

Abstract
The signal recognition particle (SRP) plays an important role in the delivery of secretory proteins to cellular membranes. Mammalian SRP is composed of six polypeptides among which SRP68 and SRP72 form a heterodimer that has been notoriously difficult to investigate. Human SRP68 was purified from overexpressing Escherichia coli cells and was found to bind to recombinant SRP72 as well as in vitro-transcribed human SRP RNA. Polypeptide fragments covering essentially the entire SRP68 molecule were generated recombinantly or by proteolytic digestion. The RNA binding domain of SRP68 included residues from positions 52 to 252. Ninety-four amino acids near the C terminus of SRP68 mediated the binding to SRP72. The SRP68–SRP72 interaction remained stable at elevated salt concentrations and engaged ∼150 amino acids from the N-terminal region of SRP72. This portion of SRP72 was located within a predicted tandem array of four tetratricopeptide (TPR)-like motifs suggested to form a superhelical structure with a groove to accommodate the C-terminal region of SRP68.
Keywords: signal recognition particle, SRP, protein secretion, RNA binding domain, protein–protein interaction
The signal recognition particle (SRP) ribonucleoprotein complex binds to ribosomes that engage in the synthesis and translocation of proteins with secretory signal sequences. SRP directs the emerging polypeptide to the lipid bilayer by first pausing translation followed by the binding of the ribosome/nascent chain-complex to the membrane-associated SRP receptor. Upon dissociation of the SRP from the membrane and the ribosome, translation continues and leads to the delivery of the secretory protein into the translocation channel (for review, see Keenan et al. 2001; Wild et al. 2002; Zwieb and Eichler 2002; Zwieb 2003; Doudna and Batey 2004; Halic et al. 2004; Luirink and Sinning 2004; Shan and Walter 2005).
The fundamental requirement of every cell to remain compartmentalized provides a rationale for the presence of an SRP in all organisms (Rosenblad et al. 2003). The mammalian SRP is composed of an ∼300-nucleotide SRP RNA molecule bound to six proteins named SRP9, SRP14, SRP19, SRP54, SRP68, and SRP72 according to their approximate molecular weights in kDa (Walter and Blobel 1983). During SRP assembly, SRP9/14 binds to the small (Alu) domain of the SRP RNA and thereby shortens the elongated particle (Andrews et al. 1987; Weichenrieder et al. 2000). The small domain is responsible for pausing the translation of the secretory proteins (Siegel and Walter 1986, 1988).
Within the large (S) domain of the SRP, SRP19 stabilizes the parallel arrangement of SRP RNA helices 6 and 8 in preparation for the binding of SRP54 (Wild et al. 2001; Hainzl et al. 2002; Kuglstatter et al. 2002; Oubridge et al. 2002). Earlier footprinting experiments (Siegel and Walter 1988) and the recent cryo-electron microscopy study of the ribosome-bound SRP (Halic et al. 2004) demonstrated that SRP68/72 is localized within the large SRP domain. However, due to the difficulty associated with obtaining sufficient amounts of pure functional SRP68, relatively little is known about the internal structure and the function of the two largest SRP proteins.
An important role for SRP68 in protein targeting was suggested first by the complete inactivation of canine SRP during the cleavage of SRP68 by elastase (Scoulica et al. 1987). In Saccharomyces cerevisiae, SRP68 was shown to be required for the stable expression of the SRP (Brown et al. 1994). Transfection of mammalian cells showed that SRP68 was present in the cytoplasm and the nucleolus implicating a role of the protein in SRP assembly (Politz et al. 2000). Experiments with 35S-labeled canine SRP68 synthesized in vitro in reticulocyte lysates suggested that the protein bound first to the SRP RNA, followed by a conformational change that permitted SRP72 to interact with SRP68 (Lütcke et al. 1993). However, recent data showed that SRP72 has the potential to bind to the SRP RNA independently of SRP68 (Iakhiaeva et al. 2005).
We have established conditions for the purification of functionally active human SRP68 and identified a relatively large RNA binding domain that involves residues between positions 52 and 252. The C-terminal region of SRP68 was found to bind to the N-terminal region of SRP72. These results, combined with consensus secondary structure predictions for SRP68 and SRP72, provide a glimpse into the spatial arrangement of SRP68 and SRP72 within the large domain of the mammalian SRP.
Results
Cloning and phylogenetic distribution of SRP68
A plasmid containing full-length human SRP68 was generated from EST clones and DNA inserts obtained by screening of a library of human HepG2 hepatoma cells as described in Materials and Methods. The gene coded for a polypeptide of 70,326 Da (620 amino acids) with a pKi of 9.22. BLAST searches (Altschul et al. 1997) identified homologs of human SRP68 in all known eukaryotic genomes including those of fungi, plants, mycetozoa (Dictyostelium), alveolata (Cryptosporidium), and euglenozoa (Trypanosoma). An alignment of representative SRP68 sequences is provided in Supplemental Materials (Supplement 1).
The phylogenetic distribution of SRP68 was shown to be shared with SRP72 (Andersen et al. 2006) indicating that SRP68 and SRP72 are functionally linked. Indeed, biochemical data demonstrated the release of SRP68/72 from the SRP as a stable heterodimer (Scoulica et al. 1987). Conserved residues were found to be distributed throughout SRP68 with the exception of a stretch of glycine residues near the N terminus of the mammalian sequences. This glycine-rich cluster was unlikely to play an important role in RNA binding (Herz et al. 1990; Steinert et al. 1991) due to the observed allelic variation of this feature among the various human SRP68 clones.
Expression and purification of human SRP68
The coding region of human SRP68 was amplified by PCR and inserted into the pET23d expression vector. Recombinant hSRP68 was synthesized in Escherichia coli BL21(DE3) cells and recovered as inclusion bodies that were washed and solubilized in phosphate buffer containing either 8 M urea or 0.5% SLS. Both procedures yielded SRP68 with a purity of >95% as determined by SDS-PAGE (Fig. 1, left panel). SRP68 could be purified further by cation-exchange chromatography on BioRex 70 (see Materials and Methods).
Figure 1.

SDS-PAGE of purified human SRP68 separated on a 10% Tricine gel (left) and of polypeptides 68a, 68a′, 68b, 68c, 68c′, 68d′, 68e, and 68e′ separated on a 12.5% Tricine gel (right). Molecular mass markers (kDa) are indicated.
Properties of recombinant SRP68
To promote the refolding of SRP68, samples that had been prepared in buffers containing urea or SLS were subjected to SRP reconstitution conditions (Walter and Blobel 1983) as described in Materials and Methods. The denaturants were gradually removed by centrifugation in 10%–40% sucrose gradients using solutions that lacked urea and detergent. Fractions were collected, analyzed by SDS-PAGE, and the intensities of the stained protein bands were measured to demonstrate that the majority of the SRP68 were recovered in the sucrose gradient fractions (Fig. 2A). The bulk of the SLS-treated SRP68 migrated more slowly than Fibrinogen (340 kDa) indicating that the samples contained oligomers estimated to be composed of three or four SRP68 molecules. Typically, samples prepared in urea (Fig. 2A, black bars) migrated as complexes that were larger than the SLS-treated SRP68 preparations.
Figure 2.
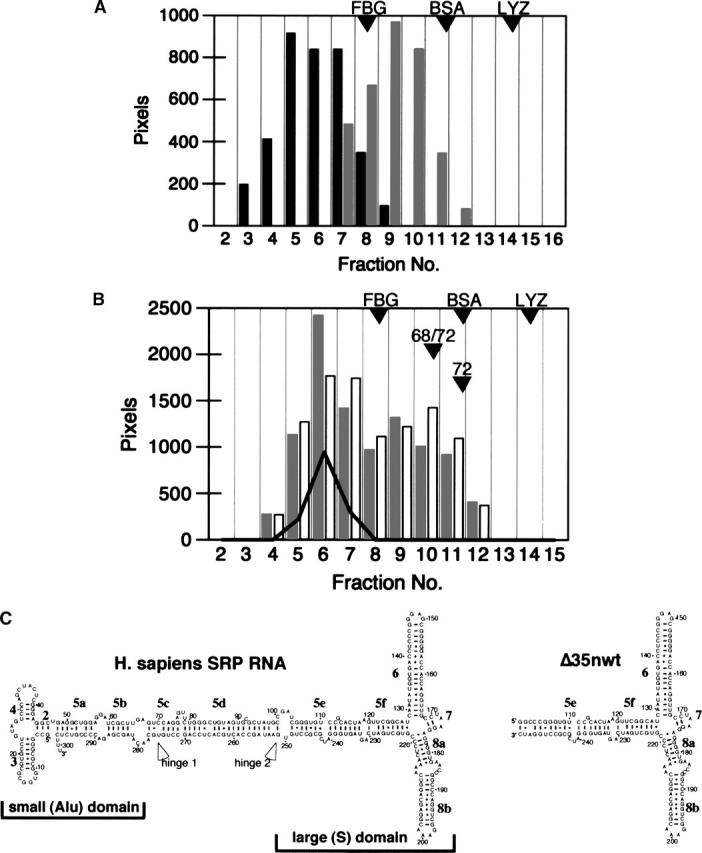
Analysis of human SRP68 on 10%–40% sucrose gradients. (A) SRP68 prepared in urea (black bars) or SLS (gray bars). (B) Comigration of SRP68 (prepared in SLS; gray bars), recombinant SRP72 (white bars), and human SRP RNA (solid line). Fractions are numbered from 2 to 16. Proteins and RNA in the various fractions were analyzed by gel electrophoresis followed by measuring the intensities of the stained bands. The positions of the molecular weight marker proteins Fibrinogen (FBG, 340 kDa), Bovine Serum Albumin (BSA, 78 kDa), and Lysozyme (LYZ, 16 kDa) are indicated. Other arrowheads show the mobilities of the free SRP68/72 heterodimer (68/72) and of purified SRP72 (72) that were analyzed in parallel. (C) Secondary structures diagrams of human SRP RNA (left) and the Δ35nwt mutant RNA (right) (Larsen and Zwieb 1991; Iakhiaeva et al. 2005). Helices are numbered from 2 to 8; letters denote helical sections. Indicated are the positions of the two hinges (Halic et al. 2004; Zwieb et al. 2005) as well as the regions corresponding to the small (Alu) and the large (S) domain. Residues are numbered in increments of 10.
To measure the potential of the recombinant SRP68 to participate in the assembly of particles, human SRP RNA (depicted as a secondary structure diagram in Fig. 2C, left panel) prepared by run-off transcription with T7 RNA polymerase (Zwieb 1991) was added during the refolding of SLS-treated SRP68. Aliquots of the sucrose gradient fractions were analyzed by SDS-PAGE (for protein content) and by agarose gel electrophoresis to determine the distribution of the RNA. A partial comigration of SRP68 with the RNA was observed, suggesting that SRP68 was able to form a complex with the RNA. In similar experiments, we not only added SRP RNA and SRP68 but also recombinant SRP72 (Iakhiaeva et al. 2005) at an approximately twofold molar excess over the RNA. As shown in Figure 2B, a significant portion of SRP68 comigrated with the RNA, indicating binding. Also protein SRP72 associated with the RNA, suggesting the formation of ternary complexes containing SRP68, SRP72, and SRP RNA. In addition, a shift of the free SRP72 to a region of the gradient was observed, which was consistent with the formation of a SRP68/72 heterodimer (marked “68/72”).
To investigate the specificity of the reactions, several control samples were prepared and analyzed in parallel reactions. Free SRP72 was shown to be in a monomeric state as judged from its migration position in the gradient. When RNA was omitted, SRP68 and SRP72 were still able to form heterodimers. Inclusion of recombinant SRP19 in the reconstitution reactions demonstrated that SRP19 bound to RNA but did not displace SRP68 or SRP72. Substituting full-length SRP RNA with an RNA fragment (Δ35nwt RNA) that corresponded to the large domain of human SRP (Fig. 2C, right panel) yielded essentially the same results. The inability to observe binding with a mutant RNA that lacked the Δ35nwt portion of the SRP RNA (ΔLD RNA, Iakhiaeva et al. 2005) demonstrated that SRP68 and SRP72 bound to the large SRP domain. Taken together, the findings proved that the recombinant SRP68 bound specifically to SRP RNA as well as to protein SRP72.
Analysis of recombinant fragments of human SRP68
To narrow down the functional regions within SRP68, we expressed and purified several recombinant fragments (Fig. 1, right panel; also listed in Table 1). The polypeptides were subjected to refolding and sucrose gradient centrifugation followed by electrophoresis of the gradient fractions as described in Materials and Methods. Fragment 68a encompassing residues 52–314 and fragment 68a′ (residues 52–270) comigrated with the Δ35nwt RNA (Fig. 3, top left panel). Both polypeptides did not form oligomers as was evident from their migration near the top of the gradient in fractions 14 and 15. Fragments that lacked the first 309 N-terminal residues of SRP68 (68c, 68c′, 68d′, 68e, and 68e′) did not comigrate with the RNA, suggesting that the region beyond position 310 lacked a detectable RNA binding activity (Fig. 3). Aggregation was observed, for example, with polypeptide 68c which migrated faster than the free Δ35nwt (Fig. 3, top right). In contrast, fragment 68e′ (residues 528–620) migrated distinctly near the top of the gradient similar to what was observed with the monomeric fragments 68a and 68a′. We were unable to express significant amounts of the small fragment 68f (residues 395–529, Table 1). These results suggested that a region of ∼120 amino acid residues (positions 380–500) was largely responsible for the formation of oligomers.
Table 1.
Recombinant fragments of human SRP68 and SRP72

Figure 3.
