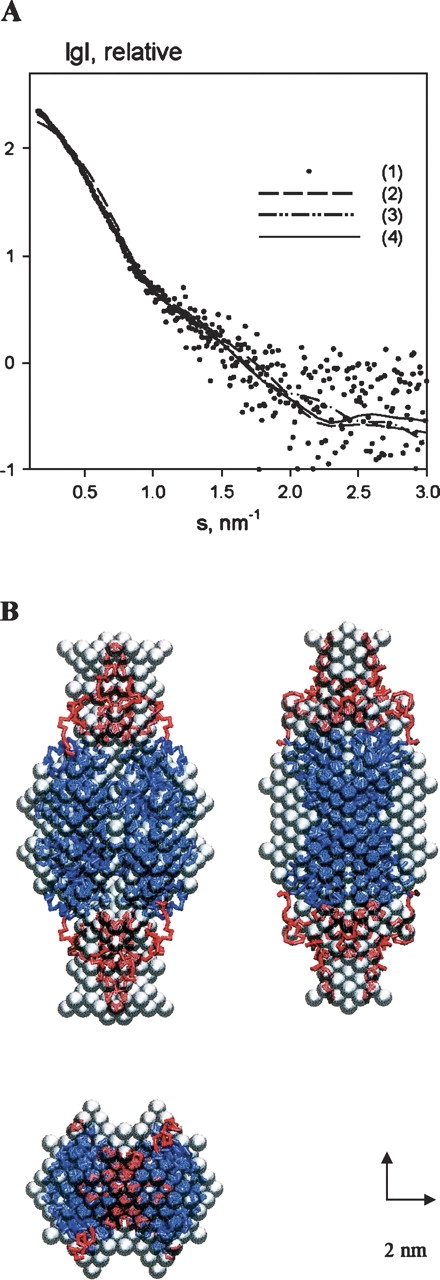
SAXS results on the full-length CtBP3. (A) Experimental scattering curve (dots, 1), and the scattering from the models: (dashed line, 2) tetrameric crystallographic model of the t-CtBP3, (dashed/dotted line, 3) ab initio bead model obtained by DAMMIN, (continuous line, 4) crystallographic model with added C-terminal loops obtained by BUNCH. The plot displays the logarithm of the scattering intensity as a function of momentum transfer s = 4π sin(θ)/λ, where θ is the scattering angle and λ = 0.15 nm is the X-ray wavelength. (B) Models of tetrameric CtBP3. Gray beads show the ab initio model of the full-length tetrameric CtBP3 obtained by DAMMIN assuming 222 symmetry of the assembly. The gray envelope is superimposed to the t-CtBP3 crystal model (blue) and to the calculated loops of C-terminal segment (red), both drawn as Cα traces.
Figure 5B presents a superposition of the ab initio reconstructed molecular shape and the tetrameric assembly derived from the crystal structure of t-CtBP3, where the monomeric truncated protein present in the asymmetric unit builds up a compact tetramer (a dimer of dimers with approximate 222 symmetry) (Nardini et al. 2003). The SAXS curve computed from the crystallographic tetramer (Fig. 5A) yielded a poor fit to the experimental data with χ = 5.4. Such discrepancy can be explained by the absence of the C-terminal region in the t-CtBP3 crystallographic model. The superposition of Figure 5B clearly shows that the compact crystallographic tetramer matches the central core of the ab initio reconstructed model and that the missing C-terminal region is rather extended, being located in the peripheral part of the tetrameric assembly. A good fitting with the experimental scattering data on the full-length CtBP3 (χ = 1.21) could be obtained by building the C-terminal region of each protein of the tetramer as a flexible chain of dummy residues, and by adding them to the known part of the structure (maintaining the 222 assembly symmetry). Several independent simulated annealing runs yielded similar conformations for the C-terminal regions; the overall shape of the final model and the location of the C termini are consistent with the low-resolution ab initio models constructed from the scattering data without a priori information (Fig. 5B). Notably, despite intermolecular contacts, the C-terminal loops appear to be much less compact than the rest of full-length CtBP3.
