

Abstract
Hydrophobicity analyses applied to databases of soluble and transmembrane (TM) proteins of known structure were used to resolve total genomic hydrophobicity profiles into (helical) TM sequences and mainly “subhydrophobic” soluble components. This information was used to define a refined “hydrophobicity”-type TM sequence prediction scale that should approach the theoretical limit of accuracy. The refinement procedure involved adjusting scale values to eliminate differences between the average amino acid composition of populations TM and soluble sequences of equal hydrophobicity, a required property of a scale having maximum accuracy. Application of this procedure to different hydrophobicity scales caused them to collapse to essentially a single TM tendency scale. As expected, when different scales were compared, the TM tendency scale was the most accurate at predicting TM sequences. It was especially highly correlated (r = 0.95) to the biological hydrophobicity scale, derived experimentally from the percent TM conformation formed by artificial sequences passing though the translocon. It was also found that resolution of total genomic sequence data into TM and soluble components could be used to define the percent probability that a sequence with a specific hydrophobicity value forms a TM segment. Application of the TM tendency scale to whole genomic data revealed an overlap of TM and soluble sequences in the “semihydrophobic” range. This raises the possibility that a significant number of proteins have sequences that can switch between TM and non-TM states. Such proteins may exist in moonlighting forms having properties very different from those of the predominant conformation.
Keywords: Kyte-Doolittle scale, hydrophobicity, hydropathy, transmembrane helix, transmembrane sequence prediction, transmembrane tendency, transmembrane propensity
Computational analysis of hydrophobicity is a useful method to identify transmembrane (TM) sequences in membrane proteins (Kyte and Doolittle 1982; von Heijne 1992; Claros and von Heijne 1994; Jones et al. 1994; Rost et al. 1995, 1996; Cserzo et al. 1997; Persson and Argos 1997; Tusnady and Simon 1998; Deber et al. 2001; Chen and Rost 2002; Ikeda et al. 2002; Lao et al. 2002). Despite the number of hydrophobicity-type scales for such predictions, it is not clear what the “ultimate”/correct scale might be. Presently used scales are of especially uncertain accuracy for sequences that have borderline hydrophobicity (von Heijne 1992; Chen and Rost 2002; Lao et al. 2002; Kall et al. 2004). This makes it difficult to fully answer questions concerning the number and nature of such “semihydrophobic” (Caputo and London 2004) sequences, which can equilibrate between TM and surface conformations (Ren et al. 1997; Rosconi and London 2002; Caputo and London 2003).
In this study, we used analysis of databases of known soluble and TM proteins in order to resolve the total genomic hydrophobicity profiles into TM and soluble sequence components. This approach allowed formulation of a scale refinement procedure that caused different scales to collapse to a single scale that approaches a theoretical limit for accuracy.
Materials and methods
Databases
To study all the protein sequences in a genome, open reading frame (ORF) databases of Escherichia coli (01/11, amino acid) (Blattner et al. 1997; Mori et al. 2000) and Saccharomyces cerevisiae (Cherry et al. 1997) in the FASTA format were downloaded from http://ecoli.aist-nara.ac.jp/gb4/FTP/ftplist.html and the Saccharomyces Genome Database (SGD) (K. Dolinski, R. Balakrishnan, K.R. Christie, M.C. Costanzo, S.S. Dwight, S.R. Engel, D.G. Fisk, J.E. Hirschman, E.L. Hong, L. Issel-Tarver, et al., ftp://ftp.yeastgenome.org/yeast/), respectively. For the other organisms studied, the databases from NCBI Genome Resources were:
ftp://ftp.ncbi.nih.gov/genomes/Bacteria/Nanoarchaeum_equitans/
ftp://ftp.ncbi.nih.gov/genomes/Bacteria/Yersinia_pestis_CO92/
ftp://flybase.net/genomes/Drosophila_melanogaster/current/fasta/
Duplicated sequences within the databases were identified and removed. Signal sequences predicted by Phobius (Kall et al. 2004) were also identified and removed from the protein sequences unless otherwise noted.
To analyze protein sequences with known TM structures, nonredundant α-helical sequences in the TransMembrane Protein DataBase (TMPDB) (Ikeda et al. 2003) version 6.3 were downloaded from http://bioinfo.si.hirosaki-u.ac.jp/~TMPDB/. Protein sequences in TMPDB were obtained, and the sequences of transmembrane (TM) helices were extracted based on the annotation in the database. The distributions of the helices’ length and hydrophobicity were calculated. To evaluate the accuracy of the boundary of the TM annotation, the lengths of the helices were either extended or shortened by one or two residues at the N and C termini, and their hydrophobicity was calculated. Correct identification of TM sequence boundaries was inferred from the observation that shortening putative TM segments did not increase their hydrophobicity, while extending them decreased it significantly (see below).
For soluble proteins, non-transmembrane proteins with known 3D structures were collected from the ASTRAL database, which as described, “is partially derived from, and augments, the Structural Classification of Proteins (SCOP) database (Murzin et al. 1995). ASTRAL SCOP genetic domain sequences with less than 95% identity to each other, based on Protein Data Bank (PDB) (Berman et al. 2000) SEQRES records,” were downloaded from the ASTRAL Compendium (Chandonia et al. 2004) at http://astral.berkeley.edu/. Abundance versus TM tendency profiles of almost identical shape were obtained when the analysis was restricted to sequences with <40% identity.
Membrane proteins under class 6 (“membrane and cell surface proteins and peptides”) of SCOP except the first (“toxins’ membrane translocation domains”) and the 36th folds (“anthrax protective antigen”) were removed. However, it should be noted that the resulting collection of “soluble” proteins still contains a residual population of proteins that were crystallized in the presence of detergents, such as pilin (PDB ID code 2PIL).
Hydrophobicity analysis
A Perl program (genome-TM) was scripted to implement a sliding-window algorithm (Kyte and Doolittle 1982; Claros and von Heijne 1994) to analyze the amino acid sequences in the protein databases. Copies of genome-TM are available from the authors. Unless otherwise noted, a sliding-window size (N) of 19 residues was used. A standard hydrophobicity plot for each protein was generated by selecting a hydrophobicity scale, which assigns a hydrophobicity index value for each of the 20 amino acids, and progressively averaging the hydrophobicity values for each and every N residues along a protein's primary sequence. The resultant averaged values were plotted versus sequence position, which was defined as the position of the residue at the center of the window being averaged. Hydrophobicity was calculated with a stepwise decreasing hydrophobicity threshold value. If the average hydrophobicity value for a specific sequence was equal to or above the threshold being used, then the program would mark the position as being an initial estimate for the sequence of a qualifying candidate. The program shifted the window for that sequence by one residue at a time until it came to a residue whose inclusion resulted in an averaged hydrophobicity value falling below the threshold, and then marked the segment minus the last residue as a qualifying candidate. If the gap between two neighboring candidates was within a certain distance M (merging factor, the default value was 4 residues), they were merged into a single candidate sequence (Deber et al. 2001). If a candidate contained segments previously identified at a higher threshold value, it was discarded, so that only the segments whose hydrophobicity value fell between the current threshold and the threshold hydrophobicity one level higher were stored as falling into the hydrophobicity range being scored. If a segment was more than 35 residues long, and thus likely to contain multiple TM sequences, it was discarded for the purpose of our calculations. However, such sequences were very rare for N ≥ 5 and M = 4.
Not every residue in a protein was scored by this approach. One reason is that sequences located between two higher hydrophobicity sequences could sometimes be less than N residues long. In addition, the probability that hydrophilic sequences are surrounded by more highly hydrophilic sequences, such that the former can be identified as being <35 residues long, decreases progressively as hydrophobicity decreases. Overall, somewhat over 50% of the total amino acid residues in a genome were in sequences within the range of hydrophobicities that were scored.
