

Abstract
The advent of the multiwavelength anomalous diffraction phasing method has significantly accelerated crystal structure determination and has become the norm in protein crystallography. This method allows researchers to take advantage of the anomalous signal from diverse atoms, but the dominant method for derivative preparation is selenomethionine substitution. Several generally applicable, high-efficiency labeling protocols have been developed for use in the bacterial, yeast, and baculovirus/insect cell expression systems but not for mammalian tissue culture. As a large number of proteins of biomedical importance can only be produced in yields sufficient for X-ray diffraction experiments in mammalian expression systems, it becomes all the more important to develop such protocols. We therefore evaluated several variables that play roles in determining incorporation levels and report here a simple protocol for selenomethionine modification of proteins in mammalian cells routinely yielding >90% labeling efficiency.
Keywords: protein labeling, protein crystallography, selenomethionine, multiple wavelength anomalous diffraction, mammalian cell culture
The multiwavelength anomalous diffraction (MAD) phasing method (Hendrickson 1991) has become the method of choice for X-ray phase determination, with >50% of the experimentally phased structures deposited in the PDB during the past year being determined by MAD. While MAD has allowed researchers to take advantage of the anomalous signal from several diverse heavy atoms, the dominant method for heavy atom derivative preparation is selenomethionine substitution. Several factors contribute to the widespread use of selenomethionine substitution, including simplicity, adaptability to different expression systems, scalability, and, in some cases, an almost quantitative replacement of methionine resulting in a homogeneous protein population. This method results in modified proteins without significant structural perturbations due to heavy atom incorporation, while eliminating the difficult and time-consuming screenings for heavy atom derivatives.
It is estimated that, for a successful MAD experiment, one selenomethionine residue is required for every ∼75–100 amino acids (Hendrickson and Ogata 1997). This corresponds to ∼80% of all proteins, which have a methionine content of 1% or more (Strub et al. 2003). There are two limitations to the method: First, the calculations above assume quantitative (or near-quantitative) methionine substitution, which often is not the case. For example, as the complexity of the expression system host increases, so does the complexity of the media required for their growth, while the efficiency of incorporation decreases concomitantly. Second, the selenomethionines must be ordered in the crystal. Methionines are often located in the hydrophobic protein core and are thus likely to be ordered, but there is no assurance that this is always the case. While it is difficult to predict a priori or to compensate for side-chain disorder, techniques can be improved to maximize the efficiency of selenomethionine incorporation.
The production of selenomethionine-labeled proteins has been reported in CHO cells (Wu et al. 1994; Lustbader et al. 1995), but up to now, no general protocol was available for the efficient preparation and accurate analysis of selenomethionine-substituted proteins produced from mammalian cell culture. Since many of our crystallographic experiments involve proteins secreted from stably transfected mammalian cell lines, we developed, optimized, and report here a protocol for the routine selenomethionine incorporation at the 90% substitution level. We also describe a method for assessment of the labeling efficiency using matrix-assisted laser desorption ionization mass spectrometry technology (MALDI-TOF).
Results
We evaluated multiple variables that could potentially contribute to the efficiency of selenomethionine incorporation, including extent of methionine depletion prior to addition of selenomethionine, length of time in the presence of selenomethionine, amount of selenomethionine in the growth media, and type of serum used during labeling. To determine the effect of each of these variables, we designed eight independent labeling experiments using a human embryonic kidney (HEK293) cell line expressing, as a secreted protein, the ligand-binding domain of the human receptor tyrosine kinase Tie2. Cells were evenly split into 15-cm culture plates and allowed to grow to ∼80%–90% confluence. The growth media were aspirated from the plates and replaced with one medium lacking methionine, and the cultures were incubated for either 6 or 12 h. Preincubation in the absence of methionine serves to deplete intracellular methionine pools (which could effectively compete for tRNA activation during selenomethionine labeling) and also permits the cell to secrete or degrade any remaining unlabeled recombinant protein that is undergoing folding or translocation through the endoplasmic reticulum and the Golgi. Following this short preincubation, the media were changed to include either 30 or 60 mg/L of selenomethionine and 10% of either dialyzed or nondialyzed fetal bovine serum. The dialyzed serum contains lower concentrations of small molecules, including amino acids such as methionine that could compete for protein incorporation. The amounts of added selenomethionine were based on the amount of methionine present in classical Dulbecco's modified Eagle's medium (DMEM). Subsequent to the addition of selenomethionine-containing media, the cells were allowed to incubate another 72 h, and then harvested.
