

Abstract
Flaviviruses cause many human diseases, including dengue fever, yellow fever, West Nile viral encephalitis, and hemorrhagic fevers, and are transmitted to their vertebrate hosts by infected mosquitoes and ticks. Domain III of the envelope protein (E-D3) is considered to be the primary viral determinant involved in the virus–host-cell receptor interaction, and thus represents an excellent target for antiviral drug development. Langat (LGT) virus is a naturally attenuated BSL-2 TBE virus and is a model for the pathogenic BSL-3 and BSL-4 viruses in the serogroup. We have determined the solution structure of LGT-E-D3 using heteronuclear NMR spectroscopy. The backbone dynamics of LGT-E-D3 have been investigated using 15N relaxation measurements. A detailed analysis of the solution structure and dynamics of LGT-E-D3 suggests potential residues that could form a surface for molecular recognition, and thereby represent a target site for antiviral therapeutics design.
Keywords: flavivirus, Langat virus, E protein, E protein domain III, NMR spectroscopy, NMR relaxation, backbone dynamics, residual dipolar coupling, protein conformation, protein–protein interactions
Viruses of the genus Flavivirus are comprised of small, single-stranded, positive-sense RNA viruses, mainly transmitted by mosquito or tick vectors, and include several human pathogens such as the four dengue viruses (DEN 1–4), yellow fever (YF), Japanese encephalitis (JE), West Nile (WN), and tick-borne encephalitis (TBE) (Heinz et al. 1994; Gritsun et al. 1995). Several flaviviruses are also recognized as potential bioweapons of mass destruction, including members of the TBE serocomplex, such as Russian spring-summer encephalitis, Kyasanur forest disease, and Omsk hemorrhagic fever. The flavivirus genome is ∼11 kb in size and is directly translated by the host cell machinery as a single polyprotein after infection. The polyprotein undergoes co- and post-translational cleavage to generate three structural proteins: capsid (C), membrane (M), and envelope (E), and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (Rey et al. 1995). The major flavivirus immunogen is the membrane-associated E-glycoprotein, which is thought to play a role in virus–host-cell receptor interactions and membrane fusion (Bhardwaj et al. 2001). The E protein is ∼500 amino acids in length and exists as a heterodimer on the surface of the virion along with the second viral surface protein, the M protein (Yu et al. 2004). The three-dimensional (3D) structures of the TBE and dengue type-2 (DEN-2) viruses have been solved by X-ray crystallography (Rey et al. 1995; Modis et al. 2003, 2004). These structures have revealed that the E protein is divided into three distinct domains, I, II, and III. Domain I of the E protein is the central domain and contains ∼120 amino acid residues, distributed into three segments (residues 1–51, 137–189, and 285–302). The two long loops between these three segments form the dimerization domain, domain II, which is believed to be the principle region of interaction between monomers in E protein dimers. Domain III (E-D3), the putative receptor-binding domain, is ∼100 amino acids in length (it varies in different species) and has been identified as the potential site for interaction with the host cell. Mutation studies on both tick-borne and mosquito-borne flaviviruses have identified several key residues on domain III that have led to escape from antibody neutralization, thereby suggesting a role for domain III in forming the surface of the viral epitope (Rey et al. 1995; Roehrig et al. 1998; Mandl et al. 2000; Crill and Roehrig 2001; Hurrelbrink and McMinn 2001; Beasley and Barrett 2002; Lin and Wu 2003; Wu et al. 2003; Volk et al. 2004). This domain forms an Ig-like β-sandwich structure composed of six to seven antiparallel β-strands and is connected by a single flexible region to domain I in the center, which folds into an eight-stranded antiparallel β-barrel.
A recent crystal structure of the TBE E protein shows that a low-pH induced conformational change reorients the E protein from a horizontal, antiparallel dimeric conformation to a vertical trimer, resulting in a parallel arrangement of the subunits (Bressanelli et al. 2004; Stiasny et al. 2004). During this conformational transition, the three domains (I, II, and III) are maintained, but their relative orientation is significantly altered. In the low-pH form, the center of mass of E-D3 is displaced by 33 Å from the neutral form, and its β-strands run roughly parallel to the strands of domain I, whereas they were nearly orthogonal in the neutral form. However, the internal structure of domain III changes the least among the three domains in both conformations. The solution structures of domain III of the mosquito-borne West Nile and Japanese Encephalitis viruses have recently been solved by NMR spectroscopy (Wu et al. 2003; Volk et al. 2004). Mutations on the surface of domain III leading to escape from antibody neutralization have been reported for JE, WN, TBE, louping ill virus, yellow fever virus, and dengue virus (Wu et al. 2003).
In this study, we report a detailed analysis of the solution structure and backbone dynamics of domain III of Langat flavivirus E protein (LGT-E-D3) by NMR spectroscopy. A comparative analysis between the LGT-E-D3 solution structure and the crystal structure (M.A.White and R.O. Fox, in prep.) and other E-D3 structures is presented. Langat (LGT) flavivirus is a tick-borne, naturally attenuated BSL-2 virus and serves as a model for the BSL-3 and BSL-4 viruses of the TBE serogroup. LGT-E-D3 bears only 40% and 41% identity to the domain III of mosquito-borne JE and WNV flaviviruses, respectively, for which the solution structures have been determined by NMR spectroscopy. LGT-E-D3 exists as a monomer in solution and demonstrates vector-specific antagonistic activity against tick-borne flavivirus binding to host cell receptors (Bhardwaj et al. 2001). This article represents the first detailed investigation of both the solution structure and backbone dynamics of domain III of any flavivirus E protein. Using the 15N relaxation results, several key residues that may be involved in surface or ligand recognition are proposed.
Results and Discussion
NMR data, secondary structure, and the assignment of resonances
The assignments and the extent of assignments were previously reported (Mukherjee et al. 2004). Backbone and side chain resonances were assigned using a combination of HNCACB, CBCA(CO)NH, C(CO)NH, HC(CO)NH, and HBHACONH spectra (Sattler et al. 1999). Residues Phe(−1)–Gly(−6) (cloning artifact) do not exhibit medium- or long-range NOEs that would be consistent with a regular secondary or tertiary structure. In general, the NOE cross-peak patterns (Kline and Wuthrich 1986; Kline et al. 1986) and chemical shift indices (Wishart et al. 1992; Wishart and Sykes 1994) for the remaining residues are consistent with no less than six β-sheet segments in the protein sequence. None of the resonances for Pro376, which precedes Pro377 in the sequence, could be assigned, which precludes the direct assignment of the conformation of the X-Pro peptide bond for Pro376 and Pro377. However, all six remaining proline residues exhibited sequential Hα-Hδ/Hδ′ NOE consistent with a trans-X-Pro peptide configuration. The Pro376–Pro377 peptide bond is found in the trans configuration in the LGT-E-D3 crystal structure (M.A. White and R.O. Fox, in prep.) and in the related TBE E protein structure (Rey et al. 1995).
Structure calculation and analysis
A total of 1292 unambiguous restraints (576 intraresidue, 310 sequential, 91 short-range, 22 medium-range, and 300 long-range) were assigned manually using NMRView. Additional NOE restraints were obtained during structure calculations by ARIA/CNS, which uses an algorithm of iterative assignment of ambiguous restraints and simulated annealing structure calculations. The ambiguous NOE restraint assignments are performed automatically on the basis of the best structures obtained at each stage of the structure calculations. Among the 20 structures calculated at each level of iteration (except for the last iteration), the best seven were used to calibrate NOE peaks and assign ambiguous restraints. Five-hundred structures were calculated in the final (eighth) iteration, and the 20 lowest-energy structures were analyzed. At the end of the eighth iteration, 86 (57 unambiguous and 29 ambiguous) additional distance restraints were assigned by ARIA. Therefore, there were 1356 unambiguous restraints at the end of ARIA/CNS iterations, with 591 intraresidue, 318 sequential, 98 short-range, 23 medium-range, and 326 long-range NOEs. There were an additional 402 ambiguous restraints that went into the structure calculations, and 132 backbone torsional angle restraints (67 ϕ and 65 ψ) were used in all structure calculations. In addition, 66 hydrogen bond restraints identified from 2D 3J-HNCO (Cornilescu et al. 1999b), and on the basis of the NOE cross-peak pattern, were added at a later stage in the calculations. The subsequent addition of the hydrogen bond restraints did not lead to an increase in the target functions, indicating that the restraints used are consistent with other experimental restraints (NOE and dihedral restraints). Structure calculations were performed with and without residual dipolar coupling data (Fig. 1B,C). A new round of structure calculations was performed by adding 80 RDC restraints along with the molecular alignment tensors (Da and R) to the previous set of restraints consisting of 1758 NOE, 66 hydrogen bond, and 132 dihedral restraints. Structural statistics and analysis results are shown in Table 1.
Figure 1.

Calculated structure of LGT-E-D3. (A) Ribbon display of the lowest-energy NMR structure of LGT-E-D3 calculated with RDC restraints. (B) Superimposition of 20 lowest-energy backbone structures calculated without RDC and (C) with RDC restraints, respectively. (D) Ribbon plot showing the three different domains of TBE envelope (dimer) protein: (red) domain I, (yellow) domain II, and (blue) domain III.
Table 1.
NMR restraints and structural statistics for the best 20 structures

Description of the structure
The 3D structure of LGT-E domain III consists of six antiparallel β-strands forming a β-barrel-type structure, resembling the immunoglobulin constant domain (Fig. 1A). The six β-strands span the residues 313–320 (β1), 326–331(β2), 341–346 (β3), 371–375 (β4), 379–385 (β5), 388–394 (β6), and a conserved disulfide bridge exists between Cys307 and Cys338. Residues 338–339 (βx), 356–358 (βy), and 363–365(βz) formed short β-strands. Table 1 summarizes the energetic and structural statistics of the 20 lowest-energy NMR structures for LGT-E-D3. The structures exhibit no distance or dihedral restraint violations greater than 0.5 Å and 5°, respectively. Two sets of structure calculations were performed; residual dipolar coupling restraints (RDC, DNH) (Zheng et al. 2004) were included in one set of structure calculations (PDB accession code 2GG1) and excluded from the other (PDB accession code 1Z66). The structure ensemble calculated without incorporating residual dipolar coupling restraints has an average pairwise atomic RMSD of 0.62 ± 0.11 Å and 1.45 ± 0.18 Å for the backbone and heavy atoms of residues 300–395, respectively. The mean backbone and heavy atom RMSD of the ensemble with respect to the average structure was 0.43 ± 0.08 and 1.00 ± 0.14 Å, respectively. The quality of the ensemble of structures was analyzed using the program PROCHECK (Laskowski et al. 1996). Ramachandran analysis indicated that ∼98.7% of all non-glycine and non-proline residues are in the favored regions of the Ramachandran plot, with no residues in the disallowed region (Table 1). The structures calculated after incorporation of the residual dipolar coupling data had minor improvements in their backbone and heavy atom RMSD. The 20 lowest-energy structures calculated after incorporation of the RDC (DNH) values had an average pairwise atomic RMSD of 0.62 ± 0.11 Å and 1.49 ± 0.17 Å for the backbone and heavy atoms, respectively. The RMSD for residues 300–395 between the 20 RDC included lowest-energy structures, and the average structure was 0.43 ± 0.08 Å for the backbone atoms and 1.03 ± 0.11 Å for the heavy atoms (Fig. 1B,C).
These secondary structure elements were essentially identical in the two sets of structures calculated with and without the RDC values. The pairwise RMSD between the two sets of structures after structural alignment over the six β-strands was 0.51 ± 0.05 Å and 1.40 ± 0.10 Å for the backbone and heavy atoms, respectively. However, the pairwise RMSD over all residues in the two sets of structures was slightly higher, with values of, 0.75 ± 0.08 Å and 1.57 ± 0.11 Å for the backbone and heavy atoms, respectively. Therefore, no substantial structural improvement was observed upon incorporating the RDC (DNH) restraints. Some residues that have very low S2 values (see Fig. 3) are not likely to be accurately refined, because of motional averaging of the RDC (Deschamps et al. 2005). These residues with S2 ≤ 0.6 mostly belong to the N-terminal (300–303, 306), loop region between β1–β2 (323), β2–β3 (334, 335), β3–β4 (348, 355, 366–368), β5–β6 (386), and in the last strand (β6) (389–391). The RMSD of the 20 lowest-energy structures calculated without RDC restraints for the rigid core (removing the residues that have S2 ≤ 0.6) drops from 0.43 ± 0.08 to 0.37 ± 0.07. Similarly, the RMSD of 20 structures (lowest energy) calculated using the RDC restraints for the rigid core changes from 0.43 ± 0.08 to 0.38 ± 0.05. In addition, the DNH values predicted by the program PALES (Zweckstetter and Bax 2001a, b) using the coordinates generated without RDC constraints fit the experimental RDC data with Pearson correlation coefficient of 0.67 ± 0.04 between the calculated and observed RDC data. A significant improvement in the correlation was observed upon fitting the experimentally determined and calculated DNH values for the RDC-refined structures as reflected by the Pearson correlation coefficient (Rp = 0.993 ± 0.001) and the Cornilescu Q factor (Cornilescu et al. 1998) (Q = 0.096 ± 0.006), thereby indicating an excellent agreement of these structures with the observed RDC data (Fig. 2B,C). The correlation coefficient (Rp = 0.995 ± 0.001) and the quality factor (Q = 0.078 ± 0.005) upon fitting the experimentally determined and the calculated DNH values for the residues in the rigid core are further improved.
Figure 3.
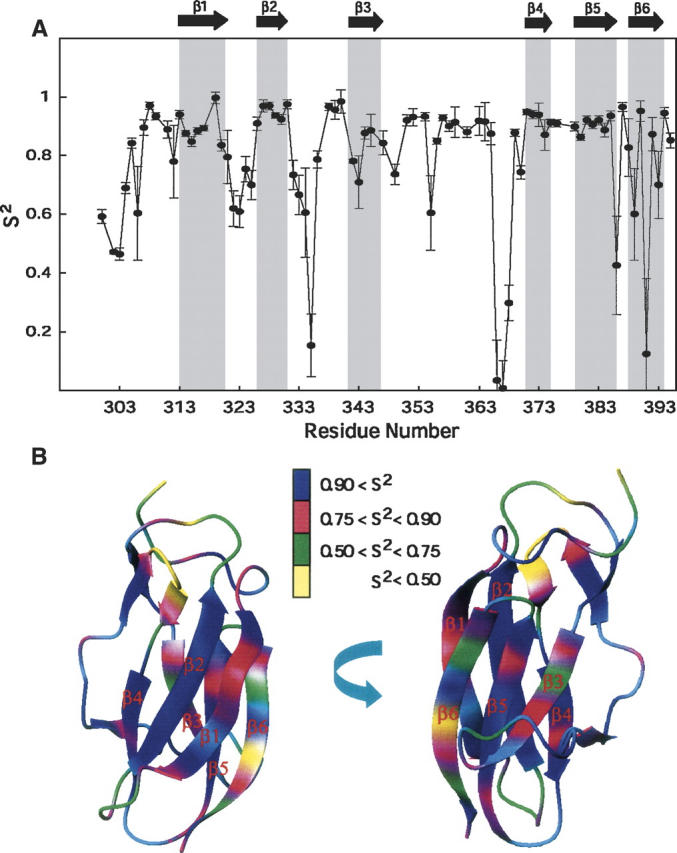
Generalized order parameters of LGT-E-D3. (A) Plot showing order parameter (S2) for LGT-E-D3. (B) S2 values painted on the surface of the lowest-energy structure of LGT-E-D3.
Figure 2.

Residual dipolar coupling analysis. (A) Histogram plot showing the distribution of the residual coupling values for LGT-E-D3 obtained from a partially aligning media (C8E5/n-octanol). (B–D) Correlation plot showing the fit between experimentally obtained RDC values and theoretically calculated RDC from the lowest energy NMR structure of LGT-E-D3 calculated without (B) and with (C) RDC using the PALES program. (D) Correlation plot of the experimental RDC and theoretical RDC values calculated from a monomer unit of the crystal structure of LGT-E-D3 using the PALES program.
Comparison of LGT-E-D3 NMR structure to its crystal structure
Although LGT-E-D3 exists as a monomer in solution (Bhardwaj et al. 2001), it forms flat pentamers in two different crystal forms under acidic conditions (pH 4.0) (M.A. White and R.O. Fox, in prep.). This structure is proposed as a model for the low-pH acid-triggered form of domain III near the virus icosahedral fivefold axis. In the X-ray structure, solved at 2.7 Å resolution, LGT-E-D3 assembles into pentameric rings that stack with 5/2 symmetry in an A-B-A-B repeat. The interface between the individual monomer units consists entirely of hydrophobic interactions, with no hydrogen bonds or salt bridges.
Although the solution structure of the LGT-E-D3 monomer is similar to the structure of each monomer unit of the LGT-E-D3 pentamer in the crystal forms, there are some important differences. Aligning the backbone atoms of all residues of one monomer unit of the LGT-E-D3 crystal structure to the RDC-refined NMR structures gives an RMSD of 2.15 Å and an RMSD of 2.32 Å for the RDC-devoid NMR structures. A closer inspection shows structural differences in the loop residues of the two aligned structures. Certain loop residues such as 321, 323, 347–349, and 367–369 are involved in packing interactions between adjacent monomer interfaces in the pentamer in the crystal structure, and may adopt a specific orientation to assume the pentamer contacts. Aligning the backbone atoms of the six major β-strands (excluding the loop residues) of the X-ray and lowest-energy NMR LGT-E-D3 monomer structures results in a significant drop in the RMSD value from 2.15 Å to 1.12 Å (for the RDC-refined NMR structure) and from 2.32 Å to 1.20 Å (for the non-RDC refined NMR structure). The experimentally measured DNH values plotted versus those calculated for one of the monomer units of the pentamer crystal structure are shown in Figure 2D. The overall quality of the fit, as indicated by the Pearson correlation coefficient (Rp) of 0.94 and Cornilescu Q factor of 0.29, indicate that the NMR structure provides a good description of the 3D structure of LGT-E-D3 in solution and agrees well with the crystal structure.
