

Abstract
The impetus for the recent development and emergence of automated function prediction methods is an exponentially growing flood of new experimental data, the interpretation of which is hindered by a shortage of reliable annotations for proteins that lack experimental characterization or significant homologs in current databases. Here we introduce PFP, an automated function prediction server that provides the most probable annotations for a query sequence in each of the three branches of the Gene Ontology: biological process, molecular function, and cellular component. Rather than utilizing precise pattern matching to identify functional motifs in the sequences and structures of these proteins, we designed PFP to increase the coverage of function annotation by lowering resolution of predictions when a detailed function is not predictable. To do this we extend a traditional PSI-BLAST search by extracting and scoring annotations (GO terms) individually, including annotations from distantly related sequences, and applying a novel data mining tool, the Function Association Matrix, to score strongly associated pairs of annotations. We show that PFP can correctly assign function using only weakly similar sequences with a significantly better accuracy and coverage than a standard PSI-BLAST search, improving it more than fivefold. The most descriptive annotations predicted by PFP (GO depth ≥8) can identify a significant subgraph in the GO with >60% accuracy and ∼100% coverage for our benchmark set. We also provide examples of the superb performance of PFP in an assessment of automated function prediction servers at the Automated Function Prediction Special Interest Group meeting at ISMB 2005 (AFP-SIG '05).
Keywords: protein function prediction, PSI-BLAST, gene ontology, low-resolution function
The fields of cell and molecular biology have as a main focus the task of clearly defining cellular roles for all proteins encoded by the DNA existing in a genome. This involves describing for each protein its biochemical function(s), cellular location(s), participation in various cellular processes, structure, interactions, etc. Recently developed technologies for molecular biology are increasingly broad, both in scope and scale. The bioinformatics community has been called upon to extract and interpret patterns in the glut of new experimental data produced by these technologies, so that they may be utilized to their full capacity.
Automated protein function prediction methods are emerging as both interpretive techniques for high-throughput experimental datasets (e.g., expression microarrays, interaction screens) and as partners to structural genomics projects (Watson et al. 2005). These algorithms can be grouped into four distinct categories (Hawkins and Kihara 2005a): evolutionary methods, which use conserved global and local sequence or structure to imply homology and motifs to assign biochemical function and binding sites (Hennig et al. 2003; Khan et al. 2003; Martin et al. 2004); comparative genomics methods, which link proteins through domain fusion events, phylogenetic profiling, conserved gene order, and common regulatory elements; cellular methods, which use large proteomics datasets to define protein–protein interaction patterns and complexes; and metabolic methods, which utilize the structured networks of biochemical pathways to match proteins to uncharacterized reactions. There are also methods that combine multiple contextual clues to assign function annotations (Pal and Eisenberg 2005).
A major limit to function prediction is its limited coverage. Typically conventional BLAST searches (Altschul et al. 1990) can only cover up to half of the genes in a genome. In order to provide functional clues that can spark analysis of large proteomics datasets, we need a method that expands coverage by lowering prediction resolution if necessary, i.e., a method that can provide accurate (but more generalized) predictions for proteins falling outside of the coverage range for current techniques.
To address this need, we have designed and implemented a public Web server for automated Protein Function Prediction, PFP (Hawkins and Kihara 2005a, b), which extends the functionality of a typical PSI-BLAST search (Altschul et al. 1997) in three distinct ways: first, we extract and score Gene Ontology (GO) annotations based on the frequency of their occurrence in highly similar sequences (Martin et al. 2004). The GO is a curated, hierarchical vocabulary describing the function of proteins in three categories: molecular function, biological process, and cellular component (Harris et al. 2004). Second, we utilize relatively weak hits produced by a PSI-BLAST query, not conventionally used for transfer of function annotation. Weakly similar, lower scoring sequences output by PSI-BLAST are not recognized as orthologs to the query sequence, but often represent proteins sharing a common functional domain. Third, we additionally consider those functions that are strongly associated with the highest scoring annotations as described previously. To score these annotations, we designed a novel data mining tool, the Function Association Matrix (FAM), which quantifies the co-occurrence of GO annotations in proteins whose sequences are included in UniProt. Thus, we can assign function using the FAM that cannot be retrieved directly from PSI-BLAST hits. The benchmark results illustrate that PFP assigns correct function even from weakly similar sequences (E-value >10) with significantly better specificity than a regular PSI-BLAST search. In the assessment of automated function prediction servers held at AFP-SIG '05, PFP performed very well, achieving the highest total score among participating servers. PFP is available as an online service at http://dragon.bio.purdue.edu/pfp.
