

Abstract
The Saccharomyces cerevisiae His6 gene codes for the enzyme phosphoribosyl-5-amino-1-phosphoribosyl-4-imidazolecarboxamide isomerase, catalyzing the fourth step in histidine biosynthesis. To get an insight into the structure and function of this enzyme, we determined its X-ray structure at a resolution of 1.30 Å using the anomalous diffraction signal of the protein's sulphur atoms at 1.77 Å wavelength. His6 folds in an (α/β)8 barrel similar to HisA, which performs the same function in bacteria and archaea. We found a citrate molecule from the buffer bound in a pocket near the expected position of the active site and used it to model the open form of the substrate (phosphoribulosyl moiety), which is a reaction intermediate. This model enables us to identify catalytic residues and to propose a reaction mechanism where two aspartates act as acid/base catalysts: Asp134 as a proton donor for ring opening, and Asp9 as a proton acceptor and donor during enolization of the aminoaldose. Asp9 is conserved in yeast His6 and bacterial or archaeal HisA sequences, and Asp134 has equivalents in both HisA and TrpF, but they occur at a different position in the protein sequence.
Keywords: isomerase, mechanism, HisF, HisA, citrate, TrpF, ProFAR, histidine biosynthesis, Amadori rearrangement, citrate, sulphur phasing
The reactions and enzymes involved in the biosynthesis of histidine have been identified in many organisms and were thoroughly reviewed (Alifano et al. 1996; Fani et al. 1997). In the yeast Saccharomyces cerevisiae, the His6 gene (YIL020c) codes for phosphoribosyl-5-amino-1-phosphoribosyl-4-imidazolecarboxamide isomerase (E.C.5.3.1.16), an enzyme that catalyzes the fourth step in the histidine pathway. This step consists in an Amadori rearrangement that isomerizes the aminoaldose moiety of ProFAR (N-(5′-phospho-D-ribosylformimino)-5-amino-1-(5″-phosphoribosyl)-4-imidazolecarboxamide) to the aminoketose of PRFAR (N-(5′-phospho-D-1′-ribulosylformimino)-5-amino-1-(5″-phospho-ribosyl)-4-imidazolecarboxamide) (Fig. 1A; Margolies and Goldberger 1967). The corresponding gene is called HisA in bacteria and archaea. The isomerization step is followed by the condensation of PRFAR with ammonia and the cleavage of the condensation product into 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) and imidazole glycerol phosphate (ImGP). Although the histidine biosynthesis pathway has been extensively studied, we still lack a detailed mechanism for many of its steps. We present here a crystal structure at 1.3 Å resolution of the product of the His6 gene of Saccharomyces cerevisiae, a monomeric protein of 29 kDa, determined using the anomalous scattering signal of sulfur atoms for phase determination. The protein contains citrate from the crystallization mixture at the expected position of the phosphoribulosyl moiety of the reaction product. We used the citrate conformation to build a model of the open form of the (phosphoribulosyl moiety) of the bound substrate, and propose a mechanism for the isomerization reaction that takes into account structural and mutation data previously obtained on Thermotoga maritima HisA (Henn-Sax et al. 2002) and incorporates elements of proposed mechanisms for HisA and TrpF.
Figure 1.
