

Abstract
Polyethylene glycol (PEG) conjugation to proteins has emerged as an important technology to produce drug molecules with sustained duration in the body. However, the implications of PEG conjugation to protein aggregation have not been well understood. In this study, conducted under physiological pH and temperature, N-terminal attachment of a 20 kDa PEG moiety to GCSF had the ability to (1) prevent protein precipitation by rendering the aggregates soluble, and (2) slow the rate of aggregation relative to GCSF. Our data suggest that PEG-GCSF solubility was mediated by favorable solvation of water molecules around the PEG group. PEG-GCSF appeared to aggregate on the same pathway as that of GCSF, as evidenced by (a) almost identical secondary structural transitions accompanying aggregation, (b) almost identical covalent character in the aggregates, and (c) the ability of PEG-GCSF to rescue GCSF precipitation. To understand the role of PEG length, the aggregation properties of free GCSF were compared to 5kPEG-GCSF and 20kPEG-GCSF. It was observed that even 5kPEG-GCSF avoided precipitation by forming soluble aggregates, and the stability toward aggregation was vastly improved compared to GCSF, but only marginally less stable than the 20kPEG-GCSF. Biological activity measurements demonstrated that both 5kPEG-GCSF and 20kPEG-GCSF retained greater activity after incubation at physiological conditions than free GCSF, consistent with the stability measurements. The data is most compatible with a model where PEG conjugation preserves the mechanism underlying protein aggregation in GCSF, steric hindrance by PEG influences aggregation rate, while aqueous solubility is mediated by polar PEG groups on the aggregate surface.
Keywords: GCSF, protein aggregation, protein stability, polyethylene glycol, solubility
Protein aggregation represents a dominant degradation pathway that is often encountered during the development of therapeutic proteins in biotechnology (Mitraki and King 1989; Mitraki et al. 1991; Clark 2001). Protein aggregates have also been shown to be intimately associated with the pathogenesis of many neurodegenerative disorders, including Alzheimer's disease, Huntington's disease, Parkinson's disease, and Retinitis Pigmentosa (Chen et al. 2002; Stefani and Dobson 2003; Li et al. 2004; Rajan and Kopito 2005). There is a growing body of evidence to suggest that protein aggregates are inherently toxic to cells, even those not specifically associated with an amyloid disease (Bence et al. 2001; Stefani and Dobson 2003). In parenterally delivered formulations, protein aggregates have the potential to cause adverse reactions in patients, such as aggregation at the site of administration and the production of neutralizing antibodies (Hermeling et al. 2004). The formation of aggregates is also a major concern during manufacturing steps, storage, and shipping of the protein drug. Due to the insoluble nature of many protein aggregates, and the hurdles in isolating intermediates, it has been inherently difficult to study the mechanisms involved in the aggregation of therapeutically important proteins until recently (Plakoutsi et al. 2005; Seefeldt et al. 2005; Williams et al. 2005). Thus, developing rational approaches to understanding and controlling protein aggregation represents a major challenge for pharmaceutical drug development.
PEG is a water-soluble, biocompatible polymer that is commonly utilized as an additive in protein formulations, and to facilitate crystallization of proteins (Cleland and Jones 1996; Kerwin et al. 2002). Historically, conjugation of protein therapeutics with PEG has been performed to improve the half-life of the protein in the blood serum because the large size of the PEG-conjugated molecule slows down renal clearance (Molineux 2002). PEG conjugation has also been used in protein encapsulation systems, where it improved the encapsulation efficiency and lowered the initial rate of release (Hora et al. 1990; Al-Azzam et al. 2005). However, there was no systematic correlation between the extent of protein aggregation and improvements in the above processes. It has been previously documented that PEG conjugation retards protein precipitation from the liquid state (Katre 1990; Kim and Park 2001); but there is an absence of systematic studies investigating the mechanism underlying this phenomenon in the literature.
In the present study, the effects of PEG conjugation to the aggregation properties of a therapeutic protein, granulocyte colony stimulating factor (GCSF) have been investigated. GCSF is a ∼19 kDa four-helix bundle protein that belongs to the family of hematopoietic cytokines (Hill et al. 1993; Chaiken and Williams 1996; Kolvenbach et al. 1997; Brems 2002; Raso et al. 2005). It regulates the growth and differentiation of hematopoietic progenitor cells to functionally activate the formation of mature neutrophils. GCSF, marketed by Amgen under the trade name of NEUPOGEN, has been widely used to treat neutropenia that is often induced by myelosuppressive chemotherapy (Metcalf et al. 1996; Buchsel et al. 2002). Neulasta is the next generation of GCSF, and has a 20-kDa PEG group attached to the N terminus of the protein. It exhibits comparable clinical benefits as GCSF, but due to its long half-life in the serum, only needs to be dosed once per chemotherapy cycle, as opposed to repeated daily administration for GCSF (Molineux 2002).
The shelf life of GCSF, commercially formulated in acidic conditions, is >2 yr at 2°C to 8°C (Herman et al. 1996). However, at higher pH, such as neutral pH, GCSF has a tendency to aggregate at moderate to high protein concentrations and elevated temperatures (Krishnan et al. 2002; Raso et al. 2005). Since the GCSF concentrations required in the blood stream for effective action are in the picomolar range (Brems 2002), it does not affect the use of GCSF as a drug, as GCSF aggregation is highly concentration dependent (Raso et al. 2005). However, aggregation can pose severe restrictions on the handling of the protein during the manufacturing process as well as for high concentration dosage forms. It is shown here that 20kPEG-GCSF has the added advantage that even at relatively high concentrations it does not precipitate out of solution. Unlike GCSF, which had a tendency to precipitate at neutral pH and 37°C (Krishnan et al. 2002; Raso et al. 2005), 20kPEG-GCSF formed soluble aggregates under the same conditions. The presence of the PEG moiety did not perturb the secondary structural transitions or covalent interactions involved in the aggregation process. Our data suggests that the pathway of PEG-GCSF soluble aggregate formation was similar to that of GCSF precipitation. Strikingly, conjugation of the PEG group to GCSF greatly retarded the rate as well as the extent of aggregate formation, for both short- and long-length PEG-GCSF. In addition to greater bioavailability, PEG conjugation thus represents a powerful approach to enhancing the inherent stability of GCSF toward aggregation while preserving the properties of the molecule.
Results
Conjugation of 20kPEG to GCSF prevented protein precipitation
To compare the solubility properties of 20kPEG-GCSF and GCSF under physiological conditions, both proteins were incubated at 37°C in 100 mM sodium phosphate at pH 6.9, for the times indicated in Figure 1A. A concentration of 5 mg/mL was chosen as the kinetics of GCSF aggregation were optimal for this study (>50% after 48 h), and lower concentrations precipitated very slowly (Raso et al. 2005). At each time point, the sample was centrifuged and the protein concentration measured using absorption at 280 nm. The percentage of insoluble protein was calculated from the difference in the protein concentration between time zero and the subsequent time points. It was observed that while GCSF rapidly precipitated, 20kPEG-GCSF stayed completely soluble. To test the effect of free PEG on the solubility of GCSF, solutions containing equimolar concentrations of GCSF and free 20kPEG were incubated at pH 6.9, 37°C for 48 h, and compared to solutions of GCSF and 20kPEG-GCSF (Fig. 1B,C). Free PEG was not able to prevent GCSF precipitation, as observed by the formation of visible precipitates in the presence of free PEG (Fig. 1C) and turbidity at 600 nm (Fig. 1B). Further, from size-exclusion chromatography, the percent of GCSF that was insoluble after a 48-h incubation (52.83 ± 1.36%) was found to be essentially the same as that obtained for GCSF in the presence of 1:1 free 20kPEG (52.1 ± 1.8%), supporting this conclusion.
Figure 1.
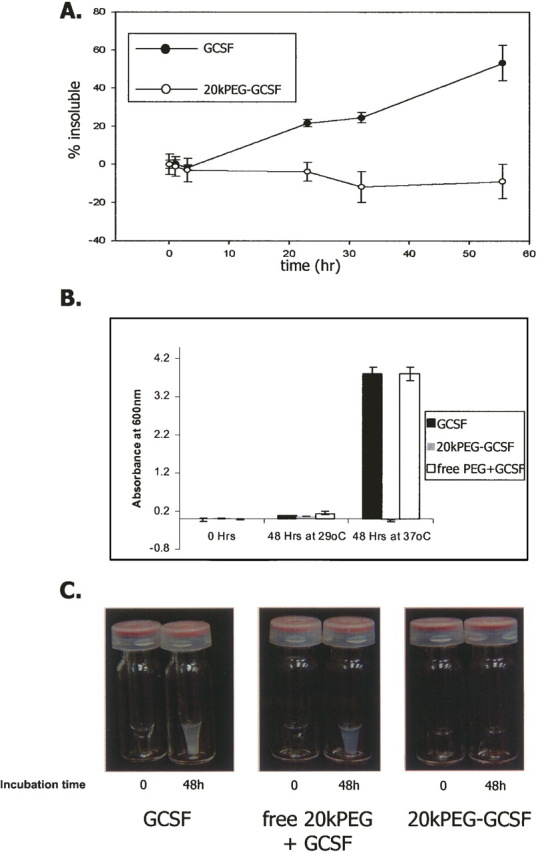
Conjugation of PEG to GCSF prevents precipitation at pH 7, 37°C. (A) GCSF (filled circles) and 20kPEG-GCSF (open circles) at 5 mg/mL and pH 6.9 were incubated at 37°C for time periods indicated, prior to measurement of protein solubility by absorbance of supernatant at 280 nm. (B) Effect of free PEG on GCSF precipitation. GCSF (black bar), 20kPEG-GCSF (gray bar), and GCSF + free 20kPEG (white bar) were incubated at 5 mg/mL, pH 6.9, and either 29°C or 37°C for 48 h. At the end of the incubation period, the samples were assessed for turbidity by measuring optical density at 600 nm. (C) Photographs of solutions of GCSF (left panel), free 20kPEG + GCSF (middle panel), and 20kPEG-GCSF after the 37°C incubation in B.
