

Abstract
Strontium is used in the treatment of osteoporosis as a ranelate compound, and in the treatment of painful scattered bone metastases as isotope. At very high doses and in certain conditions, it can lead to osteomalacia characterized by impairment of bone mineralization. The osteomalacia symptoms resemble those of hypophosphatasia, a rare inherited disorder associated with mutations in the gene encoding for tissue-nonspecific alkaline phosphatase (TNAP). Human alkaline phosphatases have four metal binding sites—two for zinc, one for magnesium, and one for calcium ion—that can be substituted by strontium. Here we present the crystal structure of strontium-substituted human placental alkaline phosphatase (PLAP), a related isozyme of TNAP, in which such replacement can have important physiological implications. The structure shows that strontium substitutes the calcium ion with concomitant modification of the metal coordination. The use of the flexible and polarizable force-field TCPEp (topological and classical polarization effects for proteins) predicts that calcium or strontium has similar interaction energies at the calcium-binding site of PLAP. Since calcium helps stabilize a large area that includes loops 210–228 and 250–297, its substitution by strontium could affect the stability of this region. Energy calculations suggest that only at high doses of strontium, comparable to those found for calcium, can strontium substitute for calcium. Since osteomalacia is observed after ingestion of high doses of strontium, alkaline phosphatase is likely to be one of the targets of strontium, and thus this enzyme might be involved in this disease.
Keywords: alkaline phosphatase, strontium, hypophosphatasia, crystal structure
Strontium, a divalent cation with a molecular weight of 87,620 Da, is the most abundant trace element in ocean water and ranks 15th in order of abundance in the Earth's crust. Strontium is an alkaline earth metal like calcium and magnesium. Physiologically, strontium and calcium are remarkably similar: They are absorbed in the gastrointestinal tract, concentrated in bone, and excreted primarily in urine; and one mechanism of strontium incorporation into bone involves ionic exchange with bone calcium. Because of these similarities, strontium is currently used in bone therapies. For instance, strontium-89 (and -85) is used for treating the scattered painful bone metastases that affect two-thirds of patients suffering from advanced and metastatic cancers (Giammarile et al. 1999; Saarto et al. 2002). In addition, stable strontium in the form of strontium–ranelate is also used in the treatment of osteoporosis (Boivin and Meunier 2003; Meunier and Reginster 2003; Reginster et al 2003).
The currently available data indicate that strontium has metabolic effects on bone in vivo. On the positive side, low doses of strontium (<4 g/L in the diet) increase both the rate of formation and the density of bone. However, very high doses can induce decreased resorption and mineral density of bone (Cabrera et al. 1999). Osteomalacia is characterized by impairment of bone mineralization, leading to accumulation of unmineralized matrix or osteoid in the skeleton (Francis and Selby 1997; Schrooten et al. 1998; D'Haese et al. 2000). More recent findings from epidemiological studies and experimental data also established a dose-related multiphasic effect of strontium on bone formation with reduced bone mineralization at high strontium doses (Schrooten et al. 2003; Verberckmoes et al. 2003).
These effects on bone mineralization thus imply that strontium is not fully equivalent to calcium, suggesting that it probably interacts with its environment in a slightly different way than calcium does. It should be noted that the defective bone mineralization symptoms of osteomalacia are quite similar to those of hypophosphatasia, a rare inherited disorder associated with mutations in the tissue-nonspecific alkaline phosphatase (AP). This disease is highly variable in its clinical expression, due to the strong allelic heterogeneity in the TNAP gene, ranging from stillbirth without mineralized bones to pathological fractures developing only late in adulthood (Whyte 1994). The similarities between both of these diseases suggest that one effect of strontium on bone mineralization could occur through a direct interaction with AP itself or through the AP pathway. In addition, an effect of high concentration of stable strontium on AP activity has been previously described (Kshirsagar 1975), and the follow-up of patients treated with 89Sr reveals a decrease in AP activity (Smeland et al. 2003). This reflects either a decrease in AP expression or a decrease in AP catalytic activity.
APs (E.C.3.1.3.1) are dimeric metalloenzymes found in many species from bacteria to humans (McComb et al. 1979M). They catalyze the hydrolysis of phosphomonoesters with release of inorganic phosphate and alcohol (Schwartz and Lipmann 1961; Harris 1990). The three-dimensional structure of the protein core is strongly conserved among species (Escherichia coli [Kim and Wyckoff 1991], shrimp [De Backer et al. 2002, 2004], human placental [Le Du et al. 2001; Llinas et al. 2005]) as well as its catalytic site with two zinc and one magnesium ions. Extensive structural studies of E. coli AP have elucidated the relationship between the catalytic-site residues and the catalytic mechanism (Hull et al. 1976; Gettins and Coleman 1983, 1984; Mandecki et al. 1991; Xu and Kantrowitz 1991; Chen et al. 1992; Matlin et al. 1992; Janeway et al. 1993; Murphy and Kantrowitz 1994; Dealwis et al. 1995a,b; Murphy et al. 1997; Holtz and Kantrowitz 1999; Stec et al 2000; Muller et al. 2001; Le Du et al. 2002). Thus, the overall catalytic mechanism is conserved between the E. coli enzyme and the mammalian enzymes (Xu and Kantrowitz 1991; Janeway et al. 1993; Murphy and Kantrowitz 1994; Kozlenkov et al. 2002). However, unique features exist in the mammalian APs, such as their allosteric properties (Hoylaerts et al. 1997) and their susceptibility to inhibition via an uncompetitive mechanism by L-amino acid, e.g., L-Phe, L-Trp, L-homoarginine, L-Leu, and levamisole (Fishman and Sie 1971; Van Belle 1976; Doellgast and Fishman 1977).
In humans, APs are differentiated into four isozymes: tissue-nonspecific AP (TNAP), found in bone, liver, and kidney; and three tissue-specific isozymes—placental (PLAP), germ cell (GCAP), and intestinal (IAP). These latter three are 90%–98% homologous, and their genes are clustered on Chromosome 2. TNAP is only 50% identical with the other three, and its gene is located on Chromosome 1 (Harris 1990). At a physiological level, the clearest evidence that APs are important in vivo has been provided by studies of human hypophosphatasia, in which a deficiency in the TNAP isozyme, caused by deactivating mutations in the TNAP gene (Weiss et al. 1988; Henthorn et al. 1992; Mornet 2000), is associated with defective bone mineralization in the form of rickets and osteomalacia (Whyte 2001). The severity and expressivity of hypophosphatasia depend on the nature of the TNAP mutation (Zurutuza et al. 1999; Di Mauro et al. 2002). The accumulated evidence indicates that the function of TNAP in bone tissue consists of hydrolyzing inorganic pyrophosphate to maintain a proper concentration of this mineralization inhibitor to ensure controlled bone mineralization (Moss et al. 1967; Meyer 1984; Whyte 2001; Hessle et al. 2002; Harmey et al. 2004). In addition to the two zinc atoms and the magnesium present at the active site, the existence of a fourth metal binding site, 10 Å away from the active site, was revealed by the crystal structure of PLAP (Le Du et al. 2001) and confirmed by X-ray fluorescence (Mornet et al. 2001).
In the present study, we show that strontium can replace calcium in APs and how this interaction affects the AP pathway. The crystal structure of PLAP in the presence of strontium chloride has been solved at 1.6 Å, a resolution comparable to that of a native PLAP (1.57 Å) with bound calcium. Given the quality of these structures, it is possible with a high degree of certainty to observe that strontium can substitute for calcium but with a different coordination. This difference in coordination involves one additional water molecule. Theoretical calculations suggest that PLAP is stabilized almost equivalently in the presence of calcium or strontium. Thus, strontium is needed at a concentration equivalent to that of calcium to achieve substitution. Although the X-ray structures are very similar in the presence of calcium or strontium, theoretical computations suggest that the stabilization of loop 210–228 in the presence of calcium or strontium is different.
Results
Structure of PLAP in complex with strontium
The structure of PLAP cocrystallized with strontium chloride [PLAP(Sr)] is virtually identical to that of the calcium-containing PLAP [PLAP(Ca)] with an RMSD of 0.24 Å. Data collection and refinement statistics are listed in Table 1. The analysis of the electron density maps shows that strontium replaces the calcium at the calcium-binding site, but not the magnesium at the active site. Compared to the structure of the native enzyme, the peak height at the calcium-binding site is twofold higher, as it would be expected for strontium. Attempts to refine a calcium atom at this position leads to a B-factor too low compared with neighboring residues and residual electron density in the Fo − Fc map (Table 1; Fig. 1). The distances between the metal and its ligands are different, with an average increase of 0.15 Å in the site containing a strontium ion (Table 2).
Table 1.
Data collection and refinement statistics

Figure 1.
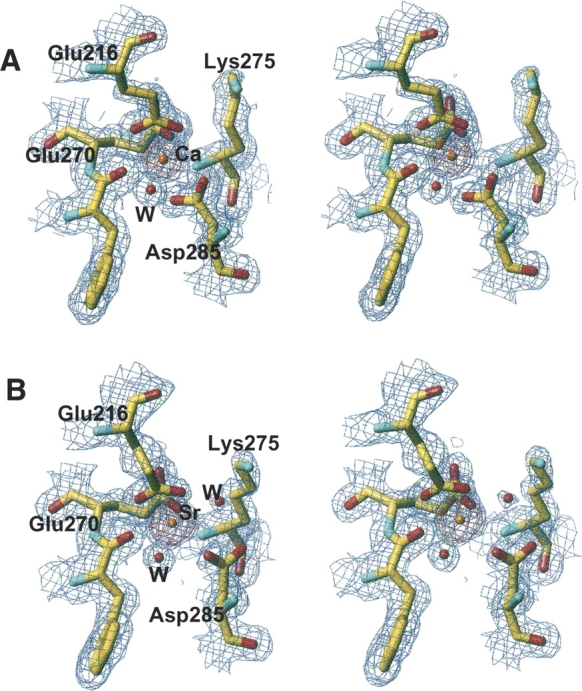
Stereo view of the 2Fo − Fc electron density map (blue) at the 1σ level and the Fo − Fc electron density map (red) at the 5σ level, at the fourth metal binding site of alkaline phosphatase in the presence of calcium (A) or strontium (B).
Table 2.
Distances between each metal and its ligand in the crystal structures

This distance variation is consistent with what has been previously observed in the case of P-selectin but higher than in the case of the oligomerization domain from rotavirus NSP4, or ribonuclease T1 (Table 3). A change in coordination was also observed in the case of ribonuclease T1, but with two additional water molecules (Table 3). The calcium coordination involves Phe269-CO, the carboxylate of Glu216, Glu270, and Asp285, and one water molecule. The interaction of Glu270 is monodentate with Ca–O distances of 2.2 Å and 4.2 Å; that of Glu216 is bidentate with Ca–O distances of 2.4 Å and 2.4 Å. The Ca–O distance is 2.3 Å and 2.5 Å between the calcium and Asp285, respectively, and the electron density is continuous between one oxygen and the calcium (Fig. 2A). In addition, the comparison of the calcium-binding site in previous structures of PLAP in complex with various ligands or of native PLAP shows distances between the carboxylate oxygens of Asp285 and the calcium of 2.5 Å and 2.5 Å, of 2.4 Å and 2.5 Å, of 2.2 Å and 2.6 Å, or of 2.2 Å and 2.7 Å (Le Du et al. 2001; Llinas et al. 2005). Thus, the distances tend to be longer than usual when distributed between the two carboxylate oxygens and adopt a more conventional length when the interaction is clearly monodentate. For this reason, we assume that the interaction with Asp285 corresponds to one bond, leading to a coordination number of the calcium of six. The strontium coordination involved an additional water molecule, and its coordination pattern is also different since Asp285 carboxylate has moved away from the metal and the monodentate character of the interaction is more pronounced with distances of 2.5 Å and 3.9 Å instead of 2.3 Å and 2.5 Å in the case of calcium (Fig. 2B). The substitution of the calcium by strontium modifies the network of interaction with the metal. The carbonyls of the Tyr269 main chain, Glu270, and the first water molecule move only by 0.05, 0.1, and 0.3 Å, respectively; the Cδ of Glu216 shifts by 0.7 Å and the interaction remains bidentate; and the Cγ of Asp285 moves by 1.1 Å and the interaction becomes monodentate (Table 3; Fig. 2). The additional water molecule interacts also with Lys275, which is added to the interaction network of the strontium, whereas it plays no role in the presence of calcium in the native enzyme. This interaction is likely to affect the chemical properties of Lys275 and, by extension, its environment. This may therefore have repercussions on the alkaline phosphatase activity since Lys275 is located in the active-site valley (Fig. 3A).
