

Abstract
Under conditions of acidic pH and elevated temperature, insulin partially unfolds and aggregates into highly structured amyloid fibrils. Aggregation of insulin leads to loss of activity and can trigger an unwanted immune response. Compounds that prevent protein aggregation have been used to stabilize insulin; these compounds generally suppress aggregation only at relatively high inhibitor concentrations. For example, effective inhibition of aggregation of 0.5 mM insulin required arginine concentrations of ≥100 mM. Here, we investigate a targeted approach toward inhibiting insulin aggregation. VEALYL, corresponding to residues B12–17 of full-length insulin, was identified as a short peptide that interacts with full-length insulin. A hybrid peptide was synthesized that contained this binding domain and hexameric arginine; this peptide significantly reduced the rate of insulin aggregation at near-equimolar concentrations. An effective binding domain and N-terminal placement of the arginine hexamer were necessary for inhibitory activity. The data were analyzed using a simple two-step model of aggregation kinetics. These results are useful not only in identifying an insulin aggregation inhibitor but also in extending a targeted protein strategy for modifying aggregation of amyloidogenic proteins.
Keywords: aggregation, amyloid, surface tension, insulin
Insulin, a small (51–amino acid) protein hormone that mediates glucose uptake, is manufactured in significant quantities for treatment of diabetes. The protein contains a 21-residue A chain and a 30-residue B chain, cross-linked by disulfide bonds (Nettleton et al. 2000; Nielsen et al. 2001a). In its native state, insulin is predominantly α-helical; the monomer self-assembles into dimers, tetramers, and hexamers, depending on the concentration and the solvent conditions (Nettleton et al. 2000; Hua and Weiss 2004). The predominant physiological form of insulin, as stored in the pancreas, is a zinc-coordinated hexamer, formed by the association of three dimers (Nielsen et al. 2001a). Similarly, recombinant insulin is formulated as a zinc-coordinated hexamer to improve its stability. Dissociation to the monomer is required for biological activity.
Dissociation of hexamers and partial unfolding of the monomer is facilitated by exposure to acidic pH and elevated temperature, agitation, or contact with hydrophobic surfaces, leading to aggregation and subsequent amyloid fibril formation (Sharp et al. 2002; Ahmad et al. 2003; Hua and Weiss 2004). Fibrillation has been observed during manufacture, purification, storage, and infusion of insulin (Nielsen et al. 2001a, b). The recently introduced lispro insulin variant is predominantly monomeric; lispro insulin is faster-acting than the hexameric stabilized form (Heinemann 2004) but has a greater tendency to aggregate. Aggregated insulin loses its activity and is not therapeutically effective; furthermore, injected protein aggregates can trigger an unwanted immune response.
Insulin fibrillation is inhibited by compounds that either prevent unfolding of the native protein or sequester partially folded aggregation-competent intermediates. Carbohydrates and glycerols increase native insulin stability (Blackshear et al. 1983; Brange and Havelund 1983; Katakam and Banga 1995); this action has been attributed to preferential exclusion of these co-solutes from the protein surface and subsequent enhancement of hydrophobic interactions within the native structure (Brange et al. 1997a). Likewise, low molecular weight compounds such as betaine, trehalose, and citrulline inhibit insulin amyloid formation, presumably by a similar mechanism (Arora et al. 2004). Other compounds such as lecithins, cyclodextrins, and polymeric surfactants reduce insulin aggregation by binding either to hydrophobic interfaces or to hydrophobic insulin domains (Thurow and Geisen 1984; Grau and Saudek 1987; Brewster et al. 1991; Brange et al. 1997a). These and other aggregation inhibitors are fairly nonspecific in action and are generally effective only at relatively high concentrations. For example, 300 mM betaine suppressed aggregation of 0.17 mM insulin, an ∼1700-fold molar excess (Arora et al. 2004).
Here, we investigate a strategy to reduce insulin aggregation at lower inhibitor concentrations by using small hybrid peptides, consisting of a recognition domain designed to specifically bind to insulin, and a disrupting domain that alters insulin aggregation. Previously, we successfully demonstrated the ability of hybrid peptides to alter aggregation kinetics of beta-amyloid (Aβ) (Ghanta et al. 1996; Pallitto et al. 1999; Lowe et al. 2001), a peptide with significant importance because its aggregation has been linked with progression of Alzheimer's disease. We postulated that a similar strategy could be used to discover compounds that can effectively stabilize insulin.
By screening proteolytic fragments of insulin, we identified short peptides as putative recognition domains. Hybrid peptides were synthesized by appending a hexameric-arginine disrupting domain to the identified recognition domain. These hybrids were evaluated for their ability to stabilize insulin under conditions (pH 2.0, 37°C) that normally lead to partial unfolding and amyloidogenesis of insulin (Brange et al. 1997a; Nielsen et al. 2001a; Hua and Weiss 2004). We show that a hybrid peptide, containing LVEALYLV as the recognition domain and an N-terminal arginine hexamer as the disrupting domain, significantly reduced the rate of insulin aggregation; we demonstrate activity of the targeted hybrid peptide in the 0.5–2 mM concentration range.