20kPEG-GCSF, but not GCSF, formed soluble aggregates
To examine the state of 20kPEG-GCSF in solution after the incubation period described above, aliquots of GCSF and 20kPEG-GCSF were analyzed by size-exclusion chromatography (SEC) (Fig. 2A,B). Both GCSF and 20kPEG-GCSF were predominantly monomeric at the beginning of incubation (t = 0). After 48 h of incubation at neutral pH and 37°C, there was a significant loss of GCSF monomer, due to conversion into insoluble forms (Fig. 2A). In contrast, at the end of the incubation period, 20kPEG-GCSF accumulated into soluble, higher order multimeric forms that eluted in the void volume of the SEC column (henceforth called “soluble aggregates”; Fig. 2B). Figure 2C quantifies the extent of soluble aggregates formed relative to time zero in 20kPEG-GCSF (open triangles), as opposed to GCSF (closed triangles), over 144 h. It was seen that there was an initial rapid increase in soluble aggregates in 20kPEG-GCSF, but appeared to plateau at ∼18% over the course of 6 d. No detectable soluble aggregates were observed in GCSF in the same time period. Further, incubation with free PEG did not cause GCSF to form soluble aggregates (data not shown). Thus, conjugation of a 20-kDa PEG group to GCSF prevented protein precipitation by rendering the aggregates soluble.
Figure 2.
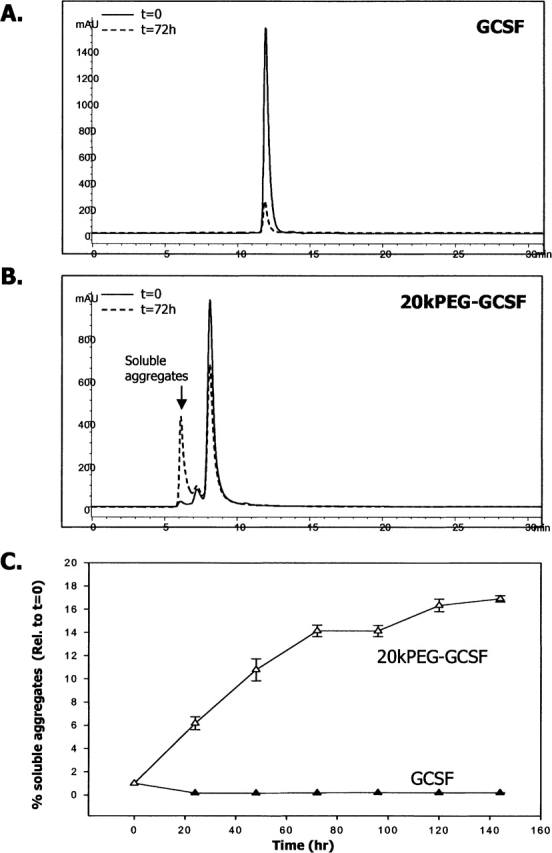
Aggregates of 20kPEG-GCSF are soluble. SEC-HPLC chromatograms of (A) GCSF and (B) 20kPEG-GCSF at t = 0 and t = 72 h of incubation at 37°C, pH 6.9. Monomer retention time of GCSF was ∼12 min, and that of 20kPEG-GCSF was ∼8.5 min. The high molecular weight material (HMW) in the 20kPEG-GCSF sample after 48 h of incubation eluted in the void volume (6–7 min). (C) Quantitation of the amount of soluble aggregates (HMW in the void volume) at each time point relative to time zero for GCSF (filled triangles) and 20kPEG-GCSF (open triangles).
20kPEG-GCSF soluble aggregates and GCSF precipitate shared a similar extent of oligomeric and covalent character
A number of experiments were carried out to investigate the mechanism underlying the formation of soluble aggregates in the case of PEG-GCSF. First, the extent of multimer formation between GCSF and 20kPEG-GCSF was investigated. One possible explanation for the appearance of a single peak near the void volume in SEC is that the “soluble aggregate” of 20kPEG-GCSF may be a single, homogenous species. On the other hand, the void volume peak may, in fact, contain several oligomeric species, which elute as one peak due to the limitations of SEC resolution. To distinguish between these possibilities, and to compare the size distribution of the soluble aggregate with respect to the insoluble precipitate of GCSF, isolated soluble aggregate species and the solubilized GCSF pellet were analyzed by SDS-PAGE (Fig. 3A, left panel). The solubilized pellet from the free PEG + GCSF combination was analyzed as a control. The monomeric form of GCSF (with or without free PEG) ran close to the predicted mass of ∼18 kDa (“GCSF,” closed arrow). On the other hand, the monomeric form of 20kPEG-GCSF ran at an apparent size of ∼55 kDa, much larger than the predicted mass of 38 kDa (“20kPEG-GCSF,” open block arrow). GCSF from solubilized pellets of either GCSF alone or GCSF + free PEG, as well as the 20kPEG-GCSF soluble aggregate migrated as an ensemble of monomer, dimer, trimer, and higher order multimers. Thus, the void volume (soluble aggregate) peak in the SEC of 20kPEG-GCSF did not represent one species, but was composed of a distribution of multimeric forms that were similar to that observed in the GCSF pellet, as assessed by SDS-PAGE.
Figure 3.
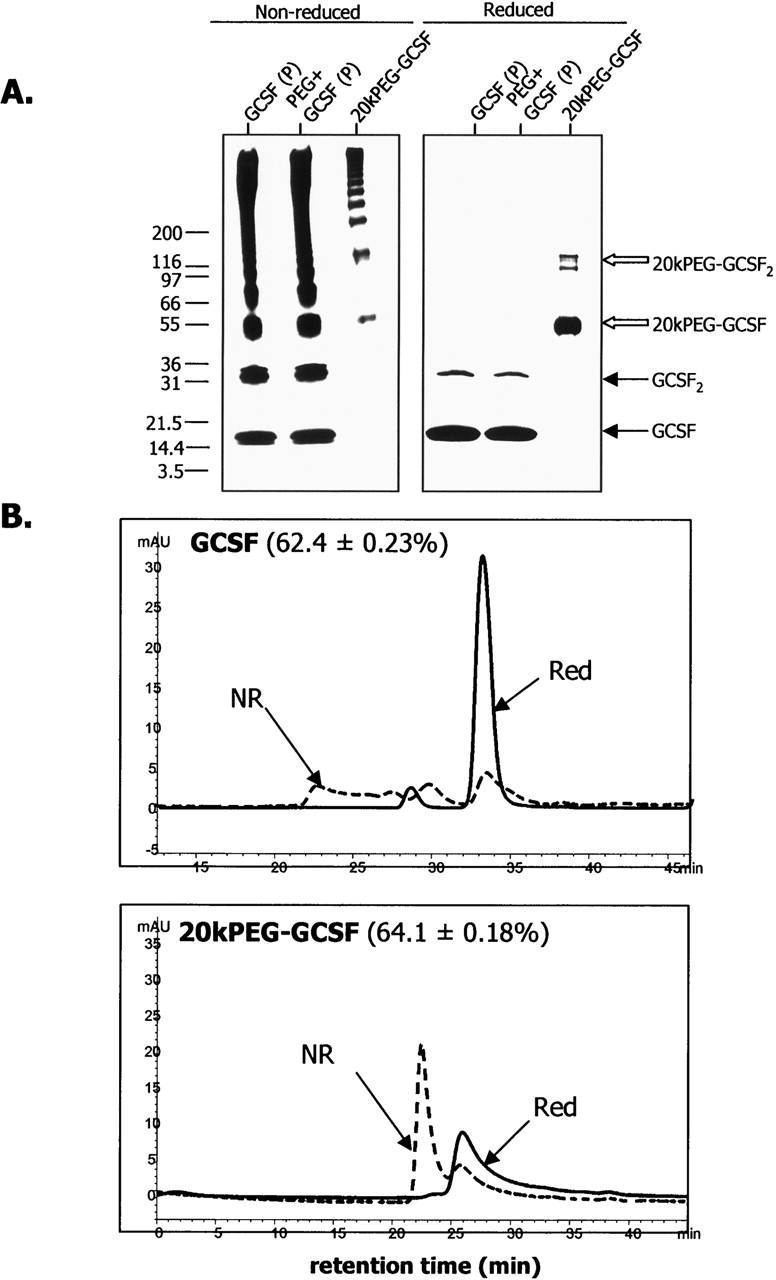
Similar extent of oligomeric and covalent character in GCSF and 20kPEG-GCSF aggregates. (A) Silver-stained SDS-PAGE gels of solubilized pellets of GCSF and GCSF + free 20kPEG, and isolated soluble aggregate of 20kPEG-GCSF. The gels were run both nonreduced (left panel) and reduced (right panel). The monomer and dimer positions for GCSF (closed arrow) and 20kPEG-GCSF (open block arrow) are indicated. (B) Denatured SEC chromatograms are shown for solubilized GCSF pellet (top panel) and isolated 20kPEG-GCSF soluble aggregates (bottom panel). Samples were either reduced and alkylated (“Red”), or run as alkylated but nonreduced (“NR”). The percent covalent character for each sample from this analysis has been indicated.