Table 3.
Average calcium-ligand distance compared to strontium distance in X-ray protein structures refined at a resolution of 2.0 Å or higher

Figure 2.
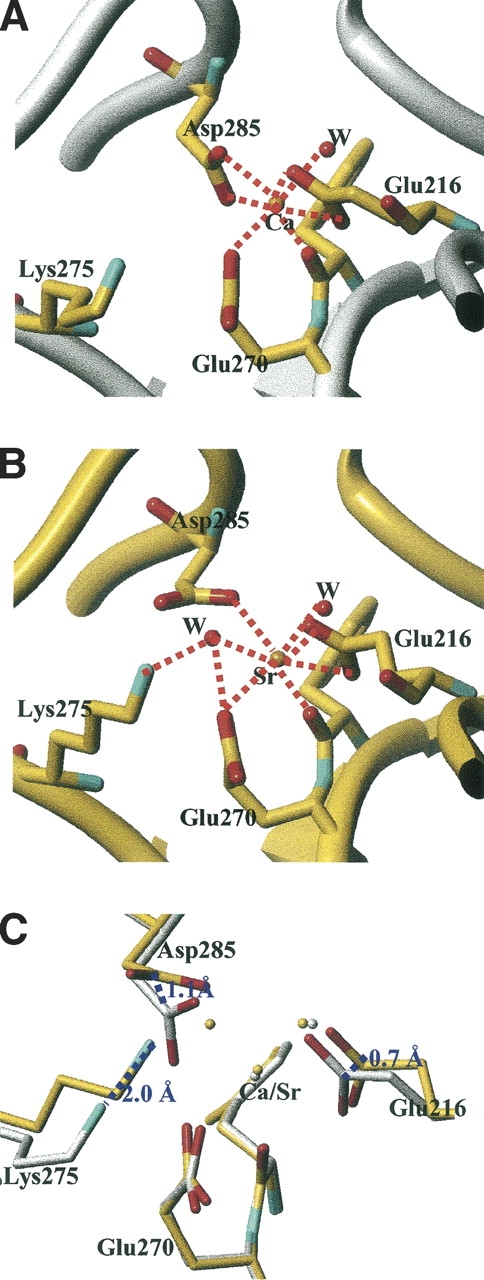
Coordination network of the calcium (A) or strontium (B) at the fourth metal binding site of alkaline phosphatase showing the residues in interaction with the metal. (C) Superimposition of the two binding sites showing the deviation between the calcium-containing AP (white structure) and the strontium-containing AP (yellow structure).
Figure 3.
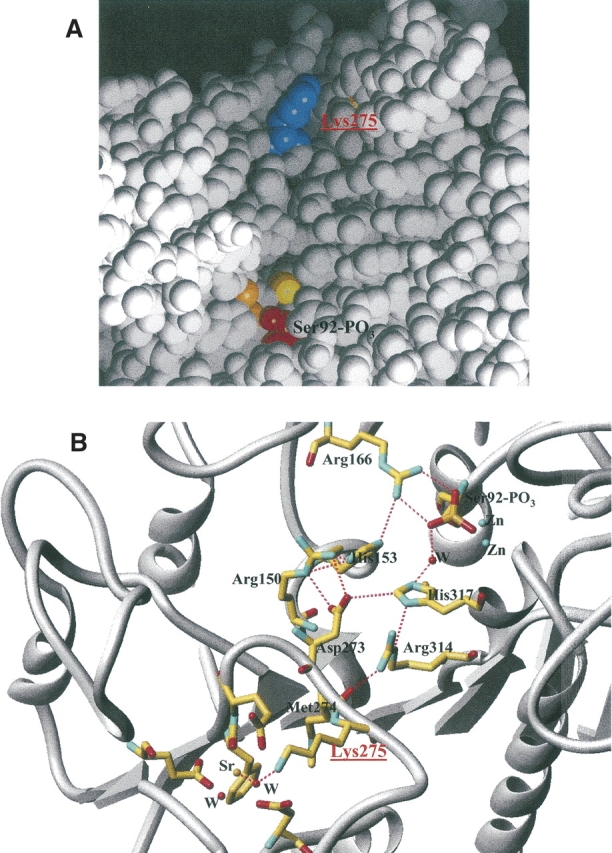
(A) Location of Lys275 (in blue) in the active-site valley of PLAP. (B) Network of interaction connecting the active site to the fourth metal binding site in the presence of strontium.
Theoretical study of the Ca/Sr competition at the fourth metal binding site
The interatomic distances corresponding to the atoms involved in the fourth PLAP binding site observed in the TCPEp relaxed structures are compared with experimental data in Table 5. The theoretical distances differ from the experimental ones by ∼0.05 Å for PLAP(Ca) and by ∼0.07 Å for PLAP(Sr), with the exception of the distance between the Glu270-δ2 oxygen and Ca in PLAP(Ca), and the distance between the Asp285-δ2 oxygen and Sr in PLAP(Sr) (those distances differ by ∼0.20 Å between theory and experiment). The experimental coordination patterns (i.e., monodentate or bidentate pattern) of the carboxylate groups involved in the fourth PLAP binding site are well conserved during the relaxation process. Our computations appear thus to be in good agreement with experiment for both Ca and Sr. The coordination network is constituted by the backbone CO group of Phe269; by the carboxylate groups of Glu216, Glu270, and Asp285; and by a water molecule for Ca and by the same groups with an additional water molecule for Sr. Thus, there is agreement between theory and experiment for the coordination numbers for Ca and Sr (7 and 8, respectively).
The RMSD values between the TCPEp structures and the experimental ones are also small for both structures (1.65 Å when considering all protein atoms and 1.30 Å when considering solely the Cα atoms). The RMSD values are even smaller when considering the atoms involved in the fourth PLAP binding site [0.71 Å for PLAP(Ca) and 0.96 Å for PLAP(Sr), respectively]. Since no constraints were imposed on the atoms belonging to that site, these results show the ability of TCPEp in describing the interactions among hard cations and proteins. It should be noticed that the TCPEp predictions regarding the fourth PLAP binding site are of better quality than those previously reported in the case of the protein calmodulin interacting with four Ca+ (Cuniasse and Masella 2003). In the latter study, the experimental structures were less carefully relaxed (e.g., only the carboxylate groups belonging to the first coordination sphere of the calcium cations were restrained to their X-ray positions during the first step of the relaxation computations).
