

Abstract
This study tested the hypothesis that high-affinity binding of macromolecular ligands to the αIIbβ3 integrin is tightly coupled to binding-site remodeling, an induced-fit process that shifts a conformational equilibrium from a resting toward an open receptor. Interactions between αIIbβ3 and two model ligands—echistatin, a 6-kDa recombinant protein with an RGD integrin-targeting sequence, and fibrinogen's γ-module, a 30-kDa recombinant protein with a KQAGDV integrin binding site—were measured by sedimentation velocity, fluorescence anisotropy, and a solid-phase binding assay, and modeled by molecular graphics. Studying echistatin variants (R24A, R24K, D26A, D26E, D27W, D27F), we found that electrostatic contacts with charged residues at the αIIb/β3 interface, rather than nonpolar contacts, perturb the conformation of the resting integrin. Aspartate 26, which interacts with the nearby MIDAS cation, was essential for binding, as D26A and D26E were inactive. In contrast, R24K was fully and R24A partly active, indicating that the positively charged arginine 24 contributes to, but is not required for, integrin recognition. Moreover, we demonstrated that priming—i.e., ectodomain conformational changes and oligomerization induced by incubation at 35°C with the ligand-mimetic peptide cHarGD—promotes complex formation with fibrinogen's γ-module. We also observed that the γ-module's flexible carboxy terminus was not required for αIIbβ3 integrin binding. Our studies differentiate priming ligands, which bind to the resting receptor and perturb its conformation, from regulated ligands, where binding-site remodeling must first occur. Echistatin's binding energy is sufficient to rearrange the subunit interface, but regulated ligands like fibrinogen must rely on priming to overcome conformational barriers.
Keywords: integrins, ligands, conformation, analytical ultracentrifugation, solid phase binding, fluorescence anisotropy, molecular modeling
Integrins, such as the heterodimeric αIIbβ3 complex, are tightly regulated signaling machines that maintain communication and contact between a cell's interior and extracellular matrix proteins (Critchley et al. 1999; Giancotti and Ruoslahti 1999; Hynes 2002). Like many integrins, resting αIIbβ3 must be activated before it can perform these functions (Hughes and Pfaff 1998; Shattil et al. 1998). Circulating human blood platelets have ∼80,000 αIIbβ3 integrins on their surface and are immersed in plasma that contains saturating concentrations of fibrinogen, their primary physiological ligand (Shattil 1999; Liddington and Bankston 2000; Plow et al. 2000).Yet αIIbβ3 and its ligands do not interact until intracellular molecules are released in response to occupancy of G-protein-coupled receptors, a process termed inside-out signaling, which enables these integrins to bind fibrinogen (Faull and Ginsberg 1996; Shattil et al. 1998; Shattil 1999). The structural changes that promote macromolecular ligand binding are called priming to distinguish them from other signaling events (Humphries et al. 2003; Humphries 2004). Then fibrinogen binding promotes integrin clustering, stabilizes receptor:ligand contacts, and activates focal adhesion kinase, in a process called outside-in signaling (Giancotti and Ruoslahti 1999; Hartwig et al. 1999; Calderwood et al. 2000).
Despite publication of crystal structures of the ectodomain of the related integrin αvβ3 in both its free and ligand-bound states (Xiong et al. 2001, 2002) and truncated αIIbβ3 constructs with tirofiban or eptifibatide bound (Xiao et al. 2004), the conformational changes that contribute to priming remain controversial (Arnaout et al. 2002; Beglova et al. 2002; Gottschalk et al. 2002; Hynes 2002; Liddington 2002; Shimaoka et al. 2002; Takagi et al. 2002; Humphries 2004; Luo et al. 2004). Beglova et al. (2002) proposed a “jackknife” model in which the resting, bent conformer observed by crystallography (Xiong et al. 2001) extends from hinges in each subunit into an elongated structure with separate stalks that resembles electron microscopy images (Carrell et al. 1985; Nermut et al. 1988; Weisel et al. 1992; Iwasaki et al. 2005). The link between cytoplasmic domain separation and bidirectional integrin signaling is supported by FRET measurements (Kim et al. 2003) and disulfide trapping studies (Luo et al. 2004) conducted on living cells. In contrast, Xiong et al. (2003) proposed the “deadbolt” model in which subtle, reversible interactions between the β-terminal domain and the βA domain in the bent αvβ3 ectodomain regulate integrin affinity (Xiong et al. 2003). Support for this minimalist priming mechanism comes from the X-ray diffraction structure of an αvβ3 ectodomain:RGD peptide complex (Xiong et al. 2002), cryo-electron microscopy that reveals a partially bent structure for full-length αIIbβ3 (Adair and Yeager 2002), and a recent three-dimensional electron microscopy (EM) structure in which αvβ3's ectodomain remained genuflexed while binding a 22-kDa fibronectin fragment (Adair et al. 2005).
The structural basis for ligand-induced outside-in signaling (Humphries et al. 2003; Mould and Humphries 2004) is still unfolding, in large measure because biophysics and EM data differ on the conformational prerequisites for macromolecular binding. The SAXS data of Mould et al. (2003) showed minimal rearrangements in α5β1’s ectodomain upon binding a fibronectin fragment, while Takagi et al.’s (2003) EM showed α5β1’s remnant stalks separated when binding a similar fragment. EM by Litvinov et al. (2004) showed that Mn++ promoted stalk separation and fibrinogen binding with native αIIbβ3 isolated in octyl glucoside micelles. However, electron tomography data recently reported by Iwasaki et al. (2005) indicate that activated αIIbβ3 integrins display a range of conformations with enhanced flexibility, especially in the β3 stalk region. Indeed, our electron microscopy observations demonstrated a broad distribution of αIIbβ3 conformers, both resting and in complex with echistatin (Hantgan et al. 2004).
Our approach focuses on defining the conformational barriers that separate synthetic RGD peptides (Hantgan et al. 1999, 2003) and peptideometic pharmaceuticals (Hantgan et al. 2001, 2002) that bind rapidly and reversibly to the initially resting αIIbβ3 complex from fibrinogen, which exhibits high-affinity interactions only with activated receptors (Shattil and Newman 2004). We have recently bridged the gap between those low-molecular-weight integrin antagonists (<1500 Da) and the 340-kDa fibrinogen molecule by demonstrating that αIIbβ3 recognition of echistatin, a 6-kDa disintegrin with an RGD integrin-targeting sequence, is tightly linked to conformational changes in the receptor's ectodomain (Hantgan et al. 2004). In fact, we found that the magnitude of those perturbations, as determined by changes in αIIbβ3’s hydrodynamic properties, are comparable to those induced by conformationally constrained peptides, such as eptifibatide and cHArGD (Hantgan et al. 2001, 2003).
Here, we applied a site-directed mutagenesis approach to define the relative contributions of electrostatic and hydrophobic interactions in promoting αIIbβ3:echistatin binding and gained new insights into the mechanisms responsible for this model protein ligand's self-priming activities. We also demonstrated that ectodomain conformational changes and oligomerization induced by incubation at 35°C with the ligand-mimetic cHarGD (Hantgan et al. 2003) result in a dramatic enhancement of αIIbβ3’s affinity for fibrinogen's 30-kDa γ-module (Medved et al. 1997), a recombinant construct that contains key residues implicated in binding the parent adhesive macromolecule.
Results
Electrostatic and hydrophobic contributions to integrin αIIbβ3:ligand interactions
We recently presented data supporting the hypothesis that echistatin's ability to modulate the structure of the αIIbβ3 integrin resides on an RGD loop, while full disintegrin activity requires an auxiliary site encompassing its carboxy-terminal nine residues (Hantgan et al. 2004). The work described in this article directly tests that hypothesis by examining the interactions of a series of well-characterized recombinant echistatin variants (rEch mutants) with point mutations designed either to modify key electrostatic interactions (R24A, R24K; D26A, D26E) or to introduce new hydrophobic residues (D27W, D27F) into echistatin's integrin-recognition site. These single-site mutations were made in the rEch (1–49) M28L construct that we have previously characterized (Hantgan et al. 2004).
The R24K and D27W mutations are of special interest because these constructs incorporate key features of the disintegrin barbourin, which displays a KGDW motif and selectively inhibits αIIbβ3 over αvβ3 (Scarborough et al. 1991). Furthermore, the D27F rEch mutant replicates an intriguing aspect of αIIbβ3’s primary physiological ligand, fibrinogen. Fibrinogen harbors an RGDF sequence in the midst of its coiled-coil domains, although the role of this site in integrin recognition is unclear (Liu et al. 1998; Rooney et al. 1998). Our RGDF and RGDW rEch mutants also display a pattern of positive, negative, and nonpolar groups that is spatially similar to those in the FDA-approved small molecule integrin antagonists, eptifibatide and tirofiban (Phillips and Scarborough 1997; Cook et al. 1999). Thus, our study provides new insights into the molecular basis of αIIbβ3’s interactions with physiological, pathological, and pharmacological ligands.
Our integrated biochemical and biophysical approach demonstrates the following points: (1) The negatively charged aspartate, the most conserved disintegrin residue, is essential for echistatin's interactions with αIIbβ3, most likely through its interactions with the MIDAS cation. (2) Echistatin's nearby arginine residue strengthens these interactions but is not required for productive complex formation. (3) Introducing nonpolar residues enhances echistatin's ability to bind αIIbβ3 but not its ability to perturb the receptor's conformation.
Subsequent sections of this article build on these concepts to examine the interactions of fibrinogen's γ-module, a recombinant 30-kDa fragment with a KQAGDV integrin-targeting sequence, with resting and primed αIIbβ3. Our goal is to determine what parameters distinguish echistatin, a self-priming ligand, from larger, regulated ligands, such as fibrinogen.
Echistatin binding to immobilized αIIbβ3
We first developed a solid-phase assay to screen rEch mutants for their ability to bind to αIIbβ3 that was captured on the wells of a microtiter plate from dilute solutions of the micellar integrin. Full-length rEch (1–49) M28L, labeled with 1–3 mol biotin/mol echistatin, exhibited specific, saturable binding to primed, immobilized αIIbβ3 with a Kd = 15 ± 6 nM (Fig. 1, concentric circles, pooled data from two experiments). However, essentially no binding was detected with resting receptors (Fig. 1, open circles, from two experiments). All subsequent solid-phase binding measurements with rEch mutants used primed receptors, i.e., those immobilized in the presence of the ligand-mimetic peptide cHarGD, which we have shown to promote the formation of open integrin conformers (Hantgan et al. 2003). cHarGD was removed by extensive washing prior to the addition of biotinylated rEch ligands.
Figure 1.

Integrin:echistatin interactions measured with a solid-phase binding assay. Data are expressed as the fraction of maximum change in the absorbance at 405 nm (Signal) following incubation with increasing concentrations of biotinylated echistatin variant ([BT-rEch] nM) to αIIbβ3 immobilized in the wells of a microtiter plate. The dashed and black lines were obtained by fitting the data to a single-site saturable binding model to determine the dissociation constant, Kd, as indicated in the text. The following symbols apply: rEch (1–49) M28L D27W (black hexagons, dashed line), primed rEch (1–49) M28L (concentric circles, black line); resting (open circles), rEch (1–49) M28L R24A (gray diamonds), rEch (1–49) D26A (black triangles, dotted line obtained by linear regression).
In contