

Abstract
The class A PBP1b from Streptococcus pneumoniae is responsible for glycosyltransferase and transpeptidase (TP) reactions, forming the peptidoglycan of the bacterial cell wall. The enzyme has been produced in a stable, soluble form and undergoes time-dependent proteolysis to leave an intact TP domain. Crystals of this TP domain were obtained, diffracting to 2.2 Å resolution, and the structure was solved by using molecular replacement. Analysis of the structure revealed an “open” active site, with important conformational differences to the previously determined “closed” apoenzyme. The active-site nucleophile, Ser460, is in an orientation that allows for acylation by β-lactams. Consistent with the productive conformation of the conserved active-site catalytic residues, adjacent loops show only minor deviation from those of known acyl-enzyme structures. These findings are discussed in the context of enzyme functionality and the possible conformational sampling of PBP1b between active and inactive states.
Keywords: penicillin-binding protein, crystal structure, peptidoglycan, conformational change, transpeptidase
The penicillin-binding proteins (PBPs) are the physiological target of the β-lactam class of anti-bacterials, the major weapon against bacterial infection. It is thus imperative that we understand this class of enzymes, and this has been assisted by high-resolution structures of PBPs, both alone and in complex with β-lactams (Wilke et al. 2005). The mode of action of β-lactams is well documented, and results from their chemical similarity to the peptidoglycan stem peptide substrate of the PBP enzymes. The lactam nucleus reacts with the PBP serine nucleophile, yielding a relatively stable acyl-enzyme intermediate. Cell lysis follows this process, resulting from inhibition of the PBP enzymes and poorly cross-linked cell wall peptidoglycan. Alongside their role as the targets for β-lactams (and subsequent problematic mutation in bacterial resistance), PBPs also participate in the important processes of cellular growth and division (Morlot et al. 2003). It has been hypothesized that different PBPs may function as part of a group of proteins forming a cell wall maintenance holoenzyme (Holtje 1996).
The final stages of peptidoglycan synthesis involve a glycosyltransferase (GT)-dependent polymerization and transpeptidase (TP)-dependent cross-linking of the N-acetylmuramic acid–β-1,4-N-acetylglucosamine (and associated stem peptide) units of the bacterial cell wall. PBPs can be subdivided into two classes: Class A enzymes harbor both GT activity and TP activity on the same polypeptide, whereas class B enzymes possess only a TP functionality, often in association with other non-penicillin-binding structural motifs (for a review of PBP structure and modularity, see Goffin and Ghuysen 1998). The class A enzymes show domain-based functionality, with the GT domain following an N-terminal transmembrane helix, and the TP domain residing in the C-terminal end of the enzyme. Both active sites are presumed to function independently, with no inhibition of either activity when inhibitors against the other functionality were used (Di Guilmi et al. 2003). TP domains have a well-characterized protein fold, observed in many class B PBP structures and also in the β-lactamase group of enzymes (Wilke et al. 2005). A structure of the GT fold, from either class A PBPs or related monofunctional GT enzymes (Wang et al. 2001), would be invaluable for novel anti-bacterial drug design—with the possibility of inhibiting an easily accessible and essential group of enzymes (Leski and Tomasz 2005).
The PBP1b protein from Streptococcus pneumoniae is unusual among class A enzymes in that constructs lacking the membrane-spanning domain remain soluble in the absence of detergent. S. pneumoniae is an important human pathogen, implicated in pneumonia, bacteremia, meningitis, and other disease states, and is responsible for in excess of 1 million fatalities yearly, particularly within developing nations. The genome of S. pneumoniae encodes at least six PBPs, and although PBP1b is not implicated in the development of β-lactam resistance (Du Plessis et al. 2000), the homologous, resistance-conferring PBP1a (45% sequence identity within the TP domain) has been solved (Contreras-Martel et al. 2006). A soluble, proteolyzed region of PBP1b provided the first structural information for any class A enzyme (Macheboeuf et al. 2005). The protein was shown to contain the classical TP fold, along with flanking N- and C-terminal domains, and surprisingly, an associated hairpin from the otherwise degraded GT domain. In contrast to the three acyl-enzyme structures, the apoenzyme reported by Macheboeuf et al. (2005) was present in a “closed” conformation. The main features of the closure of the TP domain active site were the positioning of the serine nucleophile so as to be unavailable for participation in acylation, and the movement of a loop between strands β3 and β4, causing the entrance to the active site to become occluded. Such an inactive conformation has no precedent in previously observed, β-lactam-sensitive TP structures and led to the postulation of the regulation of enzyme activity by other proteins or activation via substrate. We present here a structure for a similarly truncated PBP1b apoenzyme, observed for the first time in an “open” conformation, and discuss the implications for the functionality of the TP domain.