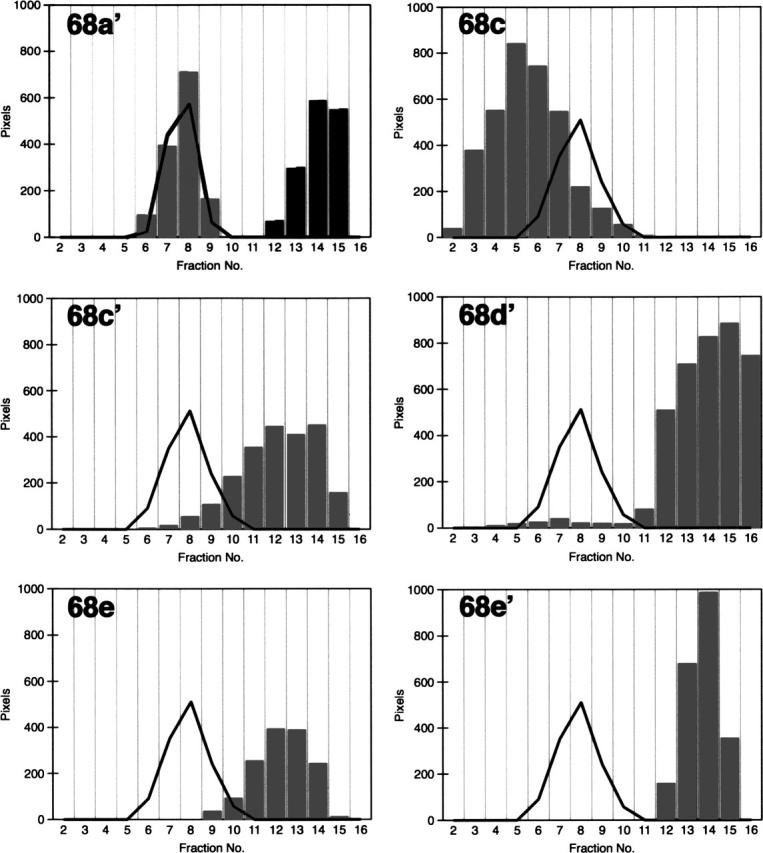
RNA binding activities of the various SRP68 fragments as measured by sucrose gradient centrifugation. The gray bars indicate the distribution of polypeptides 68a′, 68c, 68c′, 68d′, 68e, and 68e′ in the presence of the Δ35nwt RNA (solid lines). The migration of free 68a′ is shown in the top left panel by the black bars.
SRP68's RNA binding domain
SRP68 fragments 68a and 68a′ bound specifically to the large domain of the SRP RNA, whereas fragments derived from other regions of SRP68 regions were inactive. These data suggested that the RNA binding site involved residues 52–270. To determine more precisely the responsible region within the N-terminal half of SRP68, fragment 68a was treated with V8 protease using mild digestion conditions described in Materials and Methods. A proteolytic derivative of ∼24 kDa (named 68-1) and a fragment of ∼7 kDa (68-7) were generated by a primary V8 protease cleavage. Additional polypeptides designated 68-2 to 68-6 were produced at elevated protease concentrations (Fig. 4, top panel; Table 2). MALDI-TOF analysis determined the identity of the proteolytic fragments (Table 3). Consistent with the V8 protease-induced cleavage at glutamic acid residue 252, 68-1 represented a 23,824-Da fragment of 201 amino acids whereas fragment 68-7 (62 amino acid residues, 6936 Da) contained the C-terminal portion of 68a (residues 253–314 of full-length SRP68). Mild digestion of 68a′ also generated the 68-1 fragment, confirming that V8 protease had cleaved initially at position 252 (Fig. 4, top panel).
Figure 4.

The RNA binding region of SRP68. (Top panel) Mild digestion of fragments 68a (left) and 68a′ (right) with V8 protease followed by the separation of the fragments on 15% polyacrylamide Tricine gels; the migration distances of 68a and fragments 68-1 to 68-7 are indicated. The arrowhead marks the 68-7 fragment of 6.5 kDa generated by the primary V8 protease cut. (Lower left panel) Comigration of fragments 68a (black bars) and 68a-1 (gray bars) with human SRP RNA (black line) during sucrose gradient centrifugation. (Lower right panel) Collective distribution of the proteolytic fragments 68-2 to 68-7 (gray bars) generated by V8 protease. As in the lower left panel, the black line indicates the mobility of human SRP RNA.
Table 2.
Proteolytic fragments of human SRP68 and SRP72

Table 3.
Tryptic peptides identified by MALDI-TOF MS within fragments 68a and the polypeptides generated by the digestion of 68a with V8-protease

Fragments that had been partially proteolyzed by V8 were reconstituted with in vitro synthesized SRP RNA and analyzed by sucrose gradient centrifugation. Only fragments 68a and 68-1 bound to the RNA (Fig. 4, bottom left panel). In agreement with these findings, the removal of 18 amino acids from the C terminus of 68a′ (as evidenced by mass spectroscopy) did not affect the formation of a complex, whereas the shorter fragment 68b (residues 52–208; Table 1) lacked an RNA binding activity. We speculate that fragment 68b was unfolded because it was susceptible to unspecific proteolytic degradation. In summary, the data showed that residues 52–252 of SRP68 were required for the formation of a stable RNA binding domain.
Interaction between SRP68 and SRP72
Results shown in Figure 2B indicated that SRP68 and SRP72 formed heterodimers. To determine the region of human SRP72 responsible for the protein–protein interaction, the previously described fragments (Iakhiaeva et al. 2005) of human SRP72 (72a′, 72b′, 72c, and 72d; see Table 1) were purified and used in reconstitution reactions with fragment 68c. The samples were analyzed on sucrose gradients followed by SDS-PAGE of the gradient fractions (Fig. 5A). Although fragment 68c partially aggregated, fragment 72b′ bound as indicated by its comigration in fractions 7 and 8. No differences were observed in the ability of 72b′ to bind to 68c upon addition of SRP RNA or Δ35nwt RNA (data not shown), demonstrating that the RNA had no obvious role in promoting the protein–protein interaction. Furthermore, the complex was stable at salt concentrations of up to 500 mM, conditions that eliminated protein–RNA interactions within the SRP. Fragment 68c did not bind to polypeptides that lacked the N-terminal region of SRP72 (72a′, 72c, and 72d), suggesting that the first 163 amino acids of SRP72 were required for the binding to SRP68.
Figure 5.

Interactions of the protein binding site of SRP68 with fragments of SRP72. (A) The 68c polypeptide was mixed with SRP72 fragments 72a′, 72b′, 72c, or 72d and analyzed by sucrose gradient centrifugation. Fraction aliquots were analyzed by SDS-PAGE. (B) Testing the interaction between 68c′ and 72b′. (C) Testing for binding between the RNA binding domain of SRP68 (fragment 68a bound to human SRP RNA, indicated as 68a-R) and 72b′. Gradient fractions 3–15 are indicated in top of each panel.
Further analysis showed that fragment 72b′ (amino acid residues 1–163 of SRP72) did not bind to 68c′ (residues 310–554 of SRP68), indicating that the 91 amino acids encompassing the C terminus of SRP68 (residues 555–620) were important for the interaction with SRP72 (Fig. 5B). Not even trace amounts of complexes formed between 72a′ and full-length SRP68 as investigated by sensitive silverstaining of the SDS gels. Furthermore, the RNA binding domain of SRP68 (fragments 68a and 68a′) did not bind to full-length SRP72 (shown in Fig. 5C for 68a). We concluded that the C-terminal part of SRP68 and the N-terminal part of SRP72 were engaging.
Due to the relatively low molecular masses and monomeric states of 68e′ (10,450 Da) and 72b′ (18,599 Da), it was difficult to demonstrate binding between these two polypeptides. (Attempts to employ electrophoretic mobility shift assays were unsuccessful.) To generate larger polypeptides that could be analyzed by sucrose gradient centrifugation, we designed a dual-cassette vector that contained the coding regions of the two polypeptides separated by a TEV protease cleavage site and preceded by the gene for thioredoxin. Upon cleavage with TEV protease one of the two polypeptides remained as a thioredoxin-fused larger protein. Figure 6 (right panel) illustrates that all constructs contained the N-terminal region of SRP72 (72b′) physically linked to sections of protein SRP68 (68d, 68f, 68e, and 68e′) in frame with thioredoxin. The four fusions were expressed and purified as described in Materials and Methods. Digestion with TEV protease generated fragments with the expected molecular weights (Fig. 6, left panel). Analysis of the fragments by sucrose gradient centrifugation followed by SDS-PAGE revealed relatively low levels of binding of 72b′ to the Thx–68d and Thx–68f fusions (24% and 9%, respectively). In contrast, 72b′ remained associated with the Thx–68e and Thx–68e′ fusions at levels of 70% and 89%. These data confirmed that the 91 amino acids encompassing the C terminus of SRP68 were responsible for binding to SRP72.
Figure 6.

Binding of the N-terminal region of SRP72 to fragments of SRP68. (A) Fragments 68d, 68e, 68e′, 68f, fused to thioredoxin, and 72b′, separated on a 15% polyacrylamide Tris-glycine gel. Lanes labeled with plus signs indicate TEV protease digested samples. The black arrowhead marks the liberated 72b′ fragment; white arrowhead indicates the various SRP68 fragments fused to thioredoxin. Weak bands in the 28-kDa range correspond to the TEV protease. The right panel outlines the four fusion constructs indicating the start and stop codons of the plasmid as well as the position of the TEV cleavage site. Analysis of the TEV-cleaved fusion proteins by sucrose gradient centrifugation followed by the SDS-PAGE of the fractions showed degrees of binding as expressed in percent bound/free 72b′. (B) Pull-down assays with GST–72b′ and Thx–68e′. E. coli lysates analyzed on 10% Tris-Tricine SDS gels containing expressed GST–72b′ (lane 1) and Thx–68e′ (lane 2). (Lanes 3–5) Proteins in the wash of the GST-Sepharose column; (lane 6) coelution of GST–72b′ and Thx–68e′ using 20 mM glutathione; (lane 7) control reaction lacking GST–72b′ proving that Thx–68e′ has no intrinsic affinity for GST-Sepharose.
Additional support for the SRP68/72 interaction was gained from pull-down assays with 72b′ fused to GST (see Materials and Methods). GST–72b′, when bound to glutathione-Sepharose, specifically removed the Thx–68e′ polypeptide from a crude E. coli lysate (Fig. 6B).
The affinity between 68e′ and 72b′ was measured by labeling Thx–68e′ radioactively at a serine with 32P-phosphate followed by binding of low concentrations of Thx–68e′ to increasing amounts of GST–72b′ until binding was saturated (see Materials and Methods). The apparent association constant (K′a) was calculated from the GST–72b′ concentration that led to the formation of equal amounts of complexed and free Thx–68e′. Assuming that one molecule of GST–72b′ bound to one molecule of Thx–68e′, the value of K′a was determined to be ∼1.6 × 107 M−1.
Mild digestion of purified 72b′ with chymotrypsin (Materials and Methods) yielded fragments in the range of 16 to 6 kDa. The polypeptide mixture was used in reconstitution reaction with 68c followed by analysis of the sucrose gradient fractions. The results shown in Figure 7 demonstrated that removal of ∼15 terminal amino acid residues completely abolished the protein binding activity of 72b′, suggesting that nearly all amino acids from positions 1 to 163 were required for the protein–protein interaction with SRP68.
Figure 7.

The protein binding region of SRP72. Separation of a mixture of polypeptide 68c with fragments generated by mild digestion of 72b′ with chymotrypsin followed by analysis of the sucrose gradient fractions by SDS-PAGE. Fractions 4–15 are indicated on the top. Molecular weight markers in kDa are indicated on the left.
Predicted features of SRP68 and SRP72
PSIPRED consensus secondary structures predictions (McGuffin et al. 2000) reported a predominantly α-helical character for SRP68 and SRP72 (Fig. 8). Features of SRP72 included a tandem array of tetratricopeptide-like motifs as well as the previously identified RNA binding site (Iakhiaeva et al. 2005). Results shown in Figure 5 suggested that the first four TPR-like motifs in SRP72 were essential for the identified protein–protein interaction. Conserved and invariant residues were found in the RNA binding domain and clustered near the C-terminal region of SRP68 (Fig. 8; Supplement 1) within the region that bound to SRP72.
Figure 8.
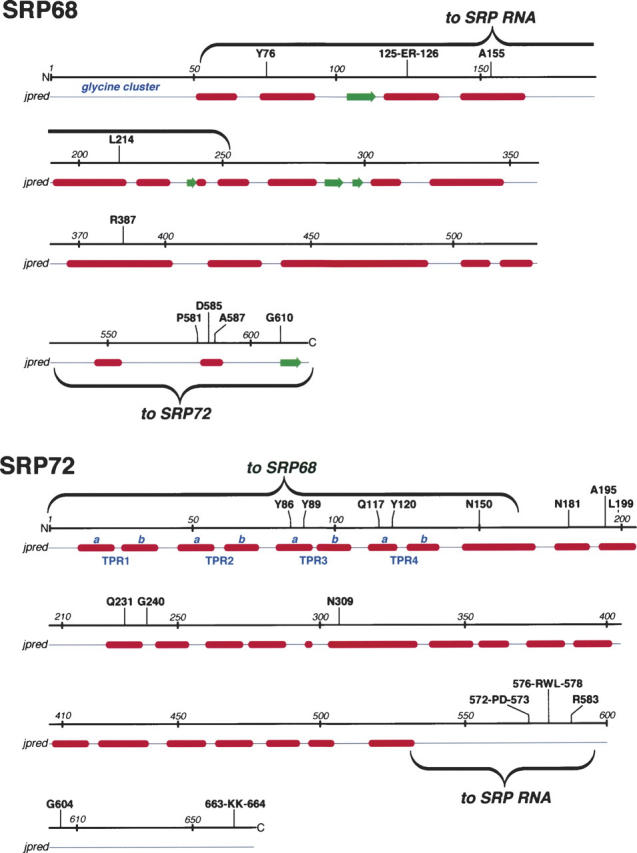
Features of SRP68/72. For both proteins, amino acid positions are given in increments of ∼50 residues. Amino acid residues that are invariant or highly conserved in the multiple sequence alignments (provided as Supplemental Material Sup1-SRP68.pdf and Sup2-SRP72.pdf) are shown above the lines. Helices (red) and β-structures (green) as predicted by jpred (Cuff and Barton 2000) are shown below the lines. Annotated in blue are the glycine cluster at the N terminus of SRP68 and the four predicted TPR-like motifs (Blatch and Lassle 1999) with their divisions into helices a and b. The wavy brackets designate the regions responsible for binding to SRP RNA and for the binding of the two proteins to each other.
Discussion
Human SRP68 was expressed in E. coli and recovered in the inclusion bodies allowing obtaining highly purified preparations. The protein could be solubilized using buffers that contained either 8 M urea or 0.5% SLS. After the gradual removal of the majority of these denaturants during centrifugation of sucrose gradients under SRP reconstitution conditions, the protein and its recombinant derivatives formed oligomers that bound to SRP RNA and SRP72. When recombinant SRP72 was added during the reconstitution, the mobility of the SRP68/72 complex was consistent with the presence of a simple heterodimer, suggesting that SRP72 disrupted the SRP68-oligomers.
In our analysis of functional domains we covered nearly all of SRP68 but did not investigate the role of the glycine-rich N-terminal region. Although similar glycine clusters were identified in proteins that bind to single-stranded RNA (Herz et al. 1990; Steinert et al. 1991), the treatment of canine SRP with elastase demonstrated that a 7-kDa fragment containing the N terminus of SRP68 was not required for binding to the SRP RNA (Lütcke et al. 1993). Furthermore, the number of glycine residues varied considerably, and this feature was found only in the mammalian SRP68 sequences, suggesting it had no role in the core functions of the SRP.
Mass spectroscopic analysis of fragments generated by mild digestion of recombinant fragment 68a (residues 52–314) with V8 protease allowed narrowing down the RNA binding domain to amino acid residue positions 52 to 252. The mass of the smallest polypeptide that still bound to RNA was 23,823 Da in agreement with the size of an about 24-kDa fragment that remained bound to the elastase-treated canine SRP (Lütcke et al. 1993). We showed that this region formed a large domain which, when truncated, became susceptible to proteases and completely lost its RNA binding capacity. No relationships of the SRP68 RNA binding region to putative RNA binding domains in Pfam (Bateman et al. 2004) or NCBI CDD (Marchler-Bauer et al. 2005) were discovered.
The relatively large RNA binding domain and earlier footprinting data (Siegel and Walter 1988) suggested that SRP68 occupied most of the sites in the large domain of the SRP which were unoccupied by SRP19 and SRP54. The crystal structures (for example, see Kuglstatter et al. 2002) indicated that portions of SRP RNA helices 5, 6, and 8 could be simultaneously available to SRP68. Indeed, systematic site-directed mutagenesis of human SRP RNA showed that almost all alterations in the large domain had a negative effect on the binding of SRP68/72 (J. Yin, E. Iakhiaeva, and C. Zwieb, unpubl.). In contrast, binding of SRP72 was affected only by mutations in SRP RNA helix 5 consistent with its relatively small RNA binding domain (Iakhiaeva et al. 2005). This lysine-rich domain was identified as Pfam B 7529 in a recently updated SRP72 alignment (Andersen et al. 2006; see also Supplement 2) and has now been incorporated into the Pfam database (Bateman et al. 2004).
Interestingly, the cryo-electron microscopy structure of canine SRP showed density attributed to SRP68/72 at helix 5 and near the RNA hinge adjoining the large SRP domain (Halic et al. 2004). However, complexes between SRP68 and the Δ35nwt RNA were as stable as complexes with a mutant RNA that contained the hinge (data not shown), suggesting that the hinge region has no role in the binding to SRP68 or SRP68/72.
The C terminus of SRP68 was suggested previously to interact with the C-terminal region of SRP72 (Lütcke et al. 1993). Using purified components in a defined system, we found no evidence for this interaction even by silver staining. Fragments 68c and 72d, and similarly polypeptides 72b′ and 68a′, did not bind to each other. Binding could not be promoted by the addition of SRP RNA or altering the buffer conditions. Instead, complexes formed in high yields between 72b′ and fragments 68c, 68e, and 68e′ in standard and high-salt conditions used previously in assembly and disassembly of canine SRP (Walter and Blobel 1983; Scoulica et al. 1987).
Mapping of the SRP68–SRP72 interaction demonstrated that all four predicted TPR-like motifs of SRP72 were required for binding. TPRs are known to form a superhelical structure with a groove that accommodates another protein. Of the two helices (named a and b, see Fig. 8) of each TPR, helix a is oriented toward the concave portion of the groove (Blatch and Lassle 1999). Since the conserved SRP72 residues Y86, Y89, Q117, and Y120 were located in helices a of TPR3 and TPR4 (Fig. 8), we propose that these residues play an important role in the binding to the C-terminal region of SRP68.
Materials and methods
Cloning, expression, and purification of human SRP68
The coding region of human SRP68 (GenBank accession no. AAF24308) were assembled from partial EST clones (Genome Systems) and from clones selected by screening of a λ-gt10 cDNA library of human HepG2 hepatoma cells obtained from the American Type Culture Collection (ATCC 77400). Screening of the cDNA library was carried out as described (Gowda et al. 1998). Probing with a 300-base pair fragment derived from EST-T48898 identified four overlapping SRP68 clones. For protein expression, the DNA was amplified with primers compatible with the NcoI and XhoI restriction sites of pET23d (Novagen). Competent E. coli DH5-α cells (Life Technologies) were transformed with the ligation mixture, and colonies were selected on LB plates containing 200 μg/mL ampicillin followed by restriction mapping and screening for expressed polypeptides with the correct size using SDS-PAGE. The full-length human SRP68 expression plasmid (phSRP68) was purified by CsCl density gradient centrifugation. The sequence was determined using Sequenase 2.0 (United States Biochemical).
To purify recombinant human SRP68, competent E. coli BL21 (DE3) cells (Stratagene) were transformed with phSRP68 and grown overnight at 37°C on LB agar plates containing 200 μg/mL ampicillin. Several colonies were transferred into 400 mL of LB containing ampicillin and grown by shaking at 37°C until the A600 reached 0.2. IPTG was added to a final concentration of 1 mM and shaking at 37°C continued for 2 h. Cells were harvested by centrifugation and stored at −70°C until further use.
The subsequent procedures were carried out at 4°C unless noted differently. Approximately 200 mg of frozen cells were resuspended in 13 mL of lysis buffer (100 mM Tris-HCl at pH 8.0, 100 mM NaCl, 1 mM EDTA, 1 mM DTT) containing 50 μL of a protease inhibitor cocktail (Sigma). The cells were sonicated using a model 300 dismembrator (Fisher Scientific) at a setting of 35% three times for 15 sec with 15-sec intervals on ice. The lysate was subjected to a low-speed centrifugation at 15,000 rpm using a Sorvall SS34 rotor for 20 min. The pellet was resuspended in 13 mL lysis buffer containing protease inhibitor cocktail and 0.5% Triton X-100 and was washed by centrifugation and removal of the supernatant. Again, the pellet was washed once with lysis buffer (lacking protease inhibitors but containing Triton X-100) followed by a wash with lysis buffer lacking Triton. The protein was solubilized in 13 mL of SLS buffer (50 mM Na-phosphate at pH 8.0, 100 mM NaCl, 5 mM EDTA, 5 mM DTT, 0.5% SLS; Sigma) and stored at 4°C. The preparation was used within 2 wk.
To achieve a higher degree of purification the recombinant SRP68 was processed further by adding four volumes of 10 mM potassium phosphate (pH 7.5), 50 mM NaCl, 1 mM EDTA, 1 mM DTT containing 8 M urea, followed by an overnight dialysis in potassium-phosphate buffer containing 6 M urea and storage at 4°C. Alternatively, 13 mL of the SLS-containing sample were subjected to a high-speed centrifugation at 40,000 rpm in a Beckman NVT65 rotor for 2 h. The supernatant was diluted 50 times with 50 mM Na-phosphate (pH 8.0), 1 mM EDTA, 5 mM DTT, 8 M urea (column buffer) and applied to a BioRex 70 (Bio-Rad Laboratories) cation-exchange column (1 mL bed volume) at room temperature equilibrated in column buffer. The column was washed with 20 mL of column buffer followed by the elution of SRP68 in the 500 mM NaCl fractions during the stepwise application of 3 mL each of 50 mM Na-phosphate (pH 8.0), 1 mM EDTA, 5 mM DTT, 6 M urea containing 200 mM, 500 mM, 700 mM, or 1 M NaCl. Protein preparations were placed at 4°C and used within 2 wk.