The experimental curve from the C-term(2) construct is shown in Figure 6A; comparison of the molecular weight of the solute measured by SAXS (56 ± 5 kDa) with that expected for the monomeric construct (26.5 kDa) indicates that C-term(2) assembles as a dimer in solution. The excluded volume (126 ± 10 × 103 Å3) is larger than the value expected for a dimeric construct, which may point to unusual flexibility and high degree of hydration of the overall molecular structure. Remarkably, the values of Rg (56.3 ± 0.7 Å) and Dmax (200 ± 10 Å) exceed those of tetrameric full-length CtBP3, indicating an extremely anisotropic shape for the C-term(2) construct. One can thus conclude that the latter construct must be at least partly unfolded. The ab initio low-resolution model, reconstructed assuming twofold symmetry, fits the experimental data with χ = 2.20 (Fig. 6A), and displays a V-like shape with dimensions about 40 × 40 × 200 Å3 (Fig. 6B).
Figure 6.
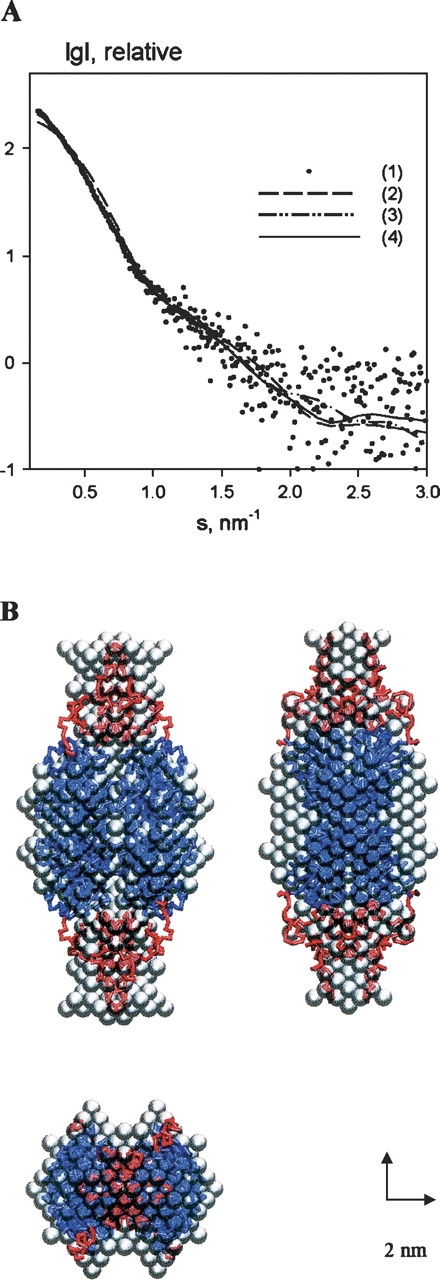
SAXS results on the CtBP3 C-term(2) construct. (A) Experimental scattering curve (dots, 1), the scattering from ab initio bead model with twofold symmetry (dashed line, 2) and from the dimeric model with added C-terminal loops (continuous line, 3). (B) Models of dimeric CtBP3 C-term(2) construct. The gray ab initio bead model of the C-term(2) dimer, obtained by DAMMIN assuming twofold symmetry, is superimposed to the t-CtBP3 crystal structure of the substrate-binding domain (blue) and with added loops calculated for the C-terminal segment (red), both drawn as Cα traces.
When the C-term(2) construct dimeric model was assembled using the proper fragments from the t-CtBP3 crystal structure, the SAXS experimental data were fitted (assuming twofold symmetry) with a discrepancy of χ = 1.55 (Fig. 6A). The C-term(2) construct model displays contacts between the globular substrate-binding domains, whereas the extended C-terminal parts occupy the periphery of the dimer (Fig. 6B). Remarkably, the C-terminal region of the C-term(2) construct appears to be more extended/unfolded than in the full-length protein, where the C termini are constrained by contacts with the globular regions within the tetramer (Fig. 5B).