Normalization of abundance versus hydrophobicity distributions for known TM and soluble sequences to genomic abundance versus hydrophobicity distributions
To normalize the abundance versus hydrophobicity distributions for sequences known to be soluble or TM to abundance versus hydrophobicity distributions for all the proteins in a genome, we scaled abundance data for known soluble and TM sequences to the height of the soluble and TM “peaks” in the genomic data. To do this, we assumed that the most highly hydrophobic sequences in a genome are all TM sequences, and the most hydrophilic sequences are all soluble (see Results for the validity of this assumption). We defined highly hydrophobic as being more hydrophobic than sequences at the peak of the TM sequence abundance versus hydrophobicity curve (Fig. 1, open circles), i.e., more hydrophobic than the most abundant hydrophobic sequences. We defined highly hydrophilic as being less hydrophobic than sequences at the peak of the soluble sequence abundance versus hydrophobicity curve (Fig. 1, open squares). Abundance values for the soluble protein and TM helix (TMPDB) databases were then normalized to have about the same number of sequences as found in genomic data over the hydrophobicity ranges in which genomic sequences were assumed to be all soluble or all TM, respectively.
Figure 1.

Abundance of sequences as a function of hydrophobicity for the sum of the E. coli and S. cerevisiae genomes as estimated by the Kyte-Doolittle and TM tendency scales. The abundance of sequences within a given hydropathy/hydrophobicity range is shown for the combined E. coli and S. cerevisiae genomes (triangles) as assessed by the Kyte-Doolittle scale (A) or the TM tendency scale (B). The abundances at each hydrophobicity value correspond to an average calculated from the sum of abundance values for E. coli and S. cerevisiae divided by two. A sliding window [N] size of 19 and a merge factor of 4 were used. Each point represents sequences with a hydrophobicity value within the range that is ≤0.1 units greater than the hydrophobicity value shown on the X-axis. To resolve curves into soluble and TM sequences, hydrophobicity distributions were calculated for soluble proteins with known structures from a soluble protein database (squares) and for TM segments from a database (TMPDB) (Ikeda et al. 2003) cataloging TM proteins (circles). The abundance values for the soluble and TM sequence databases were normalized in terms of height to fit the genomic data (see text for details). The sum of the normalized soluble and TM segment components is shown as a solid line. If not noted otherwise, in this and later calculations signal sequences predicted by the Phobius program (Kall et al. 2004) were removed from the genomic sequences. (Inset) Dependence of the percent probability that a sequence is TM upon KD hydropathy as determined from the ratio of sequence abundance in the normalized soluble database to that in the sum of the normalized soluble database and normalized TM database at each specific hydropathy value.
Accuracy of TM helix boundaries in TMP database
For membrane proteins there is some ambiguity concerning the assignment of the boundaries of hydrophobic TM helices. One reason for this uncertainty is that these structures are often derived from crystals in detergent, so that the exact boundary of the TM portion of hydrophobic helices is not certain. The most hydrophobic part of the TM helices is generally assumed to be the TM segment. To check that the boundary assignments given in the TMP database were actually demarcated in this fashion, hydrophobicity values for sequences assigned to hydrophobic helices were recalculated after arbitrarily elongating them on both ends with surrounding sequences or truncating them from both ends. Overall, truncating the sequences did not increase their hydrophobicity, while elongating them decreased it (data not shown). This means that on the average the assigned boundaries had been correctly placed at the ends of the most hydrophobic part of the TM sequences.
Calculation of genomic hydrophobicity (GH) scale
We wished to use as many TM sequences as could be identified from genomic data to develop a hydrophobicity scale based on a very large number of TM sequences. Using previous hydrophobicity scales to identify TM sequences from genomic data would bias amino acid abundances in identified sequences so as to reflect the bias in the scale being used. To avoid this circular reasoning, a naive, but effective, assumption was made: that many TM helices could be identified by a high content of the hydrophobic amino acids Ile, Leu, and Val. ILV-rich sequences were then used to investigate the relative abundance of residues other than I, L, and V. ILV-enriched sequences were identified with an algorithm assigning I, L, and V a “hydrophobicity” value of 1 and a “hydrophobicity” value of 0 to all other residues. As estimated from the distribution of Kyte-Doolittle hydropathies for sequences identified by this algorithm, ILV content had to be at least 50% for E. coli, and at least 55% for S. cerevisiae to avoid inclusion of a significant number of non-TM segments (data not shown). This procedure did not capture all TM helices but did identify 2647 E. coli and 820 S. cerevisiae sequences with 19–21 residue lengths for further analysis.
To define a genomic hydrophobicity (GH) scale, the ratio of percent abundance of residues in ILV-rich TM sequences to their percent abundance in soluble proteins (i.e., in the soluble protein database) was then calculated. The average of S. cerevisiae and E. coli TM/soluble ratios were calculated for each amino acid (except I, L, and V) and then normalized to that of Gly. Such relative TM/soluble ratios are in a format analogous to a relative “equilibrium” constant. Since the total value of the free energy of association of individual residues with membranes is likely the main parameter controlling TM insertion, an apparent “ΔGo” = −RT ln(TM/soluble) would potentially be linearly related to the tendency of residues to participate in a TM structure. Thus, the logarithm of the relative TM/soluble ratios was calculated as a parameter likely to be linearly related to TM propensities. The GH scale was then normalized so that the difference between values for the most and least hydrophobic residues was the same as for the Wimley-White scale, which is directly related to ΔGo (Jayasinghe et al. 2001). The genomic hydrophobicity value of Gly was arbitrarily set at zero. To complete the GH scale, it was necessary to define genomic hydrophobicity values for I, L, and V. This was done by using the Cowan-Whittaker (CW) scale (Cowan and Whittaker 1990), which performed well in terms of distinguishing between TM and soluble sequences (see Results), and extrapolating to derive I, L, and V GH values based on the approximately linear relationship between CW and GH scales.
Calculation of the transmembrane (TM) tendency scale
Hydrophobicity scale values from the GH scale derived above (and several other scales; see Results) were refined to improve the ability to distinguish between soluble sequences and TM helices by using the constraint that the populations of known soluble and TM sequences scored as having an equal probability of forming in TM helices should have, on the average, almost the same amino acid composition (see Results). Sequences within the hydrophobicity range in which both TM and soluble sequences were abundant were used to define the soluble/TM abundance ratio (percent abundance of each amino acid in soluble sequences)/(percent abundance in TM helices). A TM penalty was then assigned to amino acid residues that were more abundant in soluble sequences (abundance ratio >1) and a TM bonus was assigned to residues that were more abundant in TM helices (abundance ratio <1). This calculation was performed for sequences in each of a series of five narrow hydrophobicity intervals covering the range over which the initial scale being used predicted there was a 30%–80% probability that the sequences would be found in a TM helix, i.e., a 30%–40% chance of being in a TM helix, a 40%–50% chance of being in a TM helix, etc. (see Results), and then the values for different hydrophobicity intervals were averaged.
Penalty/bonus values were calculated in the form of a pseudo ΔGo = −RT ln(Kpseudo), where Kpseudo = (percent abundance in soluble sequences)/(percent abundance in TM sequences). In this format, a positive value of pseudo ΔGo is equivalent to a TM bonus and is added to the original hydrophobicity value for scales in which hydrophobic sequences have higher values than hydrophilic sequences. Pseudo-ΔGo values were added to scale values after adjusting them so that the size of the corrections would equal the same fraction of total hydrophobicity range as in the ΔG-based augmented Wimley-White scale (Jayasinghe et al. 2001). In other words, the correction equation was ΔC = (pseudo-ΔGo/ΔGomax)Cmax, where ΔC is the correction, Cmax is the difference between “hydrophobicity” values for the most and least hydrophobic residues in that scale, and ΔGomax is the difference between ΔGo values for the most and least hydrophobic residues in the augmented Wimley-White scale.