The two experimental structures of PLAP (chelating, respectively, Ca and Sr) differ by the number of water molecules and by the positions of several side chains. This explains the strong difference in the TCPEp total energies of the relaxed PLAP(M) structures: ∼1000 kcal/mol. That difference prevents a direct comparison of the latter energies in order to discuss the Ca/Sr competition at a theoretical level. Moreover, the free binding energy of a protein with an ion can be theoretically estimated by performing MD simulations at the nanosecond timescale of the complex protein/ion embedded in a solvent box. However, because of the large size of the system PLAP(M) (∼8000 atoms without accounting for the solvent molecules), such long MD simulations based on a polarizable force field are at present beyond our computational resources: An MD run in vacuo of 50 psec (with an integration time step of 0.5 fsec) of a PLAP system with the TCPEp force field requires ∼48 h on a 2 GHz Pentium IV workstation, even using the multiple time steps algorithm recently proposed for polarizable force fields based on induced dipoles (cf. Masella 2006). It is, however, possible to derive useful information from the PLAP(M) structures relaxed in vacuo with TCPEp. For instance, to estimate the strength of the binding energies of PLAP with Ca or Sr and the role of the additional water molecule surrounding Sr at the fourth PLAP binding site, we have computed the several interaction energies according to
Here, E[PLAP(M)] corresponds to the total energy of the relaxed PLAP(M) structure and E[PLAP*] to the energy of a subsystem of PLAP(M), where one of its elements (M2+ or a water molecule) is ignored during the single point energy computation. As computed from Equation 1, the interaction energies ΔE of PLAP with Ca and Sr are, respectively, −352 and −307 kcal/mol (Table 4). The difference ςE(Ca/Sr) between both the latter energies is 45 kcal/mol, which is of the same order of magnitude as the difference of hydration enthalpies between Ca and Sr: 35 kcal/mol (the ΔH0solv for the latter cations being 399 and 364 kcal/mol, respectively). Moreover, the difference ςE(Ca/Sr) increases by 10 kcal/mol if the less strongly bonded water molecule interacting with Sr at the fourth PLAP binding site is omitted during the computations. Hence, these results suggest that the interaction energies of Ca and Sr at the fourth PLAP binding site are likely to be very close when accounting for the cation desolvation process and that the additional water molecule observed in the first chelation shell of Sr reinforces its interaction energy at that binding site, by ∼10 kcal/mol.
Table 4.
Model conservation after relaxation and energy between the metal and its environment

Among the different loops that are maintained by the calcium-binding site, loop 210–228 is of particular interest. This loop is strongly bent at residues Met210–Gly211 and Gly224–Gly225, and folded back toward the fourth metal site (Fig. 4A). In addition to the interaction between the carboxylate of Glu216 and the fourth metal, the loop is maintained by hydrogen bonds between Tyr217 hydroxyl and Glu270 carboxylate, between Asp214 carboxylate and Arg204 guanidinium, and by a succession of stacking interactions that involve the ring of Tyr217, the side chain of Glu216, the ring of Trp248, and the cycle of the first carbohydrate linked to Asn249 (Fig. 4B). The crystal structures in the presence of calcium or strontium show no particular difference in the stabilization of this loop. Therefore, to evaluate the incidence of the cation nature on the interaction between that loop and the rest of the protein, the NH–CO backbone bonds have been disrupted between residues 209 and 210 and between residues 228 and 229. A hydrogen atom was then added to both the latter nitrogen and carbon atoms of each disrupted bond. The resulting structures were relaxed using a short MD run with all of the Cα atoms restrained to their starting positions. As compared to Sr, the interaction energy computed with TCPEp between that loop and the protein is stronger by ∼25 kcal/mol with Ca in the fourth metal binding site. It has to be noticed here that this difference in energy predicted by the TCPEp force field is of the same order of magnitude as that obtained when comparing the binding energies of small [(HCO2−),M2+] aggregates (M = Ca-Sr and n = 1–4) obtained from DFT quantum computations (cf. Supplemental Table 1), which range from 11 (n = 1) to 39 kcal/mol (n = 3).
Figure 4.

(A,B) Interaction of loop 210–228 (in blue) with the fourth metal binding site. (C,D) Succession of stacking interaction between loop 210–228 (in blue) and the core of PLAP structure.
Hence, even if the three-dimensional structure of PLAP is not strongly affected by the nature of the cation located in its fourth binding site, the strength of the interaction between the loop extending from residues 209 and 228 and the rest of the protein depends on the nature of this cation. This can have important consequences for the interaction between the protein PLAP and all of its possible in vivo partners, which can affect its biological activity.
Discussion
The crystal structure of PLAP in complex with strontium shows that this metal does not substitute the zinc or magnesium atoms at the active site, but substitutes the calcium at the fourth metal site located 18 Å from the active site. This fourth metal binding site has been observed for the first time in the crystal structure of PLAP (Le Du et al. 2001) and is also present in shrimp alkaline phosphatase (De Backer et al. 2002, 2004), whereas it is absent in the E. coli alkaline phosphatase (Kim and Wyckoff 1991). The high degree of conservation of the active site between mammalian APs and E. coli AP suggests that the catalytic mechanism is similar in mammalian AP and in E. coli AP. In addition, the remote location of the calcium site implies that it cannot be directly involved in the catalytic process of alkaline phosphatase. However, the concentration used in our crystallization condition is in the millimolar range, and it has been previously observed that at this concentration, strontium affects the enzymatic activity of alkaline phosphatase (Kshirsagar 1975). In addition, in the TNAP isozyme, mutation E274K or D289V (TNAP numbering) is associated with severe phenotypes, and mutation E218G (TNAP numbering) is associated with a moderate phenotype of hypophosphatasia (Mornet et al. 2001). These residues correspond, respectively, to residues Glu270, Asp285, and Glu216 in PLAP, and belong to the calcium site. From these data, we can conclude that a modification of the geometry of the calcium site due to the substitution by strontium, or the alteration of the coordination of the calcium due to the mutation of one of its ligands, affects the alkaline phosphatase enzymatic activity, and that this area is involved at some level in the regulation of the enzymatic activity.