Structural comparison with sequentially similar flaviviruses
We compared the solution structure of LGT-E-D3 (determined using the RDC data) to the domain III structures of the E protein of both tick-borne (TBE) and mosquito-borne (JEV and WNV) flaviviruses. The amino acid sequence of LGT-E-D3 is 90.3% similar to that of domain III of TBE E protein (Rey et al. 1995). The strands β1, β2, β3, β4, β5, and β6 in LGT-E-D3 correspond to strands A, B, C, E, F, and G of the TBE domain III. The residues corresponding to the extra strands Ax, Cx, and D in TBE domain III appear as short β-strands (βx, βy, and βz, respectively) in the LGT-E-D3 NMR structural ensemble. Superposition of the backbone atoms of LGT-E-D3 NMR structure on the X-ray structure of TBE domain III gave an RMSD of 2.29 Å. As observed in the comparison of the NMR and X-ray structure of LGT-E-D3, the backbone alignment of TBE domain III and LGT-E-D3 NMR structure over the six major β-strands causes a sharp decrease in the RMSD from 2.29 Å to 1.19 Å. The loop residues 323–325 and 367–369 of domain III in TBE are involved in contacts (within a 5 Å distance) with residues of domain I, while residues 321 and 322 are proximal to domain II. The loop residues 358–360 are involved in both E-D1/E-D2 contacts in the TBE structure. These interactions may result in an altered and more ordered orientation of these loop residues in the TBE crystal structure than what is observed in the LGT-E-D3 solution structure. The amino acid sequence of LGT-E-D3 bears 40% and 41% identity (obtained from ClustalW) to the amino acid sequences of the domain III of the E proteins of the mosquito-borne JE and WN flaviviruses, respectively. The global fold and secondary structural elements (e.g., the positions of the six antiparallel β-strands of LGT-E-D3) are similar to that of JEV (1PJW) and WN (1S6N) domain III. The backbone RMSDs after structural alignment of the average structure of JEV and WN with LGT-E-D3 are 1.88 and 1.85, respectively (see Fig. 6B).
Figure 6.

Sequence and structural alignment of flaviviruses. (A) Sequence alignment of domain III of the five flaviviruses (TBE, LI, JE, WN, and DEN-2) against LGT-E-D3. Mutations that have led to escape from antibody neutralization for other tick-borne (TBE and LI), mosquito-borne (WN, JE, and DEN-2) viruses are color-coded. (B) Shown in black on the ribbon plots of TBE (yellow, 1SVB), JE (red, 1PJW), West Nile (navy 1S6N), and DEN-2 (cyan, 1OAN) after structural alignment with domain III of LGT-E-D3 (blue).
NMR relaxation data: Backbone dynamics and biological implications
To better understand the influence of the internal dynamics of LGT-E-D3 on its biological function, backbone 15N relaxation data including longitudinal (R1) and transverse (R2) relaxation rates were recorded on 600 and 750 MHz spectrometers; and heteronuclear 15N–1H NOE was recorded at 750-MHz field strengths, respectively. Supplemental Figure S1 shows the R1, R2, and 15N–1H NOE data at two different field strengths (the data are provided in Supplemental Table S1). The average values of R1 at 750 MHz and 600 MHz are 1.79 ± 0.28 sec−1 (10% trimmed mean 1.77 sec−1) and 2.01 ± 0.24 sec−1 (10% trimmed mean 1.99 sec−1), respectively. The average R2 values at 750 MHz and 600 MHz were 9.10 ± 1.3 sec−1 (10% trimmed mean 9.15 sec−1) and 8.39 ± 1.14 sec−1 (10% trimmed mean 8.42 sec−1), respectively, and the average 15N–1H NOE at 750 MHz was 0.74 ± 0.15 (10% trimmed mean 0.76). Values of 15N–1H NOE exceeding 0.6 indicate that LGT-E-D3 is well-ordered in solution, except for the extreme N terminus (300–305) and certain loop residues (e.g., residue 368). An initial estimate of the rotational diffusion tensors was obtained after eliminating the residues with the R2 component having chemical exchange contributions (see Materials and Methods). The initial rotational diffusion model that best fits with the NMR data was an axially symmetric model with a (D||/D⊥) ratio of 1.17 ± 0.09 and an overall correlation time (τc) of 5.4 ± 0.40 nsec. These values were optimized to fit the complete set of relaxation data (R1, R2, and NOE at 750 MHz and R1 and R2 at 600 MHz) using the model free formalism approach (Lipari and Szabo 1982a,b; Fushman et al. 1998). The final anisotropy of the diffusion tensor and the average correlation time were found to be 1.2 (D||/D⊥) and τc = 5.75 nsec, respectively. Model selection for each residue was determined using the protocol of Mandel et al. (1995, 1996). Four residues (310, 346, 348, and 353) could not be fitted to any model.
NMR relaxation rates R1 and R2 and 15N–1H NOE values of the backbone amide 15N nucleus are influenced by dynamics on the picosecond-to-nanosecond time scale. In addition, the R2 rates are influenced by chemical exchange on the microsecond-to-millisecond time scale. Thus analysis of the R1, R2, and 15N–1H NOE provides information on the dynamics on both the picosecond-to-nanosecond as well as the microsecond-to-millisecond time scales. The generalized order parameter (S2) models the motional restriction of the amide N–H-bond vector on the picosecond-to-nanosecond time scale (Lipari and Szabo 1982a,b). As shown in Figure 3, A and B, the residues with high S2 values (average S2 > 0.8) mainly cluster around regions of well-defined secondary structure such as the β-strands, while the high internal mobility of the loop residues is indicated by their low average S2 values (<0.7). Strands β1 through β5 display average S2 values >0.8, while lower average S2 values (0.74 ± 0.27) are observed for the residues of the last β-strand, β6 (388–395). Residues 388–395 of β6 were fitted to models that included conformational exchange contributions (Rex), suggesting slow conformational exchange on the microsecond-to-millisecond time scale. Analysis of the solvent-accessible surface area (calculated using MOLMOL) (Koradi et al. 1996) for residues 388–395 in the β6 strand illustrates that the residues having solvent-exposed amide protons (389, 391, 393, and 395) have low S2 values, while alternate residues having buried amide protons (388, 390, 392, and 394) have high S2 values (Zhang and Bruschweiler 2002). The short β-strands constituted by residues 338–339 and 356–358 also exhibit high average S2 values of 0.96 ± 0.01 and 0.89 ± 0.04, respectively, thereby indicating restricted motion. The loop residues between β1 and β2 (321–325) and between β2 and β3 (332–340) exhibit low average S2 values of 0.69 ± 0.08 and 0.55 ± 0.28, respectively. A sharp drop in the S2 value for residue 335 is complemented by a large exchange term (Rex = 2.6 sec−1), indicating significant conformational exchange on the microsecond-to-millisecond time scale. The loop residues (from βy to β4) 359–370 have an average S2 value of 0.65 ± 0.38 but can be divided into two distinct regions. Within this loop, the residues 359–365 and 369–370 are flanked by regular secondary structure elements such as βy and β4, respectively, and show high average order parameters of 0.90 ± 0.02 and 0.81 ± 0.09, respectively. A large drop in S2 is observed for the more flexible region of the loop comprised of residues 366–368 (0.04, 0,01, and 0.30; average 0.11), which also show very large conformational exchange rates.
While S2 provides a measure of the rapid internal motions relative to the overall rotational diffusion, large chemical-exchange contributions (Rex) identify residues that experience conformational (or chemical) change on a microsecond-to-millisecond time scale. It has been observed that important biochemical events such as enzyme turnover rates, ligand binding and dissociation rates, and rate of conformational change often occur near the time scale of lower frequency motions (microsecond-to-millisecond). Therefore, it is possible that these slower motions observed reflect their functional activity (Epstein et al. 1995; Kristensen et al. 2000; McCoy et al. 2001; Dutta et al. 2004; Palmer 2004). Upon fitting the relaxation data to the Lipari-Szabo formalism, 22 residues were observed to show significant RLSex (>1.5 sec−1) (Camarero et al. 2001) (Fig. 4A). The residues undergoing chemical exchange were also independently determined by comparison of transverse relaxation rates and the cross-correlation rates (ηxy) between the 15N CSA and the 15N–1H dipolar relaxation (Kroenke et al. 1998). Using the above criteria of R′ex > 1.5 sec−1, 21 residues were identified as having microsecond-to-millisecond time-scale motion (Fig. 4B). The effect of site-specific variation of 15N CSA is neglected for the range of significant R′ex contributions (Fushman et al. 1998; Kroenke et al. 1998; Camarero et al. 2001). Note that 11 out of 21 residues showed significant RLSex in the Lipari-Szabo analysis. In addition, motions on the 1–10-msec time scale were also probed by a direct experiment; the relaxation compensated CPMG, using τcp values of 1 msec and 10 msec. Those residues having Rav2 (Rτcp = 10ms2,av − Rτcp = 1ms2,av) values >2 sec−1 were considered to have slow motion on the 1–10-msec time scale (Loria et al. 1999a,b; Ghose et al. 2001). As shown in Figure 4C, residues 347, 348, and 367, which are part of the loop, exhibit motions on the 1–10-msec time scale. Motion on the sub-millisecond time scale was also investigated using the R2/R1ρ ratio as described by Dutta et al. (2004), but we found no residues that undergo motion on this time scale (data not shown). Overall, there were 32 residues that show apparent motion in the microsecond-to-millisecond time scale (Rex) obtained from RLSex and R′ex analysis. These residues are colored green on the ribbon plot of LGT-E-D3 in Figure 5A. Sequence alignment (see Fig. 6A) of LGT-E-D3 and TBE-D3 demonstrates that the loop residues 321–325 (between strands β1 and β2), 359 (between βy and βz), and 366–370 (between strands βz and β4), which show a large Rex term in LGT-E-D3, are also involved in either E-D2/E-D3 or E-D1/E-D3 contacts (within 5 Å) in the corresponding TBE E protein crystal structure at neutral pH (see Fig. 5B). LGT-E-D3 and TBE-E-D3 have 90% sequence similarity and a 0.5 Å RMSD between the crystal structures of their E protein domain IIIs. Residues 323 and 325, which are conserved in both tick-borne and mosquito-borne homologs, and residues 322 and 367–369 conserved within the tick-borne flaviviruses exhibit Rex values and are also involved in packing interactions between adjacent monomers in the pentamer of LGT-E-D3 crystal structures (M.A. White and R.O. Fox, in prep.) (see Figure 5C). Residues at the N terminus (300, 303, and 306) and residues following β-strand C (351–353) in TBE-E-D3 (β3 in LGT-E-D3) are proximal (within 5 Å) to either domain I of the same or adjacent monomer unit in the crystal structure of the low-pH trimeric form of TBE, and also display a large Rex term in the LGT-E-D3 solution structure. Thus, ∼44% of the residues that undergo conformational exchange are involved in either E-D1/E-D3 or E-D2/E-D3 interactions at high and/or low pH. These results are illustrated in Figure 5.
