

Abstract
The crystal structure of Mycobacterium tuberculosis adenylate kinase (MtAK) in complex with two ADP molecules and Mg2+ has been determined at 1.9 Å resolution. Comparison with the solution structure of the enzyme, obtained in the absence of substrates, shows significant conformational changes of the LID and NMP-binding domains upon substrate binding. The ternary complex represents the state of the enzyme at the start of the backward reaction (ATP synthesis). The structure is consistent with a direct nucleophilic attack of a terminal oxygen from the acceptor ADP molecule on the β-phosphate from the donor substrate, and both the geometry and the distribution of positive charge in the active site support the hypothesis of an associative mechanism for phosphoryl transfer.
Keywords: adenylate kinase, phosphoryl transfer, X-ray crystallography, associative mechanism, Mycobacterium tuberculosis
Tuberculosis is a major world health problem, with one third of the total world population currently infected with Mycobacterium tuberculosis and more than two-million people dying annually from the disease. Sparked by the development of mycobacterial genomics (Cole et al. 1998), many efforts have been invested during the last few years in the identification and characterization of novel drug targets (Sharma et al. 2004; Andries et al. 2005). Among these, nucleoside monophosphate kinases (NMPKs) are of particular interest since they play a key role in the mainteinance of intracellular nucleotide pools in all living organisms.
Adenylate kinase (ATP:AMP phosphotransferase, EC 2.7.4.3; AK) is a ubiquitous enzyme which catalyzes the reversible, Mg2+-dependent transfer of the terminal phosphate group from ATP to AMP, releasing two molecules of ADP. A large number of biochemical and structural studies have been conducted on AKs from different sources (Yan and Tsai 1999), and several crystal structures of the enzyme have been reported, both in the unligated form as well as in complex with substrates (ATP, ADP, and AMP) or substrate analogs (AMP–PNP or diphosphoadenosine-pentaphosphate, Ap5A). These studies reveal a conserved globular architecture composed of a central core made by a parallel β-sheet surrounded by α-helices, and a P-loop motif at the N terminus that binds the phosphoryl donor (ATP). Two regions of the protein, the LID and NMP-binding regions, participate in the isolation of the substrates during catalysis and usually undergo significant conformational changes during the reaction (Yan and Tsai 1999). The AK family can be classified into two groups on the basis of their polypeptide chain length, with the long variants possessing a longer LID domain as a consequence of a 25–40-residue insert. Eukaryotic cytosolic enzymes are of the short type, while mitochondrial, yeast, and bacterial AKs generally belong to the long type. Some bacterial AKs, including the M. tuberculosis enzyme, are actually short variants with limited sequence similarity to the eukaryotic cytosolic counterparts, and have been proposed to belong to a new group of short bacterial AKs (Munier-Lehmann et al. 1999).
In M. tuberculosis, AK is codified by the gene adk (Rv0733), and is composed of 181 amino acids. The recombinant enzyme expressed in Escherichia coli has been biochemically characterized (Munier-Lehmann et al. 1999), and its 3D structure in the absence of substrates has been determined by NMR methods (Miron et al. 2004). Here we describe the crystal structure of M. tuberculosis adenylate kinase (MtAK) at 1.9 Å in complex with two molecules of ADP and Mg2+. The structure reveals significant conformational changes upon substrate binding and sheds light on the mechanism of the backward reaction. Despite the large number of available crystal structures of AKs (and those of closely related NMPKs) from different sources, no direct structural information has been reported for the ternary complex of the enzyme at the start of the backward reaction. Only the structure of uridylate kinase from Saccharomyces cerevisiae has been reported in complex with two ADP molecules (Muller-Dieckmann and Schulz 1995), but without the necessary Mg2+ ion.