To evaluate whether these multimers were covalently linked, the above samples were also treated with DTT to reduce disulfide bonds, prior to loading (Fig. 3A, right panel). It was seen that the reduced samples contained mainly the monomer, with small amounts of the dimer (“GCSF2,” closed arrow for the GCSF and GCSF + free PEG samples and “20kPEG-GCSF2,” open blocked arrow for the 20kPEG-GCSF sample). The presence of higher order multimers in the nonreduced gel suggested that they were covalent. The elimination of these higher order multimer bands indicated that they were disulfide linked, for both the GCSF pellet as well as the 20kPEG-GCSF soluble aggregate.
To quantify and compare the extent of higher order covalent oligomers present in the GCSF pellet and in the 20kPEG-GCSF soluble aggregate, these samples (the solubilized pellet of GCSF and the isolated soluble aggregate of 20kPEG-GCSF, respectively) were analyzed by denatured size-exclusion chromatography (D-SEC) (Fig. 3B), under nonreducing as well as reducing conditions. The nonreduced solubilized pellet from GCSF exhibited a monomeric species at ∼34 min, a dimer at ∼30 min, and higher order multimers at ∼22 min, near the void volume (Fig. 3B, upper panel). The nonreduced soluble aggregate from 20kPEG-GCSF eluted differently; only the monomer peak at ∼26 min and the void volume peak at ∼23 min were apparent (Fig. 3B, lower panel). Presumably, this was due to the much larger size of the 20kPEG-GCSF molecule compared to GCSF, coupled with a larger hydrodynamic radius due to solvated SDS molecules, the same forces that caused it to run larger than expected on SDS-PAGE. Treatment with DTT, as in the case of SDS-PAGE, resulted in predominantly monomeric and some dimeric forms in both samples, accompanied by complete loss of higher oligomeric forms. From this analysis, the percent of covalent forms in the GCSF pellet sample was found to be 62.4 ± 0.23%, while that for the soluble aggregate sample from 20kPEG-GCSF was 64.1 ± 0.18%. These observations revealed that the distribution of covalent, higher order multimers found in the GCSF pellet was preserved in the 20kPEG-GCSF soluble aggregate, and therefore suggested that conjugation of PEG to GCSF did not alter the forces that held the aggregates together.
Secondary structural transitions that accompany aggregation are conserved between GCSF and 20kPEG-GCSF
One hypothesis to explain why 20kPEG-GCSF did not precipitate under conditions that favored precipitation of GCSF would be that conjugation of the PEG moiety retarded the secondary structural transitions in the protein that were responsible for aggregation. As seen in the crystal structure (Hill et al. 1993), native GCSF contained mostly helical structures. This was further verified by a signal at 1656 cm−1 in second-derivative FTIR, as shown previously (Arakawa et al. 1995, Krishnan et al. 2002). On the other hand, aggregation of GCSF involved a conformational transition, as evidenced by strong intensity bands at 1620 cm−1 and 1695 cm−1, corresponding to a large extent of intermolecular β-sheet (Krishnan et al. 2002). To test whether presence of the 20-kDa PEG group influences the temperature-dependent shift in GCSF conformation from helix to β-sheet at neutral pH, FTIR spectra were recorded for GCSF and 20kPEG-GCSF with single-degree temperature increments, from 35–45°C (Fig. 4A). The protein was incubated for 1 min at each temperature prior to the spectral scan. Figure 4A (left panel) shows that GCSF at 35°C was predominantly α-helical as evidenced by the band at ∼1655 cm−1. With increasing temperature, the β-sheet content, reflected in the 1622 cm−1 and 1695 cm−1 bands, significantly increased. At 45°C, most of the α-helical structure was converted to β-sheet. Secondary structural transitions for 20kPEG-GCSF accompanied by the same increases in temperature are shown in Figure 4A (right panel). It was seen that both the starting spectral profile, as well as the changes in the spectra with temperature, were almost identical between GCSF and 20kPEG-GCSF.
Figure 4.
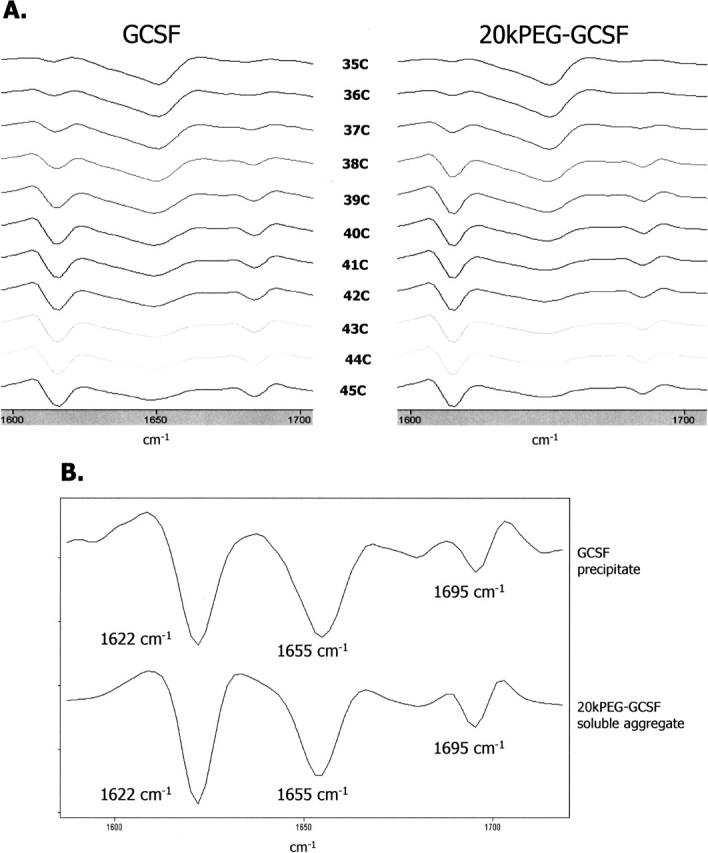
Conformational transitions and aggregate structure are preserved in PEG-GCSF. (A) PEG conjugation does not perturb the protein structure and transitions responsible for GCSF aggregation. GCSF (left panel) or PEG-GCSF (right panel) was incubated in a temperature ramp experiment from 35°–45°C. The secondary structure at each temperature was assessed by second-derivative FTIR. Helical structure was detected as a band at 1655 cm−1, while the intermolecular β-sheet was detected as a pair of bands at 1622 cm−1 and 1695 cm−1. (B) Comparison of the secondary structure of the GCSF precipitate and 20kPEG-GCSF soluble aggregate by FTIR.
Figure 4B shows a comparison of the secondary structures of the GCSF precipitate and the isolated 20kPEG-GCSF soluble aggregate as revealed by second-derivative FTIR spectroscopy. FTIR was optimal for this purpose as it could analyze secondary structure in both solid as well as liquid forms. The GCSF pellet as well as the 20kPEG-GCSF soluble aggregate possessed very similar extent of intermolecular β-sheet content, as evidenced by the overlapping peak positions and areas (for both the 1622 and 1695 cm−1 peaks). The stretching frequency and area under the curve for the 1655 cm−1 peak, corresponding to α-helical structures, was also conserved between the pellet and the soluble aggregate.
These observations suggested that conjugation of the PEG moiety to GCSF did not alter the helix to sheet transition that accompanied aggregation.
Rescue of GCSF precipitation by 20kPEG-GCSF
For proteins, it is generally regarded that the mechanism of aggregation involves the self-association of intermediates along the folding pathway (Fink 1998; Havel et al. 1998) or misfolded states (Canet et al. 1999; Stefani and Dobson 2003). The similarity in secondary structural transitions accompanying aggregation and the extent of covalent character in the aggregates of GCSF and 20kPEG-GCSF suggested a similarity between the pathway of GCSF precipitation, and the intermediates involved, to that of 20kPEG-GCSF soluble aggregate formation. To further test this, GCSF was incubated with either 1:1 20kPEG-GCSF (2.5 mg/mL each) or 2:1 20kPEG-GCSF (5 mg/mL GCSF, 2.5 mg/mL 20kPEG-GCSF), and the extent of precipitation of these samples was compared to GCSF alone or GCSF incubated with free 20kPEG (latter two at 5 mg/mL) (Fig. 5A). After 48 h of incubation at pH 6.9 and 37°C, as expected, there was significant increase in sample turbidity due to precipitation, measured as absorbance at 700 nm, in the GCSF (black bar) and GCSF + free 20kPEG (white bar) samples. However, for both GCSF + 20kPEG-GCSF samples, there was a dramatic decrease in the turbidity. Thus, co-incubation of GCSF with 20kPEG-GCSF effectively suppressed or “rescued” protein precipitation.
Figure 5.
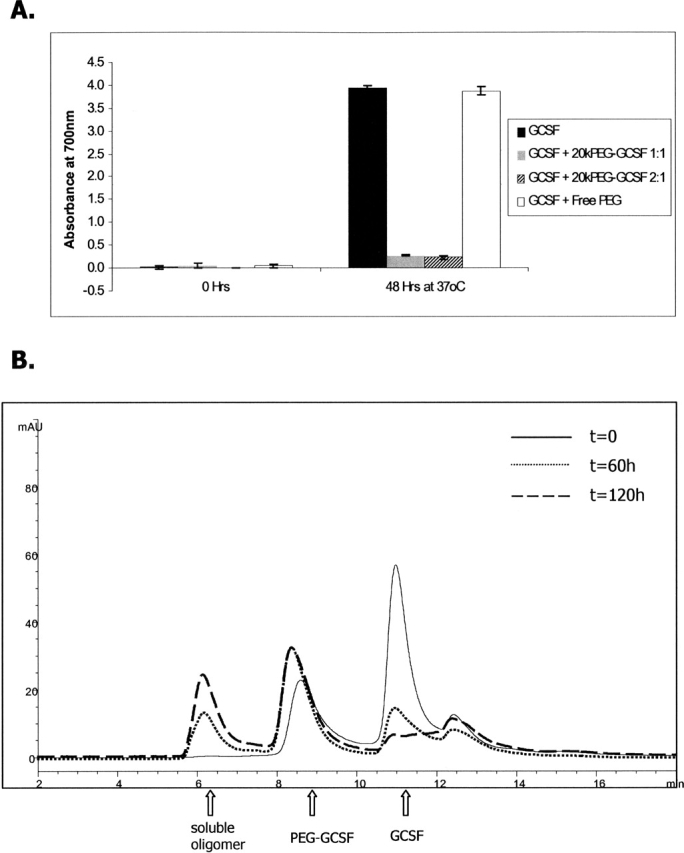
20kPEG-GCSF can rescue GCSF precipitation. (A) GCSF was incubated for 48 h at pH 6.9, 37°C either alone (black bar), or with 1:1 20kPEG-GCSF (gray bar), 2:1 20kPEG-GCSF (hatched bar), or free 20k PEG (white bar). The starting GCSF concentration was 5 mg/mL, and protein turbidity was monitored by absorbance at 700 nm. (B) GCSF was co-incubated with 1:1 20kPEG-GCSF as described above, and supernatants analyzed by SEC are shown at t = 0, t = 60 h and t = 120 h. The peaks corresponding to GCSF monomer, PEG-GCSF monomer, and soluble oligomers are shown.
To investigate the fate of GCSF and PEG-GCSF in these samples, the supernatants were analyzed by SEC, as this technique separates the monomeric forms of the two proteins. Figure 5B shows SEC chromatograms of GCSF incubated with 1:1 20kPEG-GCSF at the t = 0 h, t = 60 h, and t = 120 h time points. At t = 0, the sample contained two major peaks at ∼11 min and 8.7 min corresponding to GCSF monomer and 20kPEG-GCSF monomer, respectively. At t = 60 h and t = 120 h, there was a large decrease in the GCSF monomer peak, a shift in the peak corresponding to PEG-GCSF by ∼0.3 min, and the appearance of a soluble aggregate peak. The shift in the PEG-GCSF monomer peak suggested the formation of a GCSF:PEG-GCSF heterodimer. In the absence of precipitation, the growth of the heterodimer and soluble aggregate peaks concomitant with loss of GCSF monomer peak suggested an association between GCSF and PEG-GCSF.
Given the specificity of the aggregation process (Speed et al. 1996; Rajan et al. 2001; Krebs et al. 2004), these observations, taken together with results from Figures 3 and 4, strongly suggest that the pathway of GCSF precipitation was the same as that for 20kPEG-GCSF soluble aggregate formation.
Difference in aqueous solvation properties of GCSF and PEG-GCSF
The previous experiments indicated that the mechanism underlying solubility of the aggregates was not due to a perturbation of the nature of the GCSF protein moiety. The PEG group, however, does possess hydrophilic oxygen species in each repeat unit of the polymer. We therefore sought to determine whether the improved solubility of PEG-GCSF aggregates were due to the favorable orientation of solvent (water) molecules around PEG in spite of the increase in hydrophobicity due to aggregation. To verify this hypothesis, the ability of GCSF or PEG-GCSF to partition to an organic phase from an aqueous phase was tested. In this regard, water–octanol systems have been used extensively to investigate solvation properties of water for the molecule under consideration (Leo 1976; Wimley et al. 1996). Figure 6A shows SEC chromatograms of 0.5 mg/mL GCSF with 0.5 mg/mL free 20kPEG in the aqueous phase prior to and after overnight (16 h) incubation in a mixture of 90% aqueous phase (phosphate buffer at pH 6.9) and 10% octanol, with constant rotating to ensure that equilibrium was attained. Very little GCSF remained in the aqueous phase (2.4 ± 0.76%), suggesting that most of the GCSF had partitioned away from the aqueous phase. In contrast, the vast majority of 20kPEG-GCSF under the same conditions remained in the aqueous phase (93.2 ± 4.9%), as assessed from the SEC chromatograms shown in Figure 6B. Interestingly, the PEG-GCSF that was present in the aqueous phase after the overnight incubation was mostly in the form of soluble aggregates. Further, it was observed that the small amount of 20kPEG-GCSF that did partition into the octanol precipitated out of solution, causing turbidity. Since the only difference between the molecules was the conjugation of the PEG group, these experiments suggested that the favorable solvation of PEG-GCSF by water molecules, as opposed to GCSF, may account for the increased solubility of the PEG-GCSF aggregates.
Figure 6.

Favorable solvation of PEG-GCSF by water. Solvation differences between GCSF + free 20kPEG and 20kPEG-GCSF. Chromatograms of GCSF (A) and 20kPEG-GCSF (B) before (−octanol) and after overnight incubation in a 10% octanol–water mixture. Post incubation, the phases were separated and the aqueous phase was analyzed by SEC to determine extent of protein partitioning away from the aqueous phase.
Solubility and aggregation propensity of GCSF, 5kPEG-GCSF, and 20kPEG-GCSF
To evaluate the effect of PEG length on PEG-GCSF solubility and stability toward aggregation, a shorter, 5-kDa PEG chain conjugated to GCSF was produced (“5kPEG-GCSF”). Silver-stained SDS-PAGE profiles for GCSF, 5kPEG-GCSF, and 20kPEG-GCSF are shown in Figure 7A (inset). 5kPEG-GCSF was detected as a band around 25 kDa, consistent with its predicted mass of ∼23 kDa, which was larger than GCSF (18 kDa) and smaller than 20kPEG-GCSF (apparent mass on SDS-PAGE, ∼55 kDa; predicted mass, ∼38 kDa). The profiles of the three molecules were also compared on size-exclusion chromatography (SEC; data not shown), where the retention time of 5kPEG-GCSF (∼10 min) was in between that of GCSF (∼11 min) and 20kPEG-GCSF (∼8.5 min). When a 5 mg/mL solution of 5kPEG-GCSF was incubated at neutral pH and 37°C, it was observed that, similar to 20kPEG-GCSF, this solution stayed completely clear, indicating that even a 5-kDa PEG conjugated to GCSF was sufficient to prevent precipitation. This was confirmed by SEC analysis of GCSF, 5kPEG-GCSF, and 20kPEG-GCSF: The amount of total protein that remained in solution was unchanged relative to t = 0 for both 5kPEG-GCSF (open circles) and 20kPEG-GCSF (closed triangles), while the amount of soluble protein dropped rapidly to <10% for GCSF (Fig. 7A, closed circles).
Figure 7.
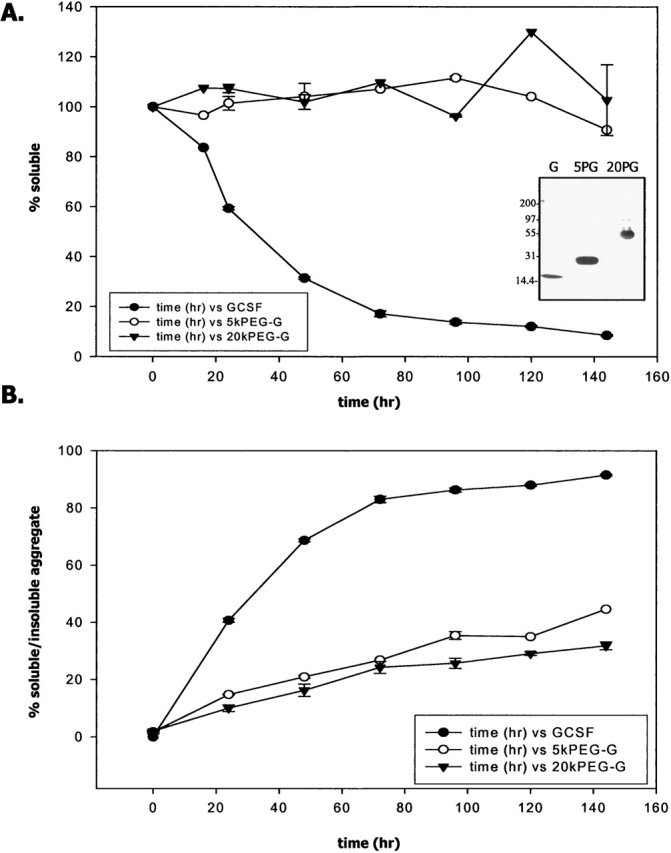
Solubility and aggregation propensity of GCSF, 5kPEG-GCSF, and 20kPEG-GCSF. (A) Percent soluble material as assessed by SEC for an incubation time course of GCSF (filled circles), 5kPEG-GCSF (open circles), and 20kPEG-GCSF (filled triangles) at pH 6.9, 37°C. (Inset) SDS-PAGE of GCSF (“G”), 5kPEG-GCSF (“5PG”), and 20kPEG-GCSF (“20PG”) to demonstrate their relative sizes. (B) From SEC measurements, the percent aggregate present in the sample at a given time point was estimated for GCSF as insoluble aggregates (from loss of main peak, closed circle), or as soluble aggregates for 5kPEG-GCSF (open circle) and 20kPEG-GCSF (closed triangle).