Discussion
The present study on the structural properties of CtBP C-term complements the crystallographic information collected in the past few years on human t-CtBP1 (Kumar et al. 2002) and rat t-CtBP3 (Nardini et al. 2003). While the crystallization of both t-CtBP1 and t-CtBP3 was readily achieved, attempts to crystallize full-length CtBPs have been unsuccessful. Moreover, we did not succeed in crystallizing both the CtBP3 C-term(1) and the C-term(2) soluble constructs, after extensive exploration of crystal growth conditions. An explanation for such behaviors lies in the high mobility, or disorder, of the C-terminal region in the full-length protein that, contrary to the substrate- and nucleotide-binding domains, may maintain an unstructured conformation, hampering the formation of productive crystal contacts. In keeping with such proposed flexibility, both CtBP1 (Kumar et al. 2002) and CtBP3 (data not shown) are proteolytically unstable, being cleaved in the region connecting the C-terminal segment to the structured protein core.
The analysis of CtBP3 primary structure, based on GlobPlot (Linding et al. 2003) and VL-XT PONDR (Li et al. 1999) approaches, points at a mainly unstructured C-terminal region (Fig. 2). Analysis of CtBP3 C-term sequence (and of corresponding regions in CtBP1 and CtBP2) highlights the presence of an amino acid composition bias, with a low content of bulky hydrophobic residues, and a high proportion of “disorder promoting” residues, particularly evident for Gly and Pro (Fig. 1). The lack of order-promoting residues and of aromatic amino acids undermines one of the basic contributions to the thermodynamic stabilization of the protein hydrophobic core, being a feature typical of natively unfolded proteins or domains (Dunker et al. 2001; Romero et al. 2001). The prediction of structural disorder for CtBP3 C-term is confirmed by the 1H-NMR correlation experiments performed on the C-term(1) construct, showing a dramatic collapse of amidic resonances in a small (1.1 ppm) range. Moreover, NOESY signals are largely overlapped, preventing the recognition of spectral signatures of secondary structure elements. Thus, 1H-NMR analysis indicates that large portions of the C-term(1) protein backbone experience dynamic fluctuations, lacking a well-defined three-dimensional structure. Such a disordered state extends to the N-terminal portion of the C-term(1) construct, covering the substrate-binding domain α5 helix in the full-length protein (Fig. 1). Since α5 (residues 310–329) is tightly packed against the rest of the substrate-binding domain in the crystal structures of both t-CtBP3 (Nardini et al. 2003) and t-CtBP1 (Kumar et al. 2002), lack of the substrate-binding domain in the C-term(1) construct is likely responsible for the observed α5 unwinding.
In order to check if the intrinsic disorder suggested by sequence analysis, and identified by the NMR experiment on the CtBP3 C-term(1) construct, is indeed a genuine property of the C-terminal segment in the full-length CtBP3, we carried over CD experiments on the full-length CtBP3 and on t-CtBP3 (Fig. 4). The difference between the two far-UV CD spectra shows as the main feature a minimum around 200 nm, indicating that the C-term (residues 351–430) contribution to the CD signal in full-length CtBP3 can be ascribed essentially to random coil conformation, typical of an unstructured protein region. Although the CD spectrum of full-length CtBP3 does not show a significant increase of ellipticity relative to t-CtBP in the region associated to α and β secondary structures, the presence of some residual secondary structure in the C-term segment, typical of a pre–molten globule state and/or induced by domain–domain interaction with the rest of the protein, cannot be excluded. Such a conclusion is supported by the results of SAXS experiments on the different protein constructs (see below). Based on the NMR and CD evidences, we can therefore conclude that the CtBP3 C-term alone cannot fold into a structured independent domain, and that it keeps its mainly disordered nature also in the full-length protein.