We found that adding the correction factor once did not tend to fully equalize the probability that populations of soluble and TM sequences scored as having the same propensity to participate in TM helices had the same average abundance of each amino acid, and thus did not minimize misidentification of TM helices. Instead, misidentification of TM helices was minimized when the correction factors were added twice or three times to the uncorrected values, and so this was done to define TM tendency values. After using the TM tendency values derived from this procedure to recalculate genomic hydrophobicity profiles, we found that a small further improvement in TM tendency values, resulting in even less misidentification, could be obtained by repeating the entire refinement technique, and then adding re-correction factors so obtained once to the preliminary TM tendency scale values. This was done to define the final TM tendency values.
Results
Estimating abundance versus hydrophobicity distribution for TM helices and soluble sequences at the genomic level
To search for the abundance of (helix-type) TM sequences, the abundance of sequences having different “hydrophobicity” values was calculated for all proteins within a genome (as identified by open reading frames; Blattner et al. 1997; Cherry et al. 1997). Figure 1 shows the abundance of such sequences for the combined genomes of E. coli and S. cerevisiae (triangles) as a function of “hydrophobicity” as defined by the widely used Kyte-Doolittle (KD) hydropathy scale (Kyte and Doolittle 1982) (Fig. 1A) and the TM tendency scale derived in this report (Fig. 1B). For both scales, the distributions show two populations. There is an abundant, hydrophilic population and a more hydrophobic population that shows up as a shoulder at high hydropathy/TM tendency values. To resolve these populations, normalized “hydrophobicity” distributions were calculated for amino acid sequences from databases of known TM helices and soluble proteins. Figure 1A shows that for the KD scale, the peak abundance for sequences from soluble proteins (squares) at a hydropathy/hydrophobicity value of ∼0.2, while that for TM sequences (circles) occurs near 1.7. For the TM tendency scale (Fig. 1B), the corresponding values are −0.1 and 0.7.
Figure 1 also shows that a mixture of the normalized TM and normalized soluble sequence distributions fits the genomic data fairly well (cf. solid lines to triangles). To a first approximation, genomic sequences represent a combination of TM and non-TM soluble segments. It is noteworthy that there is a substantial overlap between the TM and non-TM peaks at intermediate hydrophobicities. We call sequences with intermediate hydrophobicity “semihydrophobic,” and define those with hydrophobicities near that of the peak for soluble proteins “subhydrophobic.”
In the calculations above, the minimum length of potentially TM segments was defined as 19 residues. Distributions of sequence abundance versus hydrophobicity were not greatly altered when the minimum length of potentially TM segments was varied from 15 to 21 (data not shown). Increasing minimum length only shifted peaks to slightly smaller hydrophobicity values. This reflects the fact that a hydrophobic sequence too short to satisfy the minimum length value being used will be recorded as a sequence of greater length (including some flanking polar residues) but lesser hydrophobicity. In contrast, long hydrophobic sequences with ends that are less hydrophobic than their cores will tend to be recorded as being shorter sequences with a higher hydrophobicity. This means that hydrophobic sequence length and hydrophobicity cannot be defined independently, and thus this type of analysis cannot precisely define TM helix length.
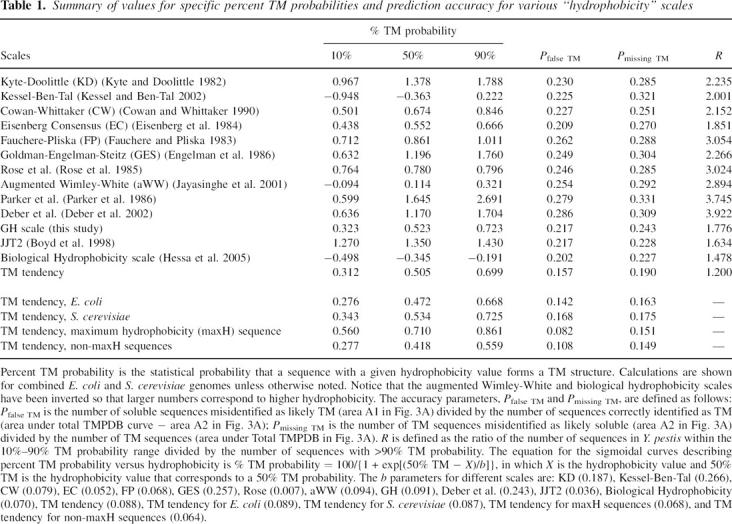
The ratio of the abundance of soluble to TM sequences can be calculated as a function of hydrophobicity value from the normalized abundance of TM and non-TM sequences versus hydrophobicity. This yields the hydrophobicity dependence of percent TM probability = the statistical percent probability that a sequence at a particular hydrophobicity value forms a TM segment. For all hydrophobicity scales tested (see below), sigmoidal curves were found to fit the percent TM probability versus hydrophobicity profile (see example in Fig. 1A, inset). The parameters that describe these sigmoidal curves for different hydrophobicity scales are given in Table 1. Percent TM probability values are useful for comparing the probability that different sequences with borderline hydrophobicity will form a TM helix.
Table 1.
Summary of values for specific percent TM probabilities and prediction accuracy for various “hydrophobicity” scales
Derivation of the TM tendency scale
Analysis of the amino acid composition of sequences at intermediate hydrophobicity values where the TM and soluble sequence peaks in Figure 1 overlap suggested a method to define an improved TM prediction scale. Using various hydrophobicity scales, we found that the population of intermediate hydrophobicity sequences from the soluble database had a different average abundance of certain amino acids from that found in the population of sequences with the same hydrophobicities from the TM database (Table 2); but a scale showing such differences cannot have maximal accuracy to predict TM helices. The reason for this is that by penalizing hydrophobicity values for residues over-represented in soluble proteins while increasing hydrophobicity values for residues over-represented in TM helices, a refined scale that more accurately distinguishes between TM and soluble sequences can be derived. In other words, a scale for predicting whether a sequence is TM has maximal accuracy only if populations of TM and soluble sequences with equal propensities to form TM structures have the same average amino acid composition (see Discussion). This is not to say that the refined scale defines better hydrophobicity values, it just allows more accurate prediction of whether sequences are TM or not (i.e., prediction of their TM tendency).
Table 2.
Frequency of various amino acids in soluble sequences (Sol) and TM sequences (TM) with about the same hydrophobicity as judged by different “hydrophobicity” scales
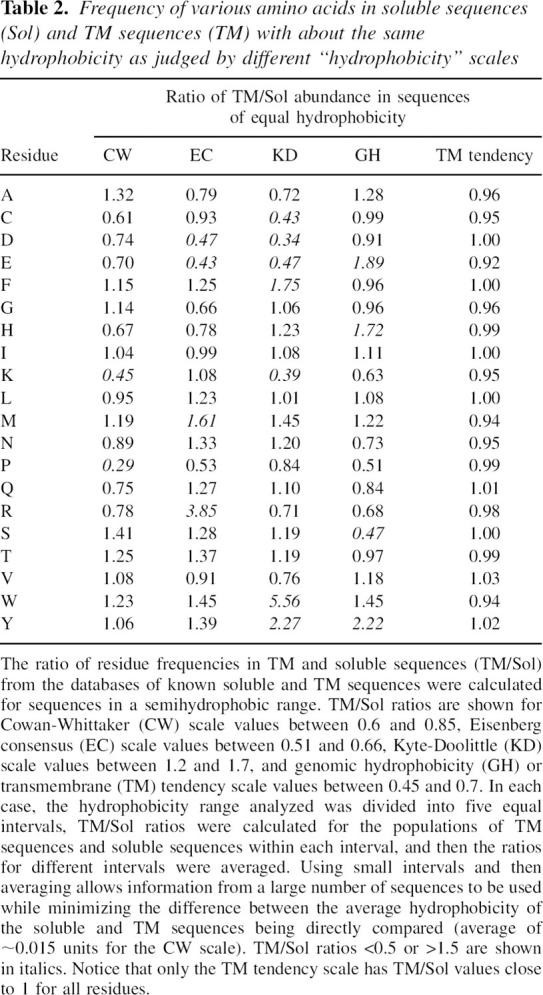
The refinement procedure outlined above was applied to a genomic hydrophobicity scale (GH scale) we derived from a database of sequences from soluble proteins and likely TM sequences defined using genomic data (see Materials and Methods). The resulting scale (Table 3) was named the TM tendency scale, and it should include any factors in addition to hydrophobicity that favor inclusion in TM sequences. (Nevertheless, because hydrophobicity is the predominant factor in TM insertion, the TM tendency scale will be referred to as a hydrophobicity-type scale.) As expected, and unlike the other scales tested, the refined TM tendency scale exhibited the expected near-equal percent abundance of different amino acids in populations of soluble and TM sequences having the same “hydrophobicity” values (Table 2).
Table 3.
TM tendency and GH scale values

Incidentally, it is not clear whether TM and soluble sequences with identical hydrophobicities should have the same average amino acid composition. However, it was found that the amino acids predicted to differ in abundance in TM and soluble populations with equal hydrophobicity depended on which hydrophobicity scale was used (Table 2). At the very least, all but one of the scales examined must be inaccurate in this regard.
The refinement procedure was also applied to several other scales that were relatively accurate in terms of predicting TM sequences (Eisenberg consensus, Cowan-Whittaker, and biological hydrophobicity; see below). The refined scales thus derived had values that were much more highly correlated than the scales from which they were derived (e.g., Fig. 2, cf. B and D), and were so highly correlated to the TM tendency scale (r > 0.99) as to indicate that refinement results in convergence to basically the same scale (Fig. 2D). This means that the scale defined by the refinement procedure is not affected by the scale chosen as the starting point, and that the refined scale represents a universal TM tendency scale. Unless otherwise noted, the results reported below are for the refined TM tendency scale derived from our GH scale.
Figure 2.

Correlation between various hydrophobicity scales. (A) Correlation between the CW and GH scales. GH scale values for I, L, and V residues (asterisks) were based on the CW scale by extrapolating from the linear correlation curve (dashed line). (B) Correlation between the EC and GH scales. (C) Correlation between the TM tendency and biological hydrophobicity scales. (D) Correlation between the refined EC and TM tendency scales. The positions of amino acid residues are designated by their one-letter codes. Correlation coefficients (r) are shown in each panel.
Estimating prediction accuracy for different hydrophobicity scales
By comparing predictions using the entire set of proteins in the normalized soluble protein and TMP databases, we adopted a comprehensive approach to the question of what scale most accurately predicts TM sequences. Prediction scales can be used to define whether sequences form TM helices on a yes/no basis using a threshold value, and the number of mistakes is the measure of the accuracy of the method. To convert the percent TM probability values we derived into a yes/no type of prediction, a 50% or greater probability of forming a TM helix was defined as the threshold for predicting that a sequence forms a TM helix (Fig. 3A).
Figure 3.

Schematic illustration of the definition of prediction accuracy as judged from the degree of overlap between hydrophobicity of soluble and TM sequences. (A) Definition of populations used for estimation of prediction errors. Soluble (solid) and TMPDB (dashed) curves schematically represent the hydrophobicity profiles of sequences in the soluble protein and TMP databases, respectively, after normalization to genomic data. A1, the population of sequences falsely predicted to be TM, is the area to the right of the 50% TM possibility line (vertical dotted line) and below the soluble protein profile (solid line). A2, the population of TM sequences that are not assigned as being TM (i.e., missing TM sequences), is the area to the left of the 50% TM possibility line and below the TMP database profile (dashed line). A1 and A2 were used to calculate the misidentification levels for hydrophobicity scales (see legend of Table 1). (B) Effect of the number of soluble sequences (relative to the number of TM sequences) upon percent TM probability versus hydrophobicity. Notice that when the relative number of soluble sequences increases (from dash-dot-dash curve to solid curve), the hydrophobicity value at which there is a 50% probability of a sequence being a TM sequence (vertical dotted line) increases.
To evaluate accuracy, abundance versus hydrophobicity distributions of the type shown in Figure 1 were calculated for a variety of hydrophobicity/hydropathy/TM probability scales (Kyte and Doolittle 1982; Fauchere and Pliska 1983; Eisenberg et al. 1984; Rose et al. 1985; Engelman et al. 1986; Cowan and Whittaker 1990; Boyd et al. 1998; Jayasinghe et al. 2001; Chen et al. 2002; Deber et al. 2002; Hessa et al. 2005). For simplicity, all of these scales will be referred to as hydrophobicity scales. These scales include the more accurate hydrophobicity-type scales (Boyd et al. 1998; Chen et al. 2002). In all cases, the basic features of distributions were similar to those shown in Figure 1 (data not shown).
Next, misidentification parameters measuring the ability of a scale to distinguish between TM helices and soluble sequences were defined. The first parameter was the fraction of sequences identified as being TM (i.e., with a TM probability >50%), but which are really soluble (Pfalse TM). The second parameter was the fraction of TM helices not correctly identified as being TM (Pmissing TM). The formal definition of these parameters is given in Table 1, with the positions of the populations of false TM (area A1) and missing TM sequences (area A2) schematically illustrated in Figure 3A. Table 1 compares the accuracy of various scales in terms of Pfalse TM and Pmissing TM. The generally similar performance of different scales reflects their moderate degree of correlation (range 0.65 < r < 0.96; average value 0.85, calculation not shown).
The most accurate scale was the TM tendency scale. Interestingly, it had, on the average, the highest correlation coefficient to the other scales (data not shown). Thus, for the scales examined, the TM tendency scale approaches most closely to a consensus scale. The second most accurate scale was the recently derived biological hydrophobicity scale (Hessa et al. 2005). Figure 2C shows that this scale is closely correlated with the TM tendency scale (r = 0.95), which is interesting because the two scales are derived from very different information (see Discussion).
The misidentification values in Table 1 seem relatively large, but this is an artifact of the way they are defined. If misidentification of soluble sequences is expressed as a fraction of the total sequences in the soluble database (rather than in the TM database), only 1% of soluble sequences are misidentified as TM by the TM tendency scale. Error rates are also exaggerated because they are calculated for an entire genome. This increases the relative amount of non-TM sequence being analyzed relative to methods in which accuracy is tested by trying to predict TM sequences in individual membrane proteins. In the latter situation, one can increase the accuracy of predicting TM helices by decreasing threshold values for defining TM sequences without significantly increasing the amount of non-TM sequence identified as being TM.
It is also noteworthy that the hydrophobicity value equivalent to a 50% probability of being TM depends slightly on the genome chosen for analysis (Table 1). The reason for this is that the hydrophobicity value corresponding to 50% TM probability depends on the relative number of soluble and TM sequences in the genome chosen for analysis (Fig. 3B).