Results
Benchmark
PFP correctly annotated the biological process at a rank of five or higher for 84% of the sequences in our benchmark set (Fig. 1) It is remarkable that the sequence coverage does not drop, but stays at the level of 50% even when only sequence hits of high E-value are used. Even when sequence hits of an E-value of <100 are ignored, the sequence coverage for biological process is 32%. This is a significant improvement (approximately fivefold or more) over a simple transfer of annotations from the single best scoring sequence retrieved by PSI-BLAST (Top PSI-BLAST) in all E-value ranges. Unlike PFP, the specificity of PSI-BLAST sharply drops when the top hit sequence is ignored. Figure 1 also shows the relative contributions of the base PFP score (PFP without FAM) and annotation scores retrieved by the FAM (PFP + FAM1000), which improves sequence coverage for biological process 5%–20% at all E-value cutoffs.
Figure 1.

Sequence coverage of PFP versus top PSI-BLAST. The sequence coverage (Y-axis) is the percentage of sequences for which a correct biological process (sharing a common parent with a target annotation at a GO depth ≥ 4) was ranked in the top five results output by PFP. The E-value cutoff value (X-axis) represents the minimum similarity for sequences used by PFP in our benchmark analysis. PFP + FAM1000 (solid black line) is PFP with associations scored by the FAM1000 matrix. PFP (w/o FAM) (broken black line) is PFP without scored associations. Top PSI-BLAST (solid gray line) transfers annotations from the most similar sequence scoring above each E-value cutoff.
The annotation-level accuracy can be much more descriptive because we can incorporate the structure of the GO (Fig. 2). Our analysis of individual annotations takes into consideration the depth of each annotation and also the edge distance between the predicted annotation and its closest target in the ontology. In Figure 2A, the overall average specificity of predicted GO terms is shown separately at each predicted GO depth. It is shown that the prediction accuracy (specificity) significantly increases when specific predictions at a deeper GO depth are made (solid line). Predictions made by PFP at a depth of 10 for benchmark sequences are all correct, indicating that when very descriptive predictions are made, they have considerable functional similarity to the target annotation and can define a consensus subgraph in the GO tree. The specificity does not drop below 25% for predictions at a depth of six or greater, even when only those sequence hits of E-value of ≥15 are used (broken line). On average, the GO depth of the common parent between a prediction and the correct annotation is five or deeper when the prediction has a GO depth of six or deeper (this can be computed by subtracting the average overprediction, i.e., the gray column, from the predicted GO depth, i.e., X-axis). Note that a function annotation of a GO depth of five is descriptive enough for many purposes of function prediction.
Figure 2.