Furthermore, we have investigated the conformational state of CtBP3 C-term in full-length CtBP3 and in the C-term(2) construct using SAXS techniques. SAXS analysis of full-length CtBP3 yields a radius of gyration and a maximum diameter compatible with a tetrameric assembly of the protein. As for the quaternary assembly observed in the crystals (Nardini et al. 2003), CtBP3 dimers assemble in solution through their nucleotide-binding domains, with 222 symmetry; the C-terminal regions of each chain in the tetramer are characterized by low globularity, providing few quaternary interactions at the dimeric interfaces (Fig. 5B).
The SAXS experiments on C-term(2) confirmed the trend for an elongated/nonglobular state in the C-terminal region (Fig. 6B), with a higher degree of unfolding relative to full-length CtBP3, and with a different global location relative to the substrate-binding domain. Therefore, the SAXS measurements strongly support the idea that the CtBP3 C-term, while maintaining high flexibility and low globularity, takes advantage of quaternary interactions to modulate its degree of compactness/structuring.
Considering the close sequence similarity among members of the CtBP family (Fig. 1), the intrinsic structural flexibility here characterized for the CtBP3 C-term is likely to be a common feature of CtBP1 and CtBP2 from different species. Such structural malleability may be justified, considering the functional role of intrinsically disordered/unstructured proteins or their domains, which have the potential to modulate the action of different partner molecules, thus enabling the fulfillment of different and often unrelated functions, as is the case of CtBP. Based on the above results, further structural studies on full-length CtBPs will focus on complexes with protein partners that could help stabilize the highly flexible C-terminal region here characterized.
Materials and methods
Expression and purification of full-length CtBP3, t-CtBP3, C-term(1) and C-term(2)
The CtBP3 coding sequence (GenBank accession no. AF067795) was amplified by PCR with the following primers: 5′-GCTCATATGTCAGGCGTCCGACCTC-3′ and 5′-GCGTGATCACTACAACTGGTCAGTCG-3′. The PCR product was digested with NDEI and BCLI, and cloned in NDEI/BCLI-digested pETlld-His derived from the addition of a sequence encoding a poly-His sequence in pETlld (Novagen), generating the pETlld-His-CtBP3 plasmid that was then transformed in the Escherichia coli BL2l-pLysS strain. The DNA coding for the t-CtBP3 (residues 1–350), the C-term(1) (residues 306–430), and the C-term(2) (residues 1–113 linked by GlyGlySer to residues 307–430) constructs were generated as reported previously (Nardini et al. 2002; Hidalgo Carcedo et al. 2004; Yang, et al. 2005). Protein expression and purification for full-length CtBP3 and t-CtBP3 were detailed elsewhere (Nardini et al. 2002; Valente et al. 2005). A similar procedure was used to purify the C-term(1) and the C-term(2) constructs, with the only changes concerning bacteria incubation overnight at 25°C, rather than for 2 h at 37°C, after IPTG addition, and additional protein purification on a Sephacryl 100 16/60 column (Amersham Biosciences) rather than a Sephacryl 200 column.
For NMR analysis 7 mg of C-term(1) were dialyzed in 10 mM potassium phosphate buffer (pH 6.6) and lyophilized. For CD measurements full-length CtBP3 and t-CtBP3 were dialyzed against PBS buffer (2.7 mM KCl, 1.5 mM KH2PO4, 136.8 mM NaCl, 8 mM Na2HPO4), 25% glycerol (pH 7.4). For SAXS experiments about 20 mg full-length CtBP3, or C-term(2), were concentrated up to 10 mg mL−1 and dialyzed twice against 1000× volumes of 5 mM Tris-HCl, 250 mM NaCl, 10 mM β-mercaptoethanol, 1 mM EDTA, 200 mM imidazole (pH 8.0).
Sequence analysis and secondary structure predictions
The sequence of CtBP3 C-terminal segment was submitted for secondary structure prediction at the JPRED server (http://on.ebi.ac.uk/servers/jpred.html). The analysis of native disorder propensity was carried out using the program GlobPlot (http://globplot.embl.de/; Linding et al. 2003) and by the Predictor of Natural Disordered Regions (PONDR) server, using the integrated predictor VL-XT (http://www.pondr.com/; Li et al. 1999; Romero et al. 2001). [Access to PONDR is provided by Molecular Kinetics, under license from the WSU Research Foundation.]