From SEC chromatograms obtained at each time point of incubation at physiological conditions of neutral pH and 37°C, the fraction of total material that existed either as a soluble aggregate (5kPEG-GCSF and 20kPEG-GCSF) or insoluble aggregate (GCSF) was calculated (Fig. 7B). It was seen that aggregate formation in 5kPEG-GCSF (open circles) was vastly reduced compared to GCSF (closed circles), and was only slightly increased compared to 20kPEG-GCSF (closed triangles). Thus, attachment of even a 5kPEG to GCSF greatly stabilized the molecule against intermolecular association leading to aggregation.
Conjugation of PEG stabilized against loss of in vitro bioactivity due to aggregation
To assess whether improvement in PEG-GCSF stability correlated with enhanced preservation of biological activity, samples of GCSF, 5kPEG-GCSF, 20kPEG-GCSF, and GCSF with free PEG as a control were monitored for biological activity after incubation at neutral pH and 37°C (Fig. 8). 32DCl3 cells were conditioned to grow in a medium that required the presence of GCSF for their proliferation. Cell proliferation was measured by assessing the extent of Alamar Blue dye metabolization. Figure 8A shows the relative activity of the four samples dosed at either 1 ng or 2 ng. Their activities were normalized to that of IL-3 (positive control) at these same doses. The cells were slightly less sensitive to 20kPEG-GCSF and GCSF + free PEG than they were to GCSF and 5kPEG-GCSF. In addition, the activity of the soluble aggregates was measured, and they were found to be completely inactive (i.e., the activity was comparable to cells and media without any protein). In all cases with GCSF and PEG-GCSF, the activity of the 2-ng dose was consistently higher than that of the 1-ng dose, demonstrating the sensitivity of the assay.
Figure 8.
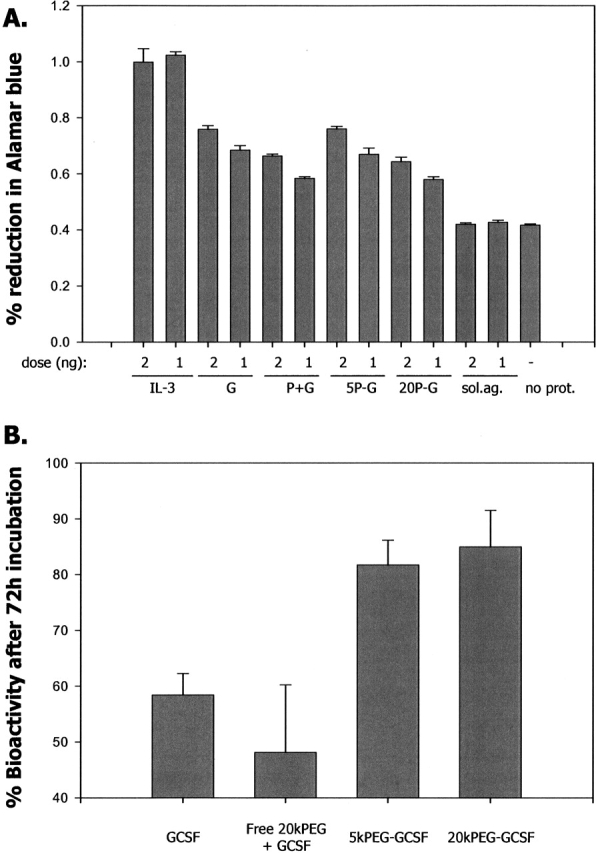
Greater preservation of in vitro biological activity by PEG conjugation. (A) Bioactivity of t = 0 GCSF (free and PEG conjugated) samples were measured from the degree of metabolism of the viability marker dye Alamar blue, for cells that had been conditioned to be dependent on GCSF for their survival. IL-3 was used as a control. The activity of the isolated 20kPEG-GCSF soluble aggregates (“sol. ag.”) was also measured. The proteins were dosed at a concentration of 2 ng or 1 ng as indicated. The activities of the proteins were compared to that of media and cells alone without protein (“no prot.”). G, GCSF; P + G, free 20kPEG plus GCSF; 5P – G, 5kPEG-GCSF; 20P-G, 20kPEG-GCSF. (B) Bioactivities of GCSF, free 20kPEG + GCSF, 5kPEG-GCSF, and 20kPEG-GCSF after 72 h of incubation at 5 mg/mL, pH 6.9, 37°C, relative to preincubation (t = 0) samples in A, whose activities were assigned to 100%.
Samples of GCSF, PEG + GCSF, 5kPEG-GCSF, and 20kPEG-GCSF that were incubated at pH 6.9 and 37°C for 72 h were then analyzed and compared to the time zero controls in Figure 8A. Figure 8B shows the bioactivity present in these samples, relative to the corresponding activity for the t = 0 samples (n = 3 measurements, averaged over both 1-ng and 2-ng doses). It was seen that the GCSF and GCSF + free PEG samples had significantly diminished activity (>30%) compared to the 5kPEG-GCSF samples and the 20kPEG-GCSF samples. Thus, resistance to aggregation in case of PEG-GCSF resulted in greater persistence of the biological activity in the molecule.
Similar principles of solubility and stability in copper-induced GCSF aggregation
We observed that addition of submillimolar quantities of copper greatly accelerated GCSF precipitation at neutral pH and 37°C. Presumably, this was due to the role of copper in facilitating intermolecular disulfide bond formation (Jensen et al. 1999). For purposes of this study, we sought to determine whether the previous observations made with PEG-GCSF aggregation followed similar principles in this scenario as well. Figure 9A shows SEC chromatograms of GCSF before and after 37°C incubation with 0.1 mM copper at a protein concentration of 0.5 mg/mL and neutral pH. It was seen that at the end of the incubation period, there was >90% precipitation (Fig. 9C), even at GCSF concentrations 10-fold lower than the earlier experiments. Consistent with this decrease in soluble material, visual precipitation of GCSF was observed. Figure 9B shows 20kPEG-GCSF SEC chromatograms under the same conditions. Similar to the earlier experiments, no visual precipitation was observed for 20kPEG-GCSF. It was noted that a large fraction of the 20kPEG-GCSF that was soluble in this experiment was monomeric, the remainder eluting in the void volume of the SEC column (“soluble aggregates”). Figure 9C shows that at the end of the incubation period, there was <20% monomer present in the GCSF sample, but >60% monomer for 20kPEG-GCSF, under the same conditions.
Figure 9.
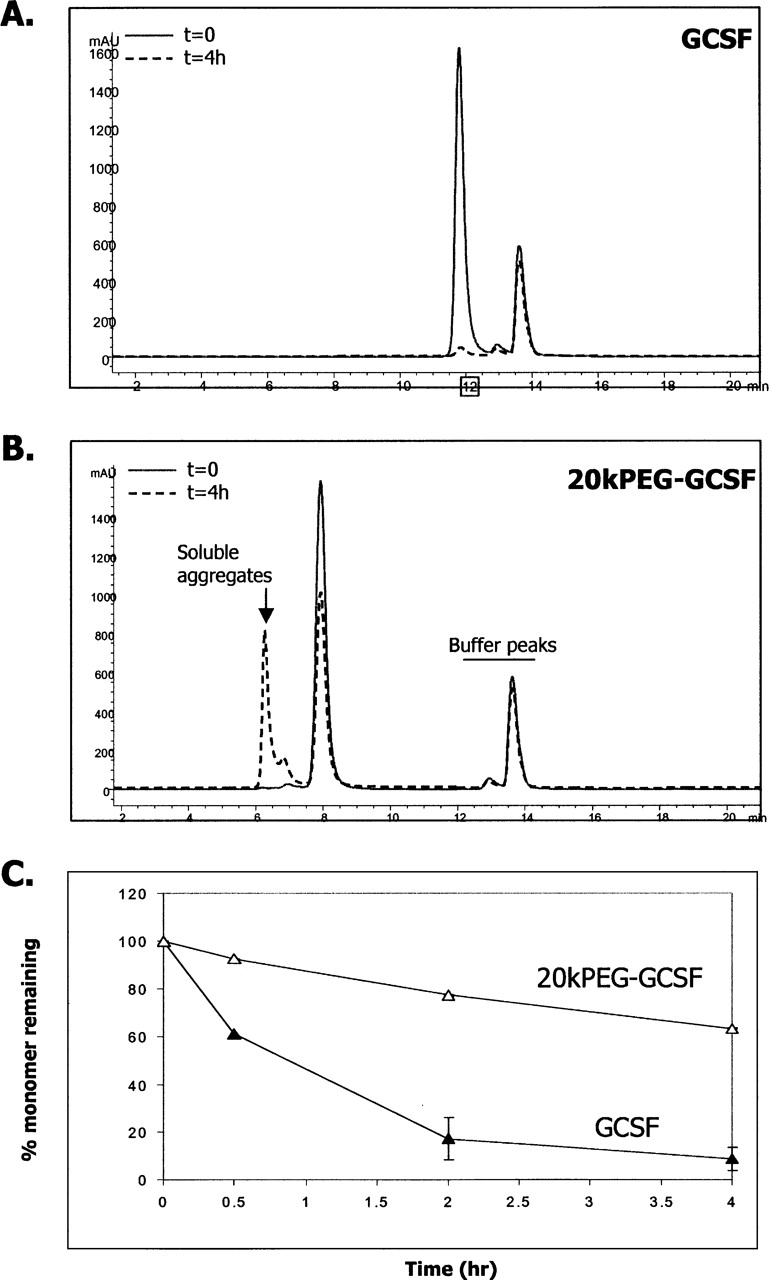
Similar principles in copper induced GCSF aggregation. SEC chromatograms of supernatant of 0.5 mg/mL GCSF solution (A) or 20kPEG-GCSF solution (B) after incubation with 0.1 mM copper (II) chloride for 4 h, in comparison to the no-copper (t = 0) control. Protein elution in the void volume (“Soluble aggregates”) and buffer peaks are indicated. (C) Quantitation of percent of GCSF (filled triangles) or 20kPEG-GCSF (open triangles) that was monomeric during the incubation period.