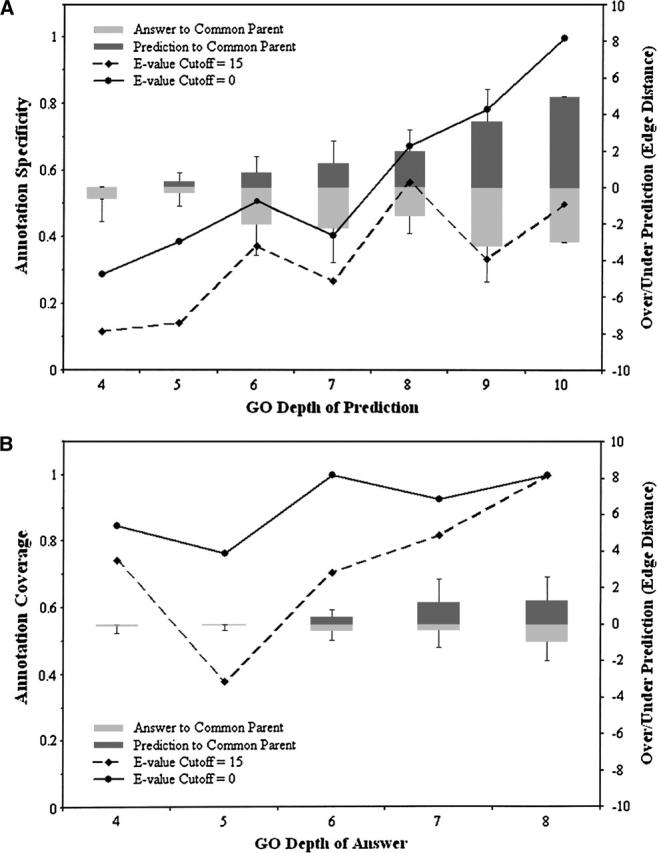
Annotation-level accuracy of PFP at different GO depths. (A) The specificity (percentage of predicted annotations sharing a common parent with a target annotation at a GO depth ≥ 4) is shown at each GO depth of predicted annotation. The overprediction distance (dark gray columns, right-hand axis) is the average edge distance between a predicted annotation and the common parent it shares with the closest target annotation; the underprediction distance is the average edge distance between a target annotation and the common parent (light gray columns). The GO depth is the edge distance between each predicted (top) or target (bottom) annotation and the category root. (B) The coverage (percentage of correctly predicted target annotations) is shown at each GO depth of the correct annotation. The overprediction distance (dark gray columns, right-hand axis) is the average edge distance between each target annotation and the common parent it shares with the closest predicted annotation; the underprediction distance is the average edge distance between the closest predicted annotation and the common parent (light gray columns). For both A and B, “E-value Cutoff = 0” (solid black line) is PFP + FAM1000 and “E-value Cutoff = 15” (broken black line) is PFP + FAM1000 using only sequence hits from PSI-BLAST with an E-value of 15 or larger.
Figure 2B shows the coverage of target annotations. For our benchmark set, it is remarkable that PFP was able to retrieve >90% of the target annotations among the top 10 predictions at a GO depth of six or greater.
Individual examples of predicted function annotations from AFP-SIG '05
PFP participated in an assessment of function prediction servers at the Automated Function Prediction Special Interest Group meeting at ISMB 2005 (AFP-SIG '05). Here we analyze our predicted annotations for each of the five target protein sequences (Table 1).
Table 1.
Summary of predictions made by PFP for AFP-SIG '05 targets

T1 was involved in thiamine biosynthesis, but its molecular function was unknown. PFP correctly identified “thiamine biosynthesis” (GO:0009228) as the top-ranked biological process. (Note: in the assessment at the AFP-SIG '05, this correct prediction was not scored, as only molecular function annotations were assessed.) This annotation was found in most of the sequences retrieved by PSI-BLAST. We additionally predicted “transferase activity, transferring glycosyl groups” (GO:0016757) and “oxidoreductase activity” (GO:0016491) as the two most probable molecular functions. Neither of these functions were annotated to similar sequences, but both were retrieved by the UniProt FAM based on strong intercategory associations to thiamine biosynthesis [P(0016757 | 0009228) = 0.176 and P(0016491 | 0009228) = 0.103].
T2 was an orphan from Thermotoga maritima (TM1622), known from structural similarity and genomic localization to be “small GTPase binding” (GO:0031267). PFP identified this sequence as “Rab GTPase binding” (GO:0017137), a child of the target annotation. Again, this annotation was not found in any of the similar sequences retrieved by PSI-BLAST, but was annotated by PFP because of strong association to “protein binding” [GO:0015301, P(0017137 | 0015301) = 0.158].