The mean net charge (R) of the protein was calculated using the program ProtParam at the EXPASY server (http://ca.expasy.org/tools) as the absolute value of the difference between the number of positively and negatively charged residues divided by the total number of amino acid residues. The mean hydrophobicity (H) is the sum of the normalized hydrophobicities of individual residues divided by the total number of amino acid residues, minus four residues (to take into account fringe effects in the calculation of hydrophobicity). Individual hydrophobicities were determined using the Protscale program at the EXPASY server, using the options “Hphob/Kyte & Doolittle,” a window size of 5, and normalizing the scale from 0 to 1. The values computed for individual residues were then summed, and divided by the total number of residues minus 4 to yield H. HBoundary was computed as HBoundary = (R + 1.15)/2.785 (Uversky 2002).
NMR spectroscopy
The C-term(1) construct was dissolved at the concentration of 10.7 mg/mL in 10 mM potassium phosphate buffer (pH 6.6) containing 8% D2O, to provide the deuterium signal for the field-frequency lock. NMR experiments were carried out at 298 K on a Bruker Avance 600 spectrometer (Bruker Biospin) operating at 14 T (corresponding to a proton Larmor frequency of 600 MHz) equipped with a triple axis-PFG probe optimized for the detection of the 1H nucleus.
The TOCSY experiment was carried out by means of the MLEV17 pulse sequence for isotropic mixing with a duration of 60 msec and a spin-locking field strength of 10 kHz. The time-proportional phase incrementation (TPPI) phase cycling method was used to obtain complex data points in the t1 dimension. The NOESY experiment was carried out by the standard pulse sequence with a mixing time of 120 msec, with the TPPI phase-cycling scheme. Solvent suppression was achieved by means of pulsed field gradients sculpting. Typical experimental settings for 2D-NMR experiments included spectral width, 7200 Hz (both F2 and F1 dimensions); recycle delay, 2.5 sec; 2048 and 512 complex data points for the t2 and t1 dimensions, respectively; 32 scans for t1 increment. The data were apodized with a square cosine window function and zero filled to a matrix of 1024 × 1024 points prior to Fourier transformation and baseline correction (Wüthrich 1986; Cavanagh et al. 1996).
CD spectroscopy
Far-UV CD spectra on full-length CtBP3 and on t-CtBP3 were collected at 20°C using a Jasco-810 spectrophotometer and a cuvette with 0.1-cm path length. All experiments were performed in PBS buffer, 25% glycerol (pH 7.4), at the protein concentration of 0.17 mg/mL. The spectra were registered from 195 to 250 nm and ran at a scan speed of 20 nm/min, time response of 2 sec, and data pitch of 0.2 nm. All the spectra were baseline-corrected. The molar mean residue ellipticity [θ] was expressed in degrees cm2 dmol−1, and calculated as [θ] = θobs MWR/(10 l c), where θobs is the observed ellipticity in degrees; MWR, the mean residue molar weight of the protein (108.44 and 109.83 for full-length CtBP3 and t-CtBP3, respectively); l, the optical path length in centimeters; and c, the protein concentration in grams per milliliters.
SAXS analyses
Synchrotron radiation X-ray scattering data were collected on the X33 beamline of the EMBL (DESY). Solutions of full-length CtBP3 were measured at protein concentrations of 2.0, 4.0, 5.2, and 8.0 mg/mL using a linear proportional gas detector, at sample-detector distance 2.4 m, and wavelength λ = 1.5 Å, covering the momentum transfer range 0.013 < s < 0.33 Å−1. Solutions of the C-term(2) construct were measured at concentrations of 2.0, 5.0, 10.0, and 20.0 mg/mL, at sample-detector distance of 2.7 m (λ = 1.5 Å), using a MAR345 Image Plate detector in the range 0.012 < s < 0.45 Å−1. To check for radiation damage, the gas detector data were collected in 15 successive 1-min frames; for the image plate, two 2-min exposures were compared; no radiation effects were observed. The data were averaged after normalization to the intensity of the incident beam, corrected for the detector response, and scattering of the buffer was subtracted using the program package PRIMUS (Konarev et al. 2003).