Further comparison of the ability to distinguish TM and non-TM sequences
Although Table 1 shows that the TM tendency scale minimizes misidentification values, these parameters probe accuracy with the soluble and TMP databases, some sequences of which were used to derive the TM tendency scale. Thus, they do not use fully independent training and testing sets and therefore are not a fully valid test of accuracy for the TM tendency scale (although a valid test for comparison of all other scales). Therefore, additional criteria were used to evaluate TM tendency scale accuracy. First, the ability to resolve TM and non-TM populations from whole genomic data was analyzed. Abundance versus “hydrophobicity” profiles were calculated for three prokaryotic and three eukaryotic genomes. Visual inspection of these profiles (Fig. 4) confirms that the TM tendency scale gives a greater degree of resolution between soluble protein and TM sequence peaks relative to the GH scale from which it was derived, with a difference in “hydrophobicity” values at the “peaks” for hydrophilic and hydrophobic proteins averaging ∼0.05–0.1 units greater for the TM tendency scale than for the GH scale. This shows that the TM tendency scale has an improved ability to discriminate between soluble sequences and TM helices relative to the GH scale (and thus also relative to all scales that Table 1 shows are not as accurate as the GH scale). A qualitatively similar improvement was found when the TM tendency scale was compared to the biological hydrophobicity scale, although the improvement was smaller, consistent with the higher accuracy of the biological hydrophobicity scale relative to the GH scale (data not shown).
Figure 4.
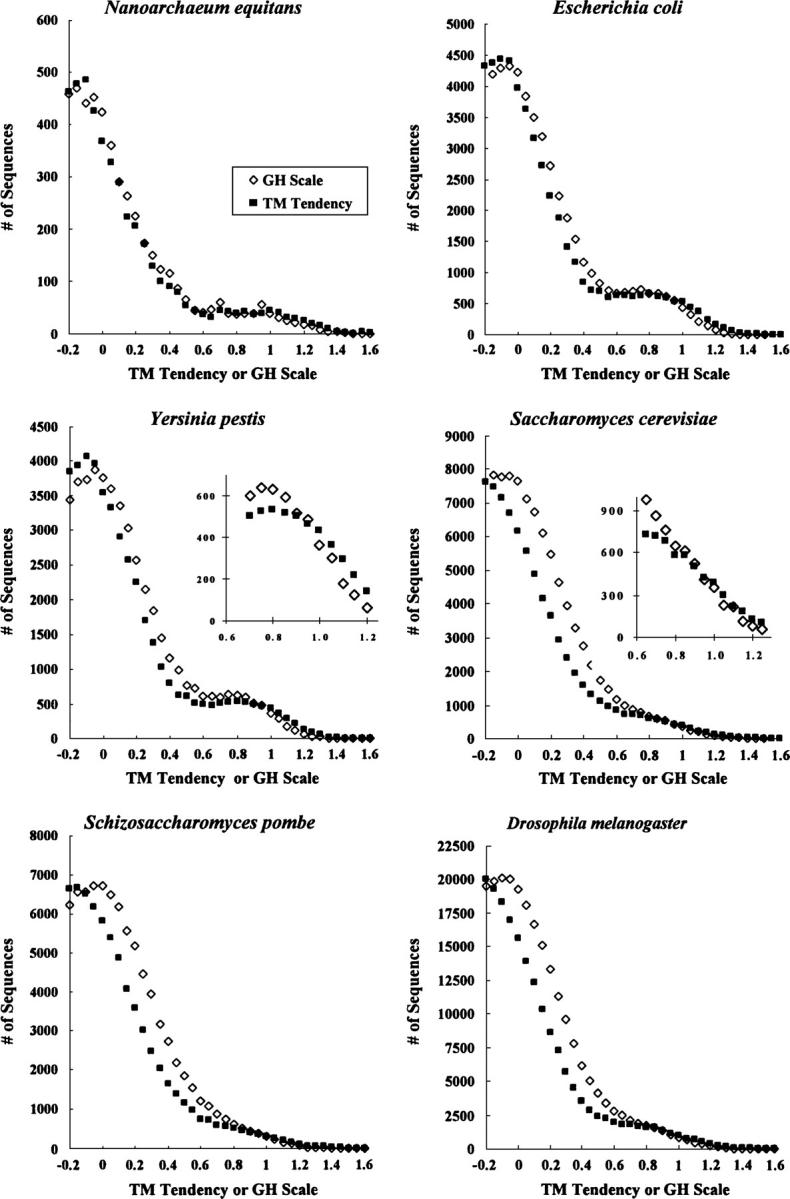
Comparison of the abundance of sequences as a function of “hydrophobicity” in various individual genomes as estimated by the GH scale and TM tendency scales. The total genomic abundance of sequences versus hydrophobicity is shown for the GH scale (open diamonds) and TM tendency (closed squares) scale for Nanoarchaeum equitans, E. coli, Y. pestis, S. cerevisiae, Schizosaccharomyces pombe, and Drosophila melanogaster. A sliding window size of 19 and a merge factor of 4 were used. Each point represents sequences with a hydrophobicity value range from equal up to <0.05 units greater than the hydrophobicity value shown on the X-axis. Insets show expanded portions of selected graphs.
Notice that conclusions based on peak resolution depend on the behavior of sequences of low and high hydrophobicities, and thus are independent of intermediate hydrophobicity sequences used for derivation of the TM tendency scale. (Even after refinement, intermediate hydrophobicity sequences remain close to the semihydrophobic range, and thus do not influence the peak resolution [data not shown].) It is also important to note that increased resolution is not an artifact of an expanded range for the TM tendency scale (X-axis expansion). The difference in “hydrophobicity” values for hydrophilic and hydrophobic residues is not expanded for the TM tendency scale relative to the GH scale (Table 3). Furthermore, the range of hydrophobicity values over which TM probability changes from 10% to 90% is slightly less for the TM tendency scale than for the GH scale (see Table 1), consistent with improved resolution for the TM tendency scale, and the opposite of what would happen if scale values were just expanded over a wider range in the case of the TM tendency scale.
Another parameter used to compare the ability of different scales to distinguish TM from non-TM sequences is the ratio of the number of sequences with intermediate hydrophobicity (defined as sequences within the 10%–90% TM probability range) to the number of likely TM sequences (>90% TM probability). The better a scale resolves intermediate hydrophobicity sequences into TM and non-TM populations, the lower this ratio (R) should be. We tested this for the Yersinia pestis genome. As shown in Table 1, this R is lowest for the TM tendency scale, and the ability of different scales to resolve TM from non-TM sequences as judged from the R parameter generally parallels that reported by Pfalse TM and Pmissing TM.
Whether a sequence is the most hydrophobic in a protein can independently illustrate the accuracy of TM tendency scale
Additional information concerning scale accuracy can be obtained by considering whether a hydrophobic sequence is the most hydrophobic in that protein (= maxH sequence) (Boyd et al. 1998) or is not (a non-maxH sequence). Figure 5A shows that for both soluble sequences in known soluble proteins and TM sequences in known TM membrane proteins the hydrophobicity of the most hydrophobic sequence in a protein (filled symbols) is on the average significantly more hydrophobic than the other sequences within that protein (open symbols). This is shown by the fact that the peaks for non-maxH sequences are at lower hydrophobicities than for maxH sequences.
Figure 5.

Hydrophobicity distributions of sequences that are the most hydrophobic in a protein (maxH) or not most hydrophobic (non-maxH) for known soluble proteins and known TM sequences and comparison to whole genomic data. (A) Abundance of the most hydrophobic (maxH) and non-maxH sequences for known soluble proteins (squares) and for known TM sequences (circles) as a function of TM tendency for sum of E. coli + S. cerevisiae genomes divided by two. (Filled symbols) maxH sequences; (open symbols) non-maxH sequences. (B) Percent probability of a soluble (squares) or TM (triangles) sequence being the most hydrophobic sequence (maxH sequence) in a soluble protein or most hydrophobic TM sequence in a TM protein, respectively, as a function of TM tendency. (C,D) Percent probability of a sequence in S. cerevisiae (C) or E. coli (D) being the most hydrophobic in a protein as a function of TM tendency (squares). Curves are for soluble sequences in soluble proteins (solid line) and TM sequences in TM proteins (dashed line) fit to genomic data by slight adjustment of the X-axis position of curves in panel B. (This adjusts for factors such as a difference in average molecular weight for proteins from databases of proteins with known structure versus proteins in a whole genome.)