The substitution of calcium by strontium modifies the network of residues that interact with the metal. The carbonyl of the Tyr269 main chain, the carboxylate of Glu270, and the first water molecule are highly stable; the carboxylate of Glu216 or Asp285 displays larger movement, and an additional water molecule is located in the coordination sphere that bridges the strontium to Lys275, associated with a 2.0 Å displacement of the Lys275 nitrogen toward the strontium (Fig. 2C). The coordination degree is 6 in the presence of calcium, and 7 in the presence of strontium, and the bonds are longer by 0.15 Å on average (Fig. 2; Table 2). These observations are in agreement with a recent systematical analysis of calcium- or strontium-containing structures, showing that the main difference between calcium and strontium is the ionic radius, as the strontium radius is roughly 0.15 Å longer than calcium, and that an increased coordination number of at least one atom is needed (Rinaldo et al. 2004). It is therefore not surprising to observe that an additional residue interacts with the metal through the additional water molecule.
The additional Lys275 is located on loop 269–285. This loop is well maintained at each extremity with the presence of Phe269 and Asp285, which both belong to the fourth metal binding site. The loop is located in the active-site valley and bridges the active-site pocket through a discrete number of hydrogen bonds (Fig. 3). Figure 3B shows the interaction network between loop 269–285 and the active site involving, in particular, Glu273 and Met274. In the presence of strontium at the fourth metal binding site, Lys275 becomes part of the metal binding site, and this additional interaction makes a bridge between the fourth metal binding site and the catalytic site. Lys275 plays the role of a switch off and on that connects the catalytic site and the fourth metal binding site. The TNAP sequence is 50% identical with that of PLAP. It is interesting to notice that residues Glu216, Glu270, and Asp285, which coordinate the calcium, are conserved. In the same way, Arg150, His153, Arg166, Asp273, Met274, Arg314, and His317, which belong to the interaction network between loop 269–285 and the active site, are conserved. Residues Arg204, Asp214, and Tyr217, which help to stabilize the interaction between loop 210–228 and the fourth metal site, are conserved, as well as Trp248 and Asn249 (and the N-glycosilation sequence Asn-X-Thr), which are also involved in the stabilization of the fourth metal site through stacking interactions. Among the previously cited residues, the only substitution observed occurred at Lys275, which is a Gln in TNAP. However, the length of the Gln side chain and its ability to form hydrogen bonds is equivalent to that of Lys, and the role of the off-and-on switch that connects the catalytic site and the fourth metal binding site is likely to also occur in TNAP.
Moreover, a peripheral binding site has been previously observed that includes residues Arg250, Met254, Ser257, Ser287, and Arg297 from the two α-helices 250–257 and 287–297 (Llinas et al. 2005). The loop between these two α-helices from Ser257 to Ser287 contains residues Phe269, Glu270, and Asp285 from the fourth metal binding site. Ser287 from the peripheral site is located only two residues from Asp285, and Arg250 is located also two residues away from Trp248, which interacts with the calcium or the strontium through a water molecule. Therefore, the large loop 250–297, which we call the “peripheral domain,” includes both the fourth metal binding site and the peripheral binding site. In addition, this peripheral domain includes Lys275, which belongs to the fourth metal site in the presence of strontium, and Glu273, which interacts with His153 and His317 from the active site. Therefore, the presence of strontium at the fourth metal site leads to an extended network of interactions that bridges the active site to the peripheral domain and that may affect the function of the protein.
At last, the interaction energy computed with TCPEp shows that the presence of calcium or strontium at the fourth metal binding site affects the strength of the interaction between loop 210–228 and the core of PLAP structure. This loop is strongly bent at Met210–Gly211 and Gly224–Gly225, and forms a flap folded back toward the fourth metal site. It locks the fourth cation and participates in the support of the peripheral domain through interaction with Trp248 and with Glu270 (Fig. 4). Therefore, the strength of the interaction between this loop and the core of PLAP structure will have important consequences for the function of the peripheral domain.
Our present data together with previous studies support a role of the site described by loops 210–228 and 250–297, including the fourth metal binding site, with loop 210–228 associated with structural support and loop 250–297 associated with a function that includes the peripheral binding site (Llinas et al. 2005). The functional role of this area remains to be determined but has probably important physiological consequences, since in the TNAP isozyme, mutations of the residues involved in calcium binding are associated with the severe phenotype of hypophosphatasia (Le Du et al. 2001).
The similarity of osteomalacia and hypophosphatasia lets us suppose that the substitution of strontium by calcium could be associated with the osteomalacia symptoms. The compensation in energy between calcium and strontium suggests an important rate of exchange of calcium and strontium. Therefore, the local concentration of strontium should be on the same order as that of calcium to be able to observe the calcium–strontium substitution. Such a concentration cannot be obtained in natural conditions, and a huge quantity of strontium should be ingested to reach the quantity of strontium compatible with high substitution at the calcium site.
Materials and methods
Purification and crystallization
Highly purified PLAP was prepared from human placenta as previously described (Le Du et al 2001). Crystals were grown at 19°C(±2°C) using sitting drop vapor diffusion, with a protein concentration of 10 mg/mL, in the presence of 10 mM p-nitrophenyl phosphate. Crystals were obtained from 12% PEG 3350, 100 mM sodium cacodylate (pH 6.5), 1 mM zinc chloride, 2 mM magnesium acetate, and 2 mM strontium chloride over a period of 5 mo.
Data collection and structure determination of PLAP complexes
Data were collected from a crystal grown in the presence of 2 mM strontium chloride in the growing solution to 1.6 Å resolution. The data were collected at the ESRF on beamline ID14-1, and processed with HKL (Otwinowski and Minor 1997). The crystals belong to the space group C2221 as the native crystals. 3Fo − 2Fc, 2Fo − Fc, and Fo − Fc maps were calculated by using the suite of programs of the Collaborative Computational Project, Number 4 (1994). The electron density maps were evaluated with the program TURBO (Roussel and Cambillau 1989), and the model was refined with REFMAC (Murshudov et al. 1997) and WARP version 5.0 (Perrakis et al. 1999). The strontium atom was positioned in the electron density map according to the peak height in the Fo − Fc electron density map. Among the different structures available of calcium-containing PLAP [PLAP(Ca)], we have selected the structure refined to the highest resolution (1.57 Å), a resolution on the same order as that of PLAP(Sr) (Llinas et al. 2005). The data collection and refinement statistics are summarized in Table 1. PROCHECK (Laskowski et al. 1993) was used to analyze the structure geometry. The three-dimensional structures of PLAP-Ca or in complex with Sr [PLAP(Sr)] were superimposed with the iterative program ALIGN (Satow et al. 1986). The coordinates have been deposited to the Protein Data Bank at Brookhaven (entry codes 1ZED, 2GLQ).