Results
Protein purification
A fragment of PBP1b from S. pneumoniae was expressed and purified, resulting in a final preparation where two bands could be observed (Fig. 1). N-terminal sequencing confirmed them both to be derived from our construct, the higher molecular weight band representing the intact construct (starting at residue MD85KVRV) and the smaller band resulting from limited proteolysis (starting at residue L184IKQQV). The protease responsible for this degradation remains unidentified. The ratio of these two bands varies from preparation to preparation and is unaffected by the presence of commercial protease inhibitors or mutation of the area around Q183 (A.L. Lovering, unpubl.). Despite this heterogeneity in the protein sample, it was deemed suitable for crystallization screening.
Figure 1.
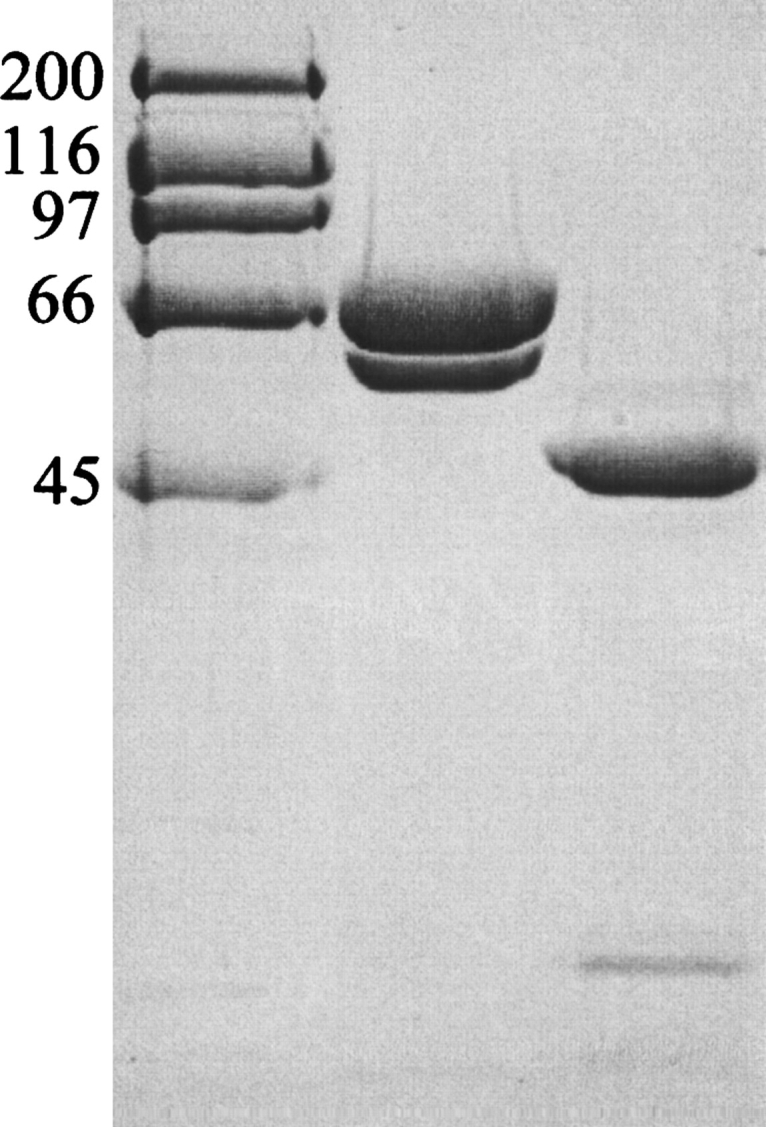
SDS-PAGE of truncated PBP1b protein samples. A 10% (w/v) acrylamide separating gel was used, with subsequent steps following standard procedure. (Lane 1) High-range molecular markers (Bio-Rad; molecular weight given in kilodaltons). (Lane 2) “Freshly prepared” truncated PBP1b protein stock. (Lane 3) Moenomycin-aged truncated PBP1b protein stocks. The aging of protein samples was carried out in an attempt to mimic any proteolysis occurring in the crystallization drop. N-terminal protein sequencing results were used to characterize the protein bands: Lane 2, upper band MD85KVRV, lower band L184IKQQV; lane 3, upper band Q323DFLPS, lower band K139AIIAT. The desired soluble truncation product of our PBP1b construct was present in freshly prepared protein samples, starting at the native sequence residue D85 (post-transmembrane helix). This sample also contains a contaminating band starting at L184, resulting from proteolysis of the GT domain. After protein aging, the larger, two-domain constructs are pared down to the linker region and TP domain (Q323 onward). The smaller, TP-associated hairpin from the GT domain (to the smallest possible extent, residues 105–119) is not visible on this gel. If the degradation observed in aged samples is comparable to that in the crystallization drop, residues 323–336 must be disordered in the electron density map. It is also possible that further proteolysis has occurred precrystallization at the protease-sensitive R336 position (Macheboeuf et al. 2005).
