

Abstract
Studies that compare proteins from thermophilic and mesophilic organisms can provide insights into ability of thermophiles to function at their high habitat temperatures and may provide clues that enable us to better define the forces that stabilize all proteins. Most of the comparative studies have focused on thermal stability and show, as expected, that thermophilic proteins have higher Tm values than their mesophilic counterparts. Although these comparisons are useful, more detailed thermodynamic analyses are required to reach a more complete understanding of the mechanisms thermophilic protein employ to remain folded over a wider range of temperatures. This complete thermodynamic description allows one to generate a stability curve for a protein that defines how the conformational stability (ΔG) varies with temperature. Here we compare stability curves for many pairs of homologous proteins from thermophilic and mesophilc organisms. Of the basic methods that can be employed to achieve enhanced thermostability, we find that most thermophilic proteins use the simple method that raises the ΔG at all temperatures as the principal way to increase their Tm. We discuss and compare this thermodynamic method with the possible alternatives. In addition we propose ways that structural alterations and changes to the amino acid sequences might give rise to varied methods used to obtain thermostability.
Keywords: protein folding, thermodynamics, protein stability curves, thermophiles, thermostability
Life exists almost everywhere on the earth, from deep-sea hydrothermal vents to the heights of the Himalayas, from boiling waters of hot springs to the cold expanses of Antarctica. The organisms that inhabit and have adapted to these extreme and diverse environments are often classified by their altered habitat, such as temperature adaptations (psychrophiles to hyperthermophiles), high salinity adaptations (halophiles), pH adaptations (acidophiles and alkaliphiles), and pressure adaptation (barophiles), to name a few groups. In general, these organisms are often called extremophiles and have been of interest to many protein chemists over the years, dating back to early studies by Perutz and colleagues (Perutz and Raidt 1975; Perutz 1978). In case of adaptations to extremes of pH, salinity, and pressure, membrane components and protective small molecules often play an important role (Jaenicke 1991) and these have been studied quite extensively (Yancey et al. 1982; van de Vossenberg et al. 1998). For temperature adaptations, however, environmental stress generally cannot be avoided by compensatory mechanisms, and thus the cellular components themselves, specifically the proteins, have to achieve thermostability (Jaenicke and Zavodszky 1990). For this reason, much interest has been directed to understanding how proteins from thermophilic organisms retain their structure and function at these elevated temperatures (Argos et al. 1979; Rees and Adams 1995; Somero 1995; Vieille and Zeikus 1996; Jaenicke 1998; Sterner and Liebl 2001).
Proteins perform important tasks in all biological systems, and they do so by maintaining a specific globular conformation. This functional state, called the native state, is marginally stabilized in a balancing act of opposing forces. The players in this balancing act have long been identified (Kauzmann 1959), although their relative contributions have been debated (Dill 1990; Creighton 1992; Rose and Wolfenden 1993; Pace et al. 1996; Honig 1999). The major stabilizing forces include the hydrophobic effect and hydrogen bonding while conformational entropy favors the unfolded state. The forces stabilizing the native state outweigh the disruptive forces marginally in a folded protein, in the range of 5–10 kcal mol−1 (Pace 1975). This balance of forces is known as the conformational stability of a protein and is defined thermodynamically as the free energy change, ΔG, for the native ↔ unfolded state transition. Measurements of and studies on protein stability have remained important over several decades owing to the central role these macromolecules play in maintaining life and their involvement in many diseases affecting humans. Studies on protein stability explore the sequence–structure–stability relationship, with stability being the measured thermodynamic quantity, since sequence defines structure, whose interactions afford stability. Sequence is also the variable that organisms alter as they evolve to adapt their proteins to the environments they inhabit. The stability of proteins is usually determined experimentally by perturbing the native state using temperature or denaturing solvent additives (urea, GuHCl) and following this “reaction” by direct (calorimetric) and indirect (spectroscopic) probes (for further details, see Lopez and Makhatadze 2002; Grimsley et al. 2003).
Since this review focuses on thermodynamics of protein stability and protein stability curves in particular, an introduction to these concepts is in order. Becktel and Schellman (1987) introduced protein stability curves, showing plots of free energy of stabilization (ΔG) as a function of temperature (Fig. 1). Such data are described by a modified version of the Gibbs–Helmholtz equation, and important thermodynamic parameters can be determined (Equation 1):
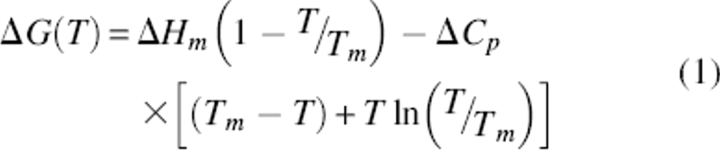 |
where ΔG(T) is the free energy at a temperature T; ΔHm is the change in enthalpy at Tm; ΔCp is the change in heat capacity associated with the unfolding of the protein; and Tm is the melting temperature or the temperature at midpoint of transition from native to denatured state. Other parameters of interest that can be calculated using modifications of Equation 1 include TS and ΔGS, where TS is the temperature of maximum stability or temperature where the change in entropy between native and denatured states is zero and ΔGS is the conformational stability at this temperature. Protein stability curves also allow the calculation of conformational stability at any temperature, including the habitat temperature of an organism (TE).
Figure 1.

A stability curve for a hypothetical protein (Becktel and Schellman 1987). The stability of a protein is plotted as a function of temperature, and the data can be explained by a modified version of the Gibbs Helmholtz equation (Equation 1). Some key thermodynamic parameters are also marked on the plot. (For an explanation of the terms used, refer to the text.)