At a particular hydrophobicity, a comparison of the number of soluble sequences from soluble proteins that are the most hydrophobic (maxH) or are not the most hydrophobic (non-maxH) allows calculation of the percent probability that a sequence with that hydrophobicity is the most hydrophobic in a soluble protein (Fig. 5B). Similarly, at a particular hydrophobicity, a comparison of the number of TM sequences from TM proteins that are the most hydrophobic (maxH) or are not the most hydrophobic (non-maxH) allows calculation of the percent probability that a sequence with this hydrophobicity is the most hydrophobic TM sequence in a TM protein (Fig. 5B). As expected, (1) the higher the hydrophobicity of a sequence, the more likely it is to be the most hydrophobic sequence in a protein, and (2) to be the most hydrophobic TM sequence in a TM protein tends to require a higher hydrophobicity than to be the most hydrophobic soluble sequence in a soluble protein (Fig. 5A,B). Thus, sequences derived from the soluble protein and TM sequence databases have very different probabilities of being the most hydrophobic sequence as a function of hydrophobicity (Fig. 5B).
This difference can be used to confirm the accuracy of the TM tendency scale. When the probability that a sequence is the most hydrophobic (maxH sequence) in a protein is assessed as a function of hydrophobicity using whole genomic data, and compared to the probability of being the most hydrophobic for soluble and TM sequences from databases of soluble and TM proteins, respectively (Fig. 5C,D), it is easy to discern the TM tendency values at which the sequences in a genome are exclusively soluble, exclusively TM, or a mixture of the two. Figure 5, C and D, shows that at low TM tendency (<0.3) the probability of a genomic sequence being the most hydrophobic fits expectations for soluble proteins. This is true because the low hydrophobicity region is dominated by sequences from soluble proteins. Similarly, at high TM tendency (>0.8), where TM sequences predominate, the probability of a sequence being the most hydrophobic in a protein fits expectations for TM sequences. The intermediate TM tendency range (0.3–0.8) does not fit the shape for either the soluble or TM sequence populations because it contains both TM and non-TM populations.
A key observation is that the intermediate hydrophobicity range defined by this method closely approximates the range in which soluble and TM sequences coexist as derived from fitting genomic data to soluble and TM protein databases (0.3–0.7 TM tendency values for 10% and 90% TM probabilities, respectively; see Table 1). In addition, the TM tendency value at which the percent TM probability is 50% in Figure 5, C and D (X-axis value ∼0.5–0.55, where the Y-axis value for the probability that a sequence is the most hydrophobic is halfway between the values for the soluble and TM curves) agrees with the 50% TM probability value for the TM tendency scale when independently derived from resolution of genomic data into soluble and TM sequence components (Table 1). This agreement is a further indication of the accuracy of the TM tendency scale.
It is noteworthy that both the overlap between (1) TM and non-TM maxH sequences and (2) the overlap between TM and non-TM non-maxH sequences (Fig. 5A) is less than the overlap between the combination of all sequences (i.e., maxH and non-maxH) (Fig. 1B). This implies that by separately calculating the percent TM probability for maxH and non-maxH sequences, it should be possible to improve prediction of TM sequences (see Table 1).
Comparison of amphiphilic, signal, and TM sequences using the TM tendency scale
Semihydrophobic helices are not equivalent to amphiphilic helices. Methods for identifying amphipathic helices (Eisenberg et al. 1984) indicate such helices represent a small fraction of the subhydrophobic sequences, and are not at all abundant in the semihydrophobic range, even when a low stringency hydrophobic moment is used (0.35). The lack of amphiphilic sequences in the semihydrophobic range is not surprising because they should have a relatively high content of polar and ionizable residues. Semihydrophobic sequences should also not be confused with signal sequences. Signal sequences (identified as described in Kall et al. 2004) were deleted from the genomic databases used in the preceding sections because they are generally not found in mature proteins, and thus would not be represented in the soluble and TMP structural databases used to resolve genomic data. When abundance versus hydrophobicity was calculated for the population of signal sequences from the E. coli and S. cerevisiae genomes, a Gaussian-like dependence of abundance on TM tendency value was found, with a peak at 0.6 units and a width at half-height of about 0.5 units. It should be noted that the omission of signal sequences does not have a major impact on the appearance of abundance versus hydrophobicity distributions for genomic data because signal sequences form only a small fraction of the total sequences (they were only 10% as abundant as sequences from mature proteins at the TM tendency value at which signal sequences show peak abundance). It should be noted that the hydrophobicity range containing signal sequences is influenced by the fact that signal sequences often have short hydrophobic cores of 7–15 residues in length (von Heijne 1985). Thus, our computations, which assigned hydrophobicities to sequences of 19 residues and longer, would tend to report the hydrophobicity of not only the signal sequence core, but also some of the surrounding hydrophilic residues.
Discussion
Why the transmembrane tendency scale approaches an upper limit to accuracy
The TM tendency scale must be close to the limit of predictive accuracy for a scale in which single values are assigned to the propensity of a particular type residue to reside in a TM sequence. As noted above, the justification for this strong statement arises from the fact that this scale fulfills a key criterion not previously recognized: Populations of known TM and soluble sequences predicted to have equal tendencies to form TM helices should have the same average contents of each amino acid. We call this the “equal average composition” criterion. Consider a scale predicting that populations of known TM and soluble sequences with an equal tendency to form TM segments do not have the same average contents of different amino acids. The predictive accuracy of such a scale can be improved by decreasing TM propensity values for residues enriched in the soluble sequence population and increasing TM propensity values for residues found in excess in the TM population. Only a scale fulfilling the “equal average composition” criterion cannot be improved by this procedure.
The possibility that there is a second, very different, scale that also fulfills the equal average composition criterion is unlikely because refinement of several different scales converged on the TM tendency scale, and because TM tendency scale values seem to correspond to consensus values for all previous hydrophobicity-type scales (see Results). However, we do not claim that the TM tendency scale is the ultimate method for predicting TM helices. Like other hydrophobicity-type, single-value scales, TM tendency ignores factors such as the identity of neighboring residues or the depth of a residue in the bilayer (Caputo and London 2004; Hessa et al. 2005). In this regard, an alternate approach to identify TM sequences involves Hidden-Markov methods. Such methods can use all differences in the pattern of sequences for TM and non-TM proteins to strengthen predictive accuracy. It is interesting to ask how TM tendency and Hidden-Markov methods compare. When we chose a sample of 36 membrane proteins of known structure that was recently used to compare scales (Chen et al. 2002), and compared the TM tendency scale to HMMTOP2, a Hidden-Markov method found to predict helices more accurately than previously reported single-value scales (Chen et al. 2002), we found little difference in the ability to correctly identify TM helices in terms of the sum of false positives, false negatives, or helix boundaries (data not shown).
Relationship of biological hydrophobicity and TM tendency scales
As noted in the Results, the biological hydrophobicity and TM tendency scales are highly correlated with each other, although they are derived from two very different approaches. The biological hydrophobicity scale was defined by a sophisticated experiment in which the ability of sequences to insert in membranes in TM form was assayed in a translocon-assisted process that mimics the insertion process in vivo (Hessa et al. 2005). It was derived for an Ala + Leu hydrophobic sequence containing at most one hydrophilic residue, and forming part of a chimera with two natural TM helices not likely to interact with the Ala + Leu sequence. Thus, biological hydrophobicity scale values are probably most accurate for an isolated TM helix in which helix–helix interactions and interactions between hydrophilic residues do not have a significant role.