Crystallization, data collection, and structure refinement
Crystals of a truncated form of PBP1b from S. pneumoniae were obtained in space group C2 with one molecule per asymmetric unit. The realization that crystals of the full length construct had not been obtained was supported directly by Matthews volume calculations, and indirectly by SDS-PAGE of protein stocks both fresh and aged (Fig. 1) and N-terminal sequencing of these gel bands (Q323DFLPS plus K139AIIAT for moenomycin-aged samples). In both the crystallization and the protein aging experiments, the bifunctional construct is pared down to the TP region and linker domain by limited proteolysis. The structure was solved by molecular replacement using a similarly truncated form of the enzyme as the search model (PDB 2BG1; Macheboeuf et al. 2005), yielding a clear solution with a correlation coefficient of 57.3%. A summary of statistics from data collection and refinement is given in Table 1. Despite several attempts, no further crystals of this form could be grown, although small crystal fragments from the original drop could be used to seed new drops, unfortunately resulting in crystals that were unsuitable for data collection (data not shown).
Table 1.
Data collection and refinement statistics

Structural features of truncated PBP1b
The electron density obtained from RESOLVE (Terwilliger 2003) Prime & Switch phase bias removal was of excellent quality, allowing the structure to be modeled with confidence (Fig. 2A). The final model has 89.4%, 9.4%, 1.2%, and 0% of residues in the core, the allowed, the generously allowed, and the disallowed regions of the Ramachandran plot, respectively. The model encompasses 468 amino acids, with 453 present in a single polypeptide that contributes the bulk of the protein fold (amino acids 337–789), and an additional 15 residues from the proteolyzed GT domain present as an associated β-hairpin (amino acids 105–119). Electron density was observed near H682 that could not be successfully modeled as a water molecule, and, in accordance with the distinctive coordination geometry and crystallization condition composition, this has been designated as a nickel ion (occ = 1, B = 68.7 Å2). The naming convention used for structural features is taken from the previously observed structure of truncated PBP1b (Macheboeuf et al. 2005), itself based on that used for class A β-lactamases. In short, the fold possesses three distinct domains (Fig. 2B)—an N-terminal “linker” domain (with a β-sheet made up of strands from the main polypeptide and the hairpin from the GT domain, and also two long, perpendicular α-helices), a functional TP domain (composed of a central, antiparallel β-sheet, flanked by three helices), and a C-terminal β-strand-rich domain that stabilizes the central fold and may also provide a scaffolding domain for interactions with other proteins in the putative cell-wall synthesizing complex.
Figure 2.
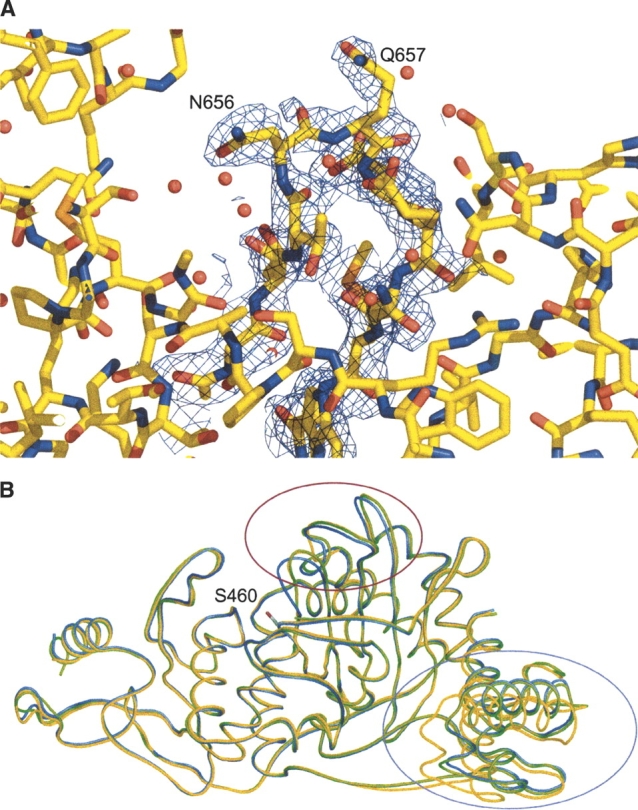
Active-site loop movement in PBP1b. (A) Electron density for active-site loop of the truncated PBP1b apoenzyme. Figure colored according to atom type (yellow indicates C; red, O; blue, N; orange, S). Map is from a 2Fo − Fc calculation omitting residues 652–662, contoured around these residues at 1σ level to 2.2 Å resolution. (B) Comparison of the Cα-traces for open, closed, and acylated PBP1b TP domains. Structures were aligned using the auto-fit procedure of SwissPDBViewer (Guex and Peitsch 1997). Open structure is of the apoenzyme reported in this study (PDB 2FFF, yellow). Closed apoenzyme structure (PDB 2BG1, blue) and nitrocefin acyl-enzyme (PDB 2BG3, green) are from results reported by Macheboeuf et al. (2005). The side chain of the active site S460 nucleophile from the open structure is shown in stick form (C atoms, gray; Oγ atom, red). Major differences between three active-site loops can be observed between the open/acyl and the closed forms (within red ellipse; residues 414–421, 653–660, and 677–687). Differences can also be observed at the mobile N-terminal domain (within magenta ellipse), especially in helices Ha and Hb (residues 337–358 and 369–384, respectively).
Comparison to previously reported PBP1b structures
As expected, the model shows close gross structural similarity to the PBP1b construct crystallized by Macheboeuf et al. (2005) with backbone RMSDs of 0.74 Å with the unacylated complex, 0.73 Å for the nitrocefin adduct structure, and 0.70/0.60 Å for chains A/B of the cefotaxime derivative. The close agreement of RMSD values from comparison of these structures belies several important shifts in active-site residues and loops and other differences present in regions Ha/Hb of the fold (Figs. 2B, 3A). A striking difference was noticeable when comparing the two apoenzyme structures; three loops present near the active site are displaced—the loop between α1 and β1 (amino acids 414–421), the loop between β3 and β4 (653–660), and the region immediately after β5 (677–687). Our apoenzyme structure shows more similarity in these regions to the previously observed acyl-enzyme complexes (Macheboeuf et al. 2005), as noted by backbone RMSDs to our coordinates for these loops of 0.77/1.07 Å (nitrocefin adduct/“closed” apoenzyme) for 414–421, 0.54/1.30 Å for 653–660, and 0.84/1.09 Å for 677–687. Differences in other regions of the structure are located in the N-terminal region of the protein fold, particularly in helices Ha and Hb and the region between strands βd and βe. This region of the structure is closer in conformation to that of the cefotaxime adducts, suggesting that this variability may be space-group-dependent (given that the cefotaxime structures were solved in a different space-group to the apoenzyme and nitrocefin adduct) (Macheboeuf et al. 2005). Unlike the conformational differences at the active site, differences in the N-terminal region are largely present as a rigid-body translation. The observation of mobility in the position of Ha and Hb is not unusual given the small contact area between the bulk of the protein fold and this domain.
Figure 3.

Detail of PBP1b active-site differences, with selected side-chain residues shown in stick form. Figure colored according to atom type (yellow indicates open PBP1b structure C; purple, closed PBP1b C; green, acyl-enzyme PBP1b C; gray, PBP1a C; , red, O; blue, N; orange, S). (A) Comparison of the open (PDB 2FFF [this study]) and closed (PDB 2BG1 [Macheboeuf et al. 2005]) apoenzyme structures. Labeling with a prime denotes residues from the closed structure. The figure shows the large shift in the polypeptide backbone of loop β3–β4 (AA 653–660) and shifts in the position of the important active-site residues T654, T655, N656, and Q657. These changes between the two apoenzyme structures result in the occlusion of the active site in the closed form, largely due to N656 blocking entry of substrate. The S460 nucleophile is unavailable for reaction in the closed form, making close contacts to the backbone region of β3 (at the oxyanion hole formed between the N atoms of S460 and T654). With the shift of the active-site loops, the antiparallel nature of β3 and β4 is disturbed in the closed form, resulting in the premature termination of strand β3. The conformations of residues R686 and R687 are also shown, differing from the glutamine side chains of the closed form construct. These mutations occur in a protease-sensitive area of the molecule and may be responsible in part for observed kinetic differences to the wild type enzyme (Macheboeuf et al. 2005). (B) Comparison of the open (this study) and nitrocefin acyl-enzyme (PDB 2BG3 [Macheboeuf et al. 2005]) structures. Nitrocefin moiety of acyl-enzyme shown as partly transparent space-fill model. The backbone atoms are in closer agreement with our structure than those of the closed apoenzyme (A), and the antiparallel nature of β3 and β4 is restored. Most of the active-site side chains are in a similar conformation between the two structures, with T654, T655, N656, and Q657 only needing minor rotametric alterations to achieve complementarity. The open form shows very little steric clash with the placement of the nitrocefin adduct, with only R686 needing a significant movement to accommodate the bulky R2 nitro group. The side chain of the active-site S460 nucleophile is also in a similar conformation between the open and the acyl-enzyme structures. Such general agreement between the two forms validates the assumption that the apoenzyme presented in this study is in a more productive conformation than that previously observed (Macheboeuf et al. 2005). (C) Comparison of the open PBP1b apoenzyme (this study) and the PBP1a (PDB 2C6W [Contreras-Martel et al. 2006]) structures. The similarity in position of the nucleophile (S460 PBP1b, S370 PBP1a) confirms the differences observed between this apoenzyme and that from previous studies (B). Residues lining the active site (T654/T655/N656 for PBP1b; T560/S561/N562 for PBP1a) also show good agreement, despite the insertion of a helix in loop b3/b4, and the change in topology of some of the active-site loops.
The conserved catalytic motifs (S460XXK, S516XN, and K651TG) of the TP fold are present in or near the regions that differ between the presented PBP1b apoenzyme structure and that reported by Macheboeuf et al. (2005). The most striking difference is the rotation of the catalytic nucleophile S460 between the two structures (Fig. 3A). In the previous structure (PDB 2BG1), the side chain of S460 is rotated toward β3 (Oγ is 2.9 Å from T654 N), in an apparently catalytically incompetent state, and unavailable to bind ligands or interact with residues from the other conserved motifs. In our structure, S460 is rotated ∼120° from its orientation in 2BG1 and is much like the conformation observed in all other β-lactam–sensitive TP structures and those of the PBP1b acyl-enzyme adducts (Fig. 3B). Unlike the interaction of S460 with T654 in 2BG1, the Oγ group is now 4.8 Å from T654 N, and a water molecule (WAT 1) sits in between the two residues. This water molecule makes close contacts with both residues, 2.4 Å from S460 Oγ and 2.6 Å from T654 N. The observation of such close contacts raises the possibility that the density may have arisen from multiple conformations of S460, but a test refinement of this scenario indicated clearly that the serine would have to move to fill this region and no density could be observed consistent with such a backbone shift. The close contacts between the water molecule and S460/T654 are likely a result of the unique electrostatic environment of this region, often referred to as the “oxyanion hole,” and indeed, similar short-range contacts have been observed in this region in related enzymes including the β-lactamases (Nukaga et al. 2003). The possibility also remains that the density arises from a chloride ion, present in the crystallization conditions at a concentration of 10 mM. The oxyanion hole promotes close contacts to stabilize the transition state during acylation of the catalytic nucleophile (Fisher et al. 2005), also promoting a hydrogen bond between T654N and the carbonyl of reacted β-lactams (2.7 Å distance in the nitrocefin adduct) (Macheboeuf et al. 2005).
In tandem to the restricted accessibility of the S460 nucleophile in 2BG1, the movement of the loop between β3 and β4 causes steric occlusion of the active site, with residues T654, T655, and N656 all in positions that would clash with the position of the reacted β-lactam in the acyl-enzyme structures. In contrast, the apoenzyme structure presented here positions the main-chain atoms of these residues much closer to those of the acyl-enzymes (Fig. 3B), and only small rotametric changes of the side chains would be necessary to accommodate the antibiotic. This is also true of residue R686 when considering the binding of the bulkier R2-group of nitrocefin. Residues Q657, E659, and N681 show some conformational differences between our structure and those from the “closed” and acyl-enzyme coordinates. These residues are present on the outer limits of the active site and appear to not play any role in ligand binding, suggesting that these differences are due to the solvent-exposed nature of this region.
The construct used in these experiments also differed from that of Macheboeuf et al. (2005) in that the native sequence of the S. pneumoniae PBP1b enzyme was used, with no mutation of the proteolysis-susceptible arginine 336, 686, and 687 residues. As no electron density was observed for R336, this may be due to proteolysis at this position. However, the electron density for the backbone atoms of R686 and R687 is excellent, suggesting that no proteolysis of this loop region has occurred for this crystal form. These residues are solvent-exposed, and density is good for R687, but only strong up to the Cγ atom of R686. The mutation of R686/687 does not appear to place any constraint upon the backbone conformation of this region or cause the shift of any neighboring amino acid side chains.
Comparison to previously reported PBP1b structures
Structures of the apo- and acylated forms of S. pneumoniae PBP1a have been solved (Contreras-Martel et al. 2006), both present in an “open” conformation. The apo- and acylated forms possess backbone RMSDs of 1.32 and 1.31 Å, respectively, with our PBP1b structure. Although there are several differences in the length and position of active-site loops when comparing PBP1b and PBP1a (Fig. 3C), there is much similarity in the position of several important residues. The availability of the S460 (S370 in PBP1a) nucleophile is constant between the two structures, further supporting the observation of differences between the coordinates presented in this study and those of the previously observed apoenzyme (Macheboeuf et al. 2005). Residues in the β3/β4 loop show good agreement (T654/T655/N656 for PBP1b, T560/S561/N562 for PBP1a) (Fig. 3C), despite the insertion of a helix in this region for PBP1a.
Discussion
The crystal structure of a truncated form of S. pneumoniae PBP1b has been solved, with important structural differences from previously reported work (Macheboeuf et al. 2005). It had been desired to obtain a complex of the soluble bifunctional enzyme with the GT inhibitor moenomycin, but it was apparent that proteolysis had pared the construct down to a GT hairpin peptide, linker region, and TP domain. Many studies have noted the fragility of the GT polypeptide region, and it has also been noted that moenomycin increases the susceptibility of this domain to proteolysis (Di Guilmi et al. 2003). Although initial indications from SDS-PAGE raised the possibility of obtaining a stable fragment from Q323 onward (Fig. 1), no electron density for the linker region was observed before D337, consistent with R336 as a protease-sensitive site (Macheboeuf et al. 2005). There is sufficient space in the crystal packing to allow for several residues before D337, and the possibility cannot be discounted that this region is present but disordered. Unfortunately, identical crystals could not be obtained to test the exact unit cell contents via mass spectrometry or protein sequencing. The associated hairpin from the GT domain is traceable to the same extent as that observed in the acyl-enzyme complexes (amino acids 105–119), although several partially ordered residues can be observed at the hairpin C terminus when lowering the sigma value of the electron density map.