The recombinant Tie2-Fc was purified from the media using the batch method with Protein A-Sepharose beads (Materials and Methods). Tie2 was cleaved off the beads with thrombin and resolved on SDS-PAGE. Under these conditions, the yield of selenomethionine-labeled protein varied from ∼60% to 80% of that of the unlabeled protein. Control (unlabeled) and selenomethionine-labeled and SDS-PAGE-purified protein samples were digested with trypsin, and peptides were analyzed by MALDI-ReTOF and TOF/TOF mass spectrometry. The N-terminal fragment (amino acid residues 1–452) of the human Tie2 receptor (NCBI no. 4,557,869) was positively identified on the basis of peptide mass fingerprint (PMF) result (36% sequence coverage; Mowse score of 79, where a score >63 is significant) and Mascot MS/MS score of 107 (protein scores >76 are significant hits) of the selected tryptic peptide at m/z = 1446.74 with sequence YIGGNLFTSAFTR. The mass spectra obtained from the unlabeled (control) and selenomethionine-labeled proteins were compared for differences. A peptide at 2534.1 amu was observed in the protein samples enriched with selenomethionine (samples 2–8) but was absent in the unlabeled control sample (Fig. 1A). This peptide maps to the predicted, selenomethionine-enriched peptide with sequence DFEALMNQHQDPLEVTQDVTR, encompassing Met-65 and assuming the selenomethionine enrichment (MH+ 2486.15 + 47.95 to account for a mass difference due to replacement of sulfur with selenium). The identity of the modified peptide was confirmed by carrying out MALDI-TOF/TOF MS/MS on selected peaks at m/z = 2534 (Fig. 1B) and 2486 (Fig. 1C), respectively. Finally, the selenium enrichment/incorporation levels of each preparation were individually calculated by integrating the areas of the peaks corresponding to the unmodified peptide at 2486.15 amu and the modified peptide at 2534.10 amu.
Figure 1.
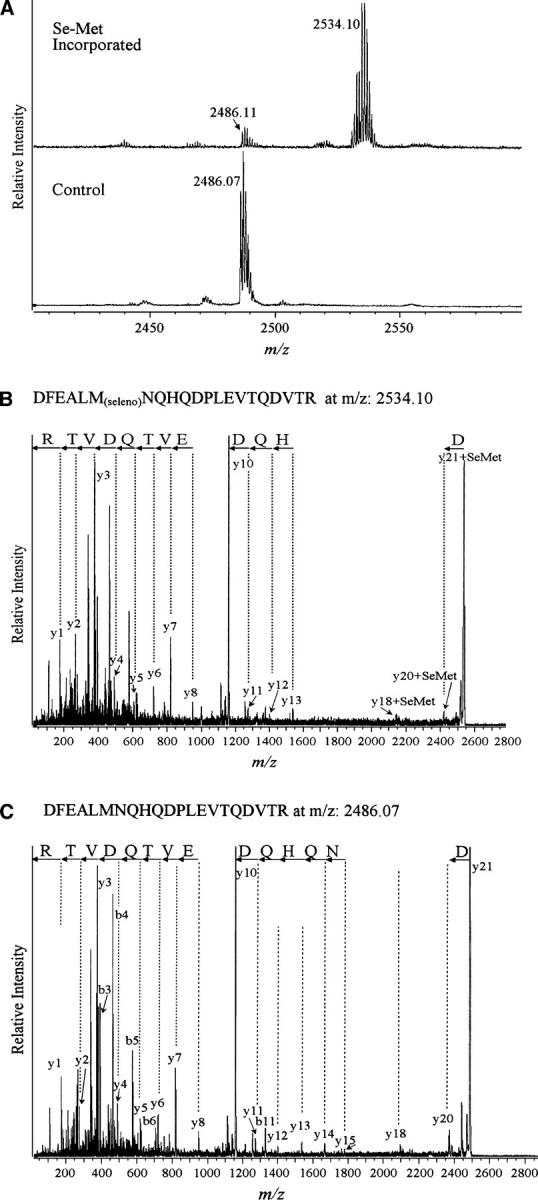
(A) Selenomethionine (Se-Met) incorporated and control Tie2 MALDI-ReTOF mass spectrometry data revealing the presence of additional peaks in Se-methionine modified, as compared to control, protein samples. (B) Mass spectrometric sequence analysis of Se-met incorporated peptide DFEALMNQHQDPLEVTQDVTR at m/z 2534.1 (C) and the control, unlabeled peptide at m/z 2486.07.