T4 was experimentally verified as a pantothenate kinase (GO) from T. maritima (TM0883). PFP was unable to identify this function as one of the ten most probable. This is because similar sequences retrieved by PSI-BLAST were erroneously annotated as a Bvg accessory factor in UniProt, which interacts with RNA polymerase to activate transcription of a toxin operon. Consequently, PFP retrieved “transcription activator activity” (GO:0016563) as the most probable function annotation. Two annotations that would be considered correct, kinase activity (GO:0016301) and ATP binding (GO:0005524) were predicted by PFP with ranks of 17 and 30, respectively. Both were annotated to putative N-acetylmannosamine kinases found by PSI-BLAST (E-value of 2.2 and 3.5), but did not have significant enough scores to be among the top 10 most probable predictions.
T5 was experimentally verified as a GDNF receptor involved in growth arrest and caspase-dependent apoptosis in humans. PFP correctly predicted this sequence to be a receptor (annotated with “protein binding” [GO:0005515] and “receptor activity” [GO:0004872]). These annotations originated in two significant PSI-BLAST hits, both GAS1 (from mouse and Caenorhabditis elegans, respectively).
T7 was experimentally verified as an aspartate dehydrogenase (GO:0015922) from T. maritima (TM1643). PFP identified the sequence as having “3′-5′-cyclic nucleotide phosphodiesterase activity” (GO:0004114), which shares a common parent of “catalytic activity” (GO:0003824) with the target molecular function annotation. T5 and T7 are examples of PFP predicting a low-resolution function when the exact function prediction cannot be made.
Discussion
Even with recent technological advances in proteomics and structural genomics methods, sequence information is by far the most readily retrieved and rich source of information available for computational analyses. Yet, as new sources of heterogeneous data are released, many in the function prediction community are quick to abandon building on simple sequence similarity methods in favor of more advanced techniques that incorporate multiple data sources. We show here that a traditional sequence similarity tool can be improved upon by using simple extensions, and that these improvements can increase the coverage of sequence-based functional inference dramatically beyond previous limits.
In expanding upon a simple PSI-BLAST search, we are able to increase sequence coverage up to 10-fold (Fig. 1, black lines). This is an improvement on previously defined limits, and is magnified when considering its effects on the use of distantly related sequences (E-value >> 1.0). When ignoring all similar sequences of E-value <50, PFP is still able to output two correct biological process annotations on average. This effect is due in large part to the scoring of individual function annotations by summing the contributions of each occurrence in multiple top hit sequences, allowing the scores of more frequently occurring features to be amplified.
We were also able to show the significance of mining association data for annotation pairs in sequence context by the FAM matrix. Annotations retrieved by simple binary association add 10%–20% coverage for lower E-values (Fig. 1). For targets T1 and T2 from AFP-SIG '05, the best scoring molecular function predictions by PFP were not found directly in sequences retrieved by PSI-BLAST, but rather were inferred based on strong associations to process annotations that were found in similar sequences (Table 1). The value of these associations is evidenced particularly in target T2, where no other participating servers were able to predict a significant annotation.
The detailed analysis of the benchmark results in Figure 2 revealed two interesting features of PFP. First, as predictions by PFP become more descriptive, the accuracy of those predictions increases (Fig. 2A). Second, although total edge distance in the GO between predicted and target annotations increases, the descriptive significance of those annotations is increased over predictions of shallower annotations. If a predicted annotation has a GO depth of six or deeper, that prediction still accurately identifies the subgraph in the GO tree with a single common parent at a depth of five, which would be considered to be highly descriptive. This is a notable advantage of PFP, that it is able to predict low-resolution function when an exact annotation cannot be inferred. Targets T5 and T7 in AFP-SIG '05 provide vivid examples of low-resolution function prediction by PFP. These low-resolution functions are difficult to predict by conventional homology/motif searches and active site tertiary structure matching methods, which use more precise pattern matching to infer specific functions and otherwise provide no functional information at all. PFP often identifies multiple annotations that share a significant common parent with a target annotation. PFP was able to retrieve nearly 100% of the stripped annotations from sequences in our benchmark set among the top 10 predictions, regardless of the depth of those annotations (Fig. 2B).