Results and Discussion
Overall structure
The crystal structure of MtAK was determined at 1.9 Å resolution by molecular replacement methods using the structure of Bacillus stearothermophilus AK (Berry and Phillips 1998; PDB entry 1ZIP) as the search probe. A summary of data collection and refinement statistics is reported in Table 1. The overall structure is very similar to those of other AKs, displaying an LID region that is reduced to a 10-residue loop (Lys125 to Asp134) as in the eukaryotic cytosolic isoforms (Fig. 1).
Table 1.
Diffraction data collection and refinement statistics

Figure 1.
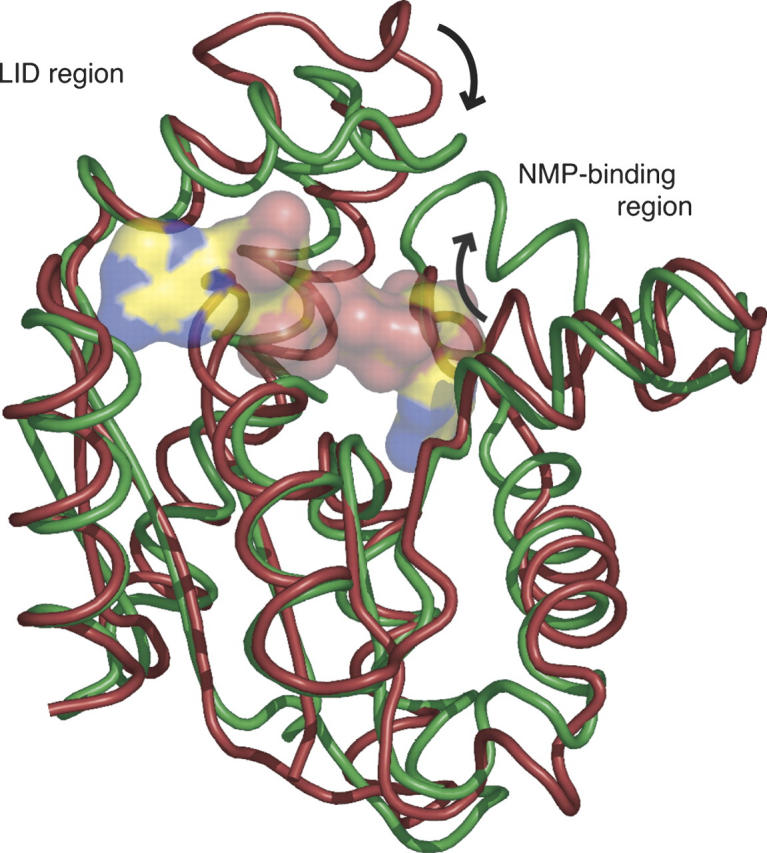
Superposition of the NMR structure of MtAK (Miron et al. 2004; PDB code 1P4S) in red, with the crystal structure of MtAK in complex with two molecules of ADP and Mg2+ (this work) in green. The ADP molecules are shown in transparent molecular surface representation. Significant movements of the LID and NMP-binding regions are indicated by arrows.
It is well established that AK undergoes significant conformational changes upon substrate binding (Yan and Tsai 1999). In the case of MtAK, the solution structure (without ligands) shows the protein in a relatively closed conformation, similar to that found for the E. coli enzyme in complex with Ap5A (Miron et al. 2004), suggesting a possible explanation for its reduced catalytic activity. However, the structural superposition of the NMR and crystal structures reveals significant conformational rearrangements of MtAK upon substrate binding. The overall root-mean-squares deviation (RMSD) is 3.0 Å for all 181 Cα positions (1.7 Å for 122 core residues). As shown in Figure 1, however, residues from the NMP-binding domain (Thr31–Val59) show an RMSD of 3.6 Å, and those from the LID region (Lys125–Asp134) reveal a still larger rearrangement (6.4 Å), in agreement with the notion that the LID and AMP-binding regions undergo large conformational rearrangements during the catalytic cycle (Gerstein et al. 1993).