The construct used in this study also retained the native protein sequence at the mutation-susceptible R336, R686, and R687 residues. It has been observed that the mutation of these amino acids to glutamines results in an acylation rate approximately three- to sixfold lower than that of the wild-type enzyme (Macheboeuf et al. 2005), possibly due to charge attraction between the negatively charged antibiotic and arginine residues. The conformations of R686 and R687 in our structure do not provide an obvious explanation for an alternate reason for the acylation rate differences and also do not appear to be responsible for the different positions of the active-site loops in the two apoenzymes.
The inactive conformation of the serine nucleophile and occluded active site of the previously determined apoenzyme were not observed in our structure (Fig. 3A). With the importance of the presented apoenzyme structure in resembling the acyl-enzyme active-site topology more closely than the previous apoenzyme structure, it must be noted that the changes from the molecular replacement model were made with no knowledge of the acyl-enzyme coordinates, which were released after those of the apoenzyme. The strong difference density from RESOLVE (Terwilliger 2003) Prime & Switch maps, good refinement statistics, and agreement with the acyl-enzyme active-site topology provide confidence in the observations presented from this study, in tandem with the nucleophile position comparison with the PBP1a enzyme (Fig. 3C). Our observed conformations of the active-site residues are also similar to those observed in other structures of soluble TP enzymes and the serine β-lactamases, presumably reflecting the low-energy form of these related active sites. The conclusions raised previously from the “closed” TP active site of PBP1b centered on the possibility of a switch between an active and an inactive enzyme that would be brought about by interaction with substrate (Macheboeuf et al. 2005). With the observation of an “open” apoenzyme in this study, and no visible interaction of the protein with external activation elements, we propose two possible interpretations: (1) that the conformations observed in the previous apoenzyme study are artifactual or (2) that both apoenzyme structures are valid and that the observable differences arise from a conformational sampling of the “open” and “closed” forms, an observation unique for TP enzymes which are usually present in a “preformed” catalytically competent state. In deference to no observable “effector” in our open structure, the possibility of conformational sampling instead of direct activation remains an attractive proposition. We can speculate that this conformational sampling may play a regulatory role in PBP1b catalytic activity. As proposed by Macheboeuf et al. (2005), there may exist a need for inactive PBP1b, where a TP functionality is inactive in nondividing cells and becomes activated during septation (Eberhardt et al. 2003). Conformational sampling of the nucleophile and active-site loops in the TP domain (particularly in the loop between β3 and β4) as the comparison of the two structures suggests, could be stabilized in a particular conformation by interaction with other proteins. Indeed, any effect on a possible equilibrium between the two forms would impose some level of control on enzyme activity. As both the GT and the TP activities are necessary for peptidoglycan formation, it would be of interest to see whether it would be necessary to regulate the GT activity of class A enzymes additionally. Some resistance-conferring mutations in PBP1a occur in regions outside the active site (Sanbongi et al. 2004; Macheboeuf et al. 2005; Contreras-Martel et al. 2006) but still have to cause a reduction in enzyme acylation. It is interesting to speculate that they may elicit part of this resistance effect through altering the ability of the protein to undergo conformational changes.
Although the unreactive state of the active-site nucleophile of 2BG1 has no parallel in β-lactam–sensitive enzymes, such a phenomenon has been observed for a β-lactam–resistant enzyme, PBP2a* of Staphylococcus aureus (Lim and Strynadka 2002). The expression of PBP2a* in methicillin-resistant S. aureus (MRSA) strains confers β-lactam insensitivity through the requirement of an energetically costly conformational change upon acylation. Due to the energetic cost of the apoenzyme to acyl-enzyme conversion, the protein is “insensitive” to regular β-lactams. Part of this conformational change occurs within α-helix 2 (analogous to PBP1b, where the catalytic nucleophile resides), but interestingly, this change also requires a twisting movement of strand β3. The movement of β3 between the acyl- and apoenzyme structures of PBP2a* is of a lesser extent than that seen here between the two apoenzyme structures of PBP1b and is limited to just a few residues, not the whole of β3-β4 and the surrounding region. However, the demonstration of such active-site plasticity in both enzymes reinforces the idea that the mobility of the β3 region may be more important in TP enzymes than previously thought, particularly in the context of enzyme activation.
The observation of two structurally different apoenzyme states also raises the question as to how they are differentially stabilized. The crystal contacts between our structure and the C2221 and P212121 forms of Macheboeuf et al. (2005) are different but appear to play no role in stabilizing the conformations of active-site loops. It is logical to attribute the lesser two active-site loop shifts to the movement of loop β3-β4, but the cause of this shift then requires explanation. Due to moderate similarity between the natural enzyme peptidoglycan stem peptide substrate or β-lactam antibiotic to any polypeptide, it may be functional for the enzyme to “close off” the active site by employing such a mobile loop. The relative populations of the open and closed states could be regulated by involvement in a cell wall multienzyme complex (Holtje 1996), with sampling of these two states present in these different crystal structures for reasons not easily observable. It will be interesting to see whether a closed enzyme form is observed in any subsequent β-lactam–sensitive PBP structures and how this state is brought about or stabilized. Increased knowledge of PBP enzymes and their functionality will help in both the understanding of their cellular role and the improvement of antibiotic design.