Theoretical computational details
Both the experimental structures of the protein AP interacting with either Ca or Sr were investigated using the flexible polarizable force field TCPEp (Masella and Cuniasse 2003), which has been shown to provide an accurate description of the interactions between proteins and hard cations (Cuniasse and Masella 2003).
For computational efficiency reasons, the two structures theoretically investigated correspond to the PLAP domain extending from residues 29 to 469, together with the water molecules present in the X-ray structures and located <3.5 Å from any protein atom. Nine counterions Cl− were added to the latter structures in order to neutralize their electrostatic charge. The TCPEp parameters for Sr2+ were defined using the same methodology used for Ca2+ (cf. Masella and Cuniasse 2003 and Supplemental Table 1, where the TCPEp results are compared to quantum DFT computation ones for small [(HCO2−)n,M2+] aggregates, where M = Ca-Sr and n = 1-4).
The PLAP(M) structures were relaxed in three steps, using low-temperature molecular dynamics (MD) simulations: (1) The structures were relaxed by performing a short MD run of 2.104 iterations with all of the atoms of the fourth PLAP binding site restrained to their X-ray positions; (2) the structures obtained at the end of the previous runs were newly relaxed with an MD run of 105 iterations and with the previous constraints removed; (3) the final structures were then finally relaxed with a quenching method, until the system kinetic temperature was <0.1 K. For these three steps, the Cαs of residues 30, 86, 250, 350, and 400 were restrained to their X-ray positions (none of them belongs to any of the PLAP binding sites).
Acknowledgments
We thank the local contacts at the ESRF BM30 and ID14 for their kind help during the data collection. We are grateful to Dr. Boivin for his reading of the manuscript and his helpful suggestions.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Marie Hélène Le Du, Laboratoire de Structure des Protéines, Département d'Ingénierie et d'Etude des Protéines, Commissariat à l'Energie Atomique, 91191 Gif sur Yvette, France; e-mail: mhledu@cea.fr; fax: +33-1-69-08-90-71.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062123806.
Abbreviations: TNAP, tissue nonspecific alkaline phosphatase; PLAP, placental alkaline phosphatase; GCAP, germ cell alkaline phosphatase; IAP, intestinal alkaline phosphatase; TCPEp, topological and classical polarization effects for proteins; AP, alkaline phosphatase.
References
- Boivin G. and Meunier P.J. 2003. The mineralization of bone tissue: A forgotten dimension in osteoporosis research. Osteoporos. Int. 14: 19–24. [DOI] [PubMed] [Google Scholar]
- Cabrera W.E., Schrooten I., De Broe M.E., D'Haese P.C. 1999. Strontium and bone. J. Bone Miner. Res. 14: 661–668. [DOI] [PubMed] [Google Scholar]
- Chen L., Neidhart D., Kohlbrenner W.M., Mandecki W., Bell S., Sowadski J., Abad-Zapatero C. 1992. 3-D structure of a mutant (Asp101 → Ser) of E. coli alkaline phosphatase with higher catalytic activity. Protein Eng. 5: 605–610. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Cuniasse P. and Masella M. 2003. A many-body model to study proteins. II. Incidence of many-body polarization effects on the interaction of the calmodulin protein with four Ca2+dications and with a target enzyme peptide. J. Chem. Phys. 119: 1874–1878. [Google Scholar]
- Dealwis C.G., Brennan C., Christianson K., Mandecki W., Abad-Zapatero C. 1995a. Crystallographic analysis of reversible metal binding observed in a mutant (Asp153 → Gly) of Escherichia coli alkaline phosphatase. Biochemistry 34: 13967–13973. [DOI] [PubMed] [Google Scholar]
- Dealwis C.G., Chen L., Brennan C., Mandecki W., Abad-Zapatero C. 1995b. 3-D structure of the D153G mutant of Escherichia coli alkaline phosphatase: An enzyme with weaker magnesium binding and increased catalytic activity. Protein Eng. 8: 865–871. [DOI] [PubMed] [Google Scholar]
- De Backer M., McSweeney S., Rasmussen H.B., Riise B.W., Lindley P., Hough E. 2002. The 1.9 Å crystal structure of heat-labile shrimp alkaline phosphatase. J. Mol. Biol. 318: 1265–1274. [DOI] [PubMed] [Google Scholar]
- De Backer M.M.E., McSweeney S., Lindley P.F., Hough E. 2004. Ligand-binding and metal-exchange crystallographic studies on shrimp alkaline phosphatase. Acta Crystallogr. D Biol. Crystallogr. 60: 1555–1561. [DOI] [PubMed] [Google Scholar]
- D'Haese P.C., Schrooten I., Goodman W.G., Cabrera W.E., Lamberts L.V., Elseviers M.M., Couttenye M.-M., De Broe M.E. 2000. Increased bone strontium levels in hemodialysis patients with osteomalacia. In Kidney Int. pp. 1107–1114. 57:. [DOI] [PubMed] [Google Scholar]
- Di Mauro S., Manes T., Hessle H., Kozlenkov A., Pizauro J.M., Hoylaerts M.F., Millán J.L. 2002. Kinetic characterization of hypophosphatasia mutations with physiological substrates. J. Bone Miner. Res. 17: 1383–1391. [DOI] [PubMed] [Google Scholar]
- Doellgast G.J. and Fishman W.H. 1977. Inhibition of human placental-type alkaline phosphatase variants by peptides containing L-leucine. Clin. Chim. Acta 75: 449–454. [DOI] [PubMed] [Google Scholar]