Preparation of SRP68 fragments
The domains of phSRP68 were expressed using PCR with suitable synthetic oligonucleotides to introduce an upstream NcoI restriction site and downstream BamHI, EcoRI, HindIII, or XhoI restriction sites. The amplified DNA fragments were cleaved with the appropriate pair of enzymes, purified by agarose gel electrophoresis, and ligated to linearized pET23d (Novagen) vector DNA. Similarly, fragments corresponding to 68d, 68e, 68e′, and 68f were amplified and cloned into to the KpnI and BamHI sites of pANKiT (S.H. Bhuiyan and C. Zwieb, unpubl.) to direct the expression of 68-domain fusion proteins. Competent E. coli DH5-α cells (Life Technologies) were transformed with the ligation mixtures and clones were selected and purified as described above. Commercial providers were used to verify the sequences.
Competent E. coli BL21 Star (DE3) pLysS cells (Invitrogen) were transformed with the plasmids directing the expression of untagged 68a, 68a′, 68c, 68c′, 68d′, or 68e′. Rosetta (DE3) pLysS cells (Novagen) were used for expression of untagged 68e and the pANKiT fusion protein constructs containing 68d, 68e, 68e′, and 68f. Transformants were selected at 37°C on LB agar plates containing 200 μg/mL ampicillin and 38 μg/mL chloramphenicol. Several colonies were transferred into 400 mL of LB containing antibiotics. Cells were grown by shaking at 37°C until the A600 reached 0.2 (or 0.6 for the fusion polypeptides), at which time IPTG was added to a final concentration of 1 mM. The cultures were incubated with continuous shaking at 37°C for 2 h. Cells were harvested by centrifugation and frozen aliquots were stored at −70°C.
Untagged fragments 68a, 68a′, and 68b were purified by resuspending 1 g of frozen cells in 25 mL of 50 mM Na-phosphate (pH 8.0), 150 mM NaCl, 5 mM EDTA, 5 mM DTT followed by sonication as described above. The pellets of the low-speed spin were resuspended in 25 mL of 50 mM Tris-HCl (pH 9.0), 300 mM NaCl, 5 mM EDTA, 5 mM DTT, and 5 M urea. The samples were solubilized by sonication and subjected to high-speed centrifugation at 40,000 rpm for 2 h in a Beckman NVT65 rotor at 4°C. The supernatants were diluted with three volumes of Tris-HCl buffer lacking salt but containing 5 M urea. The diluted samples were loaded by gravity onto a 1-mL bed volume BioRex 70 (Bio-Rad Laboratories) cation-exchange columns at 4°C that had been equilibrated in Tris-HCl buffer (pH 9.0), containing 100 mM NaCl. The columns were washed twice with 2 mL of equilibration buffer containing 5 M urea. The polypeptides were eluted with 2 mL of the urea equilibration buffer containing 500 mM NaCl and stored at 4°C.
Fragments 68c, 68c′, 68d′, 68e, and 68e′ were prepared in 0.1% SLS buffer as described above for the full-length SRP68 using ∼50 mg of cells in 3 mL of lysis buffer. Alternatively, urea-containing preparations were obtained by dissolving the washed inclusion bodies in 50 mM Na-phosphate (pH 8.0), 100 mM NaCl, 1 mM EDTA, 1 mM DTT, 8 M urea, followed by centrifugation for 20 min at 15,000 rpm (Sorvall SS34 rotor) and recovery of the polypeptides in the supernatants.
To prepare the fusion proteins about 0.3 g of cells were lysed in 20 mL of 50 mM Na-phosphate (pH 8.0), 50 mM NaCl, 1 mM EDTA, 5 mM DTT followed by sonication and centrifugation at 15,000 rpm for 20 min (Sorvall SS34 rotor). The pellets were resuspended in 20 mL of phosphate buffer containing 4 M (68e′-fusion), 5 M (68d-fusion), or 8 M (68e- and 68f-fusions) urea. Samples were subjected to another 20-min centrifugation and supernatants were centrifuged at high-speed in a SW41Ti (Beckman) rotor at 38,000 rpm for 4 h. SDS-PAGE was used to determine the purity and concentration of the polypeptides in the supernatants by comparison with known amounts of lysozyme. Prior to functional testing, 10 μg of polypeptide were digested in 450 μL of 50 mM Tris-HCl (pH 8.0), 0.5 mM EDTA, 1 mM DTT, 0.4 M urea with 10 units of TEV protease (Invitrogen) at room temperature for 2 d.
Preparation of SRP72 domains
Domains of human SRP72 were constructed as described previously (Iakhiaeva et al. 2005). For expression of the polypeptides, competent E. coli cells, either BL21-STAR-(DE3) pLysS (Invitrogen) for 72a′, 72c, and 72d or Rosetta pLysS (Novagen) for 72b′, were transformed and cell pellets were prepared.
Purification of 72a′
72a′ was prepared as described above followed by dialysis with sodium phosphate buffer containing 4 M urea.
Purification of 72b′
The supernatant of the low-speed spin (1 mL) from cells expressing the GST–72b′ fusion protein was mixed with 100 μL of glutathione Sepharose 4B suspension (Amersham) that had been equilibrated twice with 1 mL of 50 mM Na-phosphate (pH 6.5), 50 mM NaCl, 5 mM EDTA, 5 mM DTT (equilibration buffer) followed by rotation of the sample at 4°C for 30 min. The Sepharose beads were washed twice with 200 μL of equilibration buffer (30 min each). The protein was eluted after an overnight digestion at room temperature with 1.4 NIH-units of thrombin (Sigma) in 400 μL of 50 mM Na-phosphate (pH 8.0), 50 mM NaCl, 2 mM EDTA, 0.2 mM DTT.
Purification of 72c and 72d
Frozen cells (200 mg) were resuspended in 13 mL of 50 mM Na-phosphate (pH 8.0), 150 mM NaCl, 5 mM EDTA, 5 mM DTT and sonicated as described above. The pellet of the low-speed spin was resuspended in 13 mL of phosphate buffer containing 5 M urea. The samples were solubilized by sonication and subjected to high-speed centrifugation at 40,000 rpm for 2 h in a Beckman NVT65 rotor. The supernatant were diluted with two to three volumes of phosphate buffer lacking salt and containing 5 M urea. The diluted sample was loaded by gravity onto a 1-mL bed volume BioRex 70 (Bio-Rad Laboratories) cation-exchange column that had been equilibrated in phosphate buffer containing 50 mM NaCl and 5 M urea. The column was washed stepwise with two volumes of equilibration buffer containing 3 M, 2 M, 1 M, and no urea prior to elution with 500 mM, 1 M, and 1.5 M NaCl-containing buffers. The majority of the protein was recovered in the 1 M salt eluate fractions.
Synthesis of full-length and truncated human SRP RNAs
The construction of phR for synthesis of full-length human SRP RNA from the T7 RNA polymerase promoter has been described (Zwieb 1991). Deletion mutant RNAs were constructed by the digestion of phR with suitable restriction enzymes and filling of gaps with synthetic complementary oligonucleotides followed by the ligation of the fragments. Competent E. coli DH5-α cells (Life Technologies) were transformed, and clones were selected on LB plates containing 100 μg/mL ampicillin. Plasmid DNAs from individual colonies were screened by restriction mapping, purified by CsCl density gradient centrifugation, and the sequences were verified using commercial providers. RNAs were synthesized by run-off transcription in a volume of 20 μL for 2 h at 37°C containing 4 μg of BamHI- or DraI-digested plasmid DNA and components of the T7-MEGAshortscript kit (Ambion). RNA concentrations were determined from a standard curve obtained with known amounts of E. coli 5S ribosomal RNA (Sigma) by electrophoresis of sample aliquots on 2% agarose gels followed by staining with ethidium bromide and densitometry.