The forward scattering I(0) and the radius of gyration Rg were evaluated using the Guinier approximation (Guinier 1939) and by the indirect transform package GNOM (Svergun 1992), the latter providing the maximum particle dimension Dmax. Molecular masses (MM) were estimated by comparison of the forward scattering with that from reference solutions of bovine serum albumin (MM = 66 kDa). The excluded (so-called Porod) volume of the hydrated particle was computed using the equation
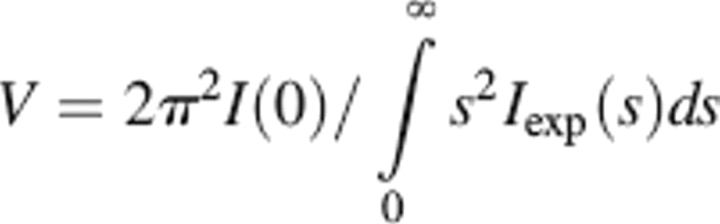 |
where Iexp(s) are the experimental data. Prior to this analysis an appropriate constant was subtracted from each data point to force the s−4 decay of the intensity at higher angles required for homogeneous particles. This “shape scattering” curve was further used to generate low-resolution ab initio models of CtBP3 full-length and C-term(2). The program DAMMIN (Svergun 1999) represents the protein as a compact interconnected assembly of beads to minimize the discrepancy
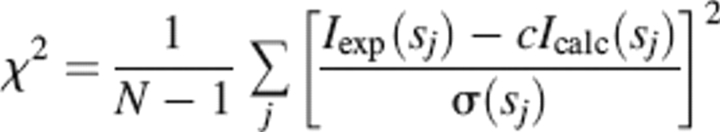 |
where N is the number of experimental points, c is a scaling factor, and Icalc(sj) and σ(sj) are the calculated intensity and the experimental error at the momentum transfer sj, respectively.
The scattering from the crystallographic model of tetrameric t-CtBP3 (Nardini et al. 2003) was calculated using CRYSOL (Svergun et al. 1995). To further characterize the missing C-terminal segment in the full-length CtBP3, the former was represented as an interconnected chain composed of dummy residues (characterized by 222 point group symmetry) (Petoukhov et al. 2002). The program BUNCH (Petoukhov and Svergun 2005) was employed to find a native-like configuration of the C-terminal fragment that fits the scattering from the full-length protein. Similar modeling was applied for the dimeric C-term(2) construct using twofold symmetry.
Acknowledgments
This work was supported by the Italian Ministry of University FIRB Grants to D.C. and M.B., and by AIRC (Italy) and by Telethon (Italy). Access to the EMBL Hamburg Outstation was supported by EU–Research Infrastructure Action under the FP6 contract RII3/CT/2004/5060008. M.B. is grateful to CIMAINA (University of Milano) and to Fondazione CARIPLO (Milano, Italy) for continuous support.
Footnotes
Reprint requests to: Martino Bolognesi, Department of Biomolecular Sciences and Biotechnology, University of Milano, Via Celoria 26 I-20131, Milano, Italy; e-mail: martino.bolognesi@unimi.it; fax: +39-02-503-14895.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062115406.
Abbreviations:BARS, brefeldin A-ADP ribosylated substrate; CD, circular dichroism; CtBP, C-terminal binding protein; CtBP C-term, C-terminal segment of CtBP; HIPK2, homeodomain interacting protein kinase 2; MM, molecular mass; NOESY, nuclear Overhauser effect spectroscopy; SAXS, small-angle X-ray scattering; SUMO, small ubiquitin-related modifier; t-CtBP3, truncated CtBP3; TOCSY, total correlation spectroscopy; TPPI, time-proportional phase incrementation.