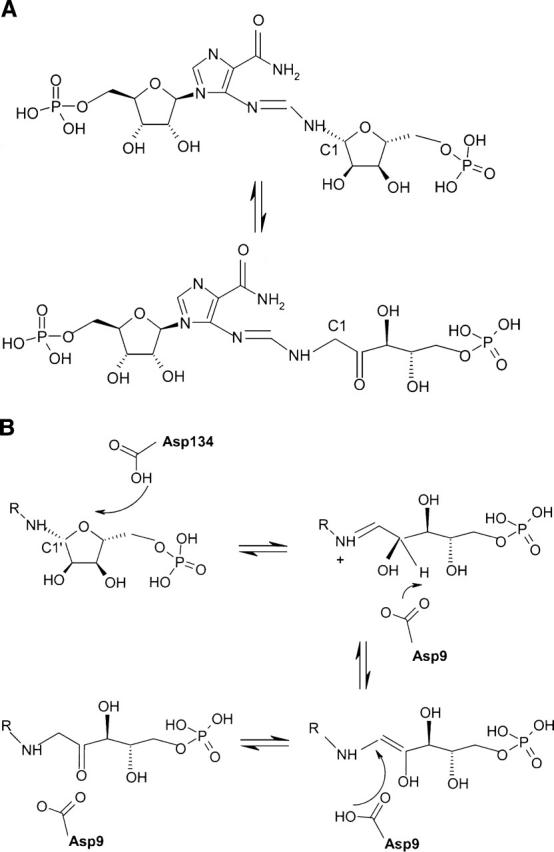
(A) Reaction catalyzed by His6: ProFAR (N-(5′-phospho-D-ribosylformimino)-5-amino-1-(5″-phosphoribosyl)-4-imidazolecarboxamide) ⇆ PRFAR (N-(5′-phospho-D-1′-ribulosylformimino)-5-amino-1-(5″-phosphoribosyl)-4-imidazolecarboxamide. (B) Proposed reaction mechanism of His6. The substrate binds as the closed form and ring opening is catalyzed by protonation mediated by Asp134 yielding the Shiff's base. After opening of the ring, the substrate is repositioned in front of the catalytic Asp9. This aspartate abstracts a proton from the C2′carbon and transfers it to the C1′position yielding the ribulose product.
Materials and methods
Cloning, expression, and purification
Cloning and expression as a His-Tagged fusion of the YIL020c gene, as well as purification of the corresponding His6 protein was essentially as described for all targets of our Yeast Structural Genomics Project (Quevillon-Cheruel et al. 2004).
Crystallization and resolution of the structure
Crystals (100–200 microns) were grown from 1:1 μL mixture of protein (10 mg/mL) with 24% PEG 4K, 0.1 M Na-citrate (pH 4.6) and 0.1 M MgCl2. For data collection, the crystals were frozen in liquid nitrogen after successive transfers in cryoprotectant buffers containing 30% ethylene glycol. Crystals of the native protein diffracted to 1.3 Å resolution on the ID14–1 beamline (ESRF, Grenoble, France). The structure was solved by sulphur SAD (single wavelength anomalous dispersion) using highly redundant data collected to 2.0 Å resolution on the BM14 beamline (ESRF) at a wavelength of 1.77 Å.
Data were processed using the program MOSFLM (Leslie 1992). The space group was C2 with one molecule per asymmetric unit (solvent content 40%). SHELXD (Schneider and Sheldrick 2002) was used to find their positions of the sulphur sites, which were then refined using the Sharp suite (Bricogne et al. 2003). From the nine identified sites, seven were from cysteine residues, one was from the unique methionine, and one was from a bound chloride ion. After phase extension, Arp/Warp (Morris et al. 2003) was able to build 80% of the model automatically. Using the high-resolution data, the model was completed by manual rebuilding using TURBO (Roussel and Cambillau 1991) and refined with REFMAC (Murshudov et al. 1997) using anisotropic B factors.
The final model contains residues 2–17, 38–179, and 187–261 and one chloride ion, one citrate molecule, three ethylene glycols, and 352 water molecules. All residues fall within the allowed regions of the Ramachandran plot as defined by the program PROCHECK (Laskowski et al. 1993). Data collection and refinement statistics of the structure are summarized in Table 1. The atomic coordinates and structure factors for the protein structure have been deposited into the Brookhaven Protein Data Bank (PDB) under accession number 2AGK.
Table 1.