Materials and methods
Cloning, expression, and purification
The gene for the truncated form of PBP1b (AA D85-P793) was PCR-amplified from total S. pneumoniae genomic DNA and cloned under the control of a T7 RNA polymerase promoter in the pET28a vector (Novagen) using standard procedures. Protein expression was induced using 0.6 mM IPTG at a temperature of 293 K for 15–16 h. Typically, 1 L of culture was pelleted and resuspended in 40 mL of ice-cold buffer, containing 50 mM glycine (pH 9.0) and 10 mM EDTA. Resuspended material was lysed in small batches of 4 mL volume, using rapid sonication (180 sec/batch). Crude lysate was centrifuged at 50,000 rpm for 35 min. The resulting supernatant was applied to Q-Sepharose FF matrix, pre-equilibrated in a buffer containing 10 mM Tris (pH 7.0), and eluted with a linear gradient of NaCl, PBP1b fractions eluting at ∼0.4 M salt. Pooled fractions were dialyzed against 10 mM K2HPO4, and then applied to a SP-Sepharose FF matrix and eluted with a linear gradient of NaCl, with PBP1b eluting at ∼1 M salt. PBP1b protein samples were subsequently cleansed of any aggregation states by size-exclusion chromatography using a Superdex-200 column, in a buffer comprised of 10 mM glycine (pH 9.0) and 0.5 M NaCl. Fractions presumed monodisperse were pooled and concentrated to a final protein concentration of 100 mg/mL. The resultant protein stock was diluted 10-fold with ultrapure H2O for crystallography trials, with the GT inhibitor moenomycin (a kind gift from Professor Hitoshi Komatsuzawa, Hiroshima University, Japan) also added to a final concentration of 2 mM.
To mimic the aging and proteolysis present in the crystallization drop, PBP1b protein stock was left in crystallization buffer containing 2 mM moenomycin for 2 mo at 277 K and then analyzed via SDS-PAGE (using a 10% [w/v] acrylamide separating gel), alongside fresh protein samples. Both intact and proteolyzed bands were blotted onto nitrocellulose and sent for N-terminal sequencing (NAPS, University of British Columbia).
Crystallization and data collection
Crystallization experiments were set up using the hanging drop vapor diffusion method (0.5 μL of protein, 0.5 μL of reservoir solution) at 291 K and a protein concentration of 10 mg/mL. A crystal appeared after several months in a droplet equilibrated against 100 mM Tris (pH 8.5), 20% (w/v) PEG MME 2000, and 10 mM NiCl2. The crystal was successfully cryoprotected by transfer to a drop of mother liquor supplemented with 20% (v/v) ethylene glycol. After 2 min of equilibration with the cryobuffer, the crystal was placed in a liquid nitrogen stream (Oxford Cryosystems) at 100 K. Diffraction data were collected on a home source (CuKα radiation, Rigaku RU300 generator), using oscillation steps of 1° and a crystal to detector distance of 200 mm.
Structure solution, model building, and refinement
Diffraction data were processed using MOSFLM, and all data manipulations were performed with the CCP4 suite (CCP4 1994). The space group was determined to be C2, with approximate cell dimensions of 132 × 78 × 60 Å and a β angle of 113°. These parameters were consistent with one molecule of a truncated form of PBP1b in the asymmetric unit. The structure was solved with MOLREP (Vagin and Teplyakov 2000), using data between 15 Å and 4 Å, and PDB 2BG1 (Macheboeuf et al. 2005) as the search model. Model bias was minimized with the Prime & Switch procedure in RESOLVE (Terwilliger 2003), using SIGMAA (Read 1986) calculated phases from the molecular replacement solution. The model was altered to fit the RESOLVE electron density map, and subsequent cycles of rebuilding and refinement were performed with REFMAC (Murshudov 1997), including TLS refinement (treating the N-terminal and TP domains as separate regions). All superpositions were performed with Swiss PDBViewer (Guex and Peitsch 1997). Structural-based figures were generated using PyMol (http://www.pymol.org).
Protein Data Bank accession codes
Coordinates and structure factors have been deposited in the RCSB Protein Data Bank with the accession code 2FFF.
Acknowledgments
This work was supported by a Michael Smith Foundation for Health Research post-doctoral fellowship to A.L.L. N.C.J.S. is a Howard Hughes Medical Institute international research scholar and is also funded by the Canadian Institutes of Health Research.
Footnotes
Reprint requests to: Natalie C.J. Strynadka, Department of Biochemistry and Molecular Biology, and the Center for Blood Research, University of British Columbia, 2350 Health Sciences Mall, Vancouver V6T 1R9, Canada; e-mail: natalie@byron.biochem.ubc.ca; fax: (604) 822-5227.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062112106.