Three-dimensional structures of proteins are often instrumental in our attempts to understand protein stability and the forces involved. Atomic resolution structures are required for enumerating stabilizing interactions like hydrogen bonds and electrostatic interactions; they are also necessary for theoretical studies attempting to correlate features like buried surface areas with magnitude of stabilizing forces like the hydrophobic effect (Pace 1992). Structures are also used to measure ΔASA or change in solvent accessible surface area upon unfolding of a protein. It has been shown that ΔASA correlates with thermodynamic quantities like ΔCp (Livingstone et al. 1991; Murphy and Freire 1992; Spolar et al. 1992) and the m-value (Myers et al. 1995), a parameter that is used to describe the denaturant-induced unfolding of a protein. Protein structures also emphasize the fact that the native state is held together by a large number of weak noncovalent interactions between constituent amino acids. The fact that proteins are only marginally stable in general makes the ability of thermophilic proteins to function particularly intriguing because, unlike membranes that show heterogeneity in their building block lipids (Russell and Fukunaga 1990), proteins are composed of the same 20 amino acids irrespective of the organism and its habitat.
There have been several studies designed to understand the thermodynamic strategies that proteins from thermophiles use to remain folded at their high habitat temperatures. Proteins from thermophiles alter their sequence such that it optimizes the interactions holding their native conformations together; these optimizations in turn alter key thermodynamic parameters like ΔCp, ΔG, and ΔH in a way that “tunes” the stability characteristics to the habitat of the organism. As protein chemists, we can measure these cardinal parameters, construct stability curves, and possibly learn about the strategies employed in thermostabilization. Nojima et al. (1977) proposed three different methods of modulating the stability curve of a protein to achieve higher thermostability (greater Tm; Fig. 2). Briefly, a hypothetical mesophilic protein can (I) raise the entire stability curve to higher ΔG so it now has a higher Tm, (II) broaden its stability curve so it now intersects the abscissa at a higher temperature, or (III) shift the entire stability curve to the right (to higher temperatures). All three methods of achieving higher thermostability have been observed in nature, some independently and others in combination. These methods have underlying thermodynamic mechanisms, for example, increasing the value of ΔHS (the change in enthalpy measured at TS) without compensating changes in ΔS will result in a similar stability curve, but with higher ΔG values at all temperatures (method I). A broadened stability curve (method II) is caused by a reduced ΔCp. Lowering the ΔS or the change in entropy for the folding transition shifts the TS to higher temperatures and has the effect of shifting the stability curve to the right (method III).
Figure 2.

Stability curves showing different methods to achieve a higher Tm. Starting with a stability curve for a hypothetical mesophilic protein (solid line), the protein may increase Tm by shifting the curve up (method I [diamonds]), by making the curve flatter (method II [circles]), or by shifting the curve to the right (method III [squares]). The thermodynamic bases and explanations for all these methods are discussed in the text.
Here we have compiled results from studies reporting thermodynamic characterization of proteins (or domains) from thermophilic species. More specifically, thermodynamic parameters have been compiled so comparisons can be made with values from mesophilic homologs where possible and conclusions drawn on the mode of thermostabilization employed in each case. Our results show which mode of thermostabilization is more commonly employed and we discuss possible reasons for the results.
Construction of the database
A literature search was performed to find experimental thermodynamic characterization of proteins from thermophilic organisms. Our search focused on studies reporting a comparison of thermodynamic data on homologous proteins from thermophiles and mesophiles. The results of the literature search were augmented with data from the Protherm database (Bava et al. 2004). In all, we found 26 sets of proteins for which conclusions concerning the thermodynamic mode of stabilization have been made or can be, based on the information provided (Table 1). Of the 26 sets of proteins, 19 make comparisons with a homologous protein from a different organism, four make comparisons with a collection of similarly sized proteins, and the remaining three do not make comparisons with other proteins. Most of the thermodynamic data are from the analyses of circular dichroism (CD) and differential scanning calorimetry (DSC) unfolding experiments, either in the presence or the absence of denaturants like urea or GuHCl; only in the case of the ferredoxin proteins was HD exchange as monitored by NMR used to estimate stability (Pfeil et al. 1997).
Table 1.
Thermodynamic parameters for homologous proteins derived from mesophiles and thermophiles

Table 1.
Continued

The transitions from native to denatured state exhibited a two-state behavior for most proteins in our compilation. Although nine proteins were dimers in solution, they were shown to follow the two-state model of folding, and the remaining 17 were monomeric and also followed the two-state model. There is one pair of proteins, namely the IPMD enzymes from Thermus thermophilus and Escherichia coli (Motono et al. 2001), where the former appears to be two-state and the latter follows a three-state unfolding model (entry 13 in Table 1).
The data show that, on average, a thermophilic protein has a Tm 31.5°C higher than its mesophilic homolog, for the 15 cases where data are available. In the case of ΔG, data were available for 17 of the 26 cases, and the average increase in ΔG of stabilization for the thermophilic homolog was 8.7 kcal mol−1 (for the ΔG values, the average reported includes ΔG values listed in column 6 of Table 1 irrespective of the temperature at which they were measured; for both ΔTm and ΔΔG, entry 13 was not included). The sequence identity for the protein homologs was also compared where sequences were available. For this purpose, we used the CLUSTALW (Thompson et al. 1994) program as implemented on the EBI server (http://www.ebi.ac.uk/clustalw/) with the default settings. For the 16 cases where alignments were possible, the average sequence identity was 51%. The alignment scores varied from 11% identity (for aspartate aminotransferases from Sulfolobus solfataricus and pig heart cytosol) to 82% (for histone proteins MfA and MfoB). The average value of 51% identity is high considering the diversity of the proteins and the sources from which they are derived.
Classification of data and example cases
The proteins compiled here have been classified based on the methods for thermostabilization first proposed by Nojima et al. (1977). However, classification into just three groups as originally proposed was not possible because we find that most proteins use different combinations of these three general methods. Also, for one of the methods proposed (method III), example cases could be found only where this method was used in combination with other methods of thermostabilization. For these reasons we have grouped proteins based on the methods of stabilization (or combinations) and arranged these groups in descending order of the number of occurrences. This scheme gives six groups of proteins; a brief description of each group with details of an example study for each is provided below.