Data collection, phasing, and refinement statistics

Results
Determination of the crystal structure by sulphur SAD phasing
His6 has only two methionines for 261 amino acids (including the N-terminal one), and we therefore incorporated methionines by site-directed mutagenesis at positions occupied by methionines in orthologous sequences. We had successfully used this approach to solve the structure of two other yeast proteins that had a low methionine content (Graille et al. 2004; Leulliot et al. 2005). It failed with His6, the mutant proteins being unstable and lost during purification (results not shown). In view of the good diffraction quality of native crystals, we attempted single wavelength phasing using the anomalous signal of the 10 sulphur atoms (eight cysteines and two methionines), on BM14 of ESRF (Gordon et al. 2001; Lartigue et al. 2004). We obtained a highly redundant 2 Å resolution data set at 1.77 Å wavelength with an excellent signal-to-noise ratio (30.3 in the last resolution shell). With these data, SHELXD (Schneider and Sheldrick 2002) unambiguously positioned nine sites corresponding to eight sulphur atoms (seven cysteines and one methionine) and a chloride ion. The ARP/wARP program (Morris et al. 2003) automatically built 80% of the residues in the solvent flattened experimental map obtained with the SHARP program (Bricogne et al. 2003). Most of the residues that could not be built by ARP/wARP are absent in the final model, probably due to flexibility.
Overall structure
The excellent quality of the sulphur phased maps resulted in a well-refined structure with an R-factor of 19.1% and an R-free of 21.6% (Table 1) at 1.3 Å resolution. Figure 2A shows a ribbon diagram of the protein. As expected, His6 is a parallel (α/β)8 barrel of overall dimensions 40 Å × 42 Å × 31 Å. It has the typical TIM barrel architecture where the β-strands are connected by long loops at their C-terminal end with short connections at their N-terminal end. A β-hairpin (β5B–β5C) is inserted in the long connection between β5 and α5. The two segments comprising residues 18–37 after β1B and residues 180–186 after β6 lack electron density.
Figure 2.
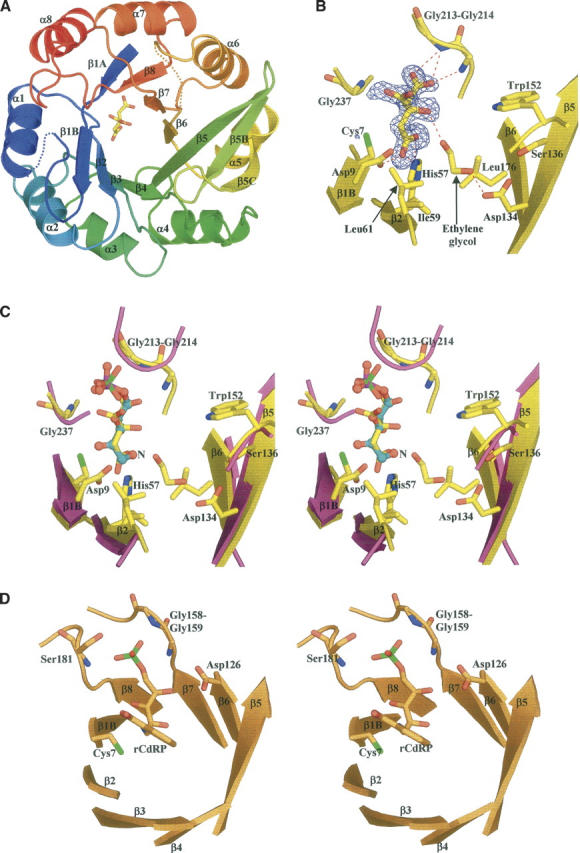
(A) Ribbon representation of His6 colored from blue (Nter) to red (Cter) with the bound citrate molecule shown as sticks. (B) View of citrate modeled into the residual Fo−Fc electron density map contoured at 3.5σ. Hydrogen bonds between citrate, ethylene glycol, and His6 are depicted by red dashed lines. (C) Stereo view representation of the active sites of Tm HisF (magenta; PDB code 1GPW) and the S. cerevisiae His6 (yellow). For clarity, only the active site is shown. The phosphate ion (phosphorus atom in green) bound to the HisF enzyme is shown as sticks. The citrate molecule bound to yeast His6 and a model for the D-ribulose-5-phosphate moiety of the His6 product, are shown in sticks (carbon atoms in yellow) and ball-and-sticks (carbon atoms, blue; phosphorus atom, magenta), respectively. The nitrogen position of the N-(5′-phospho-D-ribosylformimino) is indicated by capital N. (D) Stereo presentation of the active site of TrpF bound to the reaction product.