Results
Insulin forms fibrils at acidic pH and elevated temperatures (Hua and Weiss 2004). To confirm these findings, 3 mg/mL (0.52 mM) insulin in 1 M acetic acid (pH 2.0) was incubated at 37°C, and aliquots were taken at various time points. Amyloid fibril formation was quantified using a thioflavin T (ThT) fluorescence assay; ThT fluorescence intensity is generally taken to be proportional to the mass of amyloid fibrils (Vassar and Culling 1959). After an initial lag phase lasting 45–55 h, during which ThT fluorescence remained at background levels, we observed a rapid increase in fluorescence intensity coincident with the appearance of macroscopic aggregates. Consistent with these findings, by dynamic light scattering we observed no aggregate growth over ∼50 h followed by the sudden formation of very large aggregates. Aggregates were confirmed to have fibril morphology by transmission electron microscopy (TEM) (Fig. 1A).
Figure 1.

Fibrillogenesis of insulin. (A) TEM image of insulin fibrils, prepared after 28 h incubation at pH 2 and 37°C, followed by 2-wk storage at room temperature. The white bar represents 200 nm length. (B) ThT fluorescence of insulin alone (×) or mixed with 30 mM ArgHCl (▴), 100 mM ArgHCl (▪), or 200 mM ArgHCl (•). Insulin was dissolved at 0.52 mM in 1 M acetic acid (pH 2.0). Samples were kept at 37°C during the duration of the experiment. Lines are smoothed curves through the data points.
Arginine has been used to suppress protein aggregation in a number of studies (Buchner and Rudolph 1991; Taneja and Ahmad 1994; Qiao et al. 2001; Shiraki et al. 2002, 2004; Arakawa and Tsumoto 2003; Tsumoto et al. 2004; Xie et al. 2004). Arginine at 100–200 mM significantly inhibited insulin aggregation, increasing the lag time to ∼75 h (Fig. 1B). At 30 mM (60-fold molar excess), arginine had little effect on insulin aggregation (Fig. 1B).
We postulated that insulin aggregation could be suppressed at lower inhibitor concentrations by implementing a targeted approach. Reasoning that fibril formation requires association between specific protein domains, we first aimed to identify short insulin fragments that bind to full-length insulin. A mixture of insulin fragments was produced by limited proteolysis and incubated with immobilized insulin. Reverse-phase HPLC analysis of the mixture indicated that one peak, eluting at ∼18.8 min (∼38% acetonitrile), was reduced in size after contact with immobilized insulin (Fig. 2A). Using MALDI-TOF mass spectrometry, we determined a molecular mass of 1046 Da for the fragment. By comparing this value with expected pepsin cleavage sites, we concluded the fragment of interest to be CGSHLVEAL (1047 Da), corresponding to residues 7–15 of bovine insulin B-chain (Table 1). The reduction in the HPLC peak of CGSHLVEAL after mixture of the insulin fragments with immobilized insulin suggested that CGSHLVEAL bound to full-length insulin. However, when the insulin fragments were analyzed after contact with underivatized Sepharose beads (without the presence of immobilized insulin), a reduction in the same peak was observed (data not shown), indicating nonspecific binding or self association of CGSHLVEAL.
Figure 2.

Identification of binding domains. (A) HPLC trace of insulin fragments from pepsin cleavage before (upper trace) and after (lower trace) incubation with immobilized insulin. The arrow indicates a peak that was significantly reduced in size after contact. The peak was collected and its molecular mass determined by MALDI-TOF spectroscopy to be 1046 Da. (B) HPLC trace of a mixture of insulin-derived peptides before (upper trace) and after (lower trace) incubation with immobilized insulin. Only the peak corresponding to VEALYL changed significantly after incubation.
Table 1.
Selection of recognition domain

aPeptides corresponding to overlapping domains within the B7–B18 region of insulin were synthesized and screened for binding to immobilized insulin. +, Detectable interaction; −, no detectable interaction.
The identified fragment overlaps with an insulin domain, LVEALYLV, proposed to be involved in intermolecular contacts necessary for insulin misfolding and aggregation (Brange et al. 1997a,b). Based on this observation, we surmised that all or part of the B7–B18 region of insulin, or CGSHLVEALYLV, could serve as an effective recognition domain for an insulin hybrid compound. To narrow in on a smaller recognition domain that may demonstrate greater specificity, we synthesized overlapping sequences within this region, including GSHLV, GSHLVEAL, VEALYL, and ALYLV (Table 1). A mixture of these four peptides was incubated with immobilized insulin, and binding was detected by reverse-phase HPLC analysis. After equilibration, only the peak corresponding to VEALYL (∼23.8 min) was significantly reduced (Fig. 2B). The loss of this peak was accompanied by the appearance of a smaller peak eluting ∼1 min earlier. There was little change in the elution profile when the peptide mixture was contacted with underivatized Sepharose beads, and no peaks were observed when solvent alone was mixed with Sepharose beads containing immobilized insulin (data not shown). These results suggested that the sequence VEALYL was interacting specifically with full-length insulin; the fragment may be binding to insulin and/or undergoing conformational changes. VEALYL is entirely contained within the LVEALYLV domain proposed to be involved with insulin misfolding and aggregation (Brange et al. 1997a,b), so we hypothesized that LVEALYLV could be an effective recognition domain for insulin hybrid peptides.