Figure 4.
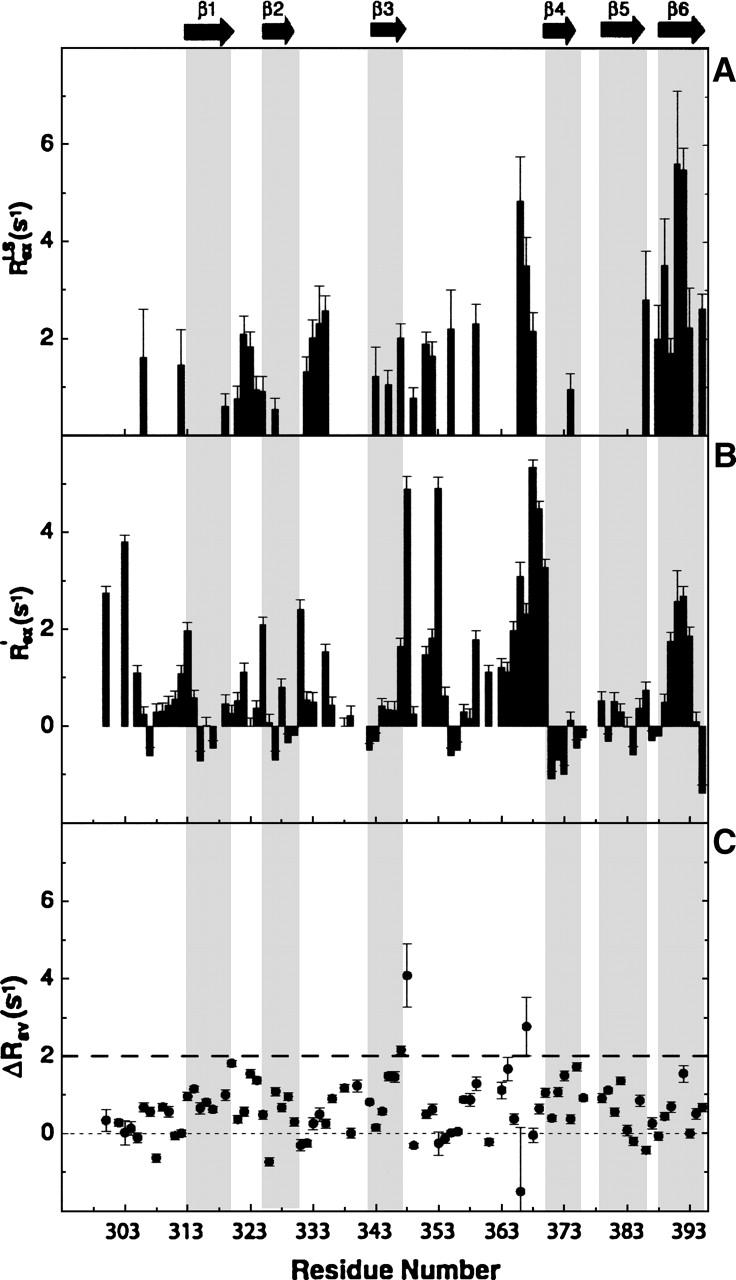
Residues showing conformational exchange. (A) Residues that show Rex value in the Lipari-Szabo analysis, and (B) those obtained from cross-correlation relaxation rates of 15N CSA and 15N–1H dipolar interactions are shown. (C) Rav2 values calculated from the relaxation compensated CPMG experiment.
Figure 5.

Plot showing slow motion. (A) Residues showing Rex value (>1.5 sec−1) obtained from RLSex and R′ex analysis are painted (as green) on the ribbon plot of the lowest-energy LGT-E-D3 NMR structure. Residues showing Rex value (B) that do not make any contact with E-D1 and E-D2 in TBE are green and those in contact with E-D1 (magenta) and E-D2 (blue) domains in TBE are yellow on the surface plot of the lowest-energy structure of LGT-E-D3 (SWISS-PROT). Contact regions were determined from the crystal structure of TBE, which bears 90% similarity to LGT (C), which are in contact with two adjacent monomer units (magenta and blue) of the LGT-E-D3 pentamer crystal structure are yellow and which do not show any contact are green. (D) Surface plot of LGT-E-D3 showing residues that show Rex in LGT-E-D3 NMR analysis (green) and residues for which mutation studies have been done (red) in TBE, LI, JE, WN, and DEN-2 and residues for which mutational studies have been done and also happen to show slow dynamics (yellow).
Relationship of apparent slow conformational fluctuations and mutational effects on infectivity
Mutations of E protein domain III that caused escape from antibody neutralization or neutralizing epitopes mapped by phage display peptide libraries have been reported for residues 310, 384, and 386 for TBE (Rey et al. 1995; Mandl et al. 2000); residues 308, 310, and 311 for louping ill virus (LI) (Wu et al. 2003); residues 307, 312, 330, 332, and 369 for WN (Beasley and Barrett 2002; Wu et al. 2003; Volk et al. 2004); residues 306, 331–333, 337, 360, and 386–390 for JE (Lin and Wu 2003; Wu et al. 2003); and residues 307, 333–351, and 383–389 for DEN-2 (Roehrig et al. 1998; Crill and Roehrig 2001; Modis et al. 2003). Figure 6A shows the sequence alignment of domain III of the five flaviviruses such as TBE, LI, JE, WN, and DEN-2 with domain III of Langat. All the mutated residues that led to escape from antibody neutralization are color-coded. On the LGT-E-D3 sequence, residues with significant microsecond-to-millisecond mobility are color-coded in green. Note that ∼34% of the residues showing slow motions were residues that when mutated led to escape from antibody neutralization. These mutations were predominantly at the distal face consisting of β3, β5, β6, and the associated loops of domain III (Fig. 6B).
A loop consisting of five residues exists between the last two β-strands, β5 and β6 in the aligned mosquito-borne flaviviruses (JE, WN, and DEN-2), but is confined to only one residue (G386) in the tick-borne flaviviruses (LGT, TBE, and LI). Escape from antibody neutralization is observed upon mutating these loop residues in JE (residues 386–390) and DEN-2 (residues 383–385) (Hiramatsu et al. 1996; Roehrig et al. 1998) and also G386 in TBE (Mandl et al. 2000). A high Rex term observed for G386 in LGT-E-D3 indicates that this residue experiences conformational exchange in the microsecond-to-millisecond time scale. A structural alignment of domain III of TBE, WN, and JE on LGT-E-D3 is shown in Figure 6B, and the mutated residues are displayed. Figure 5D displays the residues that exhibit conformational exchange in the solution structure of LGT-E-D3 (show Rex > 1.5 sec−1 values in 15N relaxation analysis) and also the residues that have been shown by mutagenesis experiments to allow escape from antibody neutralization. Analysis of Figures 5B,D and 6 clearly indicates that ∼78% of the residues that display conformational exchange on the microsecond-to-millisecond time scale are either involved in surface recognition in a homologous protein (TBE) or in the LGT-E-D3 pentamer crystal structure, or form viral epitopes for antibody recognition. In addition to the coincidence of loop flexibility and escape from antibody neutralization, significant microsecond-to-millisecond motion is seen in the β6 strand and the loop residues 347 and 348 that precede strand β3, constituting the remaining ∼22% of the residues that showed slow motion. These regions are proximal to each other and form a continuous patch in the LGT-E-D3 structure (see Fig. 5D). These residues neither are involved in contacts with E-D1 or E-D2 in the TBE crystal structure (both neutral and low-pH forms) nor have been proposed for antibody recognition by escape mutation studies on other flaviviruses (Fig. 5B,D). In DEN-2, two mutations in this region (F392Y and K393R) reduced neurovirulence for mice although it remained antigenically reactive (Hiramatsu et al. 1996). In the LGT-E-D3 pentamer crystal structure, these residues of the strand β6 and residues 347 and 348 of the proximal loop exhibit packing interactions with residues 316 and 321 in strand β1, and residues 327–329 in strand β2 of the adjacent monomer unit (M.A. White and R.O. Fox, in prep.) (Fig. 5C). In Figure 5D, the apparent surface of β6 and the residues from the proximal loop are separate from the N-terminal antigenic surface and exhibit significant conformational exchange on the microsecond-to-millisecond time scale, which may be related to the specific target for ligand or receptor binding (McCoy et al. 2001; Dutta et al. 2004). The need for these residues to interact either with ligand or receptor may explain the involvement of these residues (347, 348, and strand β6) in packing interactions at the pentamer interface of the LGT-E-D3 pentamer crystal structure. Mutation studies to monitor escape from antibody neutralization performed on residues 388–395 of strand β6 and the loop residues 347 and 348, might establish the importance of this surface as a potential viral epitope for antibody recognition in tick-borne flaviviruses. Since the E-D3s of TBE and LGT have 90% sequence identity therefore in the full-length E protein of Langat at high pH, this surface formed by residues 347, 348, and 388–395 could be available for ligand binding and/or antibody recognition and thus represent a target for potential antiviral therapeutics design. This suggestion is a qualitative hypothesis based on the observation of apparent slow motion and sequence comparison analysis. The underlying biophysical details of the interrelationship of apparent slow motion, surface accessibility, and antigenicity remain to be elucidated.
Conclusions
NMR structural and 15N dynamics studies of LGT-E-D3 were performed to characterize ligand recognition sites and understand ligand recognition properties of domain III of flavivirus E proteins from a structural perspective. Loop residues 347 and 348 and residues 388–395 of strand β6 are proximal in the 3D structure of LGT-E-D3 and form a continuous surface that shows significant conformational exchange in the slow time scale (microsecond-to-millisecond) (Rex values) in the 15N relaxation analysis. All other residues showing Rex values identify regions on the domain that are either involved in domain–domain interactions in a homologous protein such as TBE or form antibody binding sites as demonstrated by mutagenesis experiments. Therefore, loop residues 347 and 348 and residues 388–395 of strand β6 define an important region on the domain critical for molecular recognition and may form a potential viral epitope for antibody recognition and/or an as-yet-unidentified site for some other macromolecular interaction. Our results provide a structural-dynamic basis for understanding flavivirus:ligand interactions.
Materials and methods
Overexpression and purification of LGT-E-D3
Domain III of Langat flavivirus E protein (LGT-E-D3) was expressed in E. coli strain BL21(DE3) Codon Plus (Stratagene) as a C-terminal fusion to an N-terminal six histidine-MBP (maltose-binding protein) tag with a thrombin cleavage site, using the plasmid H-MBP-T, which encodes residues 300–395 of LGT-E-D3 (accession no. P29837). Details of the H-MBP-T vector and purification procedure are reported elsewhere (Alexandrov et al. 2001; Mukherjee et al. 2004). For NMR resonance assignment and structure calculations, [U-15N] (∼0.8 mM) and [U-13C-15N] samples (∼0.7 mM) were prepared in 10 mM Tris, 20 mM Bis-Tris, and 10 mM NaCl (pH 6.2).
NMR spectroscopy and data processing
NMR experiments were recorded at 25°C using Varian Inova 600 and 750 MHz spectrometers, or a Bruker Avance 500, 600, 700, and 800 MHz spectrometer (equipped with CryoProbes). All spectrometers were equipped with triple resonance probes. Spectral data were processed using NMRpipe, viewed by NMRDraw (Delaglio et al. 1995), and analyzed using NMRView (Johnson 2004). The 1H chemical shifts were referenced to water at 4.75 ppm (at 25°C), and the 13C and 15N chemical shifts were referenced indirectly using the γ13C/γ1H and γ15N/γ1H ratios, respectively. The backbone sequential assignments (Mukherjee et al. 2004) were done using 1H–15N-HSQC, 1H–13C-HSQC, 3D 15N-NOESY-HSQC, HNCO, HNCACB, CBCA(CO)NH experiments (Sattler et al. 1999). The side chain carbon and proton assignments were obtained using C(CO)NH, HC(CO)NH, HBHA(CO)NH, and HCCH-TOCSY experiments. The aromatic resonance assignments were made by analyzing 1H–13C HSQC, 3D 15N TOCSY-HSQC, and 3D 15N NOESY-HSQC data. 3JHNH coupling constants were extracted from 3D-HNHA experiments from the ratio of the diagonal and cross-peak intensities (Kuboniwa et al. 1994). The three bond HN–Hα coupling constants were used to extract the backbone ϕ angles using the Karplus equation (Karplus 1959).
The one bond NH–HN residual dipolar coupling (RDC, DNH) values were measured using the 2D IPAP (in-phase/anti-phase) 1H–15N HSQC (Ottiger et al. 1998a,b) experiment. The NH–HN splittings were obtained for partially aligned LGT-E-D3 protein (0.5 mM) both in a nonionic liquid crystalline medium (Ruckert and Otting 2000) of C8E5/n-octanol mixed in the NMR sample buffer and in nonaligned media (sample mixed in NMR buffer). The molar ratio of C8E5 to n-octanol was 0.87, and the C8E5/water ratio was 6% (w/v). Residual dipolar coupling values (DNH) were calculated by subtracting 1JNH scalar couplings obtained from the nonaligned media from 1JNH + DNH couplings obtained from the partially aligned media. The distribution of the residual dipolar couplings are plotted as a histogram in Figure 2A. The molecular alignment tensors, the axial component (Da), and the Rhombicity (R), were obtained by minimizing the difference between the experimentally measured DNH and those calculated from the monomer unit of the crystal structure using the program PALES (Zweckstetter and Bax 2000; Deep et al. 2003). The alignment tensors were also calculated using the methodology given by Clore et al. (1998) and were in good agreement with those calculated above. DNH values were also calculated from samples partially aligned in 8% polyacrylamide gels. The correlation coefficient of the DNH values obtained from two different media, PEG/Octanol and polyacrylamide gel, was 80%.
Distance, dihedral angle, and orientation restraints
The NOE-derived distance restraints were obtained from 3D 13C-edited NOESY (150 msec mixing time) and 3D 15N-edited NOESY (150 msec mixing time). NOESY cross-peak volumes/intensities were obtained using NMRView and converted into distance restraints using the symmetry ADR (ambiguous distance restraints) protocol within the ARIA program (Linge et al. 2003; Habeck et al. 2004), which accounts for the ambiguity in the NOEs arising from signal overlap and symmetry degeneracy. The ambiguous distance restraint protocol uses chemical shifts to characterize restraints. Molecular conformations are calculated from a list of ambiguous distance restraints and subsequently used to filter the assignment possibilities. The resulting iterative protocol converged to a structure ensemble with consistent NOE assignments. The NOE assignment was performed in eight iterations using the ARIA scheme, where structures generated in the previous iteration are used for the NOE analysis (calibration, partial assignment, and noise removal) of the next. In the final iteration, 20 structures with the lowest energy were further refined by molecular dynamics in explicit solvent. Structures were calculated by ARIA/CNS using a Cartesian dynamics simulated annealing protocol for NMR restrained structure refinement: (1) a high-temperature dynamics at 2000 K (10,000 steps); (2) a Cartesian dynamics cooling stage from 2000 to 1000 K (6000 steps); and (3) a second Cartesian dynamics cooling stage from 1000 K to 50 K (4000 steps). Prochiral valine and leucine methyl groups were treated using the floating chirality method (Folmer et al. 1997). The initial structure calculation runs using the ARIA/CNS protocol were done using the extended structure, whereas the subsequent structure calculations were done starting from a lowest-energy structure with good local geometry and minimum violations obtained from the previous run. The resulting NMR structures were evaluated with MOLMOL and PROCHECK (Koradi et al. 1996; Laskowski et al. 1996) (Table 1).
Backbone dihedral angle restraints (ϕ and ψ) were determined from the 3JHNH coupling constants extracted from HNHA experiment (ϕ) and also calculated by the analysis of 13Cα, 13Cβ, 13C′, and 15N chemical shifts using the TALOS program (Cornilescu et al. 1999a). TALOS predicts the backbone torsion angle intervals from the amino acid sequence and chemical shift information.
The hydrogen bond restraints, for which inter-β-strand NOEs were also observed, were extracted from the 2D 3J-HNCO experiment and added as explicit constraints during later stages of structure calculations. The hydrogen bond restraints were defined as 1.8–2.3 Å for the NH–O bond and 2.8–3.3 Å for the N–O bond.
Backbone 15N relaxation measurements and analysis
A complete set of backbone amide R1, R2 and 15N-{1H} NOE relaxation data sets were acquired on the Varian Inova 750 MHz spectrometer, and an additional set of R1 and R2 measurements was obtained on the Varian Unity 600 MHz spectrometer for LGT-E-D3 using 1H-detected pulse sequences as previously described (Kroenke et al. 1998). The R1 and R2 data sets were each collected using 16 scans and a recycle delay of 1.5 sec. For the R2 measurements, the following relaxation delays: 10, 30 (×2), 50, 70, 90, 110, 130, 150, and 170 msec, were used on the 600 MHz and on the 750 MHz spectrometers, respectively. The R1 measurements were obtained by acquiring the data using the following variable relaxation delays: 10, 30, 50 (×2), 90, 130, 230, 350, 470, 630, and 750 msec, on both the 600 MHz and 750 MHz spectrometers. R1ρ values for LGT-E-D3 were obtained at 600 MHz using a recycle delay of 1.5 sec and relaxation delays of 10, 30 (×2), 50, 70, 90, 110, 130, 150, and 170 msec. To obtain sample heating effects similar to the R2 measurements, a spin lock field (ΔωSL) of 1500 Hz was used that would allow an accurate comparison between the two rates. The measured Rapp1ρ rates were corrected for off-resonance effects by using the following equation:
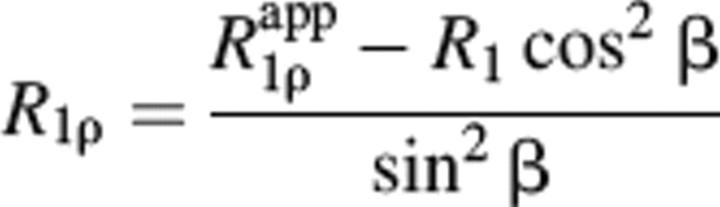 |
where β = tan−1 (ωSL/ΔωSL) and Δω is the angular frequency offset from the radiofrequency field carrier.
Relaxation-compensated CPMG experiments (RC-CPMG) (Loria et al. 1999a,b) were performed to determine the average R2 value:
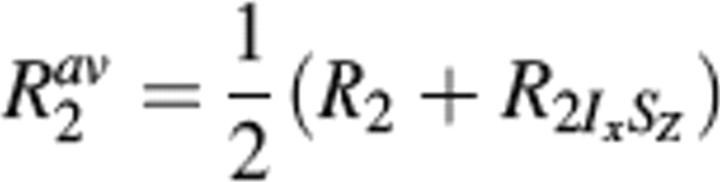 |
with τCP = 1 msec and 10 msec, where τCP is the delay between the 180° pulses of the CPMG cycle. The relaxation delays used were 0 (×2), 8, 16, 32, 48, 80 (×2), 112, 160 msec for τCP = 1 msec and 0 (×2), 40, 80, 120 (×2), 160, 200 msec for τCP = 10 msec. Recycle delays of 1.5 sec were used in all cases. The relaxation rates R1, R2, R1ρ, and Rav2 were determined by fitting the experimental data to a single exponential function given by I(t) = I0e-Rt, where R = R1, R2, R1ρ, and Rav2 using the CURVEFIT program (A.G. Palmer III, Columbia University).
To identify residues that made significant contributions of chemical exchange to the transverse relaxation rates, we used the methodology proposed by Kroenke et al. (1998), in which the chemical exchange contribution to transverse relaxation is given by:
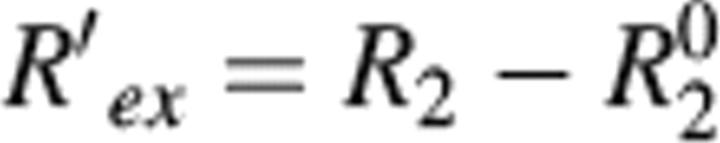 |
where R2 is measured by CPMG. R02 can be obtained by measuring the cross-correlation rates between 15N CSA and 15N–1H dipolar relaxation (CSA/DD) ηxy (Fushman et al. 1998), which is related by R02 = κηxy where κ is independent of chemical exchange and local motions (Wang et al. 2003). The cross-correlation rates (ηxy) were measured using the symmetrical re-conversion method developed by Pelupessy et al. (2003), and κ was determined iteratively as an average of <R02/ηxy= for all residues that were not subjected to chemical exchange processes, assuming that the 15N CSA is constant for all residues (Wang et al. 2001). The average value of κ at 600 MHz was found to be 1.61, and was also independently obtained using another protein.
The steady-state 15N–{1H} NOE relaxation data sets were recorded using 48 scans per point, and by recording two spectra with and without a 3.0-sec period of proton presaturation (Farrow et al. 1994). The error (σNOE) was determined using the following equation:
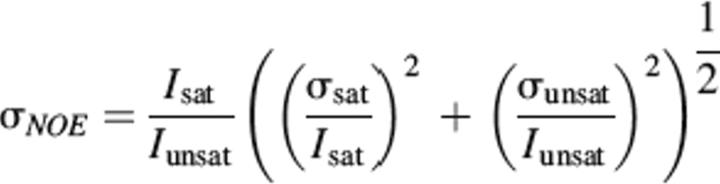 |
where, Isat and Iunsat represent the measured intensities of a particular resonance in the presence and absence of proton saturation, and σsat and σunsat represent the RMS variation in the noise in empty spectral regions of the spectra with and without proton saturation.
The internal dynamics of LGT-E-D3 were assessed by analyzing the experimental 15N relaxation parameters using the model-free formalism, and by assuming that the protein tumbles as an axially symmetric rotor. In the formalism of Lipari and Szabo, the correlation function is given by:
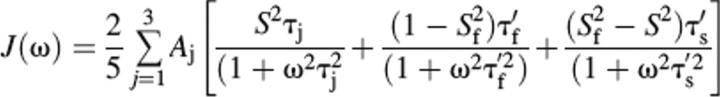 |
where S2 = S2fMS2s is the generalized order parameter, Sf and Ss are order parameters on the fast and slow time scales (Clore et al. 1990), and τf and τs are the two internal motions; τs′ = τsτc/(τs + τc); τf′ = τfτc/(τf + τc); τ1 = 1/(6D⊥), τ2 = 1/(5D|| + D⊥), and τ3 = 1/(4D|| + 5D⊥); τ′ = τj τe/(τj + τe); A1 = (3 cos2 θ − 1)2/4, A2 = 3 sin2 θ cos2 θ, A3 = ¾ sin4 θ, and θ is the angle between the N–H-bond vector and the unique axis of the diffusion tensor (Tjandra et al. 1995; Fushman and Cowburn 1999).
Rotational diffusion tensor
An initial estimation of the overall tumbling correlation time (τc) and the rotational diffusion tensors was obtained from the subset of residues with (1) low amplitude internal motion and (2) no conformational exchange. The two selection criteria used were (I) all residues having NOE ≥0.7 and (II) those residues that satisfied the following equation:
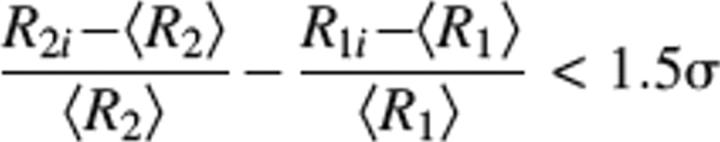 |
where <Rj> and Rji (where j = 1,2 and i is the i-th residue) are the average rates, and R1 and R2 are the individual rates of the subset of remaining residues satisfying criteria I. σ is the standard deviation of
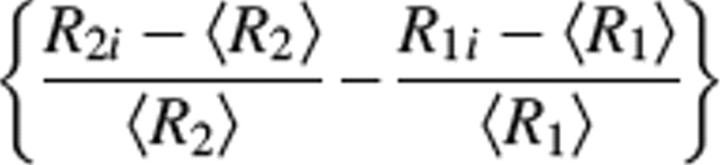 |
Thus, a total of 60 residues were used to estimate the overall correlation time and rotational diffusion tensors. Note that the lowest energy structure calculated with the RDC restraints was used for the estimation of the overall correlation time and rotational diffusion tensors. The F-test analysis was performed to choose between isotropic, axially symmetric, and fully anisotropic diffusion models. A probability factor (P%), which indicates the probability of improvement in fits when model complexity increases is coincidental, was calculated for an isotropic axially symmetric pair of models and for a fully anisotropic axially symmetric pair of models. Probability factors >5% are not statistically significant.
Relaxation data (R1, R2 at 750 MHz and R1 and R2 at 600 MHz) and the 1H–15N NOE at 750 MHz, along with the N–H vectors obtained from the lowest energy structure calculated using the RDC restraints were used to obtained the motional parameters using the program DYNAMICS (Fushman et al. 1997). These data were fitted into six models, and the selection criteria for each model were based on those of Mandel et al. (1995). The six models are: S2 (model 1); S2, τe (model 2); and S2f,S2s, τe (model 5); whereas models 3, 4, and 6 were obtained by introducing the conformational exchange contribution: S2, Rex (model 3); S2, τe, Rex (model 4); and S2f,S2s, τe, τe, Rex (model 6). The model-free parameters were optimized by minimizing χ2, defined as (Fushman et al. 1997):
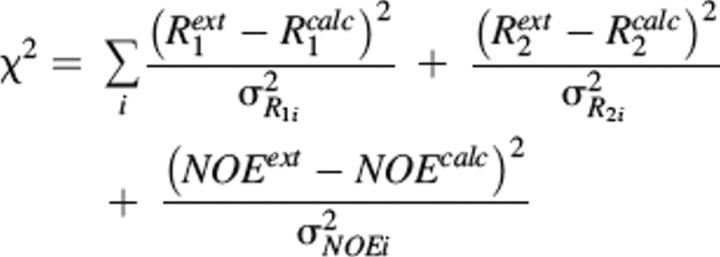 |
where calc and ext represent the calculated and experimentally determined parameters, respectively, and σR1, σR2, and σNOE are experimentally determined uncertainties.
