

Abstract
Thrombospondin-related anonymous protein, TRAP, has a critical role in the hepatocyte invasion step of Plasmodium sporozoites, the transmissible form of the parasite causing malaria. The extracellular domains of this sporozoite surface protein interact with hepatocyte surface receptors whereas its intracellular domain acts as a link to the sporozoite actomyosin motor system. Liver heparan sulfate proteoglycans have been identified as potential ligands for TRAP. Proteoglycan binding has been associated with the A- and TSR domains of TRAP. We present the solution NMR structure of the TSR domain of TRAP and a chemical shift mapping study of its heparin binding epitope. The domain has an elongated structure stabilized by an array of tryptophan and arginine residues as well as disulfide bonds. The fold is very similar to those of thrombospondin type-1 (TSP-1) and F-spondin TSRs. The heparin binding site of TRAP-TSR is located in the N-terminal half of the structure, the layered side chains forming an integral part of the site. The smallest heparin fragment capable of binding to TRAP-TSR is a tetrasaccharide.
Keywords: heparin binding, malaria, NMR spectroscopy, P. falciparum, structure, TRAP, TSR
Plasmodium parasite is the causative agent of malaria. Liver infection by sporozoites, the transmissible form of the parasite from mosquito to human, is an attractive target for malaria vaccine development. Blocking sporozoite invasion or their development in hepatocytes would stop infection before the onset of symptoms, as only the later erythrocytic phases of the infection are associated with symptoms. Two sporozoite surface proteins are known to be involved in the hepatocyte infection. Sporozoites are arrested in the liver most likely by an interaction involving circumsporozoite protein (CS) and heparan sulfate proteoglycans (HSPGs) (Rathore et al. 2001; Sinnis and Nardin 2002). At a later point, thrombospondin-related anonymous protein (TRAP) is released onto the surface of the sporozoite from specialized secretory organelles of the Apicomplexa called the micronemes. Extracellular adhesive domains of TRAP bind to host cell receptors and, powered by an interaction between the TRAP intracellular C-terminal domain and the sporozoite motor system (Kappe et al. 2004), a moving junction is formed, enabling parasite invasion of hepatocytes. HSPGs are at least partially responsible for the interaction between TRAP and hepatocytes (Matuschewski et al. 2002; Akhouri et al. 2004).
Plasmodium CS and TRAP both contain an adhesive thrombospondin type 1 domain, TSR. TSR is a small ∼60-residue domain found in extracellular proteins or in the extracellular part of transmembrane proteins that are involved in immunity, cell adhesion, and neuronal development (Tucker 2004). Structures of TSR domains from thrombospondin-1 (TSP-1; Tan et al. 2002) and F-spondin (PDB codes 1SZL and 1VEX) have been solved. These show that a TSR domain has an elongated structure consisting of an antiparallel three-stranded β-sheet. The domain core is formed by a stacked array of side chains of conserved tryptophans, arginines, and cysteines. TSRs of several proteins have been reported to mediate glycosaminoglycan (GAG) binding.
A 20-residue motif called region II of CS TSR has been implicated as the likely ligand in the adhesion of sporozoite to liver HSPGs (Sinnis and Nardin 2002; Tewari et al. 2002). Similarly, for the TSR domain of TRAP (TRAP-TSR) a role as a mediator in HSPG binding has been inferred (Robson et al. 1995; Matuschewski et al. 2002). TRAP contains a second adhesive domain N-terminal to the TSR, the A domain, which is a member of the von Willebrand factor type A protein family (vWFA). These proteins contain a metal ion-dependent adhesion site, MIDAS, implicated in ligand binding (Lee et al. 1995). In vivo studies, where either the A domain, the TSR domain, or both were mutated and the sporozoite's capability to adhere, glide, and invade a host cell were observed (Matuschewski et al. 2002), indicated that the A domain plays the major role in infectivity. The two adhesive domains were found to interact with distinct receptors, only the TSR interaction being GAG dependent. Binding studies with recombinant proteins (McCormick et al. 1999; Akhouri et al. 2004), however, suggested that both domains interact with GAGs, and that the binding is independent of MIDAS. A cluster of basic residues on the surface of the A domain was suggested to be responsible for the binding interaction (Akhouri et al. 2004).
Interestingly, P. falciparum also possesses a third TSR-containing surface antigen that is expressed at the sporozoite stage and binds to hepatocytes, PfSPATR (secreted protein with altered thrombospondin repeat; Chattopadhyay et al. 2003). Its exact function is unclear.
Potential inhibitory means for preventing infection ensues from a detailed view of the events connected to the hepatocyte invasion step of Plasmodium sporozoites. To this end, we have determined the solution NMR structure of the TSR domain of TRAP and, through chemical shift mapping, its interaction surface with heparin. We have also identified the minimum number of disaccharide units necessary for the interaction to take place. The studied TRAP-TSR domain corresponds to residues 242–288 of P. falciparum TRAP (Swiss-Prot code P16893).