We examined whether VEALYL, GSHLVEAL, or ALYLV influenced insulin aggregation. At 2 mM, none of the three peptides had a significant effect on insulin aggregation as assessed by the ThT assay (data not shown), suggesting that a binding domain alone was not sufficient for controlling insulin aggregation. We synthesized two hybrid peptides, using LVEALYLV as the recognition domain and RRRRRR attached on either the N or the C terminus as the disrupting domain. When an equimolar amount (0.5 mM) of hybrid peptide was mixed with insulin, LVEALYLVRRRRRR had little effect on insulin aggregation, while RRRRRRLVEALYLV increased the lag time for insulin aggregation, although this effect was not consistently observed (data not shown).
The concentration of hybrid peptide was increased to 2 mM to determine if consistent aggregation inhibition could be obtained. In addition to RRRRRRLVEALYLV and LVEALYLVRRRRRR, other hybrid peptides, including ALYLVRRRRRR, GSHLVEALRRRRRR, and RRRRRRGSHLVEAL, were tested. As a control, the disrupting domain alone was also tested in the ThT assay. RRRRRR and ALYLVRRRRRR had little to no effect on insulin aggregation, while both hybrid peptides with the GSHLVEAL recognition were of minor efficacy. LVEALYLVRRRRR moderately and RRRRRRLVEALYLV significantly increased the lag time and reduced the rate of aggregation (Table 2; Fig. 3). The strong inhibitory effect of 2 mM RRRRRRLVEALYLV was consistent and reproducible. RRRRRRLVEALYLV did not completely prevent aggregation over a long time; all solutions of insulin with hybrid compounds eventually precipitated, at which point monomeric insulin was no longer detected in the supernatant. However, after precipitation occurred, the aggregates formed in the presence of RRRRRRLVEALYLV did not have typical fibrillar morphology. Rather, observed aggregates were varied in morphology and included wispy sheets, ribbons, and filaments (cf. Figs. 4A and 1A).
Table 2.
Effect of compounds on insulin aggregation

aThe compound was added to insulin at the indicated concentration prior to initiation of aggregation. The putative recognition domain is indicated in bold.
bKinetics were assayed using ThT fluorescence. The lag time was calculated as the midpoint between measurements where the ThT fluorescence increased from near baseline value to at least threefold greater than baseline. The change in lag time is the difference between the measured lag time and that for insulin alone (∼50 h). Values are an average of data obtained from multiple experiments. Representative kinetic data are shown in Figures 1, 3, and 6.
Figure 3.

Kinetics of insulin fibrillogenesis in the presence of various peptides. Insulin was dissolved at 0.52 mM in 1 M acetic acid (pH 2.0), and mixed with indicated peptides at 2 mM. Samples were kept at 37°C during the duration of the experiment. ThT of insulin alone (×) or mixed with RRRRRR (○), ALYLVRRRRRR (•), GSHLVEALRRRRRR (▴), RRRRRRGSHLVEAL (△), LVEALYLVRRRRRR (▪), or RRRRRRLVEALYLV (□) was measured over time. Lines represent fitting of the data to a two-step kinetic model.
Figure 4.

TEM of insulin with RRRRRRLVEALYLV and CTAC. All samples were prepared by dissolving test compounds in 1 M acetic acid (pH 2.0) and incubating the sample at 37°C. Samples containing insulin were transferred to room temperature when visible precipitate first appeared. After 2 wk at room temperature, samples were centrifuged and the supernatant was collected. Samples were negatively stained and placed on a pioloform coating grid support film for TEM imaging. The white bars represent 200 nm length. (A) 0.52 mM insulin + 2 mM RRRRRRLVEALYLV, (B) 0.52 mM insulin + 2 mM CTAC, (C) 2 mM RRRRRRLVEALYLV, (D) 2 mM CTAC.
To quantify these results, ThT data were analyzed using a two-step kinetic model of aggregation: (1) a slow first-order conversion of nonamyloidogenic species (N) to amyloidogenic species (A), with the rate proportional to ks, and (2) a rapid second-order conversion of N to A by the recruitment of existing A, with the rate proportional to kf. ThT fluorescence was assumed to be directly proportional to the mass of A present. Complete conversion of N to A was assumed, based on reverse-phase HPLC analysis, demonstrating that all solutions of insulin with test compound were eventually depleted of monomer. Rate constants (ks and kf) were determined by nonlinear regression fit of the experimental data to the model. Because ks and kf were highly correlated and there were insufficient data points for reliable two-parameter estimation, kf was fixed at a value of 1.75×10−4 [fluorescence units]−1 h−1, and only ks was fitted to the data. Thus, ks should be considered primarily as a relative, rather than an absolute, measure of the inhibitory activity of each test compound. Holding kf constant implies that hybrid peptides inhibit insulin aggregation in the first step. This assumption would be true if, for example, inhibitors associate with native insulin and prevent unfolding, or associate with partially unfolded insulin and prevent misfolding into aggregation-competent conformers. Results of the parameter estimation are displayed in Table 3. With LVEALYLVRRRRRR present, ks decreased by ∼160-fold relative to ks for insulin alone; with RRRRRRLVEALYLV present, ks decreased by ∼1200-fold.
Table 3.
Effect of compounds on insulin aggregation

aAll compounds were tested at 2 mM. ThT fluorescence data were fit to a two-step model of aggregation kinetics, and rate constants were determined as described in the text. The rate constant ks should be considered as a relative measure of effectiveness of the inhibitory compound and not as a rigorously determined quantity.