Coordinate and data deposition
The 1H, 15N, and 13C chemical shift assignments have been deposited in the BioMagRes data bank (accession code 6800). The atomic coordinates for an ensemble of 20 structures that represent the solution structure of LGT-E-D3 have been deposited in the Protein Data Bank together with the list of restraints used for the structure calculation under accession numbers 2GG1 (including RDC restraints) and 1Z66 (excluding RDC restraints).
Electronic supplemental material
R1, R2, and NOE values and their uncertainties measured for LGT-E-D3 at 750 MHz and 600 MHz are shown in Supplemental Figure S1 and also given in Supplemental Table S1. Supplemental Table S2 shows the Lipari-Szabo model-free parameters obtained after fitting the R1, R2, and NOE data.
Acknowledgments
This work was supported by NIH grants (AI 056326 to R.O.F. and GM 47021 to D.C.), and a grant from the Robert A. Welch Foundation (H-1345 to R.O.F.). We thank the Sealy & Smith Foundation for support of the NMR Center at UTMB. Shanmin Zhang maintained the NMR spectrometers at University of Texas Medical Branch at Galveston, and we thank him for helpful advice. NMR resources at NYSBC are supported by NIH P41 GM66354 and NYSTAR. K.D. thanks Ranajeet Ghose, Fabien Ferrage, Michael Goger, and Felician Dancea for their invaluable suggestions and help. We thank David Konkel for editing the manuscript. We thank Allan Barrett for providing a clone containing the Langat E protein domain III gene and for helpful discussions. We thank Arthur Palmer for the CURVEFIT program.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Robert O. Fox, Department of Biochemistry and Molecular Biology, University of Texas Medical Branch, 301 University Blvd., Mail Route 0647, Galveston, TX 77555-0647, USA; e-mail: fox@bloch.utmb.edu; fax: (409) 747-4745.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051844006.
References
- Alexandrov A., Dutta K., Pascal S.M. 2001. MBP fusion protein with a viral protease cleavage site: One-step cleavage/purification of insoluble proteins Biotechniques 30 1194–1198. [DOI] [PubMed] [Google Scholar]
- Beasley D.W. and Barrett A.D. 2002. Identification of neutralizing epitopes within structural domain III of the West Nile virus envelope protein J. Virol. 76 13097–13100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj S., Holbrook M., Shope R.E., Barrett A.D., Watowich S.J. 2001. Biophysical characterization and vector-specific antagonist activity of domain III of the tick-borne flavivirus envelope protein J. Virol. 75 4002–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressanelli S., Stiasny K., Allison S.L., Stura E.A., Duquerroy S., Lescar J., Heinz F.X., Rey F.A. 2004. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation EMBO J. 23 728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarero J.A., Fushman D., Sato S., Giriat I., Cowburn D., Raleigh D.P., Muir T.W. 2001. Rescuing a destabilized protein fold through backbone cyclization J. Mol. Biol. 308 1045–1062. [DOI] [PubMed] [Google Scholar]
- Clore G.M., Szabo A., Bax A., Kay L.E., Driscoll P.C., Gronenborn A.M. 1990. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins J. Am. Chem. Soc. 112 4989–4991. [Google Scholar]
- Clore G.M., Gronenborn A.M., Bax A. 1998. A robust method for determining the magnitude of the fully asymmetric alignment tensor of oriented macromolecules in the absence of structural information J. Magn. Reson. 133 216–221. [DOI] [PubMed] [Google Scholar]
- Cornilescu G., Ottiger M., Bax A. 1998. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase J. Am. Chem. Soc. 120 6836–6837. [Google Scholar]
- Cornilescu G., Delaglio F., Bax A. 1999a. Protein backbone angle restraints from searching a database for chemical shift and sequence homology J. Biomol. NMR 13 289–302. [DOI] [PubMed] [Google Scholar]
- Cornilescu G., Hu S., Bax A. 1999b. Identification of the hydrogen bonding network in a protein by scalar couplings J. Am. Chem. Soc. 121 2949–2950. [Google Scholar]
- Crill W.D. and Roehrig J.T. 2001. Monoclonal antibodies that bind to domain III of dengue virus E glycoprotein are the most efficient blockers of virus adsorption to Vero cells J. Virol. 75 7769–7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deep S., Walker K.P. III, Shu Z., Hinck A.P. 2003. Solution structure and backbone dynamics of the TGFβ type II receptor extracellular domain Biochemistry 42 10126–10139. [DOI] [PubMed] [Google Scholar]
- Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Deschamps M., Campbell I.D., Boyd J. 2005. Residual dipolar couplings and some specific models for motional averaging J. Magn. Reson. 172 118–132. [DOI] [PubMed] [Google Scholar]
- Dutta K., Shi H., Cruz-Chu E.R., Kami K., Ghose R. 2004. Dynamic influences on a high-affinity, high-specificity interaction involving the C-terminal SH3 domain of p67phox Biochemistry 43 8094–8106. [DOI] [PubMed] [Google Scholar]
- Epstein D.M., Benkovic S.J., Wright P.E. 1995. Dynamics of the dihydrofolate reductase-folate complex: Catalytic sites and regions known to undergo conformational change exhibit diverse dynamical features Biochemistry 34 11037–11048. [DOI] [PubMed] [Google Scholar]
- Farrow N.A., Muhandiram R., Singer A.U., Pascal S.M., Kay C.M., Gish G., Shoelson S.E., Pawson T., Forman-Kay J.D., Kay L.E. 1994. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation Biochemistry 33 5984–6003. [DOI] [PubMed] [Google Scholar]
- Folmer R.H., Hilbers C.W., Konings R.N., Nilges M. 1997. Floating stereospecific assignment revisited: Application to an 18 kDa protein and comparison with J-coupling data J. Biomol. NMR 9 245–258. [DOI] [PubMed] [Google Scholar]
- Fushman D. and Cowburn D. 1999. The effect of noncollinearity of 15N-1H dipolar and 15N CSA tensors and rotational anisotropy on 15N relaxation, CSA/dipolar cross correlation, and TROSY J. Biomol. NMR 13 139–147. [DOI] [PubMed] [Google Scholar]
- Fushman D., Cahill S., Cowburn D. 1997. The main-chain dynamics of the dynamin pleckstrin homology (PH) domain in solution: Analysis of 15N relaxation with monomer/dimer equilibration J. Mol. Biol. 266 173–194. [DOI] [PubMed] [Google Scholar]
- Fushman D., Tjandra N., Cowburn D. 1998. Direct measurement of 15N chemical shift anisotropy in solution J. Am. Chem. Soc. 120 10947–10952. [Google Scholar]
- Ghose R., Shekhtman A., Goger M.J., Ji H., Cowburn D. 2001. A novel, specific interaction involving the Csk SH3 domain and its natural ligand Nat. Struct. Biol. 8 998–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritsun T.S., Holmes E.C., Gould E.A. 1995. Analysis of flavivirus envelope proteins reveals variable domains that reflect their antigenicity and may determine their pathogenesis Virus Res. 35 307–321. [DOI] [PubMed] [Google Scholar]
- Habeck M., Rieping W., Linge J.P., Nilges M. 2004. NOE assignment with ARIA 2.0: The nuts and bolts Methods Mol. Biol. 278 379–402. [DOI] [PubMed] [Google Scholar]
- Heinz F.X., Auer G., Stiasny K., Holzmann H., Mandl C., Guirakhoo F., Kunz C. 1994. The interactions of the flavivirus envelope proteins: Implications for virus entry and release Arch. Virol. Suppl. 9 339–348. [DOI] [PubMed] [Google Scholar]
- Hiramatsu K., Tadano M., Men R., Lai C.J. 1996. Mutational analysis of a neutralization epitope on the dengue type 2 virus (DEN2) envelope protein: Monoclonal antibody resistant DEN2/DEN4 chimeras exhibit reduced mouse neurovirulence Virology 224 437–445. [DOI] [PubMed] [Google Scholar]
- Hurrelbrink R.J. and McMinn P.C. 2001. Attenuation of Murray Valley encephalitis virus by site-directed mutagenesis of the hinge and putative receptor-binding regions of the envelope protein J. Virol. 75 7692–7702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B.A. 2004. Using NMRView to visualize and analyze the NMR spectra of macromolecules Methods Mol. Biol. 278 313–352. [DOI] [PubMed] [Google Scholar]
- Karplus M. 1959. Contact electron-spin coupling of nuclear magnetic moments J. Phys. Chem. 30 11–15. [Google Scholar]
- Kline A.D. and Wuthrich K. 1986. Complete sequence-specific 1H nuclear magnetic resonance assignments for the α-amylase polypeptide inhibitor tendamistat from Streptomyces tendae J. Mol. Biol. 192 869–890. [DOI] [PubMed] [Google Scholar]
- Kline A.D., Braun W., Wuthrich K. 1986. Studies by 1H nuclear magnetic resonance and distance geometry of the solution conformation of the α-amylase inhibitor tendamistat J. Mol. Biol. 189 377–382. [DOI] [PubMed] [Google Scholar]