In contrast, the TM tendency scale reports on the propensities of amino acid residues to participate in TM helices on a genome-wide basis, and represents a statistical average influenced by all of the parameters that complicate TM propensities. In this regard, it should be noted that the TM tendency scale does not directly measure hydrophobicity. A hydrophilic residue abundant in membrane proteins for functional reasons should have a higher TM tendency value than predicted based on hydrophobicity. Nevertheless, to some degree one can think of the TM tendency scale as the “statistical” version of biological hydrophobicity averaged over all possible sequences. Thus, the difference between these two scales is a measure of the degree to which all the interactions that occur in real membrane proteins can complicate the propensity of an isolated residue to participate in a TM helix. The relative agreement between the two scales is an indication that the effect of these interactions is relatively modest, but still significant. Once the experimental behavior of more complex sequences is defined, it will be interesting to see whether experimental data agree even more closely with the predictions of the TM tendency scale.
Relationship between TM tendency and thermodynamic tendency to form a TM state
It should be noted that the percent TM probabilities we describe in this study do not have an exact thermodynamic meaning. Percent TM probability values are statistical and are slightly dependent on the number of soluble and TM proteins in a genome (Fig. 3B). Nevertheless, percent TM probability values may not deviate far from true thermodynamic values. For the augmented Wimley-White hydrophobicity scale, the value at which there is a 50% TM probability for the sum of E. coli and S. cerevisiae sequences (0.114) is only slightly higher than the theoretical value of 0, which represents equal free energy for the TM and non-TM states (Jayasinghe et al. 2001). The slightly higher value from analysis of genomic sequences may mainly be a consequence of the fact that hydrophilic residues in natural sequences often will engage in interhelical interactions or place hydrophilic residues within the bilayer to carry out biological function. Thermodynamic scales usually evaluate the behavior of individual amino acids in a manner that does not include the effect of these factors.
In cases of post-translational membrane insertion, the folding of a protein in the soluble state can also affect the propensity to form a TM state. Equilibration of hydrophobic segments between TM and non-TM states in such cases should not be determined solely by the difference between the free energy of amino acid segments when exposed to aqueous solution relative to their free energy when part of a membrane-inserted TM helix. Instead, the appropriate equilibrium is that between the folded form of the protein in solution, and the membrane-inserted conformation. In aqueous solution, hydrophobic segments in a protein are often buried and have specific interactions with other segments that decrease free energy relative to that in the unfolded state.
Nevertheless, simple hydrophobicity is clearly the predominant factor determining membrane insertion as shown by the observation that a pure hydrophobicity scale (the augmented Wimley-White scale; Jayasinghe et al. 2001) is reasonably well correlated with the TM tendency scale (r = 0.91). It should be noted that although we normalized the TM tendency scale so that the range of values for different residues was roughly as large as the range of ΔGo values for membrane insertion given by the augmented Wimley-White scale (see Materials and Methods), we did not calibrate it so that a 50% TM tendency value of zero would be equivalent to a ΔGo of zero for TM insertion. The lack of such calibration would not affect the ability of the TM tendency scale to distinguish TM from non-TM sequences.
Semihydrophobic sequences and conversion between TM and non-TM states
One of the more important developments in this study is that there is a considerable population of sequences within the semihydrophobic range. Such sequences might form either TM or non-TM structures depending on the exact experimental or biological conditions to which they are exposed. Protein toxins such as diphtheria toxin and certain colicins, and some Bcl-class proteins (Qiu et al. 1996; Kienker et al. 1997; Ren et al. 1997; Jeong et al. 2004; Rosconi et al. 2004) have sequences already believed to switch between TM and non-TM states. These sequences often fall in the semihydrophobic (10%–90% TM probability) range. According to the TM tendency scale, helix 5, helices 6–7 (combined), helix 8, and helix 9 in the T domain of diphtheria toxin have percent TM probabilities of 15%, 35%, 63%, and 63%, respectively. In contrast, despite existing within both soluble and TM conformations, helices 8 and 9 in the channel-forming domain of colicin A have percent TM probabilities of 94% and 99.5%, respectively (calculation not shown). This suggests that if properly buried within the interior of a protein, even very hydrophobic sequences can exist in a soluble state.
The capacity to switch between TM and non-TM states is not limited to special proteins. This equilibrium has been observed for numerous simple hydrophobic helices in model membranes (Bechinger 1996; Ren et al. 1997, 1999; Lew et al. 2000; Vogt et al. 2000; Caputo and London 2004), and for simple sequences undergoing translocation via the translocon (Hessa et al. 2005). It is possible that there are additional cases in which predominantly soluble proteins have semihydrophobic sequences that allow formation of as-yet-undiscovered minor TM subpopulations, and vice versa. This could have tremendous biological significance. In some cases, the soluble and membrane-inserted conformations of such a protein might have very different tertiary structures and functions. The possibility of minor soluble or membrane-inserted populations moonlighting in this fashion is one of the more exciting possibilities revealed by this study.
Acknowledgments
We thank Steven O. Smith for helpful discussions. This work was supported by NIH grant GM31986.
Footnotes
Reprint requests to: Erwin London, Department of Biochemistry and Cell Biology, Stony Brook University, Stony Brook, NY 11794-5215, USA; e-mail: Erwin.London@stonybrook.edu; fax: (631) 632-8575.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062286306.
References
- Bechinger B. 1996. Towards membrane protein design: pH-sensitive topology of histidine-containing polypeptides. J. Mol. Biol. 263: 768–775. [DOI] [PubMed] [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner F.R., Plunkett G. III, Bloch C.A., Perna N.T., Burland V., Riley M., Collado-Vides J., Glasner J.D., Rode C.K., Mayhew G.F. et al. 1997. The complete genome sequence of Escherichia coli K-12. Science 277: 1453–1474. [DOI] [PubMed] [Google Scholar]
- Boyd D., Schierle C., Beckwith J. 1998. How many membrane proteins are there? Protein Sci. 7: 201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo G.A. and London E. 2003. Cumulative effects of amino acid substitutions and hydrophobic mismatch upon the transmembrane stability and conformation of hydrophobic α-helices. Biochemistry 42: 3275–3285. [DOI] [PubMed] [Google Scholar]
- Caputo G.A. and London E. 2004. Position and ionization state of Asp in the core of membrane-inserted α helices control both the equilibrium between transmembrane and nontransmembrane helix topography and transmembrane helix positioning. Biochemistry 43: 8794–8806. [DOI] [PubMed] [Google Scholar]
- Chandonia J.M., Hon G., Walker N.S., Lo Conte L., Koehl P., Levitt M., Brenner S.E. 2004. The ASTRAL Compendium in 2004. Nucleic Acids Res. 32:Database issue D189–D192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.P. and Rost B. 2002. State-of-the-art in membrane protein prediction. Appl. Bioinformatics 1: 21–35. [PubMed] [Google Scholar]
- Chen C.P., Kernytsky A., Rost B. 2002. Transmembrane helix predictions revisited. Protein Sci. 11: 2774–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry J.M., Ball C., Weng S., Juvik G., Schmidt R., Adler C., Dunn B., Dwight S., Riles L., Mortimer R.K. et al. 1997. Genetic and physical maps of Saccharomyces cerevisiae. Nature 387: 67–73. [PMC free article] [PubMed] [Google Scholar]
- Claros M.G. and von Heijne G. 1994. TopPred II: An improved software for membrane protein structure predictions. Comput. Appl. Biosci. 10: 685–686. [DOI] [PubMed] [Google Scholar]
- Cowan R. and Whittaker R.G. 1990. Hydrophobicity indices for amino acid residues as determined by high-performance liquid chromatography. Pept. Res. 3: 75–80. [PubMed] [Google Scholar]
- Cserzo M., Wallin E., Simon I., von Heijne G., Elofsson A. 1997. Prediction of transmembrane α-helices in prokaryotic membrane proteins: The dense alignment surface method. Protein Eng. 10: 673–676. [DOI] [PubMed] [Google Scholar]
- Deber C.M., Wang C., Liu L.P., Prior A.S., Agrawal S., Muskat B.L., Cuticchia A.J. 2001. TM Finder: A prediction program for transmembrane protein segments using a combination of hydrophobicity and nonpolar phase helicity scales. Protein Sci. 10: 212–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deber C.M., Liu L.P., Wang C., Goto N.K., Reithmeier R. 2002. The hydrophobicity threshold for peptide insertion into membranes. In Current topics in membranes: Peptide–lipid interactions (ed. McIntosh T.J.) . pp. 465–479. Academic Press, San Diego.
- Eisenberg D., Schwarz E., Komaromy M., Wall R. 1984. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 179: 125–142. [DOI] [PubMed] [Google Scholar]
- Engelman D.M., Steitz T.A., Goldman A. 1986. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu. Rev. Biophys. Biophys. Chem. 15: 321–353. [DOI] [PubMed] [Google Scholar]
- Fauchere J.-L. and Pliska V. 1983. Hydrophobic parameters of π amino acid-side chains from the partitioning of N-acetyl-amino-acid amides. Eur. J. Med. Chem. Chim. Ther. 18: 369–375. [Google Scholar]
- Hessa T., Kim H., Bihlmaier K., Lundin C., Boekel J., Andersson H., Nilsson I., White S.H., von Heijne G. 2005. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433: 377–381. [DOI] [PubMed] [Google Scholar]
- Ikeda M., Arai M., Lao D.M., Shimizu T. 2002. Transmembrane topology prediction methods: A re-assessment and improvement by a consensus method using a dataset of experimentally-characterized transmembrane topologies. In Silico Biol. 2: 19–33. [PubMed] [Google Scholar]
- Ikeda M., Arai M., Okuno T., Shimizu T. 2003. TMPDB: A database of experimentally-characterized transmembrane topologies. Nucleic Acids Res. 31: 406–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasinghe S., Hristova K., White S.H. 2001. Energetics, stability, and prediction of transmembrane helices. J. Mol. Biol. 312: 927–934. [DOI] [PubMed] [Google Scholar]
- Jeong S.Y., Gaume B., Lee Y.J., Hsu Y.T., Ryu S.W., Yoon S.H., Youle R.J. 2004. Bcl-x(L) sequesters its C-terminal membrane anchor in soluble, cytosolic homodimers. EMBO J. 23: 2146–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D.T., Taylor W.R., Thornton J.M. 1994. A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry 33: 3038–3049. [DOI] [PubMed] [Google Scholar]
- Kall L., Krogh A., Sonnhammer E.L. 2004. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338: 1027–1036. [DOI] [PubMed] [Google Scholar]
- Kessel A. and Ben-Tal N. 2002. Free energy determinants of peptide association with lipid bilayer. In Current topics in membranes: Peptide–lipid interactions (ed. McIntosh T.) . pp. 205–253. Academic Press, San Diego.
- Kienker P.K., Qiu X., Slatin S.L., Finkelstein A., Jakes K.S. 1997. Transmembrane insertion of the colicin Ia hydrophobic hairpin. J. Membr. Biol. 157: 27–37. [DOI] [PubMed] [Google Scholar]
- Kyte J. and Doolittle R.F. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157: 105–132. [DOI] [PubMed] [Google Scholar]
- Lao D.M., Arai M., Ikeda M., Shimizu T. 2002. The presence of signal peptide significantly affects transmembrane topology prediction. Bioinformatics 18: 1562–1566. [DOI] [PubMed] [Google Scholar]
- Lew S., Ren J., London E. 2000. The effects of polar and/or ionizable residues in the core and flanking regions of hydrophobic helices on transmembrane conformation and oligomerization. Biochemistry 39: 9632–9640. [DOI] [PubMed] [Google Scholar]
- Mori H., Isono K., Horiuchi T., Miki T. 2000. Functional genomics of Escherichia coli in Japan. Res. Microbiol. 151: 121–128. [DOI] [PubMed] [Google Scholar]
- Murzin A.G., Brenner S.E., Hubbard T., Chothia C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247: 536–540. [DOI] [PubMed] [Google Scholar]
- Parker J.M., Guo D., Hodges R.S. 1986. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: Correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry 25: 5425–5432. [DOI] [PubMed] [Google Scholar]
- Persson B. and Argos P. 1997. Prediction of membrane protein topology utilizing multiple sequence alignments. J. Protein Chem. 16: 453–457. [DOI] [PubMed] [Google Scholar]
- Qiu X.Q., Jakes K.S., Kienker P.K., Finkelstein A., Slatin S.L. 1996. Major transmembrane movement associated with colicin Ia channel gating. J. Gen. Physiol. 107: 313–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J., Lew S., Wang Z., London E. 1997. Transmembrane orientation of hydrophobic α-helices is regulated both by the relationship of helix length to bilayer thickness and by the cholesterol concentration. Biochemistry 36: 10213–10220. [DOI] [PubMed] [Google Scholar]
- Ren J., Lew S., Wang J., London E. 1999. Control of the transmembrane orientation and interhelical interactions within membranes by hydrophobic helix length. Biochemistry 38: 5905–5912. [DOI] [PubMed] [Google Scholar]
- Rosconi M.P. and London E. 2002. Topography of helices 5–7 in membrane-inserted diphtheria toxin T domain: Identification and insertion boundaries of two hydrophobic sequences that do not form a stable transmembrane hairpin. J. Biol. Chem. 277: 16517–16527. [DOI] [PubMed] [Google Scholar]
- Rosconi M.P., Zhao G., London E. 2004. Analyzing topography of membrane-inserted diphtheria toxin T domain using BODIPY-streptavidin: At low pH, helices 8 and 9 form a transmembrane hairpin but helices 5–7 form stable nonclassical inserted segments on the cis side of the bilayer. Biochemistry 43: 9127–9139. [DOI] [PubMed] [Google Scholar]
- Rose G.D., Geselowitz A.R., Lesser G.J., Lee R.H., Zehfus M.H. 1985. Hydrophobicity of amino acid residues in globular proteins. Science 229: 834–838. [DOI] [PubMed] [Google Scholar]
- Rost B., Casadio R., Fariselli P., Sander C. 1995. Transmembrane helices predicted at 95% accuracy. Protein Sci. 4: 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B., Fariselli P., Casadio R. 1996. Topology prediction for helical transmembrane proteins at 86% accuracy. Protein Sci. 5: 1704–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusnady G.E. and Simon I. 1998. Principles governing amino acid composition of integral membrane proteins: Application to topology prediction. J. Mol. Biol. 283: 489–506. [DOI] [PubMed] [Google Scholar]
- Vogt B., Ducarme P., Schinzel S., Brasseur R., Bechinger B. 2000. The topology of lysine-containing amphipathic peptides in bilayers by circular dichroism, solid-state NMR, and molecular modeling. Biophys. J. 79: 2644–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. 1985. Signal sequences. The limits of variation. J. Mol. Biol. 184: 99–105. [DOI] [PubMed] [Google Scholar]
- von Heijne G. 1992. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J. Mol. Biol. 225: 487–494. [DOI] [PubMed] [Google Scholar]