

Abstract
Automated function prediction (AFP) methods increasingly use knowledge discovery algorithms to map sequence, structure, literature, and/or pathway information about proteins whose functions are unknown into functional ontologies, typically (a portion of) the Gene Ontology (GO). While there are a growing number of methods within this paradigm, the general problem of assessing the accuracy of such prediction algorithms has not been seriously addressed. We present first an application for function prediction from protein sequences using the POSet Ontology Categorizer (POSOC) to produce new annotations by analyzing collections of GO nodes derived from annotations of protein BLAST neighborhoods. We then also present hierarchical precision and hierarchical recall as new evaluation metrics for assessing the accuracy of any predictions in hierarchical ontologies, and discuss results on a test set of protein sequences. We show that our method provides substantially improved hierarchical precision (measure of predictions made that are correct) when applied to the nearest BLAST neighbors of target proteins, as compared with simply imputing that neighborhood's annotations to the target. Moreover, when our method is applied to a broader BLAST neighborhood, hierarchical precision is enhanced even further. In all cases, such increased hierarchical precision performance is purchased at a modest expense of hierarchical recall (measure of all annotations that get predicted at all).
Keywords: protein function prediction, Gene Ontology, GO, prediction evaluation metrics
Recent advances in genome sequencing are creating an increasing volume of data, leading to more urgent interest in methods for automated function prediction (AFP). These methods increasingly use knowledge discovery algorithms to map sequence, structure, literature, and/or pathway information about proteins with unknown function into ontologies, such as the Gene Ontology (GO) (Gene Ontology Consortium 2000; http://www.geneontology.org), representing protein functions. We present an approach to AFP that combines knowledge of the structure of GO with BLAST e-values. While our method assumes that proteins that are similar in sequence or structure are more likely to share a function, we do not simply transfer the annotations of a similar protein to the target protein. Rather, we search for annotations that are representative of the annotations of similar proteins, based on the distribution of those annotations within the ontological structure of GO. Specifically, we use a novel knowledge discovery technique, which we call “categorization,” to automatically identify those GO nodes that most accurately represent another group of GO nodes, in this case those that are annotated to proteins similar in some respect to a target protein.
The system we have developed is an application within our POSet Ontology Laboratory Environment (POSOLE), which consists of a set of modules supporting ontology representation, mathematical analysis of those structures, categorization of nodes in an ontology, and evaluation of the predicted categorization with respect to a given set of expected answers. The system defines QueryBuilders specific to an application for mapping its relevant input to a set of ontology nodes, in this case by identifying the sequence neighborhood of the protein and associating those neighbors to GO nodes (other QueryBuilders might use bibliometric data [Verspoor et al. 2005] or structural data). The POSet Ontology Categorizer (POSOC; http://www.c3.lanl.gov/posoc)1 then categorizes this set of GO nodes to identify the most representative nodes as putative functions of the input protein.
Essential to any AFP method is the ability to measure the quality of predictions. We are keenly aware that the nature of GO as a hierarchically structured database makes traditional evaluation measures inadequate. We therefore conclude with the presentation of new evaluation metrics called “hierarchical precision” and “hierarchical recall,” which we are developing for the general task of evaluating methods for AFP into the GO.
Results
POSOC was designed to take a large set of GO nodes and identify clusters with a richer concentration of relevant information. This results directly in an increase in hierarchical precision (an extension of the standard precision measure, to be defined below). Generally in knowledge discovery algorithms, increased precision comes at the expense of decreased recall, and vice versa, as is true here.
Figure 1 shows the results of applying our method to our test data set, both of which are described in detail in Materials and Methods. Note the improvement in hierarchical precision (HP) when POSOC is included in the processing, especially on the full BLAST neighborhood as opposed to the BestBLAST neighborhood (both also described below). This comes at a modest expense of hierarchical recall (HR), resulting in relatively little variation in the hierarchical F-score.
Figure 1.

Accuracy of the top-ranked annotation predictions on the test data using only Non-IEA annotations across the three GO branches, comparing the POSOC method with specificity parameter s = 2 against the baseline. (BaseBB) BestBLAST neighborhood without POSOC; (BaseFN) full neighborhood without POSOC; (PosocBB) BestBLAST neighborhood with POSOC; (PosocFN) full neighborhood with POSOC; (HP) average hierarchical precision; (HR) average hierarchical recall; (HF) average hierarchical F-score.
We have explored the behavior of our system at different values of the POSOC parameter called specificity s, which controls whether POSOC favors annotations that are shallow or deep in the GO (see below). We have found that the increase in hierarchical precision over the baseline scenarios is most marked at the relatively low value s = 2 (data not shown). This follows from the observation that higher precision using the hierarchical measures occurs when more predictions are more general than the correct answers, and the fact that lower specificity favors more general results. However, it is not the case that simply returning the top node in each branch would give us the best results, as this would result in a large decrease in recall. Our results at s = 2 show a drop in hierarchical recall over BaselineBestBLAST, but not an unacceptable drop as evidenced by the lack of substantial change in the balanced hierarchical F-score as seen in Figure 1.
We believe that, in general, users of AFP systems would tend to value precision over recall, or false negatives over false positives. Said another way, they would prefer that annotations be accurate at the risk of not all annotations being provided. As such, the POSOC results indicate that our system provides an important boost over the alternative baseline scenarios at achieving the results in which these users are interested.
Materials and methods
A simple formulation for generic AFP into the GO can be described as follows. Assume a collection of genes or proteins and a set of GO nodes (perhaps for a particular GO branch). Then “annotation” can be regarded as assigning to each protein some collection of GO nodes. Where a known protein may have a known set of annotations, a new protein will not, and we wish to build some method that returns a predicted set of GO nodes for that target protein. Typically, we have some information about the target protein such as sequence, structure, interactions, pathways, or literature citations, and we exploit knowledge of the proteins that are “near” to it, in one or more of these ways, which do have known functions. The annotation method presented here exploits the BLAST sequence neighborhoods of target proteins, coupled with the POSOC categorizer.
In a testing situation, we start with a known protein with known annotations, and compare these against the annotations predicted by the method as if they were not known. So, while our focus below is our particular POSOC methodology, our general formulation of AFP is motivating our introduction of our novel evaluation measures, which are intended to be applicable to any AFP architecture.
POSOC method within POSOLE
Our particular architecture for AFP using sequence data, within our general POSOLE environment, is shown in Figure 2. At its heart are a QueryBuilder module associating an input query sequence with a weighted collection of GO nodes, and the POSOC module for identifying proper categorizations of that collection as GO annotation predictions. In the testing context, this process is carried on with knowledge of the known GO annotations of the sequence.
Figure 2.

Architecture of the POSOC automated ontological annotation method within the POSOLE environment.
The current query builder uses a “nearest-neighbor” approach to identify annotations of close neighbors of the input sequence in sequence space. We perform a PSI-BLAST (Position-Specific Iterated BLAST) (Altschul et al. 1997) search on the target against the NCBI nonredundant sequence database, nr (http://www.ncbi.nlm.nih.gov/BLAST/blastcgihelp.shtml#protein_databases), with five iterations, using the default e-value threshold of 10. Once the nearest neighbors have been identified, we collect GO nodes associated with these sequences using the UniProt SWISS-PROT to GO mappings. Finally, we build a weighted collection of GO nodes, where each node in the collection is weighted according to the PSI-BLAST e-value. Several near neighbors of the original target sequence may map to the same nodes, in which case each occurrence will be weighted individually according to its source.
This collection of weighted GO nodes becomes the input query to POSOC (Joslyn et al. 2004), which returns a ranked list of nodes that best “summarize” or “categorize” that collection. Note that in one extreme, returning only the top-most node of the GO branch in question is certainly an accurate categorization, covering the entire input query, but hardly precise enough to be useful. Conversely, just returning all the particular nodes in the query again is certainly as precise as is possible, but hardly does any work toward summarizing or grouping the nodes together. POSOC balances these conflicting tendencies of “specificity” and “coverage” by providing a tunable parameter “specificity” s, which for low values (s ∼ 1) returns fewer, more general categories, and for high values (s > 4) a larger collection of deep nodes.
Data sets
We evaluate our AFP performance on a “gold standard” test set of proteins comprised of a subset of SWISS-PROT proteins with both known GO mappings and PDB structures (http://www.rcsb.org/pdb). This test set was selected to enable us to compare our results with algorithms integrating structural data. Other groups have used a variety of test sets; for example, Pal and Eisenberg (2005) use a set of protein sequences from the FSSP structure library (http://www.chem.admu.edu.ph/∼nina/rosby/fssp.htm) to evaluate their ProKnow system, and Martin et al. (2004) use sequence data from seven complete genomes to test GOtcha.