Structural matches were retrieved on the MSD server of the European Bioinformatics Institute (http://www.ebi.ac.uk/msd-srv/ssm/). The match was fair, with HisA from Thermotoga maritima (PDB entry 1thf: RMSD = 2.4 Å; Z-score = 7.1), and rather better with HisF (imidazole glycerol phosphate synthase) from Pyrobaculum aerophilum (PDB entry 1h5y: RMSD = 2.1 Å; Z-score = 7.9) and Thermus thermophilus (PDB entry 1ka9: RMSD = 2.2 Å; Z-score = 7.6). In all cases, the sequence identity was low: 16% for HisA, 17%–18% for HisF. Moreover, significant differences between His6 and Thermotoga maritima HisA (TmHisA) occur in loops (data not shown). The loop after β1B is disordered in His6 but not in TmHisA; the connection between β5 and α5 has a different conformation in the two proteins; and the connection between β6 and α6 is helical in TmHisA and partly disordered in His6.
The active site
The Amadori rearrangement catalyzed by His6 involves opening the ribofuranose ring and forming a Schiff base intermediate. Proton abstraction then yields an enolamine that converts to the keto product. Each step is likely to require acid base catalysis: To protonate the ring oxygen, remove the C2′ proton for enolisation, and add one to C1′ to form the final product. The latter steps are similar to those catalyzed by TIM when converting dihydroxyacetone-phosphate into glyceraldehyde-3-phosphate. In TIM, a catalytic histidine (His95) and glutamate (Glu165) positioned at opposite faces of the substrate transfer protons from C1 to C2 in coordinated fashion (Knowles 1991). We searched for equivalents of these two catalytic residues in His6. Sequence alignment (data not shown) indicates that the only residues conserved over all HisA/His6 sequences are seven glycines, Asp9, His57, Leu61, and Trp152. Of those, Asp9 located in strand β1B and His57 in strand β2, are the obvious candidates for acid/base catalysis. The side chains of these two residues point toward the C-terminal end of the β-sheet, where enzymes with the TIM fold usually have their active site. They are adjacent in space, the His57 Nɛ atom being at 5.3 Å of the Asp9 carboxylate group (Fig. 2B,C), and surrounded by hydrophobic residues providing a nonpolar environment. In this region, the only other candidate as a catalytic residue is Cys7, which is conserved in yeast sequences but mutated into alanine in bacteria and archaea.
As shown in Figure 2B, the electron density map contained a well-defined elongated residual feature in a cleft near Asp9 and His57. It perfectly accommodates a citrate molecule, present in the purification and crystallization buffers. Two other residual density features nearby were interpreted as ethylene glycol, used as cryoprotectant. One interacts with the citrate hydroxyl (Fig. 2B), and the other is located further in the same pocket. Refinement yielded B-factors for these ligands similar to those of the surrounding side chains, suggesting full occupancy. The bound citrate adopts an extended conformation and makes contact with several conserved residues. Its C1 carboxylate group makes a bidentate hydrogen bond with Nɛ2 of His57 and is within H-bonding distance of the side-chain carboxylate of Asp9, which implies that one of the two carboxylates must be protonated. The C5 carboxylate fits in a small pocket formed by the loops connecting β7 to α7 and β8 to α8, and receives H-bonds from the main-chain NHs of the strictly conserved Gly213 and Gly214.
As the chemical instability of the substrate and product of His6/HisA precludes the structural analysis of their complexes, we used the bound citrate as a model of the open form of the phosphoribosyl part of ProFAR. The result of a superposition is shown in Figure 2C. It places the sugar C1′ and C2′ atoms in between Asp9 and His57, and the 5′-phosphate close to the position of the C5 carboxylate of citrate. This position is that of a bound phosphate ion seen in the crystal structure of Thermotoga maritima HisF (PDB code 1gpw; Douangamath et al. 2002). Like the C5 carboxylate of citrate, the modeled phosphate interacts with the NHs of Gly213, Gly214, and Gly237. These glycines are totally conserved, and substitutions introducing side chains would block this part of the pocket.