Results
Solution structure determination of TRAP-TSR
The 2D 1H, 15N-HSQC spectrum of TRAP-TSR shows 44 of the 46 possible backbone amide cross peaks that are well dispersed, except for residues W11, Q39, and V7, K26, I29. The backbone amide cross peaks of the two most N-terminal residues (G1 and S2, which come from the expression system and are not part of the native domain) are not visible in the spectrum. Additionally, cross peaks of all glutamine and tryptophan side chain N-H pairs as well as three out of four arginine side chain N-H pairs are present. A basic set of triple resonance spectra (HNCACB, CBCA(CO)NH, HNCO, CC(CO)NH, and HCCH-COSY; Sattler et al. 1999; Permi and Annila 2004) acquired from a 13C, 15N-double labeled TRAP-TSR sample resulted in a 98.8% 1H assignment, leaving only the HN, Hα protons of G1 and HN proton of S2 unassigned. The assigned chemical shifts have been deposited in the BioMagResBank with the accession code 6865.
We determined the structure of TRAP-TSR by automated iterative NOE assignment-structure calculation protocol (Herrmann et al. 2002). Except for the two termini, a well-defined ensemble of structures was obtained (Fig. ▶; Table ▶). The number of distance restraints per residue is ∼15. A typical distribution pattern is observed in which restraints in regular structures outnumber those in loops. This leads to somewhat higher root mean square deviation (RMSD) values in the loops. RMSD to the mean structure for residues 4–46 is ∼0.7 Å for backbone atoms and ∼1.2 Å for all heavy atoms. The atomic coordinates of TRAP-TSR were deposited in the PDB with the accession code 2BBX.
Figure 1.
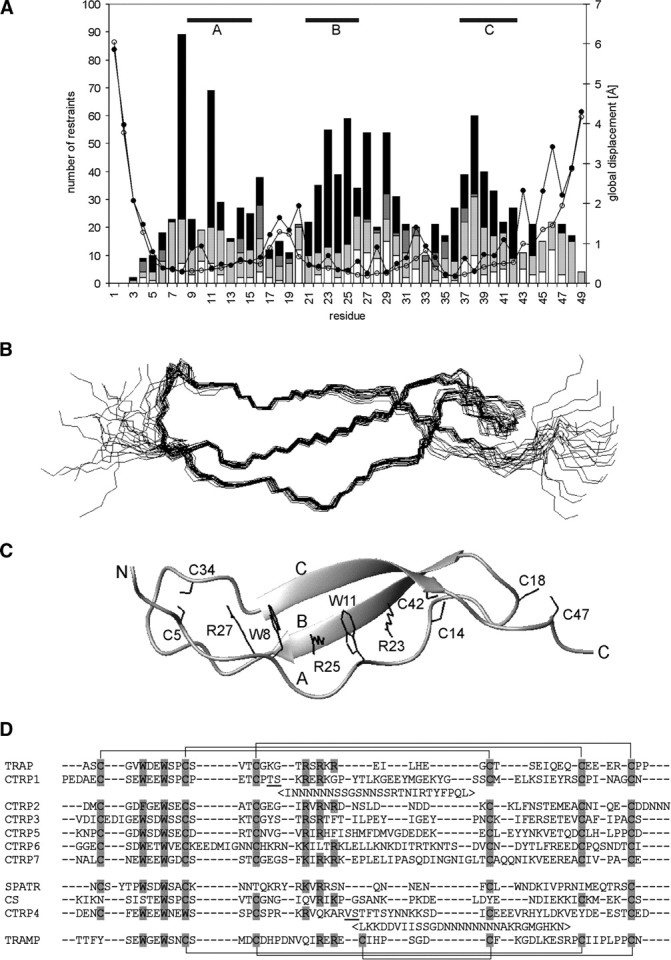
Structure of TRAP-TSR. (A) Number of NOE restraints [(black) long range, i − j ≥ 5; (dark gray) medium range, i − j < 5; (light gray) sequential, i − j = 1; (white) intraresidual, i − j = 0] and RMSD [(open circles) backbone atoms; (filled circles) all heavy atoms] per residue. The areas of regular structure are shown above. (B) Ensemble of 20 energy minimized structures of TRAP-TSR domain; (C) secondary structure together with the stabilizing stack of tryptophan, arginine, and cysteine side chains shown specifically. Protons are omitted from the figure for reasons of clarity. The three strands and the two termini are marked with capital letters. (D) Sequence alignment of P. falciparum protein domains reported to have sequence similarity to TSRs. The proteins are TRAP, CTRP (CS-protein-TRAP-related protein; Trottein et al. 1995) with seven TSR domains, SPATR (Chattopadhyay et al. 2003), CS (Dame et al. 1984), and TRAMP (thrombospondin-related apical merozoite protein; Thompson et al. 2004). TRAP and six of the CTRP TSRs have their six cysteines organized in the manner typical for Group 2 TSRs. TRAMP TSR belongs to Group 1 TSRs. The potential locations of disulfide bonds are shown above the alignment (Group 2) and below it (Group 1). SPATR, CS, and CTRP4 have a distinct pattern of conserved residues and possibly divergent structures. Potential disulfide and stack forming residues are highlighted. CTRP1 and CTRP4 have long insertions between the underlined residues. The location of these insertions is as in Tan et al. (2002). The alignment was made with ClustalW (1.83) by excluding the insertions.