Refolding, binding, and sucrose gradient analyses
Three hundred microliters of protein sample were mixed gently on ice with 100 μL of 200 mM Tris-HCl (pH 7.9), 1.2 M KOAc, 20 mM MgCl2, 4 mM DTT (fourfold concentrated SRP reconstitution buffer) containing up to 50 μg of wild-type or truncated human SRP RNA. The sample was incubated for 10 min at 37°C and loaded onto a 10%–40% sucrose gradient prepared in 50 mM potassium-phosphate (pH 7.4), 150 mM NaCl, 1 mM MgCl2, 1 mM DTT at 4°C. After centrifugation (Beckman NVT65 at 55,000 rpm for 4.5 or 6 h at 4°C), 16 fractions of ∼800 μL each were collected. The main portion of a fraction was precipitated by the addition of 160 μL 100% TCA followed by incubation on ice for 30 min and centrifugation at 15,000 rpm for 20 min at 4°C. The polypeptides in the pellet were dissolved in SDS loading buffer and analyzed by SDS-PAGE. In order to determine the RNA content, 10-μL aliquots of the sucrose gradient fractions were mixed with 2 μL of loading buffer (20 mM Tris-acetate at pH 8.0, 10 mM EDTA, 0.1% SDS, 0.25% [w/v] bromophenol blue, 0.025% [w/v] xylene cyanol, 30% glycerol) and analyzed by electrophoresis in 2% agarose gels followed by staining with ethidium bromide.
Mild proteolysis
Fragments 68a and 68a′ were digested with V8 protease from Staphylococcus aureus (Sigma) by adding 1 μg of protease to 200 μg substrate in a volume of 300 μL of 50 mM Tris-HCl (pH 9.0), 500 mM NaCl, 5 mM DTT, 5 mM EDTA buffer followed by incubation for 20 min at room temperature. About 15 μg of 72b′ were digested with 1 μg of α-chymotrypsin (Sigma, type VI) for 25 min at room temperature in a volume of 15 μL of 50 mM Na-phosphate (pH 8.0), 50 mM NaCl, 2 mM EDTA, 0.2 mM DTT.
Pull-down assays
Lysates were prepared by sonication in 50 mM Na-phosphate (pH 8.0), 50 mM NaCl, 5 mM EDTA, 5 mM DTT from Rosetta (DE3) pLysS cells containing 72b′ polypeptides fused to GST or containing 68e′ polypeptides fused to thioredoxin. The GST–72b′ lysate was bound to 100 μL of a glutathione Sepharose 4B suspension (Amersham), assembled in a small column, and incubated at 4°C for 30 min. Next, the Thx–68e′ lysate was added to the column and the incubation was repeated. The column was washed with 50 mM Na-phosphate (pH 8.0), 50 mM NaCl, 5 mM EDTA, 1 mM DTT, and bound protein was eluted by adding 20 mM glutathione to the buffer. Samples were analyzed by SDS-PAGE.
Binding experiments using 32P-labeled Thx–68e′
Purified thioredoxin-fusion 68e′ polypeptide was labeled radioactively by incubating for 30 min at room temperature with 6 μCi of 32P-γ-ATP and 40 units of protein kinase A (catalytic subunit from bovine heart, Sigma) in 30 μL of 20 mM HEPES (pH 7.9), 5 mM DTT, 10 mM MgCl2. An equal volume of 20 mM HEPES (pH 7.9), 300 mM NaCl, 1 mM EDTA, 5 mM DTT, and 50% glycerol was added prior to storage at –20°C. To measure binding, low concentrations of 32P-labeled Thx–68e′ were mixed with increasing amounts of GST–72b′ in 50 μL of 50 mM Tris-HCl (pH 7.9), 300 mM KOAc, 5 mM MgCl2, 1 mM DTT, and 0.01% Tween-20 followed by an incubation at room temperature for 1 h. Samples were added to 10-μL aerosol barrier tips (Continental Laboratory Products) containing 6-μL bed volumes of glutathione Sepharose 4B (Amersham) equilibrated in 50 mM Na-phosphate (pH 7.0), 50 mM NaCl, 5 mM DTT, 5 mM EDTA. Incubation with the beads was for 30 min at 4°C with gentle mixing every 10 min. The flowthrough and two washes with 50 μL each of 50 mM Tris-HCl (pH 7.9), 300 mM KOAc, 5 mM MgCl2, 1 mM DTT, and 0.01% Tween-20 were discarded and the bead-bound radioactivity were measured using a Beckman LS6500 scintillation counter.
Mass spectroscopy
Protein bands were excised from the polyacrylamide gels, cut into small pieces, and placed into siliconized tubes containing 250 μL of 100 mM NH4HCO3 and 50% methanol. The tubes were shaken at room temperature for 15 min. The liquid was discarded and the gel slices were washed twice with 200 μL of 5 mM NH4HCO3, 50% (v/v) acetonitrile (Sigma). The gel pieces were dried under vacuum. Ten to fifteen microliters of 12.5 μg/mL modified sequencing grade trypsin (Roche) in 50 mM NH4HCO3 were added to each sample and placed overnight in a 37°C incubator. One hundred microliters of 0.1% TFA (Sigma) were added at room temperature and the solution was transferred into new tubes. The gel pieces were extracted twice with 0.1% TFA/50% acetonitrile, and the extracts were pooled and dried completely under vacuum. The peptides were dissolved in 60 μL of 0.05% TFA/5% acetonitrile followed by the binding to C18 OMIX (Varian) pipette tips. Peptides were eluted with 5 μL of 10 mg/mL 2,5-dihydroxybenzoic acid (Fluka) and spotted onto a 384-well plate with α-cyano-4-hydroxycinammic acid in 0.1% TFA, 50% acetonitrile as matrix. Analysis was done in the reflectron mode using an AXIMA-CFR MALDI-TOF Mass Spectrometer (Kratos).
Prediction of the protein secondary structure
The secondary structures of human SRP68 and SRP72 were predicted using the PSIPRED server at http://bioinf.cs.ucl.ac.uk/psipred/ (McGuffin et al. 2000) with the default settings.
Electronic supplemental material
Supplemental material for this article includes a comparative sequence analysis of SRP68 (Supplement 1) and SRP72 (Supplement 2).
Acknowledgments
We thank Alexei Iakhiaev for critical reading of the manuscript. This work was supported by the National Institutes of Health (GM-49034) to C.Z.
Footnotes
Supplemental material see www.proteinscience.org
Reprint requests to: Christian Zwieb, Department of Molecular Biology, The University of Texas Health Science Center at Tyler, 11937 US Highway 271, Tyler, TX 75708-3154, USA; e-mail: zwieb@uthct.edu; fax: (903) 877-5731.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051861406.
Abbreviations: DTT, dithiothreitol; EDTA, ethylenediaminetetraacetic acid; EST, expressed sequence tag; ER, endoplasmic reticulum; IPTG, isopropyl-β-D-thiogalactopyranoside; LB, Luria-Bertani medium; MALDI-TOF, matrix assisted laser desorption/ionization time of flight; PAGE, polyacrylamide gel electrophoresis; PCR, polymerase chain reaction; SDS, sodium dodecyl sulfate; SLS, sodium lauryl sarcosinate; SRP, signal recognition particle; TCA, trichloroacetic acid; TEV, tobacco etch virus; TFA, trifluoroacetic acid; TPR, tetratricopeptide repeat; Tricine, N-tris(hydroxymethyl)methylglycine.