- Fishman W.H. and Sie H.G. 1971. Organ-specific inhibition of human alkaline phosphatase isoenzymes of liver, bone, intestine and placenta; L-phenylalanine, L-tryptophan and L-homoarginine. Enzymologia 41: 140–167. [PubMed] [Google Scholar]
- Francis R.M. and Selby P.L. 1997. Osteomalacia. Baillieres Clin. Endocrinol. Metab. 11: 145–163. [DOI] [PubMed] [Google Scholar]
- Gettins P. and Coleman J.E. 1983. 31P nuclear magnetic resonance of phosphoenzyme intermediates of alkaline phosphatase. J. Biol. Chem. 258: 408–416. [PubMed] [Google Scholar]
- Gettins P. and Coleman J.E. 1984. Zn(II)-113Cd(II) and Zn(II)-Mg(II) hybrids of alkaline phosphatase 31P and 113Cd NMR. J. Biol. Chem. 259: 4991–4997. [PubMed] [Google Scholar]
- Giammarile F., Mognetti T., Blondet C., Desuzinges C., Chauvot P. 1999. Bone pain palliation with 85Sr therapy. J. Nucl. Med. 40: 585–590. [PubMed] [Google Scholar]
- Harmey D., Hessle L., Narisawa S., Johnson K., Terkeltaub R., Millán J.L. 2004. Concerted regulation of inorganic pyrophosphate and osteopontin by Akp2, Enpp1 and Ank. An integrated model of the pathogenesis of mineralization disorders. Am. J. Pathol. 164: 1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris H. 1990. The human alkaline phosphatases: What we know and what we don't know. Clin. Chim. Acta 186: 133–150. [DOI] [PubMed] [Google Scholar]
- Henthorn P.S., Raducha M., Fedde K., Lafferty M.A., Whyte M.P. 1992. Different missense mutations at the tissue-nonspecific alkaline phosphatase gene locus in autosomal recessively inherited forms of mild and severe hypophosphatasia. Proc. Natl. Acad. Sci. 89: 9924–9928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessle L., Johnson K.A., Anderson H.C., Narisawa S., Sali A., Goding J.W., Terkeltaub R., Millan J.L. 2002. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc. Natl. Acad. Sci. 99: 9445–9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtz K.M. and Kantrowitz E.R. 1999. The mechanism of the alkaline phosphatase reaction: Insights from NMR, crystallography and site-specific mutagenesis. FEBS Lett. 462: 7–11. [DOI] [PubMed] [Google Scholar]
- Hoylaerts M.F., Manes T., Millán J.L. 1997. Mammalian alkaline phosphatases are allosteric enzymes. J. Biol. Chem. 272: 22781–22787. [DOI] [PubMed] [Google Scholar]
- Hull W.E., Halford S.E., Gutfreund H., Sykes B.D. 1976. 31P nuclear magnetic resonance study of alkaline phosphatase: The role of inorganic phosphate in limiting the enzyme turnover rate at alkaline pH. Biochemistry 15: 1547–1561. [DOI] [PubMed] [Google Scholar]
- Janeway C.M., Xu X., Murphy J.E., Chaidaroglou A., Kantrowitz E.R. 1993. Magnesium in the active site of Escherichia coli alkaline phosphatase is important for both structural stabilization and catalysis. Biochemistry 32: 1601–1609. [DOI] [PubMed] [Google Scholar]
- Kim E.E. and Wyckoff H.W. 1991. Reaction mechanism of alkaline phosphatase based on crystal structures. Two-metal ion catalysis. J. Mol. Biol. 218: 449–464. [DOI] [PubMed] [Google Scholar]
- Kozlenkov A., Manes T., Hoylaerts M.F., Millán J.L. 2002. Function assignment to conserved residues in mammalian alkaline phosphatases. J. Biol. Chem. 277: 22992–22999. [DOI] [PubMed] [Google Scholar]
- Kshirsagar A.G. 1975. The effect of stable strontium on the alkaline phosphatase activity of rat tissues—In vitro studies. Biochem. Pharmacol. 24: 13–20. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Le Du M.H., Stigbrand T., Taussig M.J., Ménez A., Stura E.A. 2001. Crystal structure of alkaline phosphatase from human placenta at 1.8 Å resolution. J. Biol. Chem. 276: 9158–9165. [DOI] [PubMed] [Google Scholar]
- Le Du M.H., Lamoure C., Muller B.H., Bulgakov O.V., Lajeunesse E., Menez A., Boulain J.C. 2002. Artificial evolution of an enzyme active site: Structural studies of three highly active mutants of Escherichia coli alkaline phosphatase. J. Mol. Biol. 316: 941–953. [DOI] [PubMed] [Google Scholar]
- Llinas P., Stura E.A., Ménez A., Kiss Z., Stigbrand T., Millán J.L., Le Du M.H. 2005. Structural studies of human placental alkaline phosphatase in complex with functional ligands. J. Mol. Biol. 350: 441–451. [DOI] [PubMed] [Google Scholar]
- Mandecki W., Shallcross M.A., Sowadski J., Tomazic-Allen S. 1991. Mutagenesis of conserved residues within the active site of Escherichia coli alkaline phosphatase yields enzymes with increased kcat. Protein Eng. 4: 801–804. [DOI] [PubMed] [Google Scholar]
- Masella M. 2006. The r-RESPA procedure and polarizable potentials based on induced dipoles. Mol. Phys. (in press).
- Masella M. and Cuniasse P. 2003. A many-body model to study proteins. I. Applications to MLnm+complexes, Mm+= Li+, Na+, K+, Mg2+, Ca2+ and Zn2+, L = H2O, CH3OH, HCONH2, n = 1–6, and to small hydrogen bonded systems. J. Chem. Phys. 119: 1866–1873. [Google Scholar]
- Matlin A.R., Kendall D.A., Carano K.S., Banzon J.A., Klecka S.B., Solomon N.M. 1992. Enhanced catalysis by active-site mutagenesis at aspartic acid 153 in Escherichia coli alkaline phosphatase. Biochemistry 31: 8196–8200. [DOI] [PubMed] [Google Scholar]
- McComb R.B., Bowers G.N., Posen S. In Alkaline phosphatases pp. 986–989. 1979. Plenum Press, New York.