Table 1.
NMR restraints and structural statistics for the ensemble of 20 best conformers of TRAP-TSR
TRAP-TSR folds into the general Arg-Trp layered elongated TSR structure
The TRAP-TSR domain has an elongated fold formed of an antiparallel three-stranded β-sheet (Fig. ▶). Strands B and C form a regular β-sheet (residues 21–26 and 37–42), whereas strand A is irregular with a rippled pattern (residues 9–15). Multiple hydrogen bonds between backbone and side chain atoms as well as interactions between the interleaved side chains of two tryptophans (W8, W11) and three arginines (R23, R25, R27) stabilize the structure. A stabilizing cation-π interaction is likely to form between lysine or arginine and an aromatic side chain of phenylalanine, tyrosine, or tryptophan (Gallivan and Dougherty 1999). As determined by the program CAPTURE (Gallivan and Dougherty 1999), all four possible cation-π interactions between the stacked arginines and tryptophans (R23–W11, R25–W11, R25–W8, R27–W8) are deemed energetically significant in the TRAP-TSR structure. Additional stability is brought to the structure by three disulfide bonds at the ends of the elongated structure, one on the N-terminal side (C5–C34) and two on the C-terminal side (C14-C42, C18–C47).
The fold of TRAP-TSR is very similar to those of the previously solved TSR domains (Fig. ▶), namely, thrombospondin-1 (TSP-1) TSR domains 2 and 3 (Tan et al. 2002) and F-spondin TSR domains 1 and 4 (PDB codes 1SZL and 1VEX). The three strands and the short loop 1 between strands A and B superimpose well, with RMSDs varying from approximately 1.0 Å to 2.1 Å depending on the structure. The closest similarity is found between TRAP-TSR and F-spondin TSR-4. The loop 2 between strands B and C shows more variation. TRAP-TSR has the shortest loop, with 10 residues, while the longest ones are found in TSP-1, with 19 residues in both TSR domains. A stacked array of tryptophans, arginines, and cysteines is observed in each structure. Some variation is found in the number and type of residues participating in the stack formation. TRAP-TSR has a disulfide bonding pattern similar to that observed in F-spondin TSRs. The N-terminal disulfide links the N terminus and loop 2. In TSP-1 TSRs, a link within loop 2 is observed. This does not, however, have any effect on the similarity of the fold.
Figure 2.

Comparison of TSR domain structures. Ribbon structures of F-spondin TSR-4 domain (PDB code 1VEX) (A), TRAP-TSR (B), and TSP-1 TSR-2 domain (PDB code 1LSL) (C) are shown. The structures are in the same orientation with the residues forming the stacked structure specifically shown, with tryptophans in red, arginines in blue, isoleucine in green, and cysteines in yellow. The common fold is evident even though the disulfide bonding pattern differs.
A DALI program (Holm and Sander 1993) search from the PDB revealed similar folds between TRAP-TSR and TSP-1 TSR 2 (Z-score 3.7, Cα trace RMSD 2.9 Å), F-spondin TSR-1 (Z-score 3.6, Cα trace RMSD 2.3 Å), and a minor similarity with Escherichia coli ADP-ribose pyrophosphatase (Z-score 2.2, Cα trace RMSD 3.3 Å). The N-terminal domain of the latter has a three-stranded antiparallel β-sheet fold, which is involved in homodimer stabilization (Gabelli et al. 2001). It interacts with the C-terminal domain of the other monomer through an interface of many aromatic residues. It does not contain an array of stacked Trp, Arg, and Cys side chains.
Heparin binds specifically to the N-terminal half of TRAP-TSR
A potential heparin binding site is identified on the surface of TRAP-TSR (Fig. ▶). The three stacked arginines form a positively charged region in the N-terminal half of the molecule. Heparin is a highly sulfated polysaccharide, and could form a complementary surface in an interaction with the TSR domain. NMR chemical shifts are very sensitive to changes in local electronic environment, a fact that is successfully exploited in monitoring conformational changes and mapping ligand binding interfaces. We performed chemical shift mapping by the stepwise addition of low-molecular-weight heparin (maximum Mr 3000, corresponding to 5–6 heparin disaccharides) to a 15N-labeled TRAP-TSR sample and by recording a 2D 1H, 15N-HSQC spectrum at each titration point. Selective perturbations in several TRAP-TSR peaks indicate specific binding between TRAP-TSR and heparin (Fig. ▶). Backbone amide group 1H, 15N chemical shift perturbations are confined to two regions of the sequence. The largest changes were observed for S4–G6, W8, and L30–S36 corresponding to the N terminus and loop 2. The chemical shifts of side chain Hɛ1–Nɛ1 of W8 and Hɛ–Nɛ of R27 and R23 also change significantly upon titration. Unfortunately, the R25 side chain Hɛ–Nɛ does not show a cross peak in the spectrum, so its fate upon titration could not be followed. The V7 cross peak is clustered with those of K26 and I29. One of these peaks moves significantly upon addition of heparin, most likely that of either V7 or I29. No additional peaks appeared in the spectrum during titration.
Figure 3.

Characterization of the heparin binding site of TRAP-TSR. (A) A section of the overlaid 2D 1H, 15N-HSQC spectra acquired at different heparin titration points of TRAP-TSR. The concentration of heparin grows in the order 0:1 (red), 0.2:1 (yellow), 0.4:1 (magenta), 0.6:1 (black), 0.8:1 (orange), and 1.2:1 (green). (B) Binding curves for two residues for low-molecular-weight heparin and heparin tetrasaccharide. (C) A graph representing the chemical shift changes per residue for the low-molecular-weight heparin, at a heparin to TRAP-TSR ratio of 1.2:1. Also, the changes occurring in Arg, Trp, and Gln side chains are reported. (D) Illustration of the relation of the chemical shift perturbations to surface charge distribution and to the potential binding site on heparin. At the left, two solution conformations of heparin (Mulloy et al. 1993; PDB code 1HPN) are shown. In the leftmost structure IdoA has the 2S0 conformation, and in the middle structure the 1C4 conformation. IdoA and GlcN code for α-L-iduronic acid and α-D-glucosamine. Two tetrasaccharide units are shown, separated by a dashed line. The binding epitope, formed by the negative cluster of three sulfate groups in successive monosaccharides, is circled. The largest distance between sulfate oxygens is found in the 1C4 conformation, and is ∼10.7 Å. In the upper right structure the charge distribution of the surface of TRAP-TSR is shown, where blue indicates positive and red indicates negative partial charges. The positively charged cluster has approximate dimensions of 15.3 × 13.3 Å, from the average distance in the ensemble of structures between side chain Nɛ atoms of R23 and R27 and that between side chain methyl groups of V7 and T35. In the lower right structure, residues whose Δδ ≥ 0.05 ppm at a titration ratio 1:1.2 of TRAP-TSR to heparin are colored in red, and those with 0.02 < Δδ < 0.05 ppm in orange. Residues whose amide groups were not observed due to overlap are shown in white. Residues G1–S2 and P48–P49 are omitted for clarity.
To determine the minimum length of the binding epitope on the polysaccharide, we also studied the binding of heparin fragments to TRAP-TSR. Our chemically cleaved fragments corresponded to heparin di- and tetrasaccharide units. Only the tetrasaccharide binds to TRAP-TSR. Chemical shift perturbations were observed in the same residues as in the low-molecular-weight heparin titration. However, a clear quantitative difference was seen between the two ligands (Fig. ▶). This difference is most likely associated with the number of binding sites present in the two ligands. If heparin tetrasaccharide is the shortest fragment that binds, low-molecular -weight heparin most likely contains two or three binding sites. The concentration of bound protein increases more rapidly in the low-molecular-weight heparin titration as compared to the tetrasaccharide titration, i.e., the chemical shift changes are larger with the long heparin when the same relative concentrations of the two heparin molecules are titrated.
The peak positions moved as a function of added heparin, which is a sign of fast exchange on the NMR time scale. The dissociation constant of heparin interaction with TRAP-TSR was determined from an average NMR titration curve. The curve included residues that had experienced chemical shift perturbations ≥0.05 ppm at a 1:1.2 protein to heparin ratio. Kd is ∼0.5 mM for the tetrasaccharide and 0.2 mM for low-molecular-weight heparin.
Discussion
Two highly conserved motifs have been identified as being essential for GAG binding of TSRs, namely an N-terminal motif containing tryptophan(s) (Guo et al. 1992a, b; Müller et al. 1993) and a stretch of basic residues ∼10 residues further in the amino acid sequence (Sinnis et al. 1994; Gantt et al. 1997). From our structure, it is clear that both the conserved tryptophans (8WDEW11 in P. falciparum TRAP-TSR) together with the arginines (23RSRKRE28) are essential in holding the correct fold of the domain. Their interleaved positioning enables a stabilizing electrostatic interaction. Together with the disulfide bonds at the two termini, these form the scaffold of the structure. A similar side chain array of conserved arginine and tryptophan side chains has been observed in the structures of TSP-1 TSRs (Tan et al. 2002) as well as in F-spondin TSRs (PDB codes 1SZL and 1VEX).
It has been proposed that TSRs can be divided into two groups based on linkages between the three strands (Tan et al. 2002). The major difference between the groups is the N-terminal disulfide bond. In group 1, the bond is formed between cysteines within loop 2, whereas in group 2, one cysteine resides in the loop and the other one in the N-terminal part of the sequence Figs. (▶, ▶). TRAP-TSR is a member of group 2 TSRs.