Table 1 summarizes the culture conditions that were used and the corresponding selenomethionine incorporation efficiency. Interestingly, the type of serum (dialyzed or standard serum) did not significantly change the efficiency of incorporation, indicating that fetal bovine serum may contain lower levels of free amino acids than previously thought. The length of preincubation time used to deplete intracellular methionine pools prior to addition of selenomethionine also had a relatively small effect on incorporation efficiencies. Instead, we found that the amount of selenomethionine present in the growth media had the most profound effect on the extent of selenomethionine substitution. Unfortunately, while high levels of incorporation are seen in the presence of larger concentrations of selenomethionine, toxicity also increases substantially, and protein yields decrease concurrently. Taking each of these parameters into account, we have optimized our protocol to maximize the concentration of added selenomethionine and the preincubation time in methionine-free media, while minimizing their toxicity to the mammalian cells. Specifically, we suggest as optimal a protein-labeling procedure that includes a 12-h preincubation, subsequent addition of 60 mg/L selenomethionine, and continuous cell growth in undialyzed fetal bovine serum.
Table 1.
Efficiency of selenomethionine incorporation in the recombinant Tie2 ectodomain expressed in HEK293 cells grown under varying culture conditions

As proof of principle to the applicability of this labeling method to crystallographic phase determination, we have recently determined the structure of an extracellular test protein produced in mammalian cells using SAD phases from a crystal containing ∼93% selenomethionine substitution at 1.8 Å resolution (Fig. 2). Our test protein (the receptor-binding region of Angiopoietin-2) contained four methionines in 215 amino acids with four copies of the protein monomer in the crystal asymmetric unit. The resulting electron density maps were of excellent quality (see Fig. 3) and allowed ∼90% of the structure to be built automatically via ARP/warp (CCP4 1994). Large amounts (tens of milligrams) of labeled Angiopoietin-2 were produced in mammalian cell culture using the optimized protocol outlined above. Specifically, stably transfected 293 cells were grown to ∼90% confluence in roller-bottle culture and washed briefly with a small amount of PBS prior to the addition of depletion media containing no additional methionine. Following a 12-h incubation, the media were exchanged for fresh media containing 60 mg/L selenomethionine. The culture was continued for ∼48 h, at which time the viability of the cells had decreased dramatically, and they had begun detaching from the culture vessel. The protein yield was ∼50% of that in standard DMEM. Our experiments indicate that in roller-bottle culture, the HEK293 cells are significantly more sensitive to the toxic effects of selenomethionine than in stationary monolayer cultures. We therefore suggest culturing with selenomethionine for ∼48 h in roller-bottles and for ∼72 h in dishes. Following purification, the labeled Ang2-RBD behaved similarly to the wild-type protein with respect to crystallization, and well-diffracting crystals were easily obtained. The structure was determined using a single-wavelength data set that was collected at the selenium K absorption edge.
Figure 2.

Fluorescence spectrum of crystals grown from selenium-labeled hAng2-RBD. The values of real (f′) and imaginary (f″) scattering components as a function of incident photon energy are also shown.
Figure 3.

A representative region of the solvent-flattened experimental SAD electron density map.
Discussion
As part of these studies, we evaluated several growth variables and identified the optimal conditions (based on MALDI-TOF estimates for incorporation levels) for selenomethionine incorporation into proteins produced in mammalian cell culture. The data document that efficiency of incorporation is dominated by the amount of label available. This could easily be explained by mass action rules, as the higher the concentration of selenomethionine is, the less competitive effect the intracellular methionine will have. However, selenomethionine is toxic in high concentrations, and increasing its amount in the growth media comes with a concomitant decrease in cell viability and, consecutively, protein yield. For crystallographic studies, in which a small amount of protein is sufficient for crystallization and phase determination, protein yield may not represent an issue of concern. However, if one needs to screen for crystallization conditions following labeling, lower concentrations of selenomethionine (20–30 mg/L) may need to be used to increase protein yield at the cost of having lower incorporation efficiency. The length of preincubation in the absence of methionine (methionine depletion) also plays a small but significant role, with an optimal time of 12 h. Incubations longer than 12 h decrease cell viability (data not shown) and, therefore, are not recommended.