We did not attempt to model the remainder of ProFAR. There is ample space next to the modeled C1′ atom of the modeled ribulose to fit the phosphoribosyl-4-imidazolecarboxamide moiety (Fig. 2C), and the two ethylene glycol molecules occupy part of that space. Moreover, the connecting loops after β1B and β6, which are disordered in our structure, may well become ordered when the substrate binds and interact with phosphoribosyl-4-imidazolecarboxamide.
Discussion
The example of His6 confirms that the anomalous scattering of sulphur atoms naturally present in proteins can yield high-quality X-ray structures on specialized synchrotron beam lines (Dumas et al. 1995). The 1.3 Å resolution of our X-ray data provides an accurate image of the protein showing that His6 is structurally very similar to HisA. The reaction mechanism has been analyzed in Thermotoga maritima HisA (TmHisA) by site-directed mutagenesis complementing structural studies (Henn-Sax et al. 2002). Substitution of Asp8, equivalent to His6 Asp9, yielded a protein with no measurable activity. This result is consistent with our model of the bound phosphoribosyl moiety of ProFAR binding, which is based on the observed citrate. In this model, a carboxylate oxygen of Asp9 is at 3.6 Å from both C1′ and C2′, in a position where it can abstract a proton from either carbon or give them a proton if Asp9 is protonated.
On the other hand, substituting Tm HisA His48, equivalent to His57 in His6, has only a moderate effect on TmHisA activity: kcat decreases twofold, Km increases sevenfold. Whereas the change in Km does suggest that the histidine interacts with the substrate as we see it does with citrate, the small effect on kcat excludes that residue from a role of acid/base catalyst similar to the one it has in TIM. Instead, Henn-Sax et al. (2002) point to another possibility. Substituting TmHisA Asp127 causes a 1000-fold drop in kcat. The aspartate is conserved in HisA sequences, but it is a serine in eukaryotic His6 (Ser136 in S. cerevisiae). Moreover, it is remote from the C1′ and C2′ carbons in our model. In the His6 structure, Ser136 and the Asp9 carboxylate are 12 Å apart, and they cannot both interact with adjacent carbons in the substrate. Thus, if Asp127 is a catalytic residue in HisA as the mutation data indicate, it is likely to be involved in another step than enolisation. We suggest it may be protonated and donates its proton to the sugar ring oxygen during the initial ring opening step. We also suggest that, instead of Ser136, the equivalent of Asp127 in TmHisA, Asp134 plays that role in His6. Asp134 is conserved in eukaryotic sequences, located on the same β5 strand as Ser136, and its carboxylate is 9 Å away from that of Asp9. Although we do not have a model of the bound substrate, we can assume that, when the sugar rings opens, C1′ and C2′ move from a position where the ring oxygen is close to Asp134 to the modeled position next to Asp9. Last, we suggest that, whereas TIM has two different residues acting as a base and an acid during enolization, HisA/His6 uses only one, the Asp9 carboxylate being in position both to take the proton from C2′ and to give it to C1′. Our suggestions for a reaction mechanism of His6, based on our structural observations and mutant data from the literature, are summarized in Figure 1B.