Besides being structurally important, the conserved arginine and tryptophan residues form an integral part of the binding site for heparin. Chemical shift perturbations observed in the chemical shift mapping experiments performed indicate that TRAP-TSR binds specifically to heparin at a site of interaction localized in the N-terminal half of the domain on the side of the layer of arginine and tryptophan side chains (Fig. ▶). This interaction is likely to be at least partially electrostatic by nature since the depicted site largely corresponds to a positively charged surface region found at the N-terminal half of TRAP-TSR. At the C-terminal half, several glutamate residues (E40, E43, E44, and E45) on the same face of the molecule form an oppositely charged surface. A positively charged surface is observed in the TSP-1 structure as well (Tan et al. 2002). However, other factors that modulate binding must be present, as in F-spondin TSR-1 and TSR-4 similar positive surfaces are found, but it appears that only TSR domains 5 and 6 in F-spondin are able to bind heparin, domains 1–4 not being involved (Tzarfaty-Majar et al. 2001). In F-spondin and TSP-1, differences in the degree of glycosylation between TSR domains (Gonzalez de Peredo et al. 2002) are likely to provide specificity.
Heparin has a helical extended structure in which sulfate groups form negatively charged clusters (Mulloy et al. 1993). The size and the form of the negatively charged cluster in heparin fit within the dimensions of the positively charged region on the surface of TRAP-TSR (Fig. ▶). It is thus conceivable that the binding epitope in heparin is formed by one charged cluster. A tetrasaccharide is the minimal unit resulting in an interaction of affinity sufficient to be detected in the experimental conditions used. The disaccharide lacks one sulfate group in its negative cluster as compared with the tetrasaccharide. It is possible that a stronger interaction requires the presence of a larger negative cluster. No interaction is likely to occur between TRAP-TSR domains when binding to a heparin molecule with multiple binding sites since the residues perturbed by binding were the same in the low-molecular-weight heparin and the heparin tetrasaccharide titrations, and also the dissociation constant of the low-molecular-weight heparin was approximately half that of the tetrasaccharide.
TRAP-TSR has a relatively low affinity for heparin. From the average titration curve of all residues experiencing chemical shift perturbations over a threshold value, a dissociation constant of 0.5 mM was determined for the interaction between TRAP-TSR and heparin tetrasaccharide. As has been suggested for CS TSR (Rathore et al. 2001), the TRAP-TSR domain might also act in the target cell recognition step as a low-affinity molecular probe for the sporozoite, the TRAP A-domain providing the high-affinity adhesive site(s) for specific binding. Previously, it has been shown that for the interaction between the TRAP A domain and heparin, the Kd is 80 nM (Akhouri et al. 2004), which is over three orders of magnitude smaller than observed here for TRAP-TSR.
The affinity of TRAP-TSR toward heparin observed in this study might be different in the presence of the A domain (and the rest of the extracellular part of the protein). Akhouri et al. (2004) demonstrated that the affinity for heparin of the whole extracellular domain of TRAP is twice that of the A domain alone. It is feasible that the two adhesive domains of TRAP could form a continuous binding surface for heparin. This could include the putative heparin binding site formed by the cluster of basic residues identified on the surface of the modeled structure of the TRAP A domain (Akhouri et al. 2004) and the positively charged region observed here on the surface of TRAP-TSR.
In conclusion, we have demonstrated that TRAP-TSR folds into an elongated β-sheet structure where the conserved arginine, tryptophan, and disulfide-forming cysteines form a stable stacked core. The layered side chains create a positively charged surface in the N-terminal half of TRAP-TSR that interacts with heparin. TRAP-TSR alone binds heparin with a weak affinity. However, this binding can be affected by the presence of the other extracellular domains of TRAP, namely the neighboring A domain. Structural studies with a multidomain molecule as the target will eventually reveal the roles of each domain in the interaction underlying sporozoite hepatocyte invasion as well as permit a rational structure-based approach for inhibitor design.
Materials and methods
Protein production and purification
cDNA for the TRAP-TSR domain region was constructed in a nontemplate PCR using an overlapping primer pair spanning the whole domain area. The primers used were: forward, GAGGATCCGCAAGTTGTGGTGTTTGGGACGAATGGTCTCCATGTAGTGTAACTTGTGGTAAAGGTACCAGGTCAAGAAAAAGAGAAATCTTA; reverse, AGGAATTCTATGGAGGACATCTTTCTTCTTCACATTGTTCTTGTATTTCACTTGTACATCCTTCGTGTAAGATTTCTCTTTTTCTTG.
The PCR product was cloned into pGEX-2T expression vector and sequenced to verify integrity. E. coli bacterial strain BL-21 was used for high-level expression of the GST-fusion protein. The TRAP-TSR domain was purified by thrombin-cleavage in a glutathione-sepharose column (Amersham-Biosciences) as described previously (Kilpeläinen et al. 2000).
Preparation of heparin di- and tetrasaccharides
Commercial high-molecular-weight heparin (Sigma-Aldrich) was chemically cleaved as described by Ishihara et al. (1993). After sodium borohydride reduction, size fractionation of the obtained oligosaccharides was performed on a Superdex 30 (GE Healthcare) column of size 1.6 × 60 cm, monitoring the light absorbance at ultraviolet wavelengths. Pools of fractions containing the di- and tetrasaccharides, as judged by comparison with the elution position of commercially obtained heparin hexasaccharide (Dextra Laboratories), were further purified by rechromatography separately and subjected to three rounds of lyophilization and rehydration.