The value of any gold standard depends on the accuracy of its known annotations. We use the GOA (http://www.ebi.ac.uk/GOA) UniProt (http://www.ebi.ac.uk/uniprot/index.html) annotation set augmented with a ranking for the evidence codes included in GO annotation files (e.g., IC = inferred by curator, IEA = inferred from electronic annotation), following Pal and Eisenberg (2005). For testing, we use a “Non-IEA” subset of the annotations excluding all annotations of rank 4 or below, i.e., evidence codes NAS (nontraceable author statement), IEA, and NR (no record). The purpose of this subset is to avoid the circularity of making an automated prediction using sequence similarity based on other automated predictions derived from sequence similarity, as these annotations are more likely to contain errors than the curated annotations (Gilks et al. 2002) and already incorporate the assumption we test. We then filter our gold standard set of proteins to exclude any proteins without annotations in the Non-IEA set. A total of 1282 proteins remain for testing in a leave-one-out strategy in which the protein itself is excluded from the PSI-BLAST matches.
Evaluation scenarios
We compare the behavior of our POSOC-based function prediction with two baseline scenarios. In the BaselineBestBLAST scenario, we identify the protein with the highest PSI-BLAST match value to our input protein (the “BestBLAST” protein), and simply return the annotations associated with that protein, assigning them all a rank of 1. This corresponds to the standard strategy that a biologist would use, following the assumption that two proteins close in sequence (BLAST) space will share the same functions. In the BaselineFullNeighborhood scenario, we return all annotations associated with any protein matched by PSI-BLAST within the e-value threshold, ranked by match probability e−(e-value). Due to a loss of numerical precision in the conversion to probability, we find many neighbors matching at rank 1 and see correspondingly lower (hierarchical) precision and higher (hierarchical) recall with respect to the desired answers, even at the top ranks.
POSOC itself is run in two parallel scenarios. PosocFullNeighborhood is the standard way it would be used for AFP: the annotations of each PSI-BLAST, weighted according to match probability, are submitted to POSOC for categorization. In PosocBestBLAST, only the annotations of the “BestBLAST” protein are categorized by POSOC. In each case, we expect POSOC to filter out any noise in the annotation sets to arrive at the nodes most representative of the inputs.
Evaluation measures
Let N be the set of GO nodes, either as a whole or in any particular branch or portion. Then for a given target protein x, POSOC will return a ranked list of cluster heads G(x) ⊆ N indicative of the function of the query sequence, which thereby must be compared against the set of known annotations F(x) ⊆ N. Standard evaluation measures are provided from information retrieval, including precision P, measuring the percentage of predictions which are correct; recall R, measuring the portion of annotations which we have predicted; and F-score F, combining both and reflecting their tradeoff:
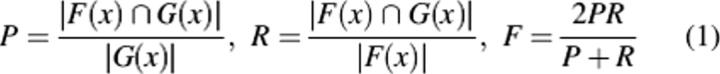 |
Each number varies between 0 and 1, where P = 0 ↔ R = 0 ↔F = 0, but P = 1 only when all predictions are correct, and R = 1 only when all correct annotations are predicted.
However, the results G(x) produced by POSOC do not form a simple set, but rather a ranked list of effectively indefinite length. Alternative measures to handle ranked lists are available (Voorhees and Tice 2000), but the measures must apply in the context where near misses are accounted for, and annotations occur into a hierarchically structured ontology. We introduce Hierarchical Precision (HP), Hierarchical Recall (HR), and Hierarchical F-score (HF) as:
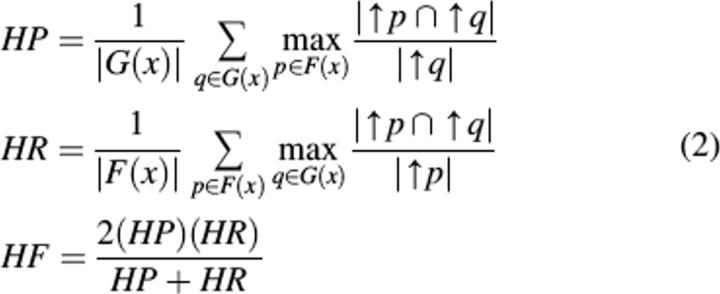 |
where ↑p indicates the set of ancestors of the GO node p ∈ P. Figure 3 shows an illustration of a situation with GO nodes GO:1–GO:7, where a single annotation F(x) = {GO:4} is compared against a single prediction G(x) = {GO:6}, so that ↑p = {1,2,4} (using just the node numbers here) and ↑q = {1,2,3,5,6}, yielding HP = 2/5 and HR = 2/3.