Stabilization by increased ΔG and reduced ΔCp (methods I and II)
The combination of increased ΔG and reduced ΔCp is the most commonly used way to achieve a higher Tm. Of the 26 sets of proteins, there are eight cases where this combination of stabilizing effects is used to increase the Tm of the thermophilic homolog. The proteins in this group are diverse with a range of sizes from the small (67 residues) nonenzymatic cold shock proteins to the large (398 residues) glycolytic enzyme phosphoglycerate kinase. The range of size and function observed points to broad applicability of this combination of methods to enhance thermostability.
A representative example from this group is the RNase H enzyme (entry 2 in Table 1) from Thermus thermophilus (Tt) and Escherichia coli (Ec) (Hollien and Marqusee 1999). RNase H is a small enzyme, which cleaves RNA from RNA–DNA hybrids; the protein from the thermophile (TtRNase H) has 166 residues and shares a 52% sequence identity with its 155-residue mesophilic homolog (EcRNase H). High-resolution structures are available for the two proteins and the structures are very similar (Katayanagi et al. 1992; Ishikawa et al. 1993). To understand the thermodynamic basis of the difference in stability between the two proteins, GuHCl denaturation experiments as a function of temperature were performed to obtain stability curves for these proteins. The data reveal that TtRNase H is indeed more thermostable because of a lowered ΔCp and a higher ΔG over a broad range of temperatures. The ΔCp for the thermophilic protein TtRNase H is 0.9 kcal mol K−1 lower than that for EcRNase H. The ΔGS is ∼5 kcal mol−1 higher than the mesophilic homolog; however, the TS are very similar for the two proteins.
Stabilization by smaller ΔCp (method II)
For the set of proteins included in this study, stabilization by reduced ΔCp is the second most common method to attain a higher Tm. Five thermophilic proteins have a smaller ΔCp compared to their mesophilic homologs, resulting in broader stability curves that allowed them to remain folded over a wider range of temperatures. Proteins in this group show some diversity in terms of their size and function, from the small DNA binding proteins like the 66-residue Sac7d to the large enzyme IPMD, which contains 345 residues. Three of the five proteins in this group are small DNA binding proteins, one is a subdomain of the enzyme phosphoglycerate kinase, and the last protein in this group is IPMD.
As a representative example of this group, consider the Sac7d protein (entry 9 in Table 1) from Sulfolobus acidocaldarius (McCrary et al. 1996). Sac7d is a small DNA binding protein that is highly basic and whose structure has been solved by NMR spectroscopy (Edmondson et al. 1995). No mesophilic homolog for Sac7d is known; hence, comparisons have been made with a number of proteins of similar size (McCrary et al. 1996). Sac7d is stable over a broad range of pH (0–10), and DSC experiments have been performed over this pH range to estimate ΔCp from a Kirchoff analysis. Solvent denaturation experiments with GuHCl were also performed and a global fit to these data provides a ΔCp. This estimate for ΔCp was found to be higher than that obtained from the DSC data, and the authors provide an excellent discussion on possible causes for this disparity (McCrary et al. 1996). In any case, either value of ΔCp produces stability curves that look very similar to those for other mesophilic proteins of similar size, and the use of either value of ΔCp does not cause significant differences in stability at 80°C, the habitat temperature of the organism. Surprisingly, estimates of free energy of at this temperature reveal that the protein is only marginally stable (1.6 kcal mol−1).
Stabilization by a higher overall ΔG (method I)
Stabilization by higher ΔG is found to be as common a method of stabilization as stabilization by reduced ΔCp. In five of the 26 cases, proteins from thermophilic organisms show a higher ΔG over a broad range of temperatures compared to their mesophilic homologs, thus shifting the stability curve up and achieving a higher Tm in the process. This group contains the three cases of HPr homologs, a pair of small archaeal histones and the enzyme cellulase subdomains. Most proteins in this group are small in size, like the archaeal histones (67 residues) or HPr homologs (88 residues), except for the cellulase catalytic domains, which have ∼270 amino acids. The composition of this group, with three of the six proteins being HPr homologs, precludes much insight into the diversity of this class in terms of both size and function.
As a representative example from this group, consider the HPr proteins from the thermophile Bacillus staerothermophilus (Bst) and the mesophile Bacillus subtilus (Bs) (entry 14 in Table 1). HPr or histidine containing protein is involved in the PEP:glycose phosphotransferase system (PTS) in bacteria (Meadow et al. 1990). The BstHPr protein is the same size (88 residues), shows high sequence identity (72%), and has a structure almost identical to that of the mesophilic homolog (Sridharan et al. 2005). The BstHPr protein, however, has a higher Tm (∼15°C) and a larger ΔGS (∼3.2 kcal mol−1). The complete analyses of stability curves reveal that the ΔCp values for the two HPr proteins are very similar at 1.3 kcal mol−1K−1 (Razvi and Scholtz 2006). The TS values for the two proteins also are similar at 24.1° and 24.8°C (Bs and BstHPr respectively). Therefore, this pair of proteins is a nearly perfect example for stabilization by method I or by a higher overall ΔG at all temperatures.
The archaeal histone proteins use different methods to gain thermostabilty
We now consider the cases of four archaeal histone homologs: Three of these homologs were derived from hyperthermophilic archaea, and the fourth, from a mesophilic archaeon (Li et al. 1998). The thermophilic histones from Methanothermus fervidus (MfA, MfB) and Pyrococcus strain GB3a (PyA1) (entries 17, 19, and 23, respectively, in Table 1) were compared to the mesophilic homolog from Methanobacterium formicicum (MfoB). The curious feature of these proteins is that each thermophilic homolog uses a different thermodynamic approach to achieve a higher Tm. The histone MfA, for example (entry 17 in Table 1), belongs to the group of proteins that utilize a higher ΔG at all temperatures (method I). The other two homologs increase their Tm by a combination of all three methods (MfB) or by combining a higher overall ΔG with a higher TS (PyA1).