Structure determination
NMR spectra for structure determination were acquired at 10°C on Varian Unity Inova spectrometers operating at 600 and 800 MHz 1H frequency, equipped with triple-resonance probeheads and actively shielded z- and triple-axis gradient systems, respectively. The sample contained 0.5 mM 13C, 15N-labeled TRAP-TSR in 20 mM bis-Tris buffer (pH 6.6) at 10°C, 94% H2O/6% D2O. The spectra were processed with Vnmr 6.1C (Varian Inc.) and analyzed with Sparky 3.106 (T.D. Goddard and D.G. Kneller, SPARKY 3, University of California, San Francisco).
3D HNCACB, CBCA(CO)NH, HNCO, CC(CO)NH, and HCCH-COSY spectra were used for the assignment of the chemical shifts (Sattler et al. 1999; Permi and Annila 2004). NOE peaks were integrated from a 3D 15N-separated NOESY-HSQC (Zhang et al. 1994) and a 13C-separated NOESY-HSQC (Muhandiram et al. 1993) spectrum modified to simultaneously excite aliphatic and aromatic carbon resonances.
Structure calculations were made automatically using the program CYANA (Herrmann et al. 2002). Peaks from the NOESY-HSQC spectra were picked and integrated manually, and the peak lists together with the chemical shift assignments were used as input for the iterative NOE assignment and structure calculation with CYANA. Three hundred conformers were generated, and the 30 conformers with lowest target function values were subjected to restrained energy minimization with AMBER 8 (D.A Case, T.A, Darden, T.E. Cheatham III, C.L. Simmerling, J. Wang, R.E. Duke, R. Luo, K.M. Merz, B. Wang, D.A. Pearlman, et al., AMBER 8, University of California, San Francisco). AMBER refinement consisted of an initial energy minimization of 2000 steps followed by a cycle of simulated annealing using the generalized Born implicit solvent model. In this cycle an initial heating to 1000 K over 4 psec was followed by 4 psec of molecular dynamics at high temperature and a slow cooling to 0 K within 12 psec. The step size was 1 fsec and the cycle consisted of 20,000 steps. The force constants associated with distance restraints and omega dihedral angles were 32 kcal mol−1 Å−1 and 80 kcal mol−1 rad−1. Structures were analyzed with PROCHECK-NMR (Laskowski et al. 1996). A final family of 20 structures with the lowest AMBER energies and best satisfying the restraints was selected to represent TRAP-TSR structure in solution. The ensemble of structures has been deposited to the Protein Data Bank with accession code 2BBX. The structure figures were generated with MOLMOL (Koradi et al. 1996) and InsightII (Accelrys Inc.) softwares.
Heparin titrations
Low-molecular-weight (average molecular weight ∼3000 Da) heparin purchased from Sigma-Aldrich, corresponding to 5–6 disaccharide units, as well as chemically cleaved heparin, corresponding to heparin di- and tetrasaccharide units, were utilized to characterize the interaction between TRAP-TSR and its substrate. Polysaccharide binding to TRAP-TSR was monitored by recording a 2D 1H, 15N-HSQC spectrum at each heparin addition point, with spectrometers operating at 500 MHz 1H (low-molecular-weight heparin) and 600 MHz 1H frequency (heparin fragments). TRAP-TSR to heparin molar ratios of 1:0, 1:0.2, 1:0.4, 1:0.6, 1:0.8, and 1:1.2 were analyzed. For low-molecular-weight heparin also the ratio of 1:3.0 and for the fragments ratios of 1:2.5 and 1:3.8 were analyzed.
Chemical shift change was calculated with 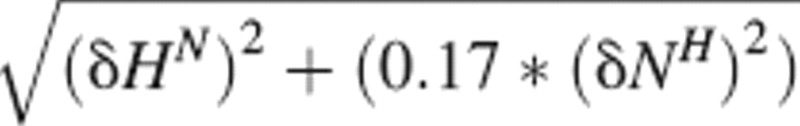 The dissociation constant was determined from an average binding curve. This curve included residues with Δδ ≥ 0.05 ppm at the titration point with a 1:1.2 TRAP-TSR to heparin ratio. Dissociation constants were determined by fitting the average chemical shift change of all nonoverlapping residues versus peptide concentration with nonlinear regression to the equation:
The dissociation constant was determined from an average binding curve. This curve included residues with Δδ ≥ 0.05 ppm at the titration point with a 1:1.2 TRAP-TSR to heparin ratio. Dissociation constants were determined by fitting the average chemical shift change of all nonoverlapping residues versus peptide concentration with nonlinear regression to the equation:
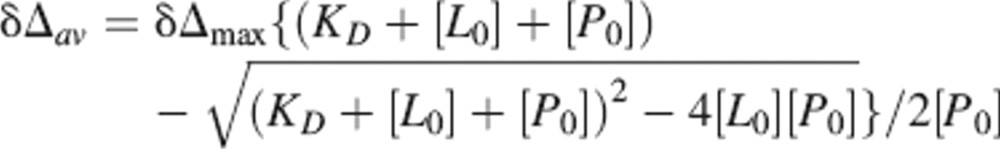 |
where δΔav is the average chemical shift change, δΔmax is the total chemical shift change at saturation, and [L0] and [P0] are the ligand and protein concentrations, respectively.