- Meunier P.J. and Reginster J.Y. 2003. Design and methodology of the phase 3 trials for the clinical development of strontium ranelate in the treatment of women with postmenopausal osteoporosis. Osteoporos. Int. 3):(Suppl. S66–S76. [DOI] [PubMed] [Google Scholar]
- Meyer J.L. 1984. Can biological calcification occur in the presence of pyrophosphate? Arch. Biochem. Biophys. 231: 1–8. [DOI] [PubMed] [Google Scholar]
- Mornet E. 2000. Hypophosphatasia: The mutations in the tissue-nonspecific alkaline phosphatase gene. Hum. Mutat. 15: 309–315. [DOI] [PubMed] [Google Scholar]
- Mornet E., Stura E., Lia-Baldini A.S., Stigbrand T., Ménez A., Ledu M.H. 2001. Structural evidence for a functional role of human tissue non-specific alkaline phosphatase in bone mineralisation. J. Biol. Chem. 276: 31171–31178. [DOI] [PubMed] [Google Scholar]
- Moss D.W., Eaton R.H., Smith J.K., Whitby L.G. 1967. Association of inorganic-pyrophosphatase activity with human alkaline-phosphatase preparations. Biochem. J. 102: 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller B.H., Lamoure C., Ledu M.H., Cattolico L., Lajeunesse E., Lemaitre F., Pearson A., Ducancel F., Menez A., Boulain J.C. 2001. Improving Escherichia coli alkaline phosphatase efficacy by additional mutations inside and outside the catalytic pocket. Chembiochem 2: 517–523. [DOI] [PubMed] [Google Scholar]
- Murphy J.E. and Kantrowitz E.R. 1994. Why are mammalian alkaline phosphatases much more active than bacterial alkaline phosphatases? Mol. Microbiol. 12: 351–357. [DOI] [PubMed] [Google Scholar]
- Murphy J.E., Stec B., Ma L., Kantrowitz E.R. 1997. Trapping and visualization of a covalent enzyme-phosphate intermediate. Nat. Struct. Biol. 4: 618–622. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin A.A., Dodson E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Perrakis A., Morris R., Lamzin V.S. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6: 458–463. [DOI] [PubMed] [Google Scholar]
- Reginster J.Y., Deroisy R., Jupsin I. 2003. Strontium ranelate: A new paradigm in the treatment of osteoporosis. Drugs Today (Barc.) 39: 89–101. [DOI] [PubMed] [Google Scholar]
- Rinaldo D., Vita C., Field M.J. 2004. Engineering strontium binding affinity in an EF-hand motif: A quantum chemical and molecular dynamics study. J. Biomol. Struct. Dyn. 22: 281–297. [DOI] [PubMed] [Google Scholar]
- Roussel A. and Cambillau C. 1989. Silicon Graphics geometry partner directory. Silicon Graphics, Mountain View, CA 77–78.
- Saarto T., Janes R., Tenhunen M., Kouri M. 2002. Palliative radiotherapy in the treatment of skeletal metastases. Eur. J. Pain 6: 323–330. [DOI] [PubMed] [Google Scholar]
- Satow Y., Cohen G.H., Padlan E.A., Davies D.R. 1986. Phosphocholine binding immunoglobulin Fab McPC603. An X-ray diffraction study at 2.7 Å. J. Mol. Biol. 190: 593–604. [DOI] [PubMed] [Google Scholar]
- Schrooten I., Cabrera W., Goodman W.G., Dauwe S., Lamberts L.V., Marynissen R., Dorriné W., De Broe M.E., D'Haese P.C. 1998. Strontium causes osteomalacia in chronic renal failure rats. Kidney Int. 54: 448–456. [DOI] [PubMed] [Google Scholar]
- Schrooten I., Behets G.J.S., Cabrera W.E., Vercauteren S.R., Lamberts L.V., Verberckmoes S.C., Bervoets A.J., Dams G., Goodman W.G., De Broe M.E.et al. 2003. Dose-dependent effects of strontium on bone of chronic renal failure rats. Kidney Int. 63: 927–935. [DOI] [PubMed] [Google Scholar]
- Schwartz J.H. and Lipmann F. 1961. Phosphate incorporation into alkaline phosphatase of E. coli. Proc. Natl. Acad. Sci. 47: 1996–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeland S., Erikssen B., Aas M., Skovlund E., Hess S.L., Fosså S.D. 2003. Role of strontium-89 as adjuvant to palliative external beam radiotherapy is questionable: Results of a double-blind randomized study. Int. J. Radiat. Oncol. Biol. Phys. 56: 1397–1404. [DOI] [PubMed] [Google Scholar]
- Stec B., Holtz K.M., Kantrowitz E.R. 2000. A revised mechanism for the alkaline phosphatase reaction involving three metal ions. J. Mol. Biol. 299: 1303–1311. [DOI] [PubMed] [Google Scholar]
- Van Belle H. 1976. Alkaline phosphatase. I. Kinetics and inhibition by levamisole of purified isoenzymes from humans. Clin. Chem. 22: 972–976. [PubMed] [Google Scholar]
- Verberckmoes S.C., De Broe M.E., D'Haese P.C. 2003. Dose-dependent effects of strontium on osteoblast function and mineralization. Kidney Int. 64: 534–543. [DOI] [PubMed] [Google Scholar]
- Weiss M., Cole D.E.C., Ray K., Whyte M., Laffeerty M.A., Mulivor R.A., Harris H. 1988. A missense mutation in the human liver/bone/kidney alkaline phosphatase gene causing a lethal form of hypophosphatasia. Proc. Natl. Acad. Sci. 85: 7666–7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte M.P. 1994. Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr. Rev. 15: 439–461. [DOI] [PubMed] [Google Scholar]
- Whyte M.P. 2001. Hypophosphatasia. In The metabolic and molecular bases of inherited disease (eds. Scriver C.R.et al.) . pp. 5313–5329. McGraw-Hill, New York.
- Xu X. and Kantrowitz E.R. 1991. A water-mediated salt link in the catalytic site of Escherichia coli alkaline phosphatase may influence activity. Biochemistry 30: 7789–7796. [DOI] [PubMed] [Google Scholar]
- Zurutuza L., Muller F., Gibrat J.F., Taillandier A., Simon-Bouy B., Serre J.L., Mornet A. 1999. Correlations of genotype and phenotype in hypophosphatasia. Hum. Mol. Genet. 8: 1039–1046. [DOI] [PubMed] [Google Scholar]