The tryptophan biosynthesis pathway includes a sugar isomerization step catalyzed by phosphoribosylanthranilate isomerase (TrpF). That step is very similar to that catalyzed by HisA, and in Streptomyces coelicolor, the same protein (PriA) carries both activities (Barona-Gomez and Hodgson 2003). HisA, TrpF, and PriA are all (α/β)8 barrels (Lang et al. 2000; Banfield et al. 2001; Henn-Sax et al. 2002). An X-ray structure of Tm TrpF has been determined in the presence of 1-[(2-carboxyphenyl)amino]-1-deoxyribulose 5-phosphate (rCdRP) (Henn-Sax et al. 2002), an analog of the reaction product. rCdRP binds at a position equivalent to that of citrate in His6, and its C2′ atom is close to the sulfhydryl group of Cys7, a residue conserved in all TrpF sequences (illustrated in Fig. 2D). As substitution to alanine destroys activity, Cys7 is likely to be a catalytic acid/base in the enolization step. Henn-Sax et al. (2002) also suggest that Asp126 of TrpF plays the role we attribute to Asp134 of His6. These aspartates are part of strand β5 in the two proteins. Asp126 is essential for activity and, in the complex with rCdRP, its carboxylate hydrogen bonds to the 4′-hydroxyl of deoxyribulose, which is the ring oxygen in the phosphoribosylanthranilate (PRA) substrate. Moreover, TrpF Asp126 and His6 Asp134 have a hydrophobic environment that should favor the neutral protonated form, needed to catalyze ring opening. The Tm TrpF-Cys7Ala mutant has no measurable activity and the TmTrpF Asp126Asn mutant has a 20,000-fold lower kcat/KmPRA value, supporting the structural observations and functional hypothesis for these two residues. TmTrpF Cys7 is at an equivalent position as Asp9 in His6 and Asp8 in Tm HisA. These residues therefore seem to adopt the same role as general base in these enzymes. Remarkably, there seems to be a great variability for the position of the catalytic acid in these enzymes. Positional variations of catalytic residues are often observed in homologous enzymes. For instance, a positional shift of the catalytic triad carboxylate was observed within the (α/β)-hydrolase fold between lipases from microbes and higher eukaryotes (Todd et al. 2001). In the absence of a structure of a complex with substrate analogs, it will be difficult to make definite statements about the identity of the catalytic acid in His6.
Acknowledgments
We thank Martin Walsh for help with data collection on the BM14 beam line at the ESRF Grenoble (France). This work was supported by grants from the Réseau National des Génopoles (CNRG).
Footnotes
Reprint requests to: Herman van Tilbeurgh, Institut de Biochimie et de Biophysique Moléculaire et Cellulaire (CNRS-UMR 8619), Université Paris-Sud, Bâtiment 430, 91405 Orsay, France; e-mail: Herman.Van-Tilbeurgh@ibbmc.u-psud.fr; fax: +33-1-69-85-37-15.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062144406.
References
- Alifano P., Fani R., Lio P., Lazcano A., Bazzicalupo M., Carlomagno M.S., Bruni C.B. 1996. Histidine biosynthetic pathway and genes: Structure, regulation, and evolution Microbiol. Rev. 60 44–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfield M.J., Lott J.S., Arcus V.L., McCarthy A.A., Baker E.N. 2001. Structure of HisF, a histidine biosynthetic protein from Pyrobaculum aerophilum Acta Crystallogr. D Biol. Crystallogr. 57 1518–1525. [DOI] [PubMed] [Google Scholar]
- Barona-Gomez F. and Hodgson D.A. 2003. Occurrence of a putative ancient-like isomerase involved in histidine and tryptophan biosynthesis EMBO Rep. 4 296–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G., Vonrhein C., Flensburg C., Schiltz M., Paciorek W. 2003. Generation, representation and flow of phase information in structure determination: Recent developments in and around SHARP 2.0 Acta Crystallogr. D Biol. Crystallogr. 59 2023–2030. [DOI] [PubMed] [Google Scholar]
- Douangamath A., Walker M., Beismann-Driemeyer S., Vega-Fernandez M.C., Sterner R., Wilmanns M. 2002. Structural evidence for ammonia tunneling across the (βα)8barrel of the imidazole glycerol phosphate synthase bienzyme complex Structure (Camb.) 10 185–193. [DOI] [PubMed] [Google Scholar]
- Dumas C., Duquerroy S., Janin J. 1995. Phasing with mercury at 1 Å wavelength Acta Crystallogr. D Biol. Crystallogr. 51 814–818. [DOI] [PubMed] [Google Scholar]
- Fani R., Tamburini E., Mori E., Lazcano A., Lio P., Barberio C., Casalone E., Cavalieri D., Perito B., Polsinelli M. 1997. Paralogous histidine biosynthetic genes: Evolutionary analysis of the Saccharomyces cerevisiae HIS6 and HIS7 genes Gene 197 9–17. [DOI] [PubMed] [Google Scholar]
- Gordon E.J., Leonard G.A., McSweeney S., Zagalsky P.F. 2001. The C1 subunit of α-crustacyanin: The de novo phasing of the crystal structure of a 40 kDa homodimeric protein using the anomalous scattering from S atoms combined with direct methods Acta Crystallogr. D Biol. Crystallogr. 57 1230–1237. [DOI] [PubMed] [Google Scholar]
- Graille M., Quevillon-Cheruel S., Leulliot N., Zhou C.Z., de la Sierra Gallay I.L., Jacquamet L., Ferrer J.L., Liger D., Poupon A., Janin J.et al. 2004. Crystal structure of the YDR533c S. cerevisiae protein, a class II member of the Hsp31 family Structure (Camb.) 12 839–847. [DOI] [PubMed] [Google Scholar]
- Henn-Sax M., Thoma R., Schmidt S., Hennig M., Kirschner K., Sterner R. 2002. Two (βα)8-barrel enzymes of histidine and tryptophan biosynthesis have similar reaction mechanisms and common strategies for protecting their labile substrates Biochemistry 41 12032–12042. [DOI] [PubMed] [Google Scholar]
- Knowles J.R. 1991. Enzyme catalysis: Not different, just better Nature 350 121–124. [DOI] [PubMed] [Google Scholar]
- Lang D., Thoma R., Henn-Sax M., Sterner R., Wilmanns M. 2000. Structural evidence for evolution of the β/α barrel scaffold by gene duplication and fusion Science 289 1546–1550. [DOI] [PubMed] [Google Scholar]
- Lartigue A., Gruez A., Briand L., Blon F., Bezirard V., Walsh M., Pernollet J.C., Tegoni M., Cambillau C. 2004. Sulfur single-wavelength anomalous diffraction crystal structure of a pheromone-binding protein from the honeybee Apis mellifera L J. Biol. Chem. 279 4459–4464. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. 1993. PROCHECK: A program to check stereochemical quality of protein structures J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Leslie A. In Joint CCP4 and EACMB newsletter on protein crystallography. . 1992. Daresbury Laboratory, Warrington, UK.
- Leulliot N., Tresaugues L., Bremang M., Sorel I., Ulryck N., Graille M., Aboulfath I., Poupon A., Liger D., Quevillon-Cheruel S.et al. 2005. High-throughput crystal-optimization strategies in the South Paris Yeast Structural Genomics Project: One size fits all? Acta Crystallogr. D Biol. Crystallogr. 61 664–670. [DOI] [PubMed] [Google Scholar]
- Margolies M.N. and Goldberger R.F. 1967. Physical and chemical characterization of the isomerase of histidine biosynthesis in Salmonella typhimurium J. Biol. Chem. 242 256–264. [PubMed] [Google Scholar]
- Morris R.J., Perrakis A., Lamzin V.S. 2003. ARP/wARP and automatic interpretation of protein electron density maps Methods Enzymol. 374 229–244. [DOI] [PubMed] [Google Scholar]
- Murshudov G., Vagin A., Dodson E. 1997. Refinement of macromolecular structures by the maximum-likelihood method Acta Crystallogr. D Biol. Crystallogr. 53 240–255. [DOI] [PubMed] [Google Scholar]
- Quevillon-Cheruel S., Liger D., Leulliot N., Graille M., Poupon A., De la Sierra-Gallay I.L., Zhou C.Z., Collinet B., Janin J., Van Tilbeurgh H. 2004. The Paris-Sud yeast structural genomics pilot-project: From structure to function Biochimie 86 617–623. [DOI] [PubMed] [Google Scholar]
- Roussel A. and Cambillau C. In TURBO-FRODO, Silicon Graphics applications directory. . 1991. Silicon Graphics, Mountain View, CA.
- Schneider T.R. and Sheldrick G.M. 2002. Substructure solution with SHELXD Acta Crystallogr. D Biol. Crystallogr. 58 1772–1779. [DOI] [PubMed] [Google Scholar]
- Todd A.E., Orengo C.A., Thornton J.M. 2001. Evolution of function in protein superfamilies, from a structural perspective J. Mol. Biol. 307 1113–1143. [DOI] [PubMed] [Google Scholar]