Acknowledgments
This work was supported in part by the Academy of Finland grants 106411 (T.P.) and 106852 (P.P.).
Footnotes
Reprint requests to: Perttu Permi, NMR Laboratory, Institute of Biotechnology, P.O. Box 65 FI-00014 University of Helsinki, Helsinki, Finland; e-mail: perttu.permi@helsinki.fi; fax: +358-9-191-59541.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.052068506.
References
- Akhouri R., Bhattacharyya R.A., Pattnaik P., Malhotra P., Sharma A. 2004. Structural and functional dissection of the adhesive domains of Plasmodium falciparum thrombospondin-related anonymous protein (TRAP). Biochem. J. 379 815–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay R., Rathore D., Fujioka H., Kumar S., de la Vega P., Haynes D., Moch K., Fryauff D., Wang R., Carucci D.J.et al. 2003. PfSPATR, a Plasmodium falciparum protein containing an altered thrombospondin type I repeat domain is expressed at several stages of the parasite life cycle and is the target of inhibitory antibodies. J. Biol. Chem. 278 25977–25981. [DOI] [PubMed] [Google Scholar]
- Dame J.B., Williams J.L., McCutchan T.F., Weber J.L., Wirtz R.A., Hockmeyer W.T., Maloy W.L., Haynes J.D., Schneider D.R., Sanders G.S.et al. 1984. Structure of the gene encoding the immunodominant surface antigen on the sporozoite of the human malaria parasite Plasmodium falciparum. Science 225 593–599. [DOI] [PubMed] [Google Scholar]
- Gabelli S.B., Bianchet M.A., Bessman M.J., Amzel L.M. 2001. The structure of ADP-ribose pyrophosphatase reveals the structural basis for the versatility of the Nudix family. Nat. Struct. Biol. 8 467–472. [DOI] [PubMed] [Google Scholar]
- Gallivan J.P. and Dougherty D.A. 1999. Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. 96 9459–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantt S.M., Clavijo P., Bai X., Esko J.D., Sinnis P. 1997. Cell adhesion to a motif shared by the malaria circumsporozoite protein and thrombospondin is mediated by its glycosaminoglycan-binding region and not by CSVTCG. J. Biol. Chem. 272 19205–19213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez de Peredo A., Klein D., Macek B., Hess D., Peter-Katalinic J., Hofsteenge J. 2002. C-Mannosylation and O-fucosylation of thrombospondin type 1 repeats. Mol. Cell. Proteomics 1 11–18. [DOI] [PubMed] [Google Scholar]
- Guo N.-H., Krutzsch H.C., Nègre E., Vogel T., Blake D.A., Roberts D.D. 1992a. Heparin- and sulfatide-binding peptides from the type I repeats of human thrombospondin promote melanoma sell adhesion. Proc. Natl. Acad. Sci. 89 3040–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo N.-H., Krutzsch H.C., Nègre E., Zabrenetzky V.S., Roberts D.D. 1992b. Heparin-binding peptides from the type I repeats of thrombospondin. J. Biol. Chem. 267 19349–19355. [PubMed] [Google Scholar]
- Herrmann T., Güntert P., Wüthrich K. 2002. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319 209–227. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233 123–138. [DOI] [PubMed] [Google Scholar]
- Ishihara M., Tyrrell D.J., Stauber G.B., Brown S., Cousens L.S., Stack R.J. 1993. Preparation of affinity-fractionated, heparin-derived oligosaccharides and their effects on selected biological activities mediated by basic fibroblast growth factor. J. Biol. Chem. 268 4675–4683. [PubMed] [Google Scholar]
- Kappe S.H.I., Buscaglia C.A., Bergman L.W., Coppens I., Nussenzweig V. 2004. Apicomplexan gliding motility and host cell invasion: Overhauling the motor model. Trends Parasitol. 20 13–16. [DOI] [PubMed] [Google Scholar]
- Kilpeläinen I., Kaksonen M., Kinnunen T., Avikainen H., Fath M., Linhardt R.J., Raulo E., Rauvala H. 2000. Heparin-binding growth-associated molecule contains two heparin-binding β-sheet domains that are homologous to the thrombospondin type 1 repeat. J. Biol. Chem. 275 13564–13570. [DOI] [PubMed] [Google Scholar]
- Koradi R., Billeter M., Wüthrich K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14 51–55. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., Rullmann J.A.C., MacArthur M.W., Kaptein R., Thornton J.M. 1996. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8 477–486. [DOI] [PubMed] [Google Scholar]
- Lee J.-O., Rieu P., Arnaout M.A., Liddington R. 1995. Crystal structure of the A domain from the α subunit of integrin CR3 (CD11b/CD18). Cell 80 631–638. [DOI] [PubMed] [Google Scholar]