References
- Bonazzi M., Spanò S., Turacchio G., Cericola C., Valente C., Colanzi A., Kweon H.-S., Hsu V.W., Polishchuck E.V., Corda D.et al. 2005. CtBP3/BARS drives membrane fission at dynamin-independent trafficking pathways Nat. Cell Biol. 7: 570–580. [DOI] [PubMed] [Google Scholar]
- Cavanagh J., Fairbrother W.J., Palmer III A.G., Skelton N.J. In Protein NMR spectroscopy. Principles and practice . 1996. Academic Press, San Diego, CA.
- Chinnadurai G. 2002. CtBP, an unconventional transcriptional corepressor in development and oncogenesis Mol. Cell 9: 213–224. [DOI] [PubMed] [Google Scholar]
- Chinnadurai G. 2003. CtBP family proteins: More than transcriptional corepressors Bioessays 25: 9–12. [DOI] [PubMed] [Google Scholar]
- Dunker A.K., Lawson J.D., Brown C.J., Williams R.M., Romero P., Oh J.S., Oldfield C.J., Campen A.M., Ratliff C.M., Hipps K.W.et al. 2001. Intrinsically disordered protein J. Mol. Graph. Model. 19: 26–59. [DOI] [PubMed] [Google Scholar]
- Fjeld C.C., Birdsong W.T., Goodman R.H. 2003. Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor Proc. Natl. Acad. Sci. 100: 9202–9207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinier A. 1939. La diffraction des rayons X aux très petits angles; application a l’étude de phenomenes ultramicroscopiques Ann. Phys. (Paris) 12: 161–237. [Google Scholar]
- Hidalgo Carcedo C., Bonazzi M., Spanò S., Turacchio G., Colanzi A., Luini A., Corda D. 2004. Golgi fragmentation during mitosis requires the membrane fissioning protein CtBP3/BARS Science 305: 93–96. [DOI] [PubMed] [Google Scholar]
- Hollósi M., Ötvös Jr. L., Urge L., Kajtár J., Perczel A., Laczkó I., Vadász Z., Fasman G.D. 1993. Ca2+-induced conformational transitions of phosphorylated peptides Biopolymers 33: 497–510. [DOI] [PubMed] [Google Scholar]
- Kagey M.H., Melhuish T.A., Wotton D. 2003. The polycomb protein Pc2 is a SUMO E3 Cell 113: 127–137. [DOI] [PubMed] [Google Scholar]
- Konarev P.V., Volkov V.V., Sokolova A.V., Koch M.H.J., Svergun D.I. 2003. PRIMUS—A Windows-PC based system for small-angle scattering data analysis J. Appl. Crystallogr. 36: 1277–1282. [Google Scholar]
- Kumar V., Carlson J.E., Ohgi K.A., Edwards T.A., Rose D.W., Escalante C.R., Rosenfeld M.G., Aggarwal A.K. 2002. Transcription corepressor CtBP is a NAD+-regulated dehydrogenase Mol. Cell 10: 857–869. [DOI] [PubMed] [Google Scholar]
- Li X., Romero P., Rani M., Dunker A.K., Obradovic Z. 1999. Predicting protein disorder for N-, C-, and internal regions Genome Inform. Ser. Workshop Genome Inform. 10: 30–40. [PubMed] [Google Scholar]
- Lin X., Sun B., Liang M., Liang Y.-Y., Gast A., Hildebrand J., Brunicardi F.C., Melchior F., Feng X.-H. 2003. Opposed regulation of corepressor CtBP by SUMOylation and PDZ binding Mol. Cell 11: 1389–1396. [DOI] [PubMed] [Google Scholar]
- Linding R., Russel R.B., Neduva V., Gibson T.J. 2003. GlobPlot: Exploring protein sequences for globularity and disorder Nucleic Acids Res. 31: 3701–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Tan H., Rost B. 2002. Loopy proteins appear conserved in evolution J. Mol. Biol. 322: 53–64. [DOI] [PubMed] [Google Scholar]
- Nardini M., Spanò S., Cericola C., Pesce A., Damonte G., Luini A., Corda D., Bolognesi M. 2002. Crystallization and preliminary X-ray diffraction analysis of brefeldin A-ADP ribosylated substrate (BARS) Acta Crystallogr. D Biol. Crystallogr. 58: 1068–1070. [DOI] [PubMed] [Google Scholar]