Stabilization by increased ΔG, smaller ΔCp, and higher TS (methods I, II, and III)
This group of thermophilic proteins achieves higher Tm by a combination of all three methods, and the archaeal histone MfB is representative of this class. This histone attains a higher Tm by combining a reduced ΔCp with higher TS and ΔG (methods I, II, and III). Three other thermophilic proteins were found to use this approach to increase their Tm; two of them are metal-cluster-containing proteins ferridoxin and rubredoxin. The third is the MGMT enzyme, which is also the largest protein in this group (174 residues).
MfB (entry 19 in Table 1) shares 80% sequence identity with MfoB, the mesophilic histone homolog, and both proteins, like other histones studied here, are dimers in solution and unfold in a two-state manner to monomers (Li et al. 1998). DSC and thermal denaturation experiments monitored by CD have been used to construct stability curves for these proteins and the ΔCp estimates are in good agreement. Also, theoretical estimates (Murphy and Freire 1992; Spolar et al. 1992; Myers et al. 1995) for the thermophilic homolog for which a structure is available are in good agreement with experimental values. The ΔCp for histone MfB, the thermophilic homolog, is 1.9 kcal mol−1K−1, which is lower than that for MfoB (2.6 kcal mol−1K−1). The ΔGS for the thermophilic MfB is 14.6 kcal mol−1, which is more than twice that for MfoB. The TS for MfB (40°C) is significantly greater than that for MfoB (32°C). Together these thermodynamic features of the MfB protein cause the Tm to be 113°C, significantly higher than that for MfoB (74.8°C).
Stabilization by increased ΔG and higher TS (methods I and III)
The third thermophilic histone homolog PyA1 represents a small group of proteins that achieve high Tm by combining a higher overall ΔG with a higher TS (methods I and III). Two other thermophilic proteins use this approach to achieve higher Tm: DHFR and cytochrome c-552 from Thermotoga maritima. PyA1 (entry 23 in Table 1) is the same size as the mesophilic homolog, MfoB (67 residues), and they share 57% sequence identity (Li et al. 1998). Analyses of DSC and CD monitored unfolding experiments provided nearly identical ΔCp estimates for the two proteins. The ΔCp for PyA1 is 2.4 kcal mol−1K−1 and for MfoB it is 2.6 kcal mol−1K−1. The ΔGS for PyA1 is, however, larger than for MfoB (17.2 kcal mol−1 vs. 7.2 kcal mol−1). The TS for PyA1 is 44°C, which is significantly higher than that for MfoB (32°C). The high ΔGS and TS for PyA1, in comparison with the mesophilic homolog MfoB, make this protein a good example for thermostabilization by a combination of methods I and III.
Stabilization by reduced ΔCp and higher TS (methods II and III)
The only case we found of a thermophilic protein using a combination of a reduced ΔCp and higher TS to achieve a higher Tm is farnesyl diphosphate/geranylgeranyl diphosphate synthase. This is a dimeric enzyme from Thermococcus kodakiensis that has 343 residues (Fujiwara et al. 2004). GuHCl denaturation experiments performed at different temperatures were combined to construct a stability curve for this enzyme. Other thermodynamic parameters, like Tm (91°C), ΔCp (2 kcal mol−1 K−1), ΔGS (3.8 kcal mol−1), and TS (60°C), were estimated from the stability curve. From comparisons with other thermostable proteins, the investigators concluded that their enzyme achieved a higher Tm by a combination of lower ΔCp with higher TS. No details of this comparison were provided.
General observations on enhanced thermostability
A comprehensive collection of thermodynamic data comparing protein homologs from thermophiles and mesophiles has been compiled. An inspection of this broad compilation lends itself to some conclusions regarding the methods of thermostabilization adopted by proteins from thermophiles and allows us to rank the different modes of thermostabilization originally proposed by Nojima et al. (1977) in terms of their occurrence.
We find that the most common way to attain a higher Tm in proteins from thermophiles is to raise the stability curve to higher values of ΔG (higher intrinsic stability) at all temperatures; 77% of the thermophilic proteins in this study use higher ΔG independently or in combination with other stabilizing effects to increase Tm. The next most popular method used to attain a higher Tm is to lower ΔCp (70% of thermophilic proteins in this study). Finally, the least number of occurrences (31% of the thermophilic proteins in this study) are reported for cases where the thermophilic protein exhibits a higher TS compared to the mesophilic homolog. However, it is pertinent to rationalize these observations based on how it might be easier to adopt one strategy rather than the other in terms of sequence changes because changes in ΔG, ΔCp, or TS are effects of sequence alterations manifested through changes in structure.
To increase the ΔG of a protein at all temperatures, many options are available at the sequence level since any number of interactions like salt bridges, hydrogen bonds, or hydrophobic interactions may be added by single amino acid changes (for example, Pace 2000; Perl et al. 2000)). Similarly, to attain a lower ΔCp, the sequence can be altered in many ways to provide for tighter core packing or by simply promoting structured clusters that persist in the denatured state, since ΔCp is strongly correlated with ΔASA for protein unfolding (Murphy and Freire 1992; Spolar et al. 1992; Myers et al. 1995). For example, it was suggested that a structured cluster in denatured TtRNase H caused the reduced ΔCp for this protein, and this effect could be reversed by a single amino acid change (Guzman-Casado et al. 2003; Robic et al. 2003).
Stabilization by shifting the stability curve to higher temperatures (higher TS) is the least common method in our data set. This might be because very specific changes to the sequence would be required to reduce ΔS or the change in entropy between the folded and the unfolded states. Since this requires that either the entropy of the denatured state be reduced relative to the folded state or the entropy of the folded state be enhanced to more closely match that of the denatured state, it will require rather precise changes in the sequence that affect one of the two states differentially. For example, constraining a certain loop in the denatured state by introduction of proline residues or introduction of glycine residues in structured regions of a protein can reduce ΔS, by decreasing the configurational entropy of the denatured state or increasing it for the native state, respectively. Specialized mutations like these will reduce the ΔS of folding, resulting in higher TS. A curious feature of proteins in this class is that a majority of them are not enzymes. This could be because nonenzymatic proteins are tolerant of more stable conformations (thus more “rigid” conformations as discussed by Jaenicke 2000) afforded by enhanced TS. Unlike a lowered ΔCp, which makes for a shallow stability curve, a high TS (method III) or high ΔG (method I) both provide for higher ΔG at high temperatures.
At the molecular level, however, each of the methods of stabilization are results of features like an increased number of hydrogen bonds, salt bridges, improved core packing, shorter and/or tighter surface loops, enhanced secondary structure propensities, or oligomerization. There have been many studies (Matthews 1993; Vogt and Argos 1997; Vogt et al. 1997; Szilagyi and Zavodszky 2000) that compare homologous proteins with known three-dimensional structures in which the number of stabilizing interactions have been compared with the aim of developing a “unifying set of rules” for thermostabilization (Petsko 2001). Although such a set of rules remains elusive, the results have been used to rationally design variants of proteins with desired properties (Bryan 2000; Eijsink et al. 2004). Computational methods have also been used with some success to rationally design proteins with enhanced thermostability (Dahiyat et al. 1997; Malakauskas and Mayo 1998; Korkegian et al. 2005). Directed evolution is another novel technique used to design protein variants with desired properties, which requires no a priori knowledge of stabilizing/destabilizing interactions like the rational design methodology. This technique has been applied to a large number of proteins with a good measure of success (Sieber et al. 1998; Wintrode and Arnold 2000; Arnold et al. 2001; Eijsink et al. 2005). It remains to be determined which thermodynamic methods have been used to achieve higher thermostability in the proteins designed by these methods. It should be especially enlightening to see the results for proteins designed by directed evolution, since this method is least constrained in terms of the sequence space explored and types of interactions that might be altered.
In conclusion, it appears that molecular studies, as well as those that characterize the thermodynamics, both fail to reveal a single cause (molecular) or effect (thermodynamic) that completely explains the ability of thermophilic proteins to survive and function at their high habitat temperatures. Instead, proteins appear to rely on combinations of stabilizing effects that manifest themselves in alterations of different thermodynamic parameters. Thus, as proteins have taught us time and time again, they are extremely adaptable and there is no single mechanism they use to maintain structure and function at high temperatures.
Acknowledgments
This work was supported by the Robert A. Welch foundation (A-1281) and the NIH (GM52483). We thank Dr. Gerald R. Grimsley and Dr. Beatrice Huyghues-Despointes for critical reading of the manuscript.
Footnotes
Reprint requests to: J. Martin Scholtz, Department of Molecular and Cellular Medicine, Texas A&M University, College Station, TX 77843-1114, USA; e-mail: jm-scholtz@tamu.edu; fax: (979) 847-9481.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062130306.
Abbreviations: Tm, melting temperature or temperature at midpoint of transition from native to denatured state in a thermal denaturation; ΔGT, free energy of stabilization at a temperature T; ΔCp, change in heat capacity associated with protein unfolding; ΔH, change in enthalpy; ΔS, change in entropy; TS, temperature of maximal stability or temperature where change in entropy between native and denatured states is zero; ΔGS, free energy of stabilization at TS; TE, environment or habitat temperature of an organism; ΔASA, change in solvent accessible surface area upon protein unfolding; GuHCl, guanidine hydrochloride; CspB, cold shock protein B; CheY, chemotactic protein Y; HPr, histidine containing phospho-carrier protein; MfA, archaeal histone A from hyperthermophile Methanothermus fervidus; MfB, archaeal histone B from hyperthermophile Methanothermus fervidus; PyA1, archaeal histone from hyperthermophile Pyrococcus strain GB3a; MfoB; archaeal histone B from mesophile Methanobacterium formicicum; IPMD, isopropyl malate dehydrogenase; MGMT, O6Methyl guanine DNA methyl transferase; DHFR, dihydrofolate reductase.
References
- Argos P., Rossman M.G., Grau U.M., Zuber H., Frank G., Tratschin J.D. 1979. Thermal stability and protein structure. Biochemistry 18: 5698–5703. [DOI] [PubMed] [Google Scholar]
- Arnold F.H., Wintrode P.L., Miyazaki K., Gershenson A. 2001. How enzymes adapt: Lessons from directed evolution. Trends Biochem. Sci. 26: 100–106. [DOI] [PubMed] [Google Scholar]
- Arnone M.I., Birolo L., Pascarella S., Cubellis M.V., Bossa F., Sannia G., Marino G. 1997. Stability of aspartate aminotransferase from Sulfolobus solfataricus. Protein Eng. 10: 237–248. [DOI] [PubMed] [Google Scholar]
- Bava K.A., Gromiha M.M., Uedaira H., Kitajima K., Sarai A. 2004. ProTherm, version 4.0: Thermodynamic database for proteins and mutants. Nucleic Acids Res. 32: D120–D121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beadle B.M., Baase W.A., Wilson D.B., Gilkes N.R., Shoichet B.K. 1999. Comparing the thermodynamic stabilities of a related thermophilic and mesophilic enzyme. Biochemistry 38: 2570–2576. [DOI] [PubMed] [Google Scholar]
- Becktel W.J. and Schellman J.A. 1987. Protein stability curves. Biopolymers 26: 1859–1877. [DOI] [PubMed] [Google Scholar]