Thus, even under conditions of accelerated GCSF precipitation, there were similar observations of enhanced solubility, “soluble aggregate” formation and greater monomer stability in the case of 20kPEG-GCSF.
Discussion
A major finding of this study was that the PEG-GCSF aggregates were soluble, and the rate and extent of their formation was vastly reduced compared to GCSF. In principle, two mechanisms could be conceived to account for this effect. One possibility is that the PEG group poses a steric hindrance to protein–protein association. Due to the presence of side-chain residues, the protein moiety has a significantly shorter backbone chain length than the PEG group. In the absence of discernable secondary structures, this means that the PEG chain possibly sweeps a large area around the protein molecule, disabling diffusional encounters with other monomers. A second possibility is that the PEG group shields hydrophobic patches on the protein on account of its long chain length, perhaps even wrapping itself around the protein. If this were the case, protection of exposed hydrophobic patches by the PEG group would reduce the rate and extent of PEG-GCSF monomer association. In this context, it is interesting to note that during the time course of incubation, soluble aggregates build up quickly in the first 3 d, then gradually level off at ∼18%. This may perhaps be linked to the steric hindrance offered by the PEG group that slows down the formation of soluble aggregates beyond a critical threshold. However, the two mechanisms may not be mutually exclusive: protection of hydrophobic patches by PEG would be expected to decrease productive collisions between monomers.
While conjugation of PEG to GCSF produced a molecule with lower aggregation propensity, the PEG group did not appear to disrupt secondary structural transitions within the molecule (Fig. 4A). Conjugation of PEG to GCSF also did not alter the free energy of unfolding of this molecule in guanidine hydrochloride unfolding experiments performed at neutral pH (data not shown). Further, the extent of covalent associations in the soluble PEG-GCSF aggregates mirrored those found in the GCSF pellet. Thus, attachment of PEG to GCSF slowed the kinetics and reduced the overall amounts of oligomers, but did not appear to perturb the nature of the interactions that led to aggregation. This notion was further supported by the co-incubation experiment of GCSF with 20kPEG-GCSF where precipitation of GCSF was almost completely suppressed. Given the specificity of protein aggregation (Speed et al. 1996; Rajan et al. 2001; Krebs et al. 2004), these data collectively suggest that the PEG-GCSF aggregation pathway mirrors that for GCSF. Interestingly, there appeared to be a difference in the aggregation properties of PEG-GCSF, depending on the method of conjugation of PEG to the N terminus: conjugation of PEG by alkylation significantly decreased the aggregation propensity compared to that obtained by acylation (Kinstler et al. 1996). This suggested a further involvement of the charge of the N-terminal amino group on the aggregation properties of PEG-GCSF.
In this study, it was observed that both 5kPEG-GCSF and 20kPEG-GCSF retained significantly higher bioactivities compared to free GCSF after 72 h of incubation at neutral pH and 37°C. However, while there was expected to be ∼20% monomer from the stability studies at the 72-h time point for free GCSF, there was still ∼50% to 60% bioactivity remaining. This phenomenon could perhaps be accounted for by the higher variability of the bioassay compared to the stability assay. If the sample size were sufficiently large, it is likely the average bioactivity values would begin to converge with the stability numbers. However, the overall trend in bioactivity measurements was completely consistent with the predictions made by the stability assay, and gave greater confidence to the use of PEG-GCSF for higher sustained bioactivity in vitro as well in vivo.
During design of the protein drug in question, the length of PEG that needs to be attached to the protein is a key consideration. Thus far, this consideration has been primarily driven by the serum half-life afforded by the size of the resulting PEG-conjugated drug molecule. This study showed that in addition to serum half-life, the aggregation propensity of the PEG-drug molecule in consideration should also be evaluated. The data with 5kPEG-GCSF indicated that a small PEG moiety attached to the drug could result in a large benefit for protein stability toward aggregation. In other words, even if a large increase in serum half-life is not desired, conjugation of PEG to a drug molecule may be considered solely from the point of view of a stability advantage toward aggregation. The improvements in monomer stability can be expected to correlate with improvements not only in storage and handling, but also in greater retention of biological activity, as shown in this study.
This study revealed that polyethylene glycol conjugated GCSF remained soluble under conditions where native GCSF precipitated. This phenomenon could possibly be explained by the very hydrophilic nature of the PEG group compared to protein moiety. The oxygen atoms in PEG group may form favorable hydrogen bond interactions with water, and make the process of solvation by water around PEG-protein more favorable than around the protein alone. In this event, in spite of protein–protein association, there would be sufficient water molecules around the PEG-protein multimers to keep the entire assembly soluble. This model would predict the existence of PEG-protein “micelles” with PEG-protein assemblies containing a hydrophobic protein core and a hydrophilic PEG exterior that is completely solvated by water. Indeed, the water/organic solvent partition experiments revealed a greater preference for PEG-GCSF to remain in the aqueous phase, supporting this model. It is possible that protein unfolding might have occurred during the transfer process, and from this experiment it would be difficult to assess the solubility of the folded versus unfolded forms of GCSF. However, the formation of soluble aggregates in this experiment by 20kPEG-GCSF is intriguing: it suggests a highly polar nature of the exterior of PEG-GCSF aggregates, perhaps by formation of PEG-protein micelles, thereby preventing transfer into the octanol phase. The presence of the oxygen groups in each repeat unit of the PEG group would potentially be a good site for solvation by water, by virtue of hydrogen bonding between the water hydrogens and the lone pair of electrons on the PEG oxygen atoms.
Controlling protein aggregation represents a major challenge during the development of protein therapeutics. Several approaches have been employed at improving protein solubility and stability. The most common one involves reengineering the protein via site-directed mutagenesis. In the case of GCSF, three groups have undertaken this challenge (Lu et al. 1999; Bishop et al. 2001; Luo et al. 2002), using the knowledge obtained by X-ray structure determination of this molecule (Hill et al. 1993; Lovejoy et al. 1993). While these approaches produced GCSF mutants with greater thermodynamic stability and biological activity than the wild-type molecule, it was difficult to predict the aggregation properties of the resulting mutant proteins. A second approach involves the addition of excipients to reduce aggregation, such as sucrose, which has been shown to inhibit the aggregation propensity of GCSF (Krishnan et al. 2002). Protein aggregation has also been suppressed by novel approaches such as application of high pressure (Foguel et al. 1999) and protein glycosylation (Sinclair and Elliott 2005). While these approaches are useful, they do not substantially alter the pharmokinetic profile of the molecule in blood serum. Protein PEG conjugation, at least in the case of GCSF, thus represents a unique solution, as it prevents protein precipitation and severely retards oligomerization, while simultaneously conferring greatly improved pharmacokinetic properties to the molecule. If this were found to be true for other molecules, this combination would greatly serve the cause of producing stable yet efficacious therapeutic protein drugs.
Materials and methods
Reagents and protein
All chemicals were of the highest purity grade available from commercial sources. Linear 5k or 20k PEG products were purchased from Shearwater polymers. Pharmaceutical quality rhGCSF was produced and purified at Amgen, Inc. using heterologous expression in Escherichia coli. PEGylation and purification of GCSF with either 5 kDa or 20 kDa PEG-aldehyde was achieved using reductive alkylation as previously described (Kinstler et al. 2002). This method has been shown to selectively conjugate a single linear PEG moiety to the N terminus of GCSF, as evidenced by endoproteinase mapping, MALDI TOF-MS, and size-exclusion HPLC with on-line light scattering (Kinstler et al. 1996, 2002). The purity of the unmodified and modified versions of GCSF were comparable and typically above 95%, as assessed by size-exclusion chromatography (SEC).
SEC
SEC was performed using a Phenomenex Biosep SEC-S 3000 4.6 mm × 3000 mm column on an Agilent 1100 system with diode array detection. The mobile phase was 50 mM sodium phosphate, 100 mM sodium chloride, 5% ethanol (pH 7.5), and the flow rate was 0.3 mL/min. It has been shown earlier that the presence of ethanol does not alter the aggregation profile as quantified by SEC, but its presence improves column performance and peak shape (Ratto et al. 1997). The total time for the method was 35 min, and the column eluate was monitored at 215 nm. Peak areas in the chromatograms were used to quantify amounts of monomer and soluble aggregate. Chromatograms for each peak were analyzed in two ways: either as percent remaining relative to the amount at time zero, or as a percent of the total protein present at that particular time point.
Aggregation conditions
For aggregation experiments involving incubation of a single protein, the final protein concentration was always 5 mg/mL, and the buffer was 100 mM sodium phosphate, pH 6.9. GCSF or PEG-GCSF was diluted into this solution from a stock solution of 50 mg/mL, kept in 20 mM sodium acetate, pH 4.0. The protein concentrations were estimated at 280 nm, where the PEG moiety has negligible absorbance. For GCSF and PEG-GCSF (5k or 20k), equal concentrations in the mg/mL units correspond to equimolar concentrations. This procedure was found to give identical results compared to a process of diluting it first in pH 4 buffer followed by exchange into pH 6.9 buffer by dialysis at 4°C. No microfiltration was carried out. Proteins were incubated at 37°C for times indicated. Prior to SEC analysis, samples were centrifuged at 14,000g for 5 min to remove any precipitate. The extent of insoluble protein was calculated as the difference between the total protein present before the incubation and the soluble protein present after the incubation, based on peak areas in the respective SEC chromatograms. For coincubation experiments, a 1:1 ratio represented a concentration of 2.5 mg/mL for each protein. Turbidity measurements were performed by reading the optical density at either 600 nm or 700 nm. For SDS-PAGE and denatured size-exclusion (D-SEC) analysis, GCSF insoluble aggregates were washed three times with 100 mM sodium phosphate buffer to remove any soluble material. The material each time was centrifuged at 14,000g for 5 min and the supernatent was removed. Pellets were solubilized using 10 mM acetate, 5% sorbitol (pH 5.2), containing 2% sodium dodecyl sulphate (SDS), and 10 mM iodoacetamide (IAA). The resulting solution was heated at 80°C for 20 min to achieve a clear solution. To prepare soluble aggregates of 20kPEG-GCSF quantitatively and rapidly, a solution of the protein at 5 mg/mL and pH 6.9 in 100 mM sodium phosphate was incubated at 50°C for 1 h. This condition was found to produce >95% soluble aggregates. For copper-induced aggregation experiments, 1 mM copper (II) chloride was diluted from a stock solution of 1 M in water. The final protein concentration was 0.5 mg/mL, diluted from an eight times GCSF stock and a 20× PEG-GCSF stock, into 100 mM sodium phosphate, pH 6.9. The pH of the final solutions was checked to ensure that the pH did not deviate from 6.9. Proteins were incubated for t = 4 h at 37°C, centrifuged, and analyzed by SEC similar to the temperature induced aggregation experiments.
SDS-PAGE
A 4% to 20% gradient gel was used (Novex/Invitrogen). Protein was visualized by silver staining. Prior to loading, samples were heated at 80°C for 20 min. For sample reduction, 10 mM DTT was added to the sample buffer prior to heating.
Denatured size-exclusion chromatography (D-SEC)
For analysis by nonreduced D-SEC, SDS and IAA were added to a final concentration of 2% and 15 mM, respectively, to the samples and the resulting solution was then incubated at 80°C for 20 min. For reduced D-SEC, SDS and DTT were added to a final concentration of 2% and 10 mM, respectively. Samples were analyzed on an Agilent 1050 HPLC with diode array detection. Two 7.8 mm × 300 mm G3000 Tosoh TSKSWxL columns were employed in series. The mobile phase was 100 mM sodium chloride, 150 mM sodium phosphate, 0.1% SDS, pH 6.9. The sample chamber was kept at room temperature and the buffer was filtered just prior to starting the HPLC method.
FTIR
Samples were analyzed by Bomem MB-series FTIR following drying using single-bounce attenuated total reflectance. Using an Omega CN76000, sample temperature was adjusted in 1°C increments with 10-min equilibration times.
Water–octanol partition experiments
One hundred micrograms of GCSF or 20kPEG-GCSF was added from a 12 mg/mL stock to obtain 180 μL total of a solution of 100 mM phosphate buffer (pH 6.9). To this solution, either 20 μL of octanol or control buffer were added to make a total of 200 μL (10% octanol). Samples were incubated overnight in a rotating container to ensure adequate mixing and equilibrium. The next morning, the phases were allowed to settle and the protein was sampled from the aqueous phase or the control sample. After centrifugation (13,000 rpm for 5 min), samples (10 μL) were analyzed by size-exclusion column as described above and the amount of protein was quantified as the area under the curve of the HPLC chromatogram.
Biological activity measurements
The bioassay was conducted in flat-bottomed 96-well plates. For each sample, 32DCl3 cells that had been previously selected for proliferative response to rh-GCSF were plated in triplicate at a concentration of 10,000 cells per well. Cells were plated in 100 μL of growth medium (Iscove's Modified Dubelcco's Medium) supplemented with 2.5% fetal bovine serum per well. Samples were diluted in the same growth medium and added to the appropriate wells in 100 μL to give a final concentration of either 1 ng/mL or 2 ng/mL. Recombinant murine IL-3 was used as a positive growth control at the same concentrations. Plates were incubated for 4 d at 37°C, 5% CO2, ambient O2, and full humidity. After 4 d of incubation, 20 μL of Alamar blue dye was added to each well. Plates were then incubated for an additional 2 d before reading absorbance of each well at wavelengths of 570 nm and 600 nm. The percent reduction of Alamar blue was calculated based on the formula provided by the manufacturer (Biosource). A higher reduction of the dye correlates with a larger number of cells per well and thus acts as an indirect measure of proliferation.
Acknowledgments
We thank Susan Hershenson for stimulating discussions and useful comments; Ravi Ali and Grace Chu for technical support; and Krishnan Sampath, Yatin Gokarn, Arnold McAuley, Vasu Dharmavaram, Dean Liu, Bruce Mason, Ramil Latypov, Tim Osslund, Carl Kolvenbach, and Bruce Kerwin for helpful feedback. We thank Amgen manufacturing and process development teams for material availability.
Footnotes
Reprint requests to: Rahul S. Rajan, Department of Pharmaceutics, Amgen Inc., Thousand Oaks, CA 91320, USA; e-mail: rrajan@amgen.com; fax: 805-375-5794; or David N. Brems, Department of Pharmaceutics, Amgen Inc., Thousand Oaks, CA 91320, USA; e-mail: dbrems@amgen.com; fax: 805-375-5794.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.052004006.
References
- Al-Azzam W., Pastrana E.A., King B., Mendez J., Griebenow K. 2005. Effect of the covalent modification of horseradish peroxidase with poly(ethylene glycol) on the activity and stability upon encapsulation in polyester microspheres J. Pharm. Sci. 94 1808–1819. [DOI] [PubMed] [Google Scholar]
- Arakawa T., Horan T.P., Leong K., Prestrelski S.J., Narhi L.O., Hu S. 1995. Structure and activity of granulocyte colony-stimulating factor derived from CHO cells containing cDNA coding for alternatively spliced sequences Arch. Biochem. Biophys. 316 285–289. [DOI] [PubMed] [Google Scholar]
- Bence N.F., Sampath R., Kopito R.R. 2001. Impairment of the ubiquitin proteasome system by protein aggregation Science 292 1552–1555. [DOI] [PubMed] [Google Scholar]
- Bishop B., Koay D.C., Sartorelli A.C., Regan L. 2001. Reengineering granulocyte colony-stimulating factor for enhanced stability J. Biol. Chem. 276 33465–33470. [DOI] [PubMed] [Google Scholar]
- Brems D.N. 2002. The kinetics of G-CSF folding Protein Sci. 11 2504–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchsel P.C., Forgey A., Grape F.B., Hamann S.S. 2002. Granulocyte macrophage colony-stimulating factor: Current practice and novel approaches Clin. J. Oncol. Nurs. 6 198–205. [DOI] [PubMed] [Google Scholar]
- Canet D., Sunde M., Last A.M., Miranker A., Spencer A., Robinson C.V., Dobson C.M. 1999. Mechanistic studies of the folding of human lysozyme and the origin of amyloidogenic behavior in its disease-related variants Biochemistry 38 6419–6427. [DOI] [PubMed] [Google Scholar]
- Chaiken I.M. and Williams W.V. 1996. Identifying structure–function relationships in four-helix bundle cytokines: Towards de novo mimetics design Trends Biotechnol. 14 369–375. [DOI] [PubMed] [Google Scholar]
- Chen S., Ferrone F.A., Wetzel R. 2002. Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation Proc. Natl. Acad. Sci. 99 11884–11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark E.D. 2001. Protein refolding for industrial processes Curr. Opin. Biotechnol. 12 202–207. [DOI] [PubMed] [Google Scholar]
- Cleland J.L. and Jones A.J. 1996. Stable formulations of recombinant human growth hormone and interferon-gamma for microencapsulation in biodegradable microspheres Pharm. Res. 13 1464–1475. [DOI] [PubMed] [Google Scholar]
- Fink A.L. 1998. Protein aggregation: Folding aggregates, inclusion bodies and amyloid Fold. Des. 3 R9–R23. [DOI] [PubMed] [Google Scholar]
- Foguel D., Robinson C.R., Caetano de Sousa Jr., P., Silva J.L., Robinson A.S. 1999. Hydrostatic pressure rescues native protein from aggregates Biotechnol. Bioeng. 63 552–558. [DOI] [PubMed] [Google Scholar]
- Havel H.A., Kauffman E.W., Plaisted S.M., Brems D.N. 1998. Reversible self-association of bovine growth hormone during equilibrium unfolding Biochemistry 25 6533–6538. [DOI] [PubMed] [Google Scholar]
- Herman A.C., Boone T.C., Lu H.S. 1996. Characterization, formulation, and stability of Neupogen (Filgrastim), a recombinant human granulocyte-colony stimulating factor In Formulation, characterization and stability of protein drugs (eds. Pearlman R. and Wang Y.J.) . pp. 303–328. Plenum Press, New York. [DOI] [PubMed]
- Hermeling S., Crommelin D.J., Schellekens H., Jiskoot W. 2004. Structure–immunogenicity relationships of therapeutic proteins Pharm. Res. 21 897–903. [DOI] [PubMed] [Google Scholar]
- Hill C.P., Osslund T.D., Eisenberg D. 1993. The structure of granulocyte-colony-stimulating factor and its relationship to other growth factors Proc. Natl. Acad. Sci. 90 5167–5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hora M.S., Rana R.K., Nunberg J.H., Tice T.R., Gilley R.M., Hudson M.E. 1990. Controlled release of interleukin-2 from biodegradable microspheres Biotechnology (N.Y.) 8 755–758. [DOI] [PubMed] [Google Scholar]
- Jensen P.Y., Bonander N., Horn N., Turner Z., Farver O. 1999. Expression, purification and copper-binding studies for the first metal-binding domain of Menkes protein Eur. J. Biochem. 264 890–896. [DOI] [PubMed] [Google Scholar]
- Katre N.V. 1990. Immunogenicity of recombinant IL-2 modified by covalent attachment of polyethylene glycol J. Immunol. 144 209–213. [PubMed] [Google Scholar]
- Kerwin B.A., Chang B.S., Gregg C.V., Gonneli M., Li T., Stambini G.B. 2002. Interactions between PEG and type I soluble tumor necrosis factor receptor: Modulation by pH and by PEGylation at the N terminus Protein Sci. 11 1825–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.J. and Park K. 2001. Glucose-binding property of pegylated concanavalin A Pharm. Res. 18 794–799. [DOI] [PubMed] [Google Scholar]
- Kinstler O.B., Brems D.N., Lauren S.L., Paige A.G., Hamburger J.B., Treuheit M.J. 1996. Characterization and stability of N-terminally PEGylated rhG-CSF Pharm. Res. 13 996–1002. [DOI] [PubMed] [Google Scholar]
- Kinstler O.B., Molineux G., Treuheit M., Ladd D., Gegg C. 2002. Characterization and stability of N-terminally PEGylated rhG-CSF Adv. Drug Deliv. Rev. 54 477–485. [DOI] [PubMed] [Google Scholar]
- Kolvenbach C.G., Narhi L.O., Philo J.S., Li T., Zhang M., Arakawa T. 1997. Granulocyte-colony stimulating factor maintains a thermally stable, compact, partially folded structure at pH 2 J. Pept. Res. 50 310–318. [DOI] [PubMed] [Google Scholar]
- Krebs M.R., Morozova-Roche L.A., Daniel K., Robinson C.V., Dobson C.M. 2004. Observation of sequence specificity in the seeding of protein amyloid fibrils Protein Sci. 13 1933–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan S., Chi E.Y., Webb J.N., Chang B.S., Shan D., Goldenberg M., Manning M.C., Randolph T.W., Carpenter J.F. 2002. Aggregation of granulocyte colony stimulating factor under physiological conditions: Characterization and thermodynamic inhibition Biochemistry 41 6422–6431. [DOI] [PubMed] [Google Scholar]
- Leo A.J. 1976. Dependence of hydrophobicity of apolar molecules on their molecular volume J. Med. Chem. 19 611–615. [DOI] [PubMed] [Google Scholar]
- Li J., Zhu M., Manning-Bog A.B., Di Monte D.A., Fink A.L. 2004. Dopamine and L-dopa disaggregate amyloid fibrils: Implications for Parkinson's and Alzheimer's disease FASEB J. 18 962–964. [DOI] [PubMed] [Google Scholar]
- Lovejoy B., Cascio D., Eisenberg D. 1993. Crystal structure of canine and bovine granulocyte-colony stimulating factor (G-CSF) J. Mol. Biol. 234 640–653. [DOI] [PubMed] [Google Scholar]
- Lu H.S., Fausset P.R., Narhi L.O., Horan T., Shingawa T., Shimamoto G., Boone T.C. 1999. Chemical modification and site-directed mutagenesis of methionine residues in recombinant human granulocyte colony-stimulating factor: Effect on stability and biological activity Arch. Biochem. Biophys. 362 1–11. [DOI] [PubMed] [Google Scholar]
- Luo P., Hayes R.J., Chan C., Stark D.M., Hwang M.Y., Jacinto J.M., Juvvadi P., Chung H.S., Kundu A., Ary M.L.et al. 2002. Development of a cytokine analog with enhanced stability using computational ultrahigh throughput screening Protein Sci. 11 1218–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf D., Robb L., Dunn A.R., Mifsud S., DiRago L. 1996. Role of granulocyte-macrophage colony-stimulating factor and granulocyte colony-stimulating factor in the development of an acute neutrophil inflammatory response in mice Blood 88 3755–3764. [PubMed] [Google Scholar]
- Mitraki A. and King J. 1989. Amino acid substitutions influencing intracellular protein folding pathways Biotechnology (N.Y.) 7 690–697. [DOI] [PubMed] [Google Scholar]
- Mitraki A., Fane B., Haase-Pettingell C., Sturtevant J., King J. 1991. Global suppression of protein folding defects and inclusion body formation Science 253 54–58. [DOI] [PubMed] [Google Scholar]
- Molineux G. 2002. Pegylation: Engineering improved pharmaceuticals for enhanced therapy Cancer Treat. Rev. Suppl A 13–16. [DOI] [PubMed] [Google Scholar]
- Plakoutsi G., Bemporad F., Calamai M., Taddei N., Dobson C.M., Chiti F. 2005. Evidence for a mechanism of amyloid formation involving molecular reorganization within native-like precursor aggregates J. Mol. Biol. 351 910–922. [DOI] [PubMed] [Google Scholar]
- Rajan R.S. and Kopito R.R. 2005. Suppression of wild-type rhodopsin maturation by mutants linked to autosomal dominant retinitis pigmentosa J. Biol. Chem. 14 1284–1291. [DOI] [PubMed] [Google Scholar]
- Rajan R.S., Illing M.E., Bence N.F., Kopito R.R. 2001. Specificity in intracellular protein aggregation and inclusion body formation Proc. Natl. Acad. Sci. 98 13060–13065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raso S.W., Abel J., Barnes J.M., Maloney K.M., Pipes G., Treuheit M.J., King J., Brems D.N. 2005. Aggregation of granulocyte-colony stimulating factor in vitro involves a conformationally altered monomeric state Protein Sci. 14 2246–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratto J.J., O'Conner S.R., Distler A.R., Wu G.-M., Hummel D., Treuheit M.J., Herman A.C., Davis J.M. 1997. Ethanol-sodium chloride-phosphate mobile phase for size-exclusion chromatography of poly(ethylene glycol) modified proteins J. Chromatogr. A 763 337–344. [Google Scholar]
- Seefeldt M.B., Kim Y.S., Tolley K.P., Seely J., Carpenter J.F., Randolph T.W. 2005. High pressure studies of aggregation of recombinant human interleukin-1 receptor antagonist: Thermodynamics, kinetics and application of accelerated formulation studies Protein Sci. 14 2258–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair A.M. and Elliott S. 2005. Glycoengineering: The effect of glycosylation on the properties of therapeutic proteins J. Pharm. Sci. 94 1626–1635. [DOI] [PubMed] [Google Scholar]
- Speed M.A., Wang D.I., King J. 1996. Specific aggregation of partially folded polypeptide chains: The molecular basis of inclusion body composition Nat. Biotechnol. 14 1283–1287. [DOI] [PubMed] [Google Scholar]
- Stefani M. and Dobson C.M. 2003. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution J. Mol. Med. 81 678–699. [DOI] [PubMed] [Google Scholar]
- Williams A.D., Sega M., Chen M., Kheterpal I., Geva M., Berthelier V., Kaleta D.T., Cook K.D., Wetzel R. 2005. Structural properties of Aβ protofibrils stabilized by a small molecule Proc. Natl. Acad. Sci. 102 7115–7120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley W.C., Creamer T.P., White S.H. 1996. Solvation energies of amino acid side chains and backbone in a family of host–guest pentapeptides Biochemistry 35 5109–5124. [DOI] [PubMed] [Google Scholar]