We were able to show with PFP that sequence similarity-based function inference is a much more powerful tool than previously expected. However, the nature of incorrect predictions output by PFP indicates that even our extensions of PSI-BLAST fall victim to the shortcomings of sequence-based function prediction, i.e., if no annotated sequences are included in PSI-BLAST hits at all, or if similar sequences to a query share no common function, PFP is unable to retrieve an accurate annotation. It will be increasingly important in the near future to utilize heterogeneous contextual functional data, including structural similarity and fold recognition scores, to further expand coverage of automated function prediction tools.
Materials and methods
The current implementation of the PFP server uses PSI-BLAST (blastpgp version: 2.2.10) to predict the top 10 most probable functions in each of the three GO categories (biological process, molecular function, and cellular component). Rather than transferring annotations directly from a single highly similar sequence retrieved by PSI-BLAST to a query sequence, PFP uses a scoring scheme to rank GO annotations assigned to all of the most similar sequences according to (1) their frequency of occurrence in those sequences and (2) the degree of similarity of the originating sequence to the query. This is similar to the scoring basis for the R-value used by the GOtcha method to score annotations from pairwise alignment matches (Martin et al. 2004). A GO term, fa, is scored as follows:
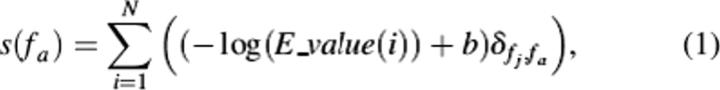 |
where s(fa) is the final score assigned to the GO term, fa, N is the number of the similar sequences retrieved by PSI-BLAST, E_value(i) is the E-value given to the sequence i, and fj is a GO term assigned to the sequence i. δfj,fa returns 1 when fj equals fa, and 0 otherwise. To maintain the integrity of the PSI-BLAST search, we use the default E-value threshold for inclusion in multiple iterations (−h 0.005) and set the maximum number of iterations to three (−j 3). By shifting the scoring space by a constant (b), individual annotations from weakly similar (E-value > 1) can be considered and scored. Here we use b = 2 (or log10[100]) to allow the use of sequence matches to an E-value of 100.
PFP also incorporates a novel data mining tool for quantifying association between function annotations (i.e., GO terms), the Function Association Matrix (FAM), to find and score GO terms which are strongly associated with those retrieved by the PSI-BLAST search. The FAM describes the frequency at which two functions occur together in the same context by quantifying the co-occurrence of each pair of annotations within UniProt sequences. This allows the FAM to associate function annotations from different GO categories, e.g., the biological process “positive regulation of transcription, DNA-dependent” (GO:0045893) is strongly associated with the molecular function “DNA binding activity” (GO:0003677) and the cellular component “nucleus” (GO:0005634). Associations can describe parallel functions that may be defined in multiple categories or complementary functions that are defined in one or more categories. Sixty-three percent of the sequence-based associations mined from UniProt bridge two GO categories. While these relationships may be intuitive to most molecular biologists, the FAM is the first tool to define them probabilistically.
Including associations precalculated by the FAM, the score given to a function fa is modified as follows:
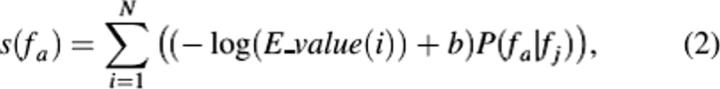 |
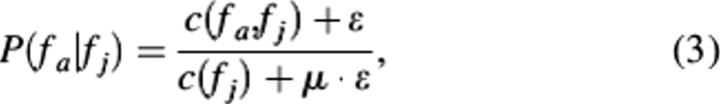 |
where P(fa | fj) is the conditional probability that fa is associated with fj, c(fa, fj) is number of times fa and fj are assigned simultaneously to each sequence in UniProt, and c(fj) is the total number of times fj appeared in UniProt, μ is the size of one dimension of the FAM (i.e., the total number of unique GO terms), and ɛ is the pseudo-count. A pseudo-count is added to each association under the assumption that the annotated proteins used to generate our matrices represent only a subset of all proteins.