- Koradi R., Billeter M., Wuthrich K. 1996. MOLMOL: A program for display and analysis of macromolecular structures J. Mol. Graph. 14 51–55. [DOI] [PubMed] [Google Scholar]
- Kristensen S.M., Siegal G., Sankar A., Driscoll P.C. 2000. Backbone dynamics of the C-terminal SH2 domain of the p85α subunit of phosphoinositide 3-kinase: Effect of phosphotyrosine-peptide binding and characterization of slow conformational exchange processes J. Mol. Biol. 299 771–788. [DOI] [PubMed] [Google Scholar]
- Kroenke C., Loria P., Lee K., Rance M., Palmer A.G. III. 1998. Longitudinal and transverse 1H-15N dipolar/15N chemical shift anisotropy relaxation interference: Unambiguous determination of rotational diffusion tensors and chemical exchange effects in biological macromolecules J. Am. Chem. Soc. 120 7905–7915. [Google Scholar]
- Kuboniwa H., Grzesiek S., Delaglio F., Bax A. 1994. Measurement of HN-H α J couplings in calcium-free calmodulin using new 2D and 3D water-flip-back methods J. Biomol. NMR 4 871–878. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., Rullmannn J.A., MacArthur M.W., Kaptein R., Thornton J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR J. Biomol. NMR 8 477–486. [DOI] [PubMed] [Google Scholar]
- Lin C.W. and Wu S.C. 2003. A functional epitope determinant on domain III of the Japanese encephalitis virus envelope protein interacted with neutralizing-antibody combining sites J. Virol. 77 2600–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linge J.P., Habeck M., Rieping W., Nilges M. 2003. ARIA: Automated NOE assignment and NMR structure calculation Bioinformatics 19 315–316. [DOI] [PubMed] [Google Scholar]
- Lipari G. and Szabo A. 1982a. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity J. Am. Chem. Soc. 104 4546–4559. [Google Scholar]
- Lipari G. and Szabo A. 1982b. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results J. Am. Chem. Soc. 104 4559–4570. [Google Scholar]
- Loria J.P., Rance M., Palmer A.G. III. 1999a. A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy J. Am. Chem. Soc. 121 2331–2332. [Google Scholar]
- Loria J.P., Rance M., Palmer A.G. III. 1999b. A TROSY CPMG sequence for characterizing chemical exchange in large proteins J. Biomol. NMR 15 151–155. [DOI] [PubMed] [Google Scholar]
- Mandel A.M., Akke M., Palmer A.G. III. 1995. Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme J. Mol. Biol. 246 144–163. [DOI] [PubMed] [Google Scholar]
- Mandel A.M., Akke M., Palmer A.G. III. 1996. Dynamics of ribonuclease H: Temperature dependence of motions on multiple time scales Biochemistry 35 16009–16023. [DOI] [PubMed] [Google Scholar]
- Mandl C.W., Allison S.L., Holzmann H., Meixner T., Heinz F.X. 2000. Attenuation of tick-borne encephalitis virus by structure-based site-specific mutagenesis of a putative flavivirus receptor binding site J. Virol. 74 9601–9609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy M.A., Senior M.M., Gesell J.J., Ramanathan L., Wyss D.F. 2001. Solution structure and dynamics of the single-chain hepatitis C virus NS3 protease NS4A cofactor complex J. Mol. Biol. 305 1099–1110. [DOI] [PubMed] [Google Scholar]
- Modis Y., Ogata S., Clements D., Harrison S.C. 2003. A ligand-binding pocket in the dengue virus envelope glycoprotein [see comment] Proc. Natl. Acad. Sci. 100 6986–6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modis Y., Ogata S., Clements D., Harrison S.C. 2004. Structure of the dengue virus envelope protein after membrane fusion [see comment] Nature 427 313–319. [DOI] [PubMed] [Google Scholar]
- Mukherjee M., Dutta K., Pascal S.M., Fox R.O. 2004. Backbone and side chain resonance assignments of domain III of the tick-borne Langat flavivirus envelope protein J. Biomol. NMR 29 535–536. [DOI] [PubMed] [Google Scholar]
- Ottiger M., Delaglio F., Bax A. 1998a. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra J. Magn. Reson. 131 373–378. [DOI] [PubMed] [Google Scholar]
- Ottiger M., Delaglio F., Marquardt J.L., Tjandra N., Bax A. 1998b. Measurement of dipolar couplings for methylene and methyl sites in weakly oriented macromolecules and their use in structure determination J. Magn. Reson. 134 365–369. [DOI] [PubMed] [Google Scholar]
- Palmer A.G. III. 2004. NMR characterization of the dynamics of biomacromolecules Chem. Rev. 104 3623–3640. [DOI] [PubMed] [Google Scholar]
- Pelupessy P., Espallargas G.M., Bodenhausen G. 2003. Symmetrical reconversion: Measuring cross-correlation rates with enhanced accuracy J. Magn. Reson. 161 258–264. [DOI] [PubMed] [Google Scholar]
- Rey F.A., Heinz F.X., Mandl C., Kunz C., Harrison S.C. 1995. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution Nature 375 291–298. [DOI] [PubMed] [Google Scholar]
- Roehrig J.T., Bolin R.A., Kelly R.G. 1998. Monoclonal antibody mapping of the envelope glycoprotein of the dengue 2 virus, Jamaica Virology 246 317–328. [DOI] [PubMed] [Google Scholar]
- Ruckert M. and Otting G. 2000. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments J. Am. Chem. Soc. 122 7793–7797. [Google Scholar]
- Sattler M., Schleucher J., Griesinger C. 1999. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulse field gradients Prog. Nucl. Magn. Reson. Spectrosc. 34 93–158. [Google Scholar]
- Stiasny K., Bressanelli S., Lepault J., Rey F.A., Heinz F.X. 2004. Characterization of a membrane-associated trimeric low-pH-induced form of the class II viral fusion protein E from tick-borne encephalitis virus and its crystallization J. Virol. 78 3178–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjandra N., Kuboniwa H., Ren H., Bax A. 1995. Rotational dynamics of calcium-free calmodulin studied by 15N-NMR relaxation measurements Eur. J. Biochem. 230 1014–1024. [DOI] [PubMed] [Google Scholar]
- Volk D.E., Beasley D.W., Kallick D.A., Holbrook M.R., Barrett A.D., Gorenstein D.G. 2004. Solution structure and antibody binding studies of the envelope protein domain III from the New York strain of West Nile virus J. Biol. Chem. 279 38755–38761. [DOI] [PubMed] [Google Scholar]
- Wang C., Grey M.J., Palmer A.G. III. 2001. CPMG sequences with enhanced sensitivity to chemical exchange J. Biomol. NMR 21 361–366. [DOI] [PubMed] [Google Scholar]
- Wang C., Rance M., Palmer A.G. III. 2003. Mapping chemical exchange in proteins with MW > 50 kD J. Am. Chem. Soc. 125–8968. [DOI] [PubMed]
- Wishart D.S. and Sykes B.D. 1994. The 13C chemical-shift index: A simple method for the identification of protein secondary structure using 13C chemical-shift data J. Biomol. NMR 4 171–180. [DOI] [PubMed] [Google Scholar]
- Wishart D.S., Sykes B.D., Richards F.M. 1992. The chemical shift index: A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy Biochemistry 31 1647–1651. [DOI] [PubMed] [Google Scholar]
- Wu K.P., Wu C.W., Tsao Y.P., Kuo T.W., Lou Y.C., Lin C.W., Wu S.C., Cheng J.W. 2003. Structural basis of a flavivirus recognized by its neutralizing antibody: Solution structure of the domain III of the Japanese encephalitis virus envelope protein J. Biol. Chem. 278 46007–46013. [DOI] [PubMed] [Google Scholar]
- Yu S., Wuu A., Basu R., Holbrook M.R., Barrett A.D., Lee J.C. 2004. Solution structure and structural dynamics of envelope protein domain III of mosquito- and tick-borne flaviviruses Biochemistry 43 9168–9176. [DOI] [PubMed] [Google Scholar]
- Zhang F. and Bruschweiler R. 2002. Contact model for the prediction of NMR N-H order parameters in globular proteins J. Am. Chem. Soc. 124 12654–12655. [DOI] [PubMed] [Google Scholar]
- Zheng D., Aramini J.M., Montelione G.T. 2004. Validation of helical tilt angles in the solution NMR structure of the Z domain of Staphylococcal protein A by combined analysis of residual dipolar coupling and NOE data Protein Sci. 13 549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweckstetter M. and Bax A. 2000. Prediction of sterically induced alignment in a dilute liquid crystalline phase: Aid to protein structure determination by NMR J. Am. Chem. Soc. 122 3791–3792. [Google Scholar]
- Zweckstetter M. and Bax A. 2001a. Characterization of molecular alignment in aqueous suspensions of Pf1 bacteriophage J. Biomol. NMR 20 365–377. [DOI] [PubMed] [Google Scholar]
- Zweckstetter M. and Bax A. 2001b. Single-step determination of protein substructures using dipolar couplings: Aid to structural genomics J. Am. Chem. Soc. 123 9490–9491. [DOI] [PubMed] [Google Scholar]