References
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen E.S., Rosenblad M.A., Larsen N., Westergaard J.C., Burks J., Wower I.K., Wower J., Gorodkin J., Samuelsson T., Zwieb C. 2006. The tmRDB and SRPDB resources Nucleic Acids Res. 34 D163–D168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews D.W., Walter P., Ottensmeyer F.P. 1987. Evidence for an extended 7SL RNA structure in the signal recognition particle EMBO J. 6 3471–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A., Coin L., Durbin R., Finn R.D., Hollich V., Griffiths-Jones S., Khanna A., Marshall M., Moxon S., Sonnhammer E.L.et al. 2004. The Pfam protein families database Nucleic Acids Res. 32 D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatch G.L. and Lassle M. 1999. The tetratricopeptide repeat: A structural motif mediating protein–protein interactions Bioessays 21 932–939. [DOI] [PubMed] [Google Scholar]
- Brown J.D., Hann B.C., Medzihradszky K.F., Niwa M., Burlingame A.L., Walter P. 1994. Subunits of the Saccharomyces cerevisiae signal recognition particle required for its functional expression EMBO J. 13 4390–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuff J.A. and Barton G.J. 2000. Application of multiple sequence alignment profiles to improve protein secondary structure prediction Proteins 40 502–511. [DOI] [PubMed] [Google Scholar]
- Doudna J.A. and Batey R.T. 2004. Structural insights into the signal recognition particle Annu. Rev. Biochem. 73 539–557. [DOI] [PubMed] [Google Scholar]
- Gowda K., Black S.D., Moeller I., Sakakibara Y., Liu M.-C., Zwieb C. 1998. Protein SRP54 of human signal recognition particle: Cloning, expression, and comparative analysis of functional sites Gene 207 197–207. [DOI] [PubMed] [Google Scholar]
- Hainzl T., Huang S., Sauer-Eriksson A.E. 2002. Structure of the SRP19 RNA complex and implications for signal recognition particle assembly Nature 417 767–771. [DOI] [PubMed] [Google Scholar]
- Halic M., Becker T., Pool M.R., Spahn C.M., Grassucci R.A., Frank J., Beckmann R. 2004. Structure of the signal recognition particle interacting with the elongation-arrested ribosome Nature 427 808–814. [DOI] [PubMed] [Google Scholar]
- Herz J., Flint N., Stanley K., Frank R., Dobberstein B. 1990. The 68 kDa protein of signal recognition particle contains a glycine-rich region also found in certain RNA-binding proteins FEBS Lett. 276 103–107. [DOI] [PubMed] [Google Scholar]
- Iakhiaeva E., Yin J., Zwieb C. 2005. Identification of an RNA-binding domain in human SRP72 J. Mol. Biol. 345 659–666. [DOI] [PubMed] [Google Scholar]
- Keenan R.J., Freymann D.M., Stroud R.M., Walter P. 2001. The signal recognition particle Annu. Rev. Biochem. 70 755–775. [DOI] [PubMed] [Google Scholar]
- Kuglstatter A., Oubridge C., Nagai K. 2002. Induced structural changes of 7SL RNA during the assembly of human signal recognition particle Nat. Struct. Biol. 9 740–744. [DOI] [PubMed] [Google Scholar]
- Larsen N. and Zwieb C. 1991. SRP-RNA sequence alignment and secondary structure Nucleic Acids Res. 19 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luirink J. and Sinning I. 2004. SRP-mediated protein targeting: Structure and function revisited Biochim. Biophys. Acta 1694 17–35. [DOI] [PubMed] [Google Scholar]
- Lütcke H., Prehn S., Ashford A.J., Remus M., Frank R., Dobberstein B. 1993. Assembly of the 68- and 72-kD proteins of signal recognition particle with 7S RNA J. Cell Biol. 121 977–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A., Anderson J.B., Cherukuri P.F., DeWeese-Scott C., Geer L.Y., Gwadz M., He S., Hurwitz D.I., Jackson J.D., Ke Z.et al. 2005. CDD: A conserved domain database for protein classification Nucleic Acids Res. 33 D192–D196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin L.J., Bryson K., Jones D.T. 2000. The PSIPRED protein structure prediction server Bioinformatics 16 404–405. [DOI] [PubMed] [Google Scholar]
- Oubridge C., Kuglstatter A., Jovine L., Nagai K. 2002. Crystal structure of SRP19 in complex with the S domain of SRP RNA and its implication for the assembly of the signal recognition particle Mol. Cell 9 1251–1261. [DOI] [PubMed] [Google Scholar]
- Politz J.C., Yarovoi S., Kilroy S.M., Gowda K., Zwieb C., Pederson T. 2000. Signal recognition particle components in the nucleolus Proc. Natl. Acad. Sci. 97 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblad M.A., Gorodkin J., Knudsen B., Zwieb C., Samuelsson T. 2003. SRPDB: Signal recognition particle database Nucleic Acids Res. 31 363–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoulica E., Krause E., Meese K., Dobberstein B. 1987. Disassembly and domain structure of the proteins in the signal-recognition particle Eur. J. Biochem. 163 519–528. [DOI] [PubMed] [Google Scholar]
- Shan S.O. and Walter P. 2005. Co-translational protein targeting by the signal recognition particle FEBS Lett. 579 921–926. [DOI] [PubMed] [Google Scholar]
- Siegel V. and Walter P. 1986. Removal of the Alu structural domain from signal recognition particle leaves its protein translocation activity intact Nature 320 81–84. [DOI] [PubMed] [Google Scholar]
- Siegel V. and Walter P. 1988. Binding sites of the 19-kDa and 68/72-kDa signal recognition particle (SRP) proteins on SRP RNA as determined in protein-RNA “footprinting” Proc. Natl. Acad. Sci. 85 1801–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert P.M., Mack J.W., Korge B.P., Gan S.Q., Haynes S.R., Steven A.C. 1991. Glycine loops in proteins: Their occurrence in certain intermediate filament chains, loricrins and single-stranded RNA binding proteins Int. J. Biol. Macromol. 13 130–139. [DOI] [PubMed] [Google Scholar]
- Walter P. and Blobel G. 1983. Disassembly and reconstitution of signal recognition particle Cell 34 525–533. [DOI] [PubMed] [Google Scholar]
- Weichenrieder O., Wild K., Strub K., Cusack S. 2000. Structure and assembly of the Alu domain of the mammalian signal recognition particle Nature 408 167–173. [DOI] [PubMed] [Google Scholar]
- Wild K., Sinning I., Cusack S. 2001. Crystal structure of an early protein-RNA assembly complex of the signal recognition particle Science 294 598–601. [DOI] [PubMed] [Google Scholar]
- Wild K., Weichenrieder O., Strub K., Sinning I., Cusack S. 2002. Towards the structure of the mammalian signal recognition particle Curr. Opin. Struct. Biol. 12 72–81. [DOI] [PubMed] [Google Scholar]
- Zwieb C. 1991. Interaction of protein SRP19 with signal recognition particle RNA lacking individual RNA-helices Nucleic Acids Res. 19 2955–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwieb C. 2003. Signal recognition particle-mediated protein targeting Recent Res. Dev. Mol. Biol. 1 205–224. [Google Scholar]
- Zwieb C. and Eichler J. 2002. Getting on target: The archaeal signal recognition particle Archaea 1 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwieb C., Van Nues R.W., Rosenblad M.A., Brown J.D., Samuelsson T. 2005. A nomenclature for all signal recognition particle RNAs RNA 11 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]