- Matuschewski K., Nunes A.C., Nussenzweig V., Ménard R. 2002. Plasmodium sporozoite invasion into insect and mammalian cells is directed by the same dual binding system. EMBO J. 21 1597–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick C.J., Tuckwell D.S., Crisanti A., Humphries M.J., Hollingdale M.R. 1999. Identification of heparin as a ligand for the A-domain of Plasmodium falciparum thrombospondin-related adhesion protein. Mol. Biochem. Parasitol. 100 111–124. [DOI] [PubMed] [Google Scholar]
- Muhandiram D.R., Farrow N.A., Xu G.-Y., Smallcombe S.H., Kay L.E. 1993. A gradient C-13 NOESY-HSQC experiment for recording NOESY spectra of C-13-labeled proteins dissolved in H2O. J. Magn. Reson. B. 102 317–321. [Google Scholar]
- Müller H.-M., Reckmann I., Hollingdale M.R., Bujard H., Robson K.J.H., Crisanti A. 1993. Thrombospondin related anonymous protein (TRAP) of Plasmodium falciparum binds specifically to sulfated glycoconjugates and to HepG2 hepatoma cells suggesting a role for this molecule in sporozoite invasion of hepatocytes. EMBO J. 12 2881–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulloy B., Forster M.J., Jones C., Davies D.B. 1993. N.m.r. and molecular-modelling studies of the solution conformation of heparin. Biochem. J. 293 849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Permi P. and Annila A. 2004. Coherence transfer in proteins. Prog. Nucl. Magn. Reson. Spectrosc. 44 97–137. [Google Scholar]
- Rathore D., McCutchan T.F., Garboczi D.N., Toida T., Hernáiz M.J., LeBrun L.A., Lang S.C., Linhardt R.J. 2001. Direct measurement of the interactions of glycosaminoglycans and a heparin decasaccharide with the malaria circumsporozoite protein. Biochemistry 40 11518–11524. [DOI] [PubMed] [Google Scholar]
- Robson K., Frevert U., Reckmann I., Cowan G., Beier J., Scragg I.G., Takehara K., Bishop D.H.L., Pradel G., Sinden R.et al. 1995. Expression during sporozoite ontogeny and binding to human hepatocytes. EMBO J. Plasmodium falciparum 14 3883–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M., Schleucher J., Griesinger C. 1999. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectrosc. 34 93–158. [Google Scholar]
- Sinnis P. and Nardin E. 2002. Sporozoite antigens: Biology and immunology of the circumsporozoite protein and thrombospondin-related anonymous protein. Chem. Immunol. 80 70–96. [DOI] [PubMed] [Google Scholar]
- Sinnis P., Clavijo P., Fenyö D., Chait B.T., Cerami C., Nussenzweig V. 1994. Structural and functional properties of region II-plus of the malaria circumsporozoite protein. 1994. J. Exp. Med. 180 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan K., Duquette M., Liu J., Dong Y., Zhang R., Joachimiak A., Lawler J., Wang J. 2002. Crystal structure of the TSP-1 type 1 repeats: A novel layered fold and its biological implication. J. Cell Biol. 159 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari R., Spaccapelo R., Bistoni F., Holder A.A., Crisanti A. 2002. Function of region I and II adhesive motifs of Plasmodium falciparum circumsporozoite protein in sporozoite motility and infectivity. J. Biol. Chem. 277 47613–47618. [DOI] [PubMed] [Google Scholar]
- Thompson J., Cooke R.E., Moore S., Anderson L.F., Janse C.J., Waters A.P. 2004. PTRAMP; a conserved Plasmodium thrombospondin-related apical merozoite protein. Mol. Biochem. Parasitol. 134 225–232. [DOI] [PubMed] [Google Scholar]
- Trottein F., Triglia T., Cowman A.F. 1995. Molecular cloning of a gene from Plasmodium falciparum that codes for a protein sharing motifs found in adhesive molecules from mammals and Plasmodia. Mol. Biochem. Parasitol. 74 129–141. [DOI] [PubMed] [Google Scholar]
- Tzarfaty-Majar V., López-Alemany R., Feinstein Y., Gombau L., Goldsmith O., Soriano E., Muñoz-Cánoves P., Klar A. 2001. Plasmin-mediated release of the guidance molecule F-spondin from the extracellular matrix. J. Biol. Chem. 276 28233–28241. [DOI] [PubMed] [Google Scholar]
- Tucker R.P. 2004. The thrombospondin type 1 repeat family. Int. J. Biochem. Cell Biol. 36 969–974. [DOI] [PubMed] [Google Scholar]
- Zhang O., Kay L.E., Olivier J.P., Forman-Kay J. 1994. Backbone 1H and 15N resonance assignments of the N-terminal SH3 domain of drk in folded and unfolded states using enhanced-sensitivity pulsed field gradient NMR techniques. J. Biomol. NMR 4 845–858. [DOI] [PubMed] [Google Scholar]