- Bryan P.N. 2000. Protein engineering of Subtilisin. Biochim. Biophys. Acta 1543: 203–222. [DOI] [PubMed] [Google Scholar]
- Consalvi V., Chiaraluce R., Giangiacomo L., Scandurra R., Christova P., Karshikoff A., Knapp S., Ladenstein R. 2000. Thermal unfolding and conformational stability of the recombinant domain II of glutamate dehydrogenase from the hyperthermophile Thermotoga maritima. Protein Eng. 13: 501–507. [DOI] [PubMed] [Google Scholar]
- Creighton T.E. 1992. Folding and binding. Curr. Opin. Struct. Biol. 2: 1–5. [Google Scholar]
- Dahiyat B.I., Sarisky C.A., Mayo S.L. 1997. De novo protein design: Towards fully automated sequence selection. J. Mol. Biol. 273: 789–796. [DOI] [PubMed] [Google Scholar]
- Dams T. and Jaenicke R. 1999. Stability and folding of dihydrofolate reductase from the hyperthermophilic bacterium Thermotoga maritima. Biochemistry 38: 9169–9178. [DOI] [PubMed] [Google Scholar]
- Deutschman W.A. and Dahlquist F.W. 2001. Thermodynamic basis for the increased thermostability of CheY from the hyperthermophile Thermotoga maritima. Biochemistry 40: 13107–13113. [DOI] [PubMed] [Google Scholar]
- Dill K.A. 1990. Dominant forces in protein folding. Biochemistry 29: 7133–7155. [DOI] [PubMed] [Google Scholar]
- Edmondson S.P., Qiu L., Shriver J.W. 1995. Solution structure of the DNA-binding protein Sac7d from the hyperthermophile Sulfolobus acidocaldarius. Biochemistry 34: 13289–13304. [DOI] [PubMed] [Google Scholar]
- Eijsink V.G., Bjork A., Gaseidnes S., Sirevag R., Synstad B., van den Burg B., Vriend G. 2004. Rational engineering of enzyme stability. J. Biotechnol. 113: 105–120. [DOI] [PubMed] [Google Scholar]
- Eijsink V.G., Gaseidnes S., Borchert T.V., van den Burg B. 2005. Directed evolution of enzyme stability. Biomol. Eng. 22: 21–30. [DOI] [PubMed] [Google Scholar]
- Fujiwara S., Yamanaka A., Hirooka K., Kobayashi A., Imanaka T., Fukusaki E. 2004. Temperature-dependent modulation of farnesyl diphosphate/geranylgeranyl diphosphate synthase from hyperthermophilic archaea. Biochem. Biophys. Res. Commun. 325: 1066–1074. [DOI] [PubMed] [Google Scholar]
- Grattinger M., Dankesreiter A., Schurig H., Jaenicke R. 1998. Recombinant phosphoglycerate kinase from the hyperthermophilic bacterium Thermotoga maritima: Catalytic, spectral and thermodynamic properties. J. Mol. Biol. 280: 525–533. [DOI] [PubMed] [Google Scholar]
- Grimsley G.R., Huyghues-Despointes B.M.P., Pace C.N., Scholtz J.M. 2003. Measuring the conformational stability of a protein. In Purifying proteins for proteomics: A laboratory manual (ed. Simpson R.J.) . pp. 535–566. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Guzman-Casado M., Parody-Morreale A., Robic S., Marqusee S., Sanchez-Ruiz J.M. 2003. Energetic evidence for formation of a pH-dependent hydrophobic cluster in the denatured state of Thermus thermophilus ribonuclease H. J. Mol. Biol. 329: 731–743. [DOI] [PubMed] [Google Scholar]
- Hernandez G. and LeMaster D.M. 2001. Reduced temperature dependence of collective conformational opening in a hyperthermophile rubredoxin. Biochemistry 40: 14384–14391. [DOI] [PubMed] [Google Scholar]
- Hiller R., Zhou Z.H., Adams M.W., Englander S.W. 1997. Stability and dynamics in a hyperthermophilic protein with melting temperature close to 200°C. Proc. Natl. Acad. Sci. 94: 11329–11332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J. and Marqusee S. 1999. A thermodynamic comparison of mesophilic and thermophilic ribonucleases H. Biochemistry 38: 3831–3836. [DOI] [PubMed] [Google Scholar]
- Honig B. 1999. Protein folding: From the Levinthal paradox to structure prediction. J. Mol. Biol. 293: 283–293. [DOI] [PubMed] [Google Scholar]
- Hu C.Q. and Sturtevant J.M. 1989. A differential scanning calorimetric study of the binding of sulfate ion and of Cibacron blue F3GA to yeast phosphoglycerate kinase. Biochemistry 28: 813–818. [DOI] [PubMed] [Google Scholar]
- Ionescu R.M., Smith V.F., O'Neill Jr. J.C., Matthews C.R. 2000. Multistate equilibrium unfolding of Escherichia coli dihydrofolate reductase: Thermodynamic and spectroscopic description of the native, intermediate, and unfolded ensembles. Biochemistry 39: 9540–9550. [DOI] [PubMed] [Google Scholar]
- Ishikawa K., Okumura M., Katayanagi K., Kimura S., Kanaya S., Nakamura H., Morikawa K. 1993. Crystal structure of ribonuclease H from Thermus thermophilus HB8 refined at 2.8 Å resolution. J. Mol. Biol. 230: 529–542. [DOI] [PubMed] [Google Scholar]
- Jacob M., Holtermann G., Perl D., Reinstein J., Schindler T., Geeves M.A., Schmid F.X. 1999. Microsecond folding of the cold shock protein measured by a pressure-jump technique. Biochemistry 38: 2882–2891. [DOI] [PubMed] [Google Scholar]
- Jaenicke R. 1991. Protein stability and molecular adaptation to extreme conditions. Eur. J. Biochem. 202: 715–728. [DOI] [PubMed] [Google Scholar]
- Jaenicke R. 1998. What ultrastable globular proteins teach us about protein stabilization. Biochemistry (Mosc.) 63: 312–321. [PubMed] [Google Scholar]