Protein–ligand interactions
In the crystal structure, a molecule of ADP binds to the ATP-binding site, while the NMP-binding site hosts either ADP or AMP with similar (50%) occupancy (Fig. 2A). An Mg2+ ion is also positioned at the center of the cleft, between the β-phosphates of each ADP ligand. The structure therefore represents the enzyme–substrate ternary complex immediately before the backward reaction, which leads to the phosphorylation of the ADP molecule in the ATP site, and the formation of AMP in the NMP site. As observed in other AK structures, the adenosine moiety of ADP in the ATP site interacts with the enzyme through a single hydrogen bond between the exocyclic amino group of adenine and the carbonyl oxygen of Gly166 (Fig. 2B), in agreement with the relatively poor specifity of AKs toward the nucleotide occupying this site. On the other hand, the adenosine moiety at the NMP site makes several hydrogen bonding interactions with protein residues (Thr31, Val59, Gly85, and Gln92; Fig. 2B). In particular, the carboxamide side chain of Gln92, which is hydrogen-bonded to the backbone amide of Arg88 and the hydroxyl group of Ser61, is equivalent to Gln101 in eukaryotic AK1, whose role in the discrimination of the NMP substrate has already been demonstrated by site-directed mutagenesis (Yan and Tsai 1999).
Figure 2.

MtAK–substrate interactions. Panel A shows a close view of the substrate-binding cleft. The 2Fo − Fc electron density map at 1.9 Å resolution (contoured at 1 σ) is shown for the nucleotide substrates and the cofactor Mg2+ (green sphere). Water molecules are drawn as red spheres. Panel B shows a schematic representation of enzyme–substrate hydrogen bonding interactions (dashed lines), with the interatomic distances given in angstroms.
The ADP phosphate groups participate in an extended network of interactions. The conserved P-loop is fully closed over the ADP molecule in the ATP site (i.e., the acceptor substrate in the backward reaction), with all its phosphate oxygen atoms making hydrogen bonding interactions with backbone amide groups of P-loop residues Gly10, Gly12, Lys13, Gly14, and Thr15 (Fig. 2B). Moreover, the ɛ-amino group of Lys13 (the invariant lysine of the P-loop) is hydrogen bonded to the terminal oxygens of both ADP molecules. In contrast, the phosphate moiety of ADP from the NMP site interacts with the guanidinium groups of five arginine residues: Arg36 and Arg88 are hydrogen-bonded to α-phosphate oxygens, and Arg127, Arg129, and Arg140 to β-phosphate oxygens (Fig. 2B). The conserved Arg88 is a critical residue in the E. coli enzyme, where the R88G mutation lowers the activity of the backward reaction to 0.1% of the wild type, and leads to a 85-fold higher Km value for AMP (Reinstein et al. 1989).
Although part of the LID loop, Arg129 interacts with the ADP bound in the NMP site (Fig. 2). This observation is consistent with the recent finding that the equivalent residue (Arg156) in the E. coli enzyme binds the AMP phosphate (Berry et al. 2006), and not the terminal phosphate of ATP as inferred from the previously reported structure of E. coli AK in complex with AMP and AMP-PNP (Berry et al. 1994). The above observations suggest that, in the M. tuberculosis enzyme, Arg129 may trigger the closure of the LID loop upon nucleotide binding to the NMP site, as proposed for the E. coli (Berry et al. 2006) and the Methanococcus voltae (Criswell et al. 2003) enzymes. It is worth noting that these observations are in contrast with the currently accepted view of an iso-random bi-bi mechanism for adenylate kinases (Sheng et al. 1999), which would require independent motions of the LID and NMP-binding regions.
The Mg2+ ion is in contact with the β-phosphates from both ADP substrates. The divalent ion is coordinated by the O2B oxygen atoms belonging to each β-phosphate, forming a bidentate complex, and two water molecules (Fig. 2). One of these water molecules interacts with the carboxylic oxygens of Asp84, another strictly conserved residue known to be necessary for Mg2+ binding (Rose et al. 1991), and with two terminal oxygens from the β-phosphate of the donor ADP. The second water molecule is positioned within hydrogen bonding distance to the hydroxyl group of Ser30 and to the guanidinium group of Arg36, which is also involved in fixing the α-phosphate of the donor ADP.