In addition to Ang2 and Tie2 discussed above, we have used the protocol presented here to produce several other selenomethionine-labeled secreted proteins in our laboratory, including netrin-1, plexinB3, and the ectodomains of ADAM10 and EphA3 (data not shown). In all cases, incorporation efficiencies of 85% or higher were achieved, with expression yields sufficient for crystallographic studies. The protocol is also highly suitable for high-throughput/structural-genomics applications as it is portable, scalable, and flexible, and adaptable to the growth of different cell types under a variety of cell growth conditions. For example, efficient labeling can be performed in CHO, HEK293, COS, or any other mammalian cell type grown in serum-free media or in serum-supplemented media.
To increase even further the repertoire of proteins amenable to selenomethionine-based MAD phasing, it should be possible to expand the methodology reported here to include a complementary dual labeling approach that has recently been applied in Escherichia coli (Strub et al. 2003). More specifically, proteins could be labeled with a combination of selenomethionine and selenocysteine, thereby increasing the potential number of heavy atom scatterers within the crystal. As does selenomethionine incorporation, selenocysteine labeling often results in a highly isomorphous protein, allowing even disulfide-bond formation. Strub et al. (2003) suggest that 88%–93% of all proteins would be open to MAD phasing using dual labeling.
Materials and methods
Cell growth and labeling
All culture media and supplements were obtained from GIBCO. L-Selenomethionine was obtained from Anatrace Corp. The stable human embryonic kidney (293-HEK) cell line (Invitrogen) expressing the Tie2 ectodomain (1–452) fused to the constant domain of IgG (Fc) was maintained in high-glucose DME supplemented with 10% fetal bovine serum, penicillin, streptomycin, and 150 μg/mL hygromycin. For experiments assessing selenomethionine incorporation, cells were grown to ∼80%–90% confluence in 15-cm dishes using media lacking hygromycin.
Mass spectrometry
SDS-PAGE-resolved proteins were digested with trypsin, and peptides were purified and fractionated on a Poros 50 R2 reverse phase micro-tip as described (Erdjument-Bromage et al. 1998). The resulting peptide pools were analyzed by matrix-assisted laser desorption/ionization reflectron time-of-flight (MALDI-ReTOF) mass spectrometry using a Bruker UltraFlex TOF/TOF instrument (Bruker Daltonik GmbH) as described (Winkler et al. 2002). Selected experimental masses (m/z) were then taken to search a nonredundant protein database (NR) (3,245,378 entries on January 28, 2006; National Center for Biotechnology Information, Bethesda, MD) using the PeptideSearch algorithm (Matthias Mann, Max-Planck Institute for Biochemistry, Martinsried, Germany). (An updated version of this program is currently available as PepSea from Applied Biosystems/MDS Sciex.) A molecular weight range twice the predicted weight was covered, with mass accuracy restriction better than 40 ppm and a maximum of one missed cleavage site allowed per peptide. Mass spectrometric sequencing of selected peptides was done by MALDI-TOF/TOF (MS/MS) analysis on the same prepared samples, before and after selenomethionine labeling, using the LIFT mode of the instrument. Fragment ion spectra were taken to search the NR database using the MASCOT MS/MS Ion Search program, version 2.0.04 for Windows (Matrix Science Ltd.) (Perkins et al. 1999). Any identification thus obtained was verified by comparing the computer-generated fragment ion series of the predicted tryptic peptide with the experimental MS/MS data.
Large-scale protein production
For large-scale protein production of human Angiopoietin-2 receptor-binding domain (hAng2), stable HEK293 cells were grown to 80%–90% confluence in roller-bottle culture. Intracellular methionine pools were depleted with a 12-h incubation in DME (lacking L-methionine or L-cysteine) supplemented with 30 mg/L L-cystine-2HCl, and 10% FBS.
It is important to note that all cell media up to this point can be pooled, and the secreted unlabeled protein purified and used for structural or other studies. Following the preincubation, the media were replaced with DME supplemented with 60 mg/L L-selenomethionine, 30 mg/L L-cystine-2HCl, and 10% FBS. The cells were cultured for an additional 48 h prior to media harvest. The media were clarified by filtration, and recombinant secreted Angiopoietin-2-Fc was purified by affinity chromatography on rProteinA-Sepharose (GE Healthcare). The Fc tag was removed with thrombin. Additional purification on an SD200 gel filtration column (GE Healthcare) removed the fusion tag as well as residual contaminants. N-terminal sequencing confirmed the identity of the product, and mass spectrometry documented a 93% substitution rate for the methionines in the Angiopoietin-derived ATTMMIRPADFGS peptide.