Abbreviations: ASU, asymmetric unit; EDTA, ethylenediaminetetraacetic acid; GT, glycosyltransferase; IPTG, isopropyl thio-β-d-galactoside; MRSA, methicillin-resistant Staphylococcus aureus; PEG MME, polyethylene glycol monomethyl ether; PBP, penicillin binding protein; TP, transpeptidase; RMSD, root mean square deviation.
References
- Collaborative Computational Project, Number 4 (CCP4). 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Contreras-Martel C., Job V., Di Guilmi A.M., Vernet T., Dideberg O., Dessen A. 2006. Crystal structure of penicillin-binding protein 1a (PBP1a) reveals a mutational hotspot implicated in β-lactam resistance in Streptococcus pneumoniae. J. Mol. Biol. 355: 684–696. [DOI] [PubMed] [Google Scholar]
- Di Guilmi A.M., Dessen A., Dideberg O., Vernet T. 2003. Functional characterization of penicillin-binding protein 1b from Streptococcus pneumoniae. J. Bacteriol. 185: 1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Plessis M., Smith A.M., Klugman K.P. 2000. Analysis of penicillin-binding protein 1b and 2a genes from Streptococcus pneumoniae. Microb. Drug Resist. 6: 127–131. [DOI] [PubMed] [Google Scholar]
- Eberhardt C., Kuerschner L., Weiss D.S. 2003. Probing the catalytic activity of a cell division–specific transpeptidase in vivo with β-lactams. J. Bacteriol. 185: 3726–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher J.F., Meroueh S.O., Mobashery S. 2005. Bacterial resistance to β-lactam antibiotics: Compelling opportunism, compelling opportunity. Chem. Rev. 105: 395–424. [DOI] [PubMed] [Google Scholar]
- Goffin C. and Ghuysen J.M. 1998. Multimodular penicillin-binding proteins: An enigmatic family of orthologs and paralogs. Microbiol. Mol. Biol. Rev. 62: 1079–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N. and Peitsch M.C. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18: 2714–2723. [DOI] [PubMed] [Google Scholar]
- Holtje J.V. 1996. A hypothetical holoenzyme involved in the replication of the murein sacculus of Escherichia coli. Microbiology 142: 1911–1918. [DOI] [PubMed] [Google Scholar]
- Leski T.A. and Tomasz A. 2005. Role of penicillin-binding protein 2 (PBP2) in the antibiotic susceptibility and cell wall cross-linking of Staphylococcus aureus: Evidence for the cooperative functioning of PBP2, PBP4, and PBP2A. J. Bacteriol. 187: 1815–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D. and Strynadka N.C. 2002. Structural basis for the β lactam resistance of PBP2a from methicillin-resistant Staphylococcus aureus. Nat. Struct. Biol. 9: 870–876. [DOI] [PubMed] [Google Scholar]
- Macheboeuf P., Di Guilmi A.M., Job V., Vernet T., Dideberg O., Dessen A. 2005. Active site restructuring regulates ligand recognition in class A penicillin-binding proteins. Proc. Natl. Acad. Sci. 102: 577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlot C., Zapun A., Dideberg O., Vernet T. 2003. Growth and division of Streptococcus pneumoniae: Localization of the high molecular weight penicillin-binding proteins during the cell cycle. Mol. Microbiol. 50: 845–855. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Nukaga M., Mayama K., Hujer A.M., Bonomo R.A., Knox J.R. 2003. Ultrahigh resolution structure of a class A β-lactamase: On the mechanism and specificity of the extended-spectrum SHV-2 enzyme. J. Mol. Biol. 328: 289–301. [DOI] [PubMed] [Google Scholar]
- Read R.J. 1986. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. A 42: 140–149. [Google Scholar]
- Sanbongi Y., Ida T., Ishikawa M., Osaki Y., Kataoka H., Suzuki T., Kondo K., Ohsawa F., Yonezawa M. 2004. Complete sequences of six penicillin-binding protein genes from 40 Streptococcus pneumoniae clinical isolates collected in Japan. Antimicrob. Agents Chemother. 48: 2244–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C. 2003. SOLVE and RESOLVE: Automated structure solution and density modification. Methods Enzymol. 374: 22–37. [DOI] [PubMed] [Google Scholar]
- Vagin A. and Teplyakov A. 2000. An approach to multi-copy search in molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 56: 1622–1624. [DOI] [PubMed] [Google Scholar]
- Wang Q.M., Peery R.B., Johnson R.B., Alborn W.E., Yeh W.K., Skatrud P.L. 2001. Identification and characterization of a monofunctional glycosyltransferase from Staphylococcus aureus. J. Bacteriol. 183: 4779–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke M.S., Lovering A.L., Strynadka N.C. 2005. β-Lactam antibiotic resistance: A current structural perspective. Curr. Opin. Microbiol. 8: 525–533. [DOI] [PubMed] [Google Scholar]