Catalytic mechanism
In the crystal structure, one terminal oxygen of the acceptor ADP directly points to the phosphorus atom of the donor β-phosphate, at a distance of 3 Å and in line with the Pβ–Oα,β bond (Fig. 2A). The distance between the two Pβ atoms is 4 Å, a shorter value than the medium range of 4.5–5 Å calculated in a recent molecular dynamics simulation of the E. coli AK in complex with ATP, AMP, and Mg2+ (Krishnamurthy et al. 2005). This geometry strongly suggests a direct nucleophilic attack of O1B on the Pβ atom of the donor substrate, following an associative mechanism for the reaction. The distribution of positive charges in the active site further supports this model. In particular, the side chains of Arg127 and Arg140 bind the O3B oxygen of the transferable phosphate from opposite sides, and together with the ɛ-amino group of Lys13 and the Mg2+ ion, are well positioned to neutralize the negative charge developed on the trigonal bipyramidal transition state (Reinstein et al. 1990). Moreover, Arg129 makes a hydrogen bond with the α−β bridging oxygen of the donor ADP; this interaction, although compatible with a dissociative mechanism, may also be required for an associative mechanism to enhance the positive polarization of the phosphorus atom to be transferred, facilitating the nucleophilic attack by the incoming oxygen. A similar role has been proposed for the equivalent Arg93 in Dictyostelium discoideum UMP/CMP kinase (Schlichting and Reinstein 1997). It is worth noting that Arg127, Arg129, and Arg140 of the M. tuberculosis enzyme correspond respectively to Arg132, Arg138, and Arg149 in the E. coli enzyme, whose important role for the stabilization of the transition state has been demonstrated by mutagenesis (Yan and Tsai 1999).
Materials and methods
Protein production
The gene adk from M. tuberculosis was cloned into the expression vector pET-28a (Novagen). The recombinant plasmid was introduced in E. coli BL21(DE3) pLysS (Novagen) and the tranformed strain was grown at 30°C in LB medium containing 50 μg/mL kanamycin and 30 μg/mL chloramphenicol until the value of A600 = 2.0 was reached. Expression of MtAK was induced by adding 1 mM IPTG (isopropyl-thio-β-D-galactopiranoside), and the heterologous protein was purified as described (Munier-Lehmann et al. 1999).
Crystallization and data collection
The native protein was crystallized using the hanging drop vapor diffusion method by mixing 1.5 μL of protein solution (18 mg/mL) containing 5 mM AMP and 5 mM ADP with 1.5 μL of the reservoir solution containing 20% (w/v) PEG4000, 5 mM MgCl2, 0.1 M Na-HEPES (pH 6.5), 0.2 M sodium acetate. Rod-like crystals (0.1 × 0.1 × 0.4 mm) grew within a week at 18°C. The crystals were transferred to a cryoprotectant solution containing 20% (v/v) PEG4000, 25% (v/v) glycerol, and 0.1 M Na-HEPES (pH 6.5), and flash-frozen in liquid nitrogen. Diffraction data were collected at 100 K on a single frozen crystal at the ESRF beamline ID29 (λ = 0.976 Å). Data processing and reduction were carried out using the programs MOSFLM, SCALA and TRUNCATE from the CCP4 software package (Collaborative Computational Project Number 4 1994). The crystals are monoclinic, space group P21, with cell dimensions a = 36.54 Å, b = 64.91 Å, c = 39.77 Å, β = 110.12° and one enzyme monomer in the asymmetric unit. The 3D structure was determined by molecular replacement methods using the program AMoRe (Navaza 1994) and the coordinates of B. stearothermophilus AK (Berry and Phillips 1998; PDB code 1ZIP) as the search model. The initial Fourier difference maps revealed unambiguous electron density corresponding to the two ADP ligands. Crystallographic refinement was carried out by alternate cycles of model building with the program O (Jones et al. 1991) and refinement with the programs REFMAC5 (Murshudov et al. 1999) and ARP/wARP (Perrakis et al. 1999). The parameters after the final refinement cycles are shown in Table 1. The complete polypeptide chain is present in the model (residues 1–181), although the lateral chains of Glu22 and Arg51 are not visible due to weak electron density. According to PROCHECK (Laskowski et al. 1993), 95.6% of non-Gly and non-Pro residues lie in the most favored regions of the Ramachandran plot, while the remaining 4.4% lie in additional allowed regions. Structural superpositions of the X-ray and NMR structures were carried out using the program MASS (Dror et al. 2003). Figures 1 and 2A were prepared using PyMol v0.98 (http://pymol.sourceforge.net).