- Nardini M., Spanò S., Cericola C., Pesce A., Massaro A., Millo E., Luini A., Corda D., Bolognesi M. 2003. CtBP/BARS: A dual-function protein involved in transcription co-repression and Golgi membrane fission EMBO J. 22: 3122–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield C.J., Cheng Y., Cortese M.S., Brown C.J., Uversky V.N., Dunker A.K. 2005. Comparing and combining predictors of mostly disordered proteins Biochemistry 44: 1989–2000. [DOI] [PubMed] [Google Scholar]
- Petoukhov M.V. and Svergun D.I. 2005. Global rigid body modelling of macromolecular complexes against small-angle scattering data Biophys. J. 89: 1237–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov M.V., Eady N.A., Brown K.A., Svergun D.I. 2002. Addition of missing loops and domains to protein models by x-ray solution scattering Biophys. J. 83: 3113–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero P., Obradovic Z., Li X., Garner E.C., Brown C.J., Dunker A.K. 2001. Sequence complexity of disordered protein Proteins 42: 38–48. [DOI] [PubMed] [Google Scholar]
- Shi Y., Sawada J.-I., Sui G., Affar, El.B., Whetstine J.R., Lan F., Ogawa H., Luke M.P.-S., Nakatani Y., Shi Y. 2003. Coordinated histone modifications mediated by a CtBP co-repressor complex Nature 422: 735–738. [DOI] [PubMed] [Google Scholar]
- Svergun D.I. 1992. Determination of the regularization parameter in indirect transform methods using perceptual criteria J. Appl. Crystallogr. 25: 495–503. [Google Scholar]
- Svergun D.I. 1999. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing Biophys. J. 76: 2879–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D.I., Barberato C., Koch M.H.J. 1995. CRYSOL—A program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates J. Appl. Crystallogr. 28: 768–773. [Google Scholar]
- Turner J. and Crossley M. 2001. The CtBP family: Enigmatic and enzymatic transcriptional co-repressors Bioessays 23: 683–690. [DOI] [PubMed] [Google Scholar]
- Uversky V.N. 2002. Natively unfolded proteins: A point where biology waits for physics Protein Sci. 11: 739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V.N., Gillespie J.R., Fink A.L. 2000. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 41: 415–427. [DOI] [PubMed] [Google Scholar]
- Valente C., Spanò S., Luini A., Corda D. 2005. Purification and functional properties of the membrane fissioning protein CtBP/BARS Methods Enzymol. 404: 296–316. [DOI] [PubMed] [Google Scholar]
- Weigert R., Silletta M.G., Spanò S., Turacchio G., Cericola C., Colanzi A., Senatore S., Mancini R., Polishchuk E.V., Salmona M.et al. 1999. CtBP/BARS induces fission of Golgi membranes by acylating lysophosphatidic acid Nature 402: 429–433. [DOI] [PubMed] [Google Scholar]
- Wüthrich K. In NMR of proteins and nucleic acids . 1986. Wiley, New York.
- Yang J.-S., Lee S.Y., Spanò S., Gad H., Zhang L., Nie Z., Bonazzi M., Corda D., Luini A., Hsu V.W. 2005. A role for BARS at the fission step of COPI vesicle formation from Golgi membrane EMBO J. 24: 4133–4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Piston D.W., Goodman R.H. 2002. Regulation of corepressor function by nuclear NADH Science 295: 1895–1897. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Yoshimatsu Y., Hildebrand J., Frisch S.M., Goodman R.H. 2003. Homeodomain interacting protein kinase 2 promotes apoptosis by downregulating the transcriptional corepressor CtBP Cell 115: 177–186. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Nottke A., Goodman R.H. 2005. Homeodomain-interacting protein kinase-2 mediates CtBP phosphorylation and degradation in UV-triggered apoptosis Proc. Natl. Acad. Sci. 102: 2802–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]