- Jaenicke R. 2000. Do ultrastable proteins from hyperthermophiles have high or low conformational rigidity? Proc. Natl. Acad. Sci. 97: 2962–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenicke R. and Bohm G. 2001. Thermostability of proteins from Thermotoga maritima. Methods Enzymol. 334: 438–469. [DOI] [PubMed] [Google Scholar]
- Jaenicke R. and Zavodszky P. 1990. Proteins under extreme physical conditions. FEBS Lett. 268: 344–349. [DOI] [PubMed] [Google Scholar]
- Kanaya S. and Itaya M. 1992. Expression, purification, and characterization of a recombinant ribonuclease H from Thermus thermophilus HB8. J. Biol. Chem. 267: 10184–10192. [PubMed] [Google Scholar]
- Katayanagi K., Miyagawa M., Matsushima M., Ishikawa M., Kanaya S., Nakamura H., Ikehara M., Matsuzaki T., Morikawa K. 1992. Structural details of ribonuclease H from Escherichia coli as refined to an atomic resolution. J. Mol. Biol. 223: 1029–1052. [DOI] [PubMed] [Google Scholar]
- Kauzmann W. 1959. Some factors in the interpretation of protein denaturation. Adv. Protein Chem. 14: 1–63. [DOI] [PubMed] [Google Scholar]
- Knapp S., Karshikoff A., Berndt K.D., Christova P., Atanasov B., Ladenstein R. 1996. Thermal unfolding of the DNA-binding protein Sso7d from the hyperthermophile Sulfolobus solfataricus. J. Mol. Biol. 264: 1132–1144. [DOI] [PubMed] [Google Scholar]
- Knapp S., Mattson P.T., Christova P., Berndt K.D., Karshikoff A., Vihinen M., Smith C.I., Ladenstein R. 1998. Thermal unfolding of small proteins with SH3 domain folding pattern. Proteins 31: 309–319. [DOI] [PubMed] [Google Scholar]
- Korkegian A., Black M.E., Baker D., Stoddard B.L. 2005. Computational thermostabilization of an enzyme. Science 308: 857–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.F., Allen M.D., Bycroft M., Wong K.B. 2005. Electrostatic interactions contribute to reduced heat capacity change of unfolding in a thermophilic ribosomal protein l30e. J. Mol. Biol. 348: 419–431. [DOI] [PubMed] [Google Scholar]
- Li W.T., Grayling R.A., Sandman K., Edmondson S., Shriver J.W., Reeve J.N. 1998. Thermodynamic stability of archaeal histones. Biochemistry 37: 10563–10572. [DOI] [PubMed] [Google Scholar]
- Livingstone J.R., Spolar R.S., Record Jr. M.T. 1991. Contribution to the thermodynamics of protein folding from the reduction in water-accessible nonpolar surface area. Biochemistry 30: 4237–4244. [DOI] [PubMed] [Google Scholar]
- Lopez M.M. and Makhatadze G.I. 2002. Differential scanning calorimetry. Methods Mol. Biol. 173: 113–119. [DOI] [PubMed] [Google Scholar]
- Malakauskas S.M. and Mayo S.L. 1998. Design, structure and stability of a hyperthermophilic protein variant. Nat. Struct. Biol. 5: 470–475. [DOI] [PubMed] [Google Scholar]
- Matthews B.W. 1993. Structural and genetic analysis of protein stability. Annu. Rev. Biochem. 62: 139–160. [DOI] [PubMed] [Google Scholar]
- McCrary B.S., Edmondson S.P., Shriver J.W. 1996. Hyperthermophile protein folding thermodynamics: Differential scanning calorimetry and chemical denaturation of Sac7d. J. Mol. Biol. 264: 784–805. [DOI] [PubMed] [Google Scholar]
- Meadow N.D., Fox D.K., Roseman S. 1990. The bacterial phosphoenolpyruvate: Glycose phosphotransferase system. Annu. Rev. Biochem. 59: 497–542. [DOI] [PubMed] [Google Scholar]
- Milla M.E., Brown B.M., Sauer R.T. 1994. Protein stability effects of a complete set of alanine substitutions in Arc repressor. Nat. Struct. Biol. 1: 518–523. [DOI] [PubMed] [Google Scholar]
- Motono C., Oshima T., Yamagishi A. 2001. High thermal stability of 3-isopropylmalate dehydrogenase from Thermus thermophilus resulting from low ΔCp of unfolding. Protein Eng. 14: 961–966. [DOI] [PubMed] [Google Scholar]
- Murphy K.P. and Freire E. 1992. Thermodynamics of structural stability and cooperative folding behavior in proteins. Adv. Protein Chem. 43: 313–361. [DOI] [PubMed] [Google Scholar]
- Myers J.K., Pace C.N., Scholtz J.M. 1995. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4: 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima H., Ikai A., Oshima T., Noda H. 1977. Reversible thermal unfolding of thermostable phosphoglycerate kinase. Thermostability associated with mean zero enthalpy change. J. Mol. Biol. 116: 429–442. [DOI] [PubMed] [Google Scholar]
- Nojima H., Hon-Nami K., Oshima T., Noda H. 1978. Reversible thermal unfolding of thermostable cytochrome c-552. J. Mol. Biol. 122: 33–42. [DOI] [PubMed] [Google Scholar]
- Ohmae E., Kurumiya T., Makino S., Gekko K. 1996. Acid and thermal unfolding of Escherichia coli dihydrofolate reductase. J. Biochem. 120: 946–953. [DOI] [PubMed] [Google Scholar]
- Pace C.N. 1975. The stability of globular proteins. CRC Crit. Rev. Biochem. 3: 1–43. [DOI] [PubMed] [Google Scholar]
- Pace C.N. 1992. Contribution of the hydrophobic effect to globular protein stability. J. Mol. Biol. 226: 29–35. [DOI] [PubMed] [Google Scholar]
- Pace C.N. 2000. Single surface stabilizer. Nat. Struct. Biol. 7: 345–346. [DOI] [PubMed] [Google Scholar]
- Pace C.N., Shirley B.A., McNutt M., Gajiwala K. 1996. Forces contributing to the conformational stability of proteins. FASEB J. 10: 75–83. [DOI] [PubMed] [Google Scholar]