Accession number
The atomic coordinates and the structure factors have been deposited with the Protein Data Bank (PDB code 2CDN).
Acknowledgments
We thank V. Bondet and J. Bellalou (Institut Pasteur) for help with protein production. This work was partially supported by grants from the Institut Pasteur (GPH-Tuberculose), the ANRS, France (Contract No. 2003/004), the National Genopole Network (Contract RG-2002-08), and the European Commission (SPINE, Contract No. QLG2-CT-2002-00988, and X-TB, Contract No. QLK2-CT-2001-02018).
Footnotes
Reprint requests to: Pedro M. Alzari, Unité de Biochimie Structurale, Institut Pasteur, 25 rue du Dr. Roux, F-75724 Paris Cedex 15, France; e-mail: alzari@pasteur.fr; fax: +33-1-45688604.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062163406.
References
- Andries K., Verhasselt P., Guillemont J., Gohlmann H.W., Neefs J.M., Winkler H., Van Gestel J., Timmerman P., Zhu M., Lee E.et al. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis Science 307 223–227. [DOI] [PubMed] [Google Scholar]
- Berry M.B. and Phillips G.N. 1998. Crystal structures of Bacillus stearothermophilus adenylate kinase with bound Ap5A, Mg2+Ap5A, and Mn2+Ap5A reveal an intermediate lid position and six coordinate octahedral geometry for bound Mg2+ and Mn2+ Proteins 32 276–288. [DOI] [PubMed] [Google Scholar]
- Berry M.B., Meador B., Bilderback T., Liang P., Glaser M., Phillips G.N. Jr. 1994. The closed conformation of a highly flexible protein: The structure of E. coli adenylate kinase with bound AMP and AMPPNP Proteins 19 183–198. [DOI] [PubMed] [Google Scholar]
- Berry M.B., Bae E., Bilderback T.R., Glaser M., Phillips G.N. Jr. 2006. Crystal structure of ADP/AMP complex of Escherichia coli adenylate kinase Proteins 62 555–556. [DOI] [PubMed] [Google Scholar]
- Cole S.T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S.V., Eiglmeier K., Gas S., Barry C.E. 3rdet al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence Nature 393 537–544. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4. 1994. The CCP4 (Collaborative Computational Project 4) suite: Programs for protein crystallography Acta Crystallogr. D Biol. Crystallogr 50 760–763. [DOI] [PubMed] [Google Scholar]
- Criswell A.R., Bae E., Stec B., Konisky J., Phillips G.N. Jr. 2003. Structure of thermophilic and mesophilic adenylate kinases from the genus Methanococcus J. Mol. Biol. 330 1087–1099. [DOI] [PubMed] [Google Scholar]
- Dror O., Benyamini H., Nussinov R., Wolfson H.J. 2003. Multiple structural alignment by secondary structures: Algorithm and applications Protein Sci. 12 2492–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M., Schulz G., Chothia C. 1993. Domain closure in adenylate kinase. Joints on either side of the two helices close like neighboring fingers J. Mol. Biol. 229 494–501. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou J.Y., Cowan S.W., Kjeldgaard M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy H., Lou H., Kimple A., Vieille C., Cukier R.I. 2005. Associative mechanism for phosphoryl transfer: A molecular dynamics simulation of Escherichia coli adenylate kinase complexed with its substrates Proteins 58 88–100. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. 1993. Procheck: A program to check the stereochemical quality of protein structures J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Miron S., Munier-Lehmann H., Craescu C.T. 2004. Structural and dynamic studies on ligand-free adenylate kinase from Mycobacterium tuberculosis revealed a closed conformation that can be related to the reduced catalytic activity Biochemistry 43 67–77. [DOI] [PubMed] [Google Scholar]
- Muller-Dieckmann H.J. and Schulz G.E. 1995. Substrate specificity and assembly of the catalytic center derived from two structures of ligated uridylate kinase J. Mol. Biol. 246 522–530. [DOI] [PubMed] [Google Scholar]
- Munier-Lehmann H., Burlacu-Miron S., Craescu C.T., Mantsch H.H., Schultz C.P. 1999. A new subfamily of short bacterial adenylate kinase with the Mycobacterium tuberculosis enzyme as a model: A predictive and experimental study Proteins 36 238–248. [PubMed] [Google Scholar]
- Murshudov G.N., Vagin A.A., Lebedev A., Wilson K.S., Dodson E.J. 1999. Efficient anisotropic refinement of macromolecular structures using FFT Acta Crystallogr. D Biol. Crystallogr. 55 247–255. [DOI] [PubMed] [Google Scholar]
- Navaza J. 1994. AMoRe: An automated package for molecular replacement Acta Crystallogr. A 50 157–163. [Google Scholar]
- Perrakis A., Morris R., Lamzin V.S. 1999. Automated protein model building combined with iterative structure refinement Nat. Struct. Biol. 6 458–463. [DOI] [PubMed] [Google Scholar]
- Reinstein J., Gilles A.M., Rose T., Wittinghofer A., Saint Girons I., Barzu O., Surewicz W.K., Mantsch H.H. 1989. Structural and catalytic role of arginine 88 in Escherichia coli adenylate kinase as evidenced by chemical modification and site-directed mutagenesis J. Biol. Chem. 264 8107–8112. [PubMed] [Google Scholar]
- Reinstein J., Schlichting I., Wittinghofer A. 1990. Structurally and catalytically important residues in the phosphate binding loop of adenylate kinase of Escherichia coli Biochemistry 29 7451–7459. [DOI] [PubMed] [Google Scholar]
- Rose T., Glaser P., Surewicz W.K., Mantsch H.H., Reinstein J., Le Blay K., Gilles A.M., Barzu O. 1991. Structural and functional consequences of amino acid substitutions in the second conserved loop of Escherichia coli adenylate kinase J. Biol. Chem. 266 23654–23659. [PubMed] [Google Scholar]
- Schlichting I. and Reinstein J. 1997. Structures of active conformations of UMP kinase from Dictyostelium discoideum suggest phosphoryl transfer is associative Biochemistry 36 9290–9296. [DOI] [PubMed] [Google Scholar]
- Sharma K., Chopra P., Singh Y. 2004. Recent advances towards identification of new drug targets for Mycobacterium tuberculosis Expert Opin. Ther. Targets 8 79–93. [DOI] [PubMed] [Google Scholar]
- Sheng X.R., Xia L., Xian M.P. 1999. An iso-random Bi Bi mechanism for adenylate kinase J. Biol. Chem. 274 22238–22242. [DOI] [PubMed] [Google Scholar]
- Yan H. and Tsai M.D. 1999. Nucleoside monophosphate kinases: Structure, mechanism, and substrate specificity Adv. Enzymol. Relat. Areas Mol. Biol. 73 103–134. [DOI] [PubMed] [Google Scholar]