Protein crystallization and structure determination
The purified hAng2 was concentrated to ∼15 mg/mL in a buffer containing 10 mM HEPES (pH 7.0), 200 mM NaCl and crystallized at room temperature by hanging-drop vapor diffusion against a reservoir containing 0.2 M ammonium sulfate, 0.1 M sodium acetate (pH 5.2), and 22% PEG-4000. Crystals were transferred to mother liquor with 20% glycerol for flash-freezing. A 1.8 Å SAD data set was collected at NSLS beamline X9A. All data were processed using DENZO and SCALEPACK (Otwinowski and Minor 1997). The selenium atom positions were determined using SnB and input into autoSHARP (Evans and Bricogne 2002) and used to calculate a solvent flattened map of excellent quality. The space group is C2 with a = 140.28 Å, b = 94.56 Å, c = 84.60 Å, β = 94.81°, and four independent molecules in the asymmetric unit. Model building was initially performed using Arp/Warp (CCP4 1994) and later proceeded through an iterative process of building in O and refinement of the model in CNS (Jones et al. 1991; Brunger et al. 1998). Stereochemical analysis of the refined models using PROCHECK of the CCP4 Package (CCP4 1994) revealed main-chain and side-chain parameters better than or within the typical range of values for protein structures determined at corresponding resolutions.
Acknowledgments
We thank Laura Fabrizio for help with mass spectrometry and Lynne Lacomis for help with figures. This study was supported by the NCI Cancer Center Support Grant P30 CA08748 (to P.T.) and the National Heart Lung and Blood Institute HL077249 (to D.B.N.).
Footnotes
Reprint requests to: Dimitar B. Nikolov, Structural Biology Program, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, NY 10021, USA; e-mail: nikolovd@mskcc.org; fax: (212) 717-3135; or William A. Barton, Department of Biochemistry, Virginia Commonwealth University, Richmond, VA 23298, USA; e-mail: wabarton@vcu.edu; fax: (804) 828-1473.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062244206.
References
- Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S. et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (CCP4). 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Erdjument-Bromage H., Lui M., Lacomis L., Grewal A., Annan R.S., MacNulty D.E., Carr S.A., Tempst P. 1998. Micro-tip reversed-phase liquid chromatographic extraction of peptide pools for mass spectrometric analysis. J. Chromatogr. A 826: 167–181. [DOI] [PubMed] [Google Scholar]
- Evans G. and Bricogne G. 2002. Triiodide derivatization and combinatorial counter-ion replacement: Two methods for enhancing phasing signal using laboratory Cu Kα X-ray equipment. Acta Crystallogr. D Biol. Crystallogr. 58: 976–991. [DOI] [PubMed] [Google Scholar]
- Hendrickson W.A. 1991. Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science 254: 51–58. [DOI] [PubMed] [Google Scholar]
- Hendrickson W.A. and Ogata C.M. 1997. Phase determination from multiwavelength anomalous diffraction measurements. Methods Enzymol. 276: 494–523. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou J.Y., Cowan S.W., Kjeldgaard M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47: 110–119. [DOI] [PubMed] [Google Scholar]
- Lustbader J.W., Wu H., Birken S., Pollak S., Gawinowicz Kolks M.A., Pound A.M., Austen D., Hendrickson W.A., Canfield R.E. 1995. The expression, characterization, and crystallization of wild-type and selenomethionyl human chorionic gonadotropin. Endocrinology 136: 640–650. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Perkins D.N., Pappin D.J., Creasy D.M., Cottrell J.S. 1999. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20: 3551–3567. [DOI] [PubMed] [Google Scholar]
- Strub M.-P., Hoh F., Sanchez J.-F., Strub J.M., Bock A., Aumelas A., Dumas C. 2003. Selenomethionine and selenocysteine double labeling strategy for crystallographic phasing. Structure 11: 1359–1367. [DOI] [PubMed] [Google Scholar]
- Winkler G.S., Lacomis L., Philip J., Erdjument-Bromage H., Svejstrup J.Q., Tempst P. 2002. Isolation and mass spectrometry of transcription factor complexes. Methods 26: 260–269. [DOI] [PubMed] [Google Scholar]
- Wu H., Lustbader J.W., Liu Y., Canfield R.E., Hendrickson W.A. 1994. Structure of human chorionic gonadotropin at 2.6 Å resolution from MAD analysis of the selenomethionyl protein. Structure 2: 545–558. [DOI] [PubMed] [Google Scholar]