Benchmark
The benchmark analysis described here (Figs. 1, 2) was performed on a random set of 2000 nonredundant protein sequences selected from UniProt. To test the appropriateness of using weakly similar sequences retrieved by PSI-BLAST, we ran PFP ignoring the most significant hits using several E-value cutoffs (E-values > 0.01, 0.1, 1, 2, 3, 5, 10, 15, 20, 25, 50, 100). Three separate tests were performed: (1) transfer of annotations from the top sequence hit from PSI-BLAST above each cutoff, (2) PFP as described above without scoring associated annotations (w/o FAM), and (3) PFP using a filtered FAM, FAM1000. FAM1000 removes the lowest scoring binary associations, increasing the significance of higher scoring associations. For each of the annotation predictions, we determined the shared common parent between the predicted annotation and the answer, along with the depth in the GO tree for each of those terms, the edge distance between all predictions to the common parent, and also from answers to the common parent. All queries of the GO were performed on a local copy of the GO database (mySQL version: go_20050710).
We measured the sequence coverage (number of sequences for which correct BP annotations were predicted in the top five divided by total number of sequences queried) (Fig. 1) and the specificity (number of predictions which correctly match a given annotation divided by total number of predictions) and coverage (number of given annotations which were correctly matched by a prediction divided by total number of given annotations) per predicted annotation (Fig. 2). A correct annotation prediction was considered to be one sharing a common parent at least four edges deeper than the root node of each GO category. A correct sequence prediction was considered to be one for which at least one correct annotation prediction was made.
Server
The PFP server (http://dragon.bio.purdue.edu/pfp) is maintained on a dual Intel Xeon processor (3.00 GHz) Linux station with 1.0-GB system memory at the Kihara Lab, Department of Biological Sciences, Purdue University.
Acknowledgments
D.K. acknowledges the support from the National Institute of General Medical Sciences of the National Institutes of Health (Grant Number R01 GM-075004).
Footnotes
Reprint requests to: Diasuke Kihara, Department of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA; e-mail: dkihara@purdue.edu; fax: (765) 496-1189.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062153506.
References
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215 403–410. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M.A., Clark J., Ireland A., Lomax J., Ashburner M., Foulger R., Eilbeck K., Lewis S., Marshall B., Mungall C.et al. 2004. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32 D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins T. and Kihara D. 2005a. PFP: Automatic annotation of protein function by relative GO association in multiple functional contexts. The 13th Annual International Conference on Intelligent Systems for Molecular Biology. Detroit, MI. –117.
- Hawkins T. and Kihara D. 2005b. The use of context-based functional association in automated protein function prediction methods. The 13th Annual International Conference on Intelligent Systems for Molecular Biology, Automatic Function Prediction—Special Interest Group. Detroit, MI. pp. 16–17.
- Hennig S., Groth D., Lehrach H. 2003. Automated Gene Ontology annotation for anonymous sequence data. Nucleic Acids Res. 31 3712–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S., Situ G., Schmidt C.J. 2003. GoFigure: Automated Gene Ontology annotation. Bioinformatics 19 2484–2485. [DOI] [PubMed] [Google Scholar]
- Martin D.M.A., Barriman M., Barton G.J. 2004. GOtcha: A new method for prediction of protein function assessed by the annotation of several genomes. BMC Bioinformatics 5 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal D. and Eisenberg D. 2005. Inference of protein function from protein structure. Structure (Camb.) 13 121–130. [DOI] [PubMed] [Google Scholar]
- Watson J.D., Laskowski R.A., Thornton J.M. 2005. Predicting protein function from sequence and structural data. Curr. Opin. Struct. Biol. 15 275–284. [DOI] [PubMed] [Google Scholar]