- Perl D., Mueller U., Heinemann U., Schmid F.X. 2000. Two exposed amino acid residues confer thermostability on a cold shock protein. Nat. Struct. Biol. 7: 380–383. [DOI] [PubMed] [Google Scholar]
- Perutz M.F. 1978. Electrostatic effects in proteins. Science 201: 1187–1191. [DOI] [PubMed] [Google Scholar]
- Perutz M.F. and Raidt H. 1975. Stereochemical basis of heat stability in bacterial ferredoxins and in haemoglobin A2. Nature 255: 256–259. [DOI] [PubMed] [Google Scholar]
- Petsko G.A. 2001. Structural basis of thermostability in hyperthermophilic proteins, or “there's more than one way to skin a cat”. Methods Enzymol. 334: 469–478. [DOI] [PubMed] [Google Scholar]
- Pfeil W., Gesierich U., Kleemann G.R., Sterner R. 1997. Ferredoxin from the hyperthermophile Thermotoga maritima is stable beyond the boiling point of water. J. Mol. Biol. 272: 591–596. [DOI] [PubMed] [Google Scholar]
- Razvi A. and Scholtz J.M. 2006. A thermodynamic comparison of HPr proteins from extremophilic organisms. Biochemistry 45: 4084–4092. [DOI] [PubMed] [Google Scholar]
- Rees D.C. and Adams M.W. 1995. Hyperthermophiles: Taking the heat and loving it. Structure 3: 251–254. [DOI] [PubMed] [Google Scholar]
- Robic S., Guzman-Casado M., Sanchez-Ruiz J.M., Marqusee S. 2003. Role of residual structure in the unfolded state of a thermophilic protein. Proc. Natl. Acad. Sci. 100: 11345–11349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose G.D. and Wolfenden R. 1993. Hydrogen bonding, hydrophobicity, packing, and protein folding. Annu. Rev. Biophys. Biomol. Struct. 22: 381–415. [DOI] [PubMed] [Google Scholar]
- Ruiz-Sanz J., Filimonov V.V., Christodoulou E., Vorgias C.E., Mateo P.L. 2004. Thermodynamic analysis of the unfolding and stability of the dimeric DNA-binding protein HU from the hyperthermophilic eubacterium Thermotoga maritima and its E34D mutant. Eur. J. Biochem. 271: 1497–1507. [DOI] [PubMed] [Google Scholar]
- Russell N.J. and Fukunaga N. 1990. A comparison of thermal adaptation of membrane lipids in psychrophilic and thermophilic bacteria. FEMS Microbiol. Rev. 75: 171–182. [Google Scholar]
- Schindler T. and Schmid F.X. 1996. Thermodynamic properties of an extremely rapid protein folding reaction. Biochemistry 35: 16833–16842. [DOI] [PubMed] [Google Scholar]
- Shiraki K., Nishikori S., Fujiwara S., Hashimoto H., Kai Y., Takagi M., Imanaka T. 2001. Comparative analyses of the conformational stability of a hyperthermophilic protein and its mesophilic counterpart. Eur. J. Biochem. 268: 4144–4150. [DOI] [PubMed] [Google Scholar]
- Sieber V., Pluckthun A., Schmid F.X. 1998. Selecting proteins with improved stability by a phage-based method. Nat. Biotechnol. 16: 955–960. [DOI] [PubMed] [Google Scholar]
- Somero G.N. 1995. Proteins and temperature. Annu. Rev. Physiol. 57: 43–68. [DOI] [PubMed] [Google Scholar]
- Spolar R.S., Livingstone J.R., Record Jr. M.T. 1992. Use of liquid hydrocarbon and amide transfer data to estimate contributions to thermodynamic functions of protein folding from the removal of nonpolar and polar surface from water. Biochemistry 31: 3947–3955. [DOI] [PubMed] [Google Scholar]
- Sridharan S., Razvi A., Scholtz J.M., Sacchettini J.C. 2005. The HPr proteins from the thermophile Bacillus stearothermophilus can form domain-swapped dimers. J. Mol. Biol. 346: 919–931. [DOI] [PubMed] [Google Scholar]
- Sterner R. and Liebl W. 2001. Thermophilic adaptation of proteins. Crit. Rev. Biochem. Mol. Biol. 36: 39–106. [DOI] [PubMed] [Google Scholar]
- Szilagyi A. and Zavodszky P. 2000. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: Results of a comprehensive survey. Struct. Fold. Des. 8: 493–504. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Vossenberg J.L., Driessen A.J., Konings W.N. 1998. The essence of being extremophilic: The role of the unique archaeal membrane lipids. Extremophiles 2: 163–170. [DOI] [PubMed] [Google Scholar]
- Vieille C. and Zeikus J.G. 1996. Thermozymes: Identifying molecular determinants of protein structural and functional stability. Trends Biotechnol. 14: 183–190. [Google Scholar]
- Vogt G. and Argos P. 1997. Protein thermal stability: Hydrogen bonds or internal packing? Fold. Des. 2: S40–S46. [DOI] [PubMed] [Google Scholar]
- Vogt G., Woell S., Argos P. 1997. Protein thermal stability, hydrogen bonds, and ion pairs. J. Mol. Biol. 269: 631–643. [DOI] [PubMed] [Google Scholar]
- Wassenberg D., Welker C., Jaenicke R. 1999. Thermodynamics of the unfolding of the cold-shock protein from Thermotoga maritima. J. Mol. Biol. 289: 187–193. [DOI] [PubMed] [Google Scholar]
- Wintrode P.L. and Arnold F.H. 2000. Temperature adaptation of enzymes: Lessons from laboratory evolution. Adv. Protein Chem. 55: 161–225. [DOI] [PubMed] [Google Scholar]
- Xu S., Qin S., Pan X.M. 2004. Thermal and conformational stability of Ssh10b protein from archaeon Sulfolobus shibattae. Biochem. J. 382: 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancey P.H., Clark M.E., Hand S.C., Bowlus R.D., Somero G.N. 1982. Living with water stress: Evolution of osmolyte systems. Science 217: 1214–1222. [DOI] [PubMed] [Google Scholar]