Figure 3.

Illustration of hierarchical precision and recall calculations.
Ranked evaluation results
For each test protein, we must compare an unranked set of correct annotations to the ranked list returned by POSOC. We therefore calculated HP and HR separately, although cumulatively, at each rank, considering only the predictions up to a given rank against the full set of correct annotations. This allows us to assess the impact of rank on our predictions: how steeply does hierarchical precision drop off and hierarchical recall increase as we move down the ranks? To assess this across the full set of test proteins, we average HP and HR at each rank.
Space limitations preclude showing results for all ranks. Moreover, the number of test proteins that have predictions drops sharply and unevenly at lower ranks, and so these averaged values become less reliable as we move down to ranks generally >4. Thus results for rank = 1 only are provided in Figure 1, but this allows us to most directly compare BaselineBestBLAST with the other scenarios, since that baseline only has predictions at rank 1.
Discussion
There are several methodological limitations of current AFP methods that our hierarchical evaluations measures are trying to address. First, AFP should deal fundamentally with the need to accommodate and measure not just “exact matches” but also “near misses” of different sorts. If a particular annotation is wrong, can we say more about how far off it is? Thus, there is a first need to generalize classical precision and recall measures to accommodate a sense of distance among annotations.
In the particular context of the GO, errors are introduced if it is considered to be a simple list of functional categories. The hierarchical structure of the GO represents the interaction between specific and general categories that are either low or high in the structure, respectively. Moreover, annotations to low nodes are considered as annotations to high nodes as well, what Eisner et al. (2005) describe as the “true path rule.” This results in a mathematical structure of an ordered set (Joslyn et al. 2004), which must be taken into account when measuring how well an AFP method performs. In particular, an annotation to a parent, grandparent, or other ancestor of a true annotation must also be considered as a true, albeit less than ideally specific, annotation.
Moreover, in many cases predicting a parent, grandparent, or sibling of a correct annotation may be acceptable, or even preferable to an exact match. In the example in Figure 3, if exact matches were required, traditional precision and recall from Equation 1 would both be 0, despite the fact that GO:1 and GO:2 are both correct, albeit more general, annotations on which both GO:6 and GO:4 agree.
This issue has been attended to only very little in the literature. Kiritchenko et al. (2005) and Eisner et al. (2005) have proposed an explicitly hierarchical extension of precision and recall with respect to the subgraph containing the predicted node and all of its ancestors (the “node subgraph”) and the node subgraph of the correct node. Pal and Eisenberg (2005) consider precision at various ontology depths, hierarchically matching nodes in the node subgraph of the predicted node and nodes in the node subgraph of the correct node. Both solutions, however, require methodological completion, and neither explicitly addresses the primary case of comparing a set of node predictions with a set of answers.
In prior work (Joslyn et al. 2004; Verspoor et al. 2005), we have measured performance with respect to direct hits, “nuclear family” (parent, child, sibling) and “extended family” (grandparent, uncle, cousin, etc.) relations between nodes. Our approach now aims to extend these ideas by placing precision and recall in a metric space context to generally account for near misses (Pekalska 2005), and adopting metrics specifically appropriate for hierarchical structures cast as partially ordered sets (Monjardet 1981). While this work is ongoing, we have immediately here extended Kiritchenko et al.'s (2005) approach from single-node comparisons to sets of nodes, producing Equation 2, an approach that is similar to that of Eisner et al. (2005).
HP captures the property that errors at higher levels of the hierarchy are punished more severely, and more distant errors are punished more heavily than a near miss. The use of the sum of maxima in Equation 2 captures the intuition that for each prediction, we must find the closest match to any of the possible answers as defined by the gold standard. This is easiest to understand by considering the most extreme case of exact matches: if all predictions exactly match an element of the expected answer set, this results in HP = 1. These predictions are no less correct simply because there are other possible answers (which would be the case, for instance, if hierarchical precision were averaged across the elements of the set).
In combination with ranked assessments, it is possible for hierarchical precision to increase as we go down the ranks. In particular, a new prediction at a lower rank might be closer to one of the correct answers than any prediction up to that point. In this case, when we use the sum of maxima in Equation 2, we will find that hierarchical precision increases.
The need for an evaluation measure including some form of “partial credit” for near misses is demonstrated by Table 1, which shows the case when the BestBLAST neighborhood of non-IEA annotations is used, without POSOC, to induce predictions that are then compared to exact matches into GO using the standard precision and recall measures. This is the most straightforward AFP process, and shows very poor performance because of the lack of consideration of near neighbors in the evaluation. Note that the inclusion of near misses in the measure means that HP and HR will always be higher than the corresponding P and R values for a given test set (cf. Table 1 and Fig. 1, BaselineBestBLAST results), thus they are not directly comparable measures.
Table 1.
Regular (nonhierarchical) average Precision (P), Recall (R), and F-score (F) for the BaselineBestBLAST neighborhood using the non-IEA annotation set calculated only for exact matches to GO annotations in the gold standard data set (also only considering non-IEA annotations)

The hierarchical measures are able to give credit for predicted answers even when they are not exact. In the case of considering a single prediction against a single correct answer, when the prediction is a successor of the actual answer, then HR = 1, while HP < 1, with HP larger in deeper parts of the ontology, and decreasing with distance between the two nodes. When a prediction is an ancestor of the actual answer, then HP = 1, while HR < 1, with HR larger for more specific nodes, and again decreasing with distance between the two nodes. An overall high hierarchical precision is indicative of most predictions being ancestors of the actual answers and more general. Higher hierarchical recall indicates that more predictions are successors of the actual and are more specific. We note that similar observations have been advanced by Eisner et al. (2005).
Given that hierarchical precision is enhanced for matches higher in the hierarchy, our increased hierarchical precision could indicate that the gold standard answers are actually distributed at a moderately high level in GO. Similarly, the relatively low precision value s = 2 used here will tend to produce higher predictions, thus explaining part of the HP/HR tradeoff shown in Figure 1. Deeper consideration of these issues, including independent measurement of the depth of sets of predictions and correct answers, awaits future work.
Acknowledgments
This work was also supported by the Department of Energy under contract W-7405-ENG-36 to the University of California. We particularly thank the Protein Function Inference Group at the Los Alamos National Laboratory for the motivation to conduct this study and extensive discussion during its development.
Footnotes
Reprint requests to: Karin Verspoor, Los Alamos National Laboratory, PO Box 1663, M.S. B256, Los Alamos, NM 87545, USA; e-mail: verspoor@lanl.gov; fax: (505) 667-1126.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062184006.
References
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner R., Poulin B., Szafron D., Lu P., Greiner R. 2005. Improving protein function prediction using the hierarchical structure of the Gene Ontology. 2005 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology. San Diego, CA.
- The Gene Ontology Consortium. 2000. Gene Ontology: Tool for the unification of biology. Nat. Genet. 25 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilks W., Audit B., De Angelis D., Tsoka S., Ouzounis C.A. 2002. Modeling the percolation of annotation errors in a database of protein sequences. Bioinformatics 18 1641–1649. [DOI] [PubMed] [Google Scholar]
- Joslyn C., Mniszewski S., Fulmer A., Heaton G. 2004. The Gene Ontology Categorizer. Bioinformatics 20(Suppl. 1), i169–i177. [DOI] [PubMed] [Google Scholar]
- Kiritchenko S., Matwin S., Famili A.F. 2005. Functional annotation of genes using hierarchical text categorization. Proc. BioLINK SIG Meeting on Text Data Mining at ISMB'05. Detroit, MI.
- Martin D., Berriman M., Barton G. 2004. GOtcha: A new method for prediction of protein function assessed by the annotation of seven genomes. BMC Bioinformatics 5 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monjardet B. 1981. Metrics on partially ordered sets—A survey. Discrete Math. 35 173–184. [Google Scholar]
- Pal D. and Eisenberg D. 2005. Inference of protein function from protein structure. Structure 13 121–130. [DOI] [PubMed] [Google Scholar]
- Pekalska E. “Dissimilarity representations in pattern recognition. Concepts, theory and applications.” Ph.D. thesis 2005. Delft University of Technology, Delft, The Netherlands ASCI Dissertation Series, 109.
- Verspoor K., Cohn J., Joslyn C., Mniszewski S., Rechtsteiner A., Rocha L.M., Simas T. 2005. Protein annotation as term categorization into the Gene Ontology using word proximity networks. BMC Bioinformatics 6(Suppl. 1), S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees E. and Tice D.M. 2000. The TREC-8 question answering track evaluation. In The Eighth Text Retrieval Conference (TREC-8) (eds. Voorhees E. and Harman D.K.) . pp. 83–106. NIST Special Publication 500–246.