We previously demonstrated that the ability of hybrid compounds to accelerate Aβ aggregation correlated with their ability to increase the solvent surface tension (Kim et al. 2003; Gibson and Murphy 2005). Therefore, we measured the effect of RRRRRRLVEALYLV on solvent surface tension. Interestingly, while the peptide slightly increased surface tension at low concentrations (0.14 mM), it reduced surface tension in the concentration range (0.5–2 mM) where we demonstrated its ability to inhibit insulin aggregation (Fig. 5). A decrease in surface tension above a critical constant is characteristic of micelle-forming compounds. RRRRRRLVEALYLV self-associated into large oligomers of significant size and variable morphology, as determined from dynamic light scattering (data not shown) and TEM (Fig. 4C). These aggregates formed rapidly, and were soluble and were not ThT-positive (data not shown). In preliminary light-scattering studies we observed no substantial change in size over a 10-h period, suggesting that the aggregates are relatively stable.
Figure 5.

Surface tension of RRRRRRLVEALYLV in 1 M acetic acid, as measured using the pendant drop method.
RRRRRRGSHLVEAL reduced surface tension and formed oligomers similar to RRRRRRLVEALYLV (data not shown), although it was not an effective inhibitor of insulin aggregation (Tables 2, 3; Fig. 3). To test whether the inhibitory activity of RRRRRRLVEALYLV could be replicated by a generic surfactant, we tested the effect of the cationic surfactant cetyltrimethylammonium chloride (CTAC) on insulin aggregation. CTAC reportedly has a critical micelle concentration in water of 1.4 mM (Tofani et al. 2004). CTAC at 2 mM formed large oligomeric species (Fig. 4D) and reduced solvent surface tension even more than did RRRRRRLVEALYLV (to ∼35 dyne/cm). Despite the reduction in surface tension, CTAC accelerated the onset of insulin aggregation (Table 2; Fig. 6). Interestingly, the maximum ThT fluorescence intensity was reduced in the presence of CTAC (Fig. 6). Despite this reduction in ThT fluorescence, insulin aggregates formed in the presence of CTAC were numerous and of fibrillar morphology (Fig. 4C). However, these samples appeared to be more gel-like than typical insulin fibrils. Thus, CTAC appears to affect solvation of insulin fibrils and to reduce their ThT binding, but not to inhibit fibril nucleation or growth.
Figure 6.

Kinetics of insulin fibrillogenesis in the presence of hybrid peptide or surfactant. ThT of insulin alone (×) or mixed with 2 mM RRRRRRLVEALYLV (▪) or 2 mM CTAC (▴) was measured over time. Insulin was dissolved at 0.52 mM in 1 M acetic acid (pH 2.0). Samples were kept at 37°C during the duration of the experiment. Lines are smoothed curves and do not represent fitting of the data to a kinetic model.
Taken together, these results suggest that surface tension effects alone are not sufficient to cause inhibition of insulin amyloid formation and indicate that inhibition of aggregation by hybrid peptides requires a specific recognition domain affinity.
Discussion
We previously described a strategy in which hybrid peptides, containing a recognition domain and a binding domain, are used to change Aβ aggregation kinetics (Ghanta et al. 1996). Stronger inhibition of Aβ toxicity by hybrid peptides correlated with higher binding affinity of the recognition domain (Cairo et al. 2002) and with greater acceleration of aggregation (Pallitto et al. 1999; Lowe et al. 2001). The active hybrid peptides contained charged amino acids in the disrupting domain (Lowe et al. 2001); acceleration of Aβ aggregation by hybrid peptides correlated strongly with their ability to increase the solvent surface tension (Kim et al. 2003; Gibson and Murphy 2005). While many co-solutes, such as betaine, arginine, or trehalose, influence protein aggregation through their effect on the protein–solvent interface, they are typically only active at higher concentrations because they have a global rather than a targeted effect. A significant benefit of hybrid peptides is that they have activity at low equimolar concentration; we theorize that this is because recognition domain affinity localizes the hybrid peptide near the aggregating protein.
Insulin and Aβ share a common characteristic: Under appropriate conditions, they both aggregate into amyloid fibrils. However, in extending the Aβ hybrid peptide strategy to the design of insulin hybrid peptides, some differences between Aβ and insulin aggregation had to be considered. First, insulin is structured in its native monomeric state while Aβ is not. Hybrid peptides with kosmotropic disrupting domains accelerated Aβ aggregation (Gibson and Murphy 2005), presumably because kosmotropes stabilize structured protein, and the most structured state of Aβ is the amyloid fibril. For insulin, it is less obvious whether the monomer or the fibril is more “structured” and therefore more favored in the presence of kosmotropes. Second, the kinetics of aggregation differ under the experimental conditions used: With insulin, a long initial lag time preceded a rapid conversion to insoluble aggregates, while with Aβ, aggregates formed almost immediately but grew slowly into fully formed fibrils (Pallitto and Murphy 2001).
A recognition domain was first identified by observing that VEALYL (B12–B17 of insulin) interacted with immobilized full-length insulin (Fig. 2B). VEALYL is fully contained within LVEALYLV, identified as a domain involved in insulin-misfolding contacts (Brange et al. 1997a,b). Attaching an arginine hexamer disrupting domain to the N terminus resulted in a hybrid peptide, RRRRRRLVEALYLV, that strongly inhibited the rate of insulin aggregation at 2 mM as effectively as 100 mM arginine. Attaching the arginine hexamer to the C terminus of LVEALYLV produced a moderately effective inhibitor. When ALYLV or GSHLVEAL was linked to RRRRRR, the hybrid peptides did not significantly reduce aggregation (Table 2; Fig. 3). The recognition domain or disrupting domain alone (VEALYLV or RRRRRR) were ineffective inhibitors. These results indicate that a specific recognition domain, together with a properly oriented disrupting domain, are required for effective inhibition of insulin aggregation at low inhibitor concentration.
Similarly, we previously determined that a peptide containing a recognition domain derived from an Aβ sequence (KLVFF) coupled to an arginine hexamer provided a targeted means to alter Aβ aggregation kinetics (Gibson and Murphy 2005). In that case, C-terminal placement of the disrupting domain was used because N-terminal placement produced a peptide that, although initially soluble, rapidly aggregated and precipitated (Ghanta et al. 1996).
RRRRRRLVEALYLV exhibited micelle-like behavior in that the peptide reduced solvent surface tension above a critical concentration of ∼0.2–0.4 mM (Fig. 5), which is near the minimum concentration required for inhibition of insulin aggregation. This micellar behavior likely arises from the amphiphilic nature of the hybrid peptide. Interestingly, surfactants have been demonstrated to increase the stability of insulin formulations, presumably by shielding hydrophobic domains from involvement in intermolecular protein interactions (Lougheed et al. 1983). The interaction of surfactants with proteins has been described by the “beads-necklace model,” in which it is hypothesized that a surfactant forms micelle-like clusters on precise sites of a protein (Chen and Teixeira 1986; Guo and Chen 1990; Tofani et al. 2004). To test whether the inhibitory activity of RRRRRRLVEALYLV could be explained by its surfactant-like property, we examined the effect of the cationic surfactant CTAC on insulin aggregation. We chose to use CTAC because, similar to the hybrid peptide, it has cationic polar groups, and it decreases surface tension to nearly the same extent as RRRRRRLVEALYLV. Unlike RRRRRRLVEALYLV, CTAC did not lengthen the lag time required for the onset of insulin aggregation (Fig. 6), nor did it prevent formation of fully formed fibrils (Fig. 4B), although it did decrease the maximum intensity of ThT fluorescence (Fig. 6). Thus, it seems unlikely that the inhibitory activity of RRRRRRLVEALYLV is attributable directly to its micellar-like behavior. However, the micellar behavior may be indirectly responsible for inhibition of aggregation for the following reason: Hybrid peptides targeted to Aβ and containing hexameric arginine as a disrupting domain increased solvent surface tension and accelerated Aβ aggregation. The maximum concentration of Aβ-targeted hybrid peptide tested was 0.4 mM; over this concentration range the surface tension of the solvent increased linearly (Kim et al. 2003). Because of some combination of a change in recognition domain, a switch from C- to N-terminal placement of the disrupting domain, and/or an increase in concentration, the insulin-targeted hybrid peptide associated into structures that reduced solvent surface tension at concentrations above a critical concentration. The reduction in solvent surface tension may have decreased the unfavorable interactions between water and exposed hydrophobic patches on insulin, interactions which otherwise would drive aggregation. Since reducing surface tension inhibited amyloid fibril formation, this suggests that insulin fibrils are more “structured” than insulin monomers, at least at the experimental conditions (pH 2.0, 37°C) used in these studies.
GSHLVEAL did not bind to immobilized insulin, and the hybrid peptide RRRRRRGSHLVEAL did not inhibit insulin aggregation, even though it reduced solvent surface tension. This result indicates that an effective binding domain is required for inhibition of aggregation of insulin.
In summary, work reported here extends our previous studies in exploring a strategy for designing compounds that are targeted toward altering aggregation of specific proteins. In both cases, a combination of an effective recognition domain and a disrupting domain with strong effects on solvent properties was required. The ability to specifically target a protein with an aggregation-modulating compound presents the opportunity to develop novel materials for controlling aggregation-related disorders as well as new methods for processing and stabilizing manufactured proteins.
Materials and methods
Peptides
Bovine pancreatic insulin and arginine (L-arginine monohydrochloride) were purchased from Sigma Aldrich. Protected amino acids and resins were purchased from Novabiochem. All other reagents were purchased from Sigma Aldrich or Fisher Scientific. Peptides were synthesized by Fmoc solid-phase peptide synthesis using an Applied Biosystems Model 432A “Synergy” automated synthesis instrument. Resins were washed with ethanol and dried. Resin cleavage was accomplished with 95% trifluoroacetic acid, and the resulting peptides were precipitated and washed with diethyl ether. The cleaved peptides were purified by reverse-phase HPLC (C18 column) using a 5%–50% acetonitrile/water gradient over 35 min. Purified peptides were stored as lyophilized powders at –70°C.
Insulin cleavage with pepsin
Ethylenediaminetetraacetic acid (EDTA) and dithiothreitol (DTT) were purchased from Acros Organics (Geel, Belgium). All other reagents were purchased from Sigma Aldrich or Fisher Scientific. Insulin was dissolved to 0.6 mg/mL by adding MilliQ H2O, reducing the pH with the addition of 1 M HCl dropwise until the insulin was fully soluble (pH ∼2), and then adjusting the pH with dropwise addition of 1 M NaOH to pH ∼3.5. The solution was then diluted to 0.5 mg/mL with Gly-HCl buffer (0.2 M at pH 3.5), with 1 mM EDTA added to chelate any Zn (Liu et al. 1991) and 5 mM DTT added to reduce disulfides (Holmgren 1979). The solution was mixed for 2–3 h; 25 mg pepsin-agarose was suspended and rinsed two times with 2 mL MilliQ H2O, followed by two rinses with 2 mL Gly–HCl buffer. The resin was then suspended in 5 mL Gly–HCl buffer, and 30 mL of the insulin solution was added. Reaction was allowed to proceed with continuous mixing for 5 h. Cleavage was monitored by HPLC (C18 column) using an acetonitrile/water gradient.
Analysis of fragment binding to insulin
Bovine pancreatic insulin was immobilized to the surface of CnBr-Sephrose beads as follows: A solution of 1 mg/mL bovine insulin was prepared by adding phosphate buffered saline (PBS, 10 mM at pH 7.0), 1 M HCl to solubilize insulin (pH ∼2), then 1 M NaOH to adjust back to pH 7.0 (insulin remained soluble). EDTA (1 mM) was added to the solution. CnBr-Sepharose beads (60 mg) were rinsed 10 times with 1 mL of 1 mM HCl. The beads were then suspended briefly in 0.5 mL PBS. Immediately, the PBS was removed and replaced with 1 mL of the insulin solution. The mixture was vortexed briefly and placed on a rotator at 4°C. After 8 h, the insulin solution was removed and replaced with 0.4 mL of 1 M ethanolamine (pH ∼7.5) for 12 h with rotation at 4°C to block remaining reactive sites. Insulin immobilization was confirmed by measuring a significant reduction in the absorbance at 280 nm.
Next, insulin fragments were screened for binding to the immobilized insulin. The immobilized insulin was rinsed three times with 1 mL Gly–HCl buffer (0.2 M at pH 3.5), then 1 mL of ∼0.5 mg/mL test solution (either pepsin-cleaved insulin fragments or synthesized fragments) was added to the immobilized insulin and the solution was rotated at 4°C, allowing sufficient time for fragments to bind. Fragment binding was monitored by HPLC (C18 column) using a 5%–50% acetonitrile/water gradient over 35 min. Reduction of an elution peak suggested possible binding of a fragment that eluted with that peak.
Thioflavin T (ThT) fluorescence
A stock solution was prepared by dissolving bovine insulin at 6 mg/mL into filtered (0.22 μm) 1 M acetic acid (100 mM NaCl, 1 mM EDTA at pH 2.3). The stock solution was immediately aliquoted into 1 M acetic acid or 1 M acetic acid containing a test compound, and each sample was adjusted to pH 2.0 using 1 M HCl to afford a final insulin concentration of 3 mg/mL (0.52 mM). Samples were mixed briefly by hand and then placed at 37°C. A stock solution of 1 mM ThT in water was prepared and filtered (0.45 μm) into a test tube, which was wrapped in aluminum foil and stored at 4°C. Immediately before performing a ThT measurement, the stock solution was diluted 20-fold with filtered (0.22 μm) MilliQ H2O. Samples (10 μL) were added to each well of a 96-well plate, followed by 200 μL diluted ThT solution. Fluorescence intensity was measured with a BioTek Instruments, Inc. FLx800 Microplate Fluorescence Reader using an excitation wavelength of 440/30 nm and an emission wavelength of 485/20 nm.
Fitting of ThT data to aggregation kinetic model
ThT data were analyzed using a kinetic model of protein aggregation based on the following simple two-step mechanism
 |
where N is nonamyloidogenic species and A is amyloidogenic species. Note that this model is a classic example of an autocatalytic reaction, and we implicitly assume that each species converted from nonamyloidogenic to amyloidogenic retains its autocatalytic function whether or not it is incorporated into an aggregate. Assuming complete conversion of N to A at long time, the resulting set of differential equations was integrated analytically to obtain
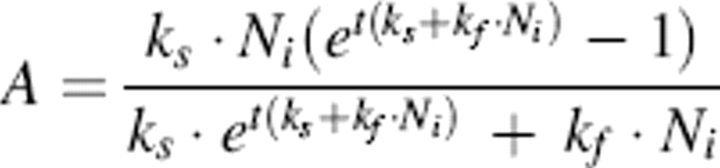 |
where Ni is initial concentration of N and t is the time of aggregation. ThT fluorescence was assumed to be directly proportional to the mass of A present. Since the rate constants (ks and kf) are highly correlated and there were not enough data points to obtain a confident two-parameter estimation, kf was fixed and ks was estimated using nonlinear regression to fit experimental data, using the software program Athena.
Surface tension
The equilibrium surface tension of test compounds was measured using a FTÅ200 pendant drop tensiometer (First Ten Angstroms). Test compounds were dissolved in 1 M acetic acid (100 mM NaCl, 1 mM EDTA at pH 2.3). A droplet of the resulting solution was then formed at the end of a blunt 22-gauge stainless steel needle, and the shape of the droplet was imaged. The surface tension at the liquid interface was calculated from γ = ΔρgR20/β, where γ is the surface tension, Δρ is the difference in density between fluids at the interface (assuming that the droplet has the density of water), g is the gravitational constant, R0 is the radius of drop curvature at apex, and β is the shape factor which can be defined through the Young-Laplace equation of droplet shape (Hansen and Rodsrud 1991). Surface tension values were determined as the average of duplicate measurements.
Transmission electron microscopy (TEM)
Samples were imaged using a Philips CM120 Transmission Electron Microscope (FEI Corp.). Aggregated samples used for ThT experiments were collected when visible precipitate first appeared, and then samples were stored for 2 wk at room temperature. After centrifuging the samples at 16,000g for 5 min, most of the supernatant was discarded. MilliQ H2O (50 μL) was added to the remaining pellet, and the samples were vortexed rapidly for 20 sec. Samples were stained with Nano-W (methylamine tungstate) negative stain (Nanoprobes.com) and were placed on a pioloform coating grid support film (Ted Pella Inc.).
Acknowledgments
Funding for this work was provided by the National Science Foundation (BES-0330537).
Footnotes
Reprint requests to: Regina M. Murphy, Department of Chemical and Biological Engineering, University of Wisconsin, Madison, 1415 Engineering Drive, Madison, WI 53706, USA; e-mail: regina@engr.wisc.edu; fax: (608) 262-5434.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051879606.
References
- Ahmad A., Millett I.S., Doniach S., Uversky V.N., Fink A.L. 2003. Partially folded intermediates in insulin fibrillation Biochemistry 42 11404–11416. [DOI] [PubMed] [Google Scholar]
- Arakawa T. and Tsumoto K. 2003. The effects of arginine on refolding of aggregated proteins: Not facilitate refolding, but suppress aggregation Biochem. Biophys. Res. Commun. 304 148–152. [DOI] [PubMed] [Google Scholar]
- Arora A., Ha C., Park C.B. 2004. Inhibition of insulin amyloid formation by small stress molecules FEBS Lett. 564 121–125. [DOI] [PubMed] [Google Scholar]
- Blackshear P.J., Rohde T.D., Palmer J.L., Wigness B.D., Rupp W.M., Buchwald H. 1983. Glycerol prevents insulin precipitation and interruption of flow in an implantable insulin infusion pump Diabetes Care 6 387–392. [DOI] [PubMed] [Google Scholar]
- Brange J. and Havelund S. 1983. Insulin pumps and insulin quality—Requirements and problems Acta Med. Scand. Suppl. 671 135–138. [DOI] [PubMed] [Google Scholar]
- Brange J., Andersen L., Laursen E.D., Meyn G., Rasmussen E. 1997a. Toward understanding insulin fibrillation J. Pharm. Sci. 86 517–525. [DOI] [PubMed] [Google Scholar]
- Brange J., Dodson G.G., Edwards J., Holden P.H., Whittingham J.L. 1997b. A model of insulin fibrils derived from the x-ray crystal structure of a monomeric insulin (despentapeptide insulin) Proteins 27 507–516. [PubMed] [Google Scholar]
- Brewster M.E., Hora M.S., Simpkins J.W., Bodor N. 1991. Use of 2-hydroxypropyl-β-cyclodextrin as a solubilizing and stabilizing excipient for protein drugs Pharm. Res. 8 792–795. [DOI] [PubMed] [Google Scholar]
- Buchner J. and Rudolph R. 1991. Renaturation, purification and characterization of recombinant Fab-fragments produced in Escherichia coli Biotechnology (N.Y.) 9 157–162. [DOI] [PubMed] [Google Scholar]
- Cairo C.W., Strzelec A., Murphy R.M., Kiessling L.L. 2002. Affinity-based inhibition of β-amyloid toxicity Biochemistry 41 8620–8629. [DOI] [PubMed] [Google Scholar]
- Chen S.H. and Teixeira J. 1986. Structure and fractal dimension of protein–detergent complexes Phys. Rev. Lett. 57 2583–2586. [DOI] [PubMed] [Google Scholar]
- Ghanta J., Shen C.-L., Kiessling L.L., Murphy R.M. 1996. A strategy for designing inhibitors of β-amyloid toxicity J. Biol. Chem. 271 29525–29528. [DOI] [PubMed] [Google Scholar]
- Gibson T.J. and Murphy R.M. 2005. Design of peptidyl compounds that affect β-amyloid aggregation: Importance of surface tension and context Biochemistry 44 8898–8907. [DOI] [PubMed] [Google Scholar]
- Grau U. and Saudek C.D. 1987. Stable insulin preparation for implanted insulin pumps. Laboratory and animal trials Diabetes 36 1453–1459. [DOI] [PubMed] [Google Scholar]
- Guo X.H. and Chen S.H. 1990. The structure and thermodynamics of protein–SDS complexes in solution and the mechanism of their transports in gel electrophoresis process Chem. Phys. 149 129–139. [Google Scholar]
- Hansen F.K. and Rodsrud G. 1991. Surface-tension by pendant drop. 1. A fast standard instrument using computer image-analysis J. Colloid Interface Sci. 141 1–9. [Google Scholar]
- –T27; discussion T29–T30Heinemann L. 2004. Overcoming obstacles: New management options. Eur. J. Endocrinol 151 (Supp. 2) T23. [DOI] [PubMed] [Google Scholar]
- Holmgren A. 1979. Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide J. Biol. Chem. 254 9627–9632. [PubMed] [Google Scholar]
- Hua Q.X. and Weiss M.A. 2004. Mechanism of insulin fibrillation: The structure of insulin under amyloidogenic conditions resembles a protein-folding intermediate J. Biol. Chem. 279 21449–21460. [DOI] [PubMed] [Google Scholar]
- Katakam M. and Banga A.K. 1995. Aggregation of insulin and its prevention by carbohydrate excipients PDA J. Pharm. Sci. Technol. 49 160–165. [PubMed] [Google Scholar]
- Kim J.R., Gibson T.J., Murphy R.M. 2003. Targeted control of kinetics of β-amyloid self-association by surface tension-modifying peptides J. Biol. Chem. 278 40730–40735. [DOI] [PubMed] [Google Scholar]
- Liu F.-Y., Kildsig D.O., Mitra A.K. 1991. Insulin aggregation in aqueous media and its effect on alpha-chymotrypsin-mediated proteolytic degradation Pharm. Res. 8 925–929. [DOI] [PubMed] [Google Scholar]
- Lougheed W.D., Albisser A.M., Martindale H.M., Chow J.C., Clement J.R. 1983. Physical stability of insulin formulations Diabetes 32 424–432. [DOI] [PubMed] [Google Scholar]
- Lowe T.L., Strzelec A., Kiessling L.L., Murphy R.M. 2001. Structure–function relationships for inhibitors of β-amyloid toxicity containing the recognition sequence KLVFF Biochemistry 40 7882–7889. [DOI] [PubMed] [Google Scholar]
- Nettleton E.J., Tito P., Sunde M., Bouchard M., Dobson C.M., Robinson C.V. 2000. Characterization of the oligomeric states of insulin in self-assembly and amyloid fibril formation by mass spectrometry Biophys. J. 79 1053–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen L., Frokjaer S., Brange J., Uversky V.N., Fink A.L. 2001a. Probing the mechanism of insulin fibril formation with insulin mutants Biochemistry 40 8397–8409. [DOI] [PubMed] [Google Scholar]
- Nielsen L., Frokjaer S., Carpenter J.F., Brange J. 2001b. Studies of the structure of insulin fibrils by Fourier transform infrared (FTIR) spectroscopy and electron microscopy J. Pharm. Sci. 90 29–37. [DOI] [PubMed] [Google Scholar]
- Pallitto M.M. and Murphy R.M. 2001. A mathematical model of the kinetics of β-amyloid fibril growth from the denatured state Biophys. J. 81 1805–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallitto M.M., Ghanta J., Heinzelman P., Kiessling L.L., Murphy R.M. 1999. Recognition sequence design for peptidyl modulators of β-amyloid aggregation and toxicity Biochemistry 38 3570–3578. [DOI] [PubMed] [Google Scholar]
- Qiao Z.-S., Guo Z.-Y., Feng Y.-M. 2001. Putative disulfide-forming pathway of porcine insulin precursor during its refolding in vitro Biochemistry 40 2662–2668. [DOI] [PubMed] [Google Scholar]
- Sharp J.S., Forrest J.A., Jones R.A.L. 2002. Surface denaturation and amyloid fibril formation of insulin at model lipid–water interfaces Biochemistry 41 15810–15819. [DOI] [PubMed] [Google Scholar]
- Shiraki K., Kudou M., Fujiwara S., Imanaka T., Takagi M. 2002. Biophysical effect of amino acids on the prevention of protein aggregation J. Biochem. 132 591–595. [DOI] [PubMed] [Google Scholar]
- Shiraki K., Kudou M., Kudou M., Nishikori S.X., Kitagawa S., Imanaka T., Takagi M. 2004. Arginine ethylester prevents thermal inactivation and aggregation of lysozyme Eur. J. Biochem. 271 3242–3247. [DOI] [PubMed] [Google Scholar]
- Taneja S. and Ahmad F. 1994. Increased thermal stability of proteins in the presence of amino acids Biochem. J. 303 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurow H. and Geisen K. 1984. Stabilisation of dissolved proteins against denaturation at hydrophobic interfaces Diabetologia 27 212–218. [DOI] [PubMed] [Google Scholar]
- Tofani L., Feis A., Snoke R.E., Berti A.D., Baglioni P., Smulevich G. 2004. Spectroscopic and interfacial properties of myoglobin/surfactant complexes Biophys. J. 87 1186–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsumoto K., Umetsu M., Kumagai I., Ejima D., Philo J.S., Arakawa T. 2004. Role of arginine in protein refolding, solubilization, and purification Biotechnol. Prog. 20 1301–1308. [DOI] [PubMed] [Google Scholar]
- Vassar P.S. and Culling C.F. 1959. Fluorescent stains, with special reference to amyloid and connective tissues Arch. Pathol. 68 487–498. [PubMed] [Google Scholar]
- Xie Q., Guo T., Lu J., Zhou H.-M. 2004. The guanidine like effects of arginine on aminoacylase and salt-induced molten globule state Int. J. Biochem. Cell Biol. 36 296–306. [DOI] [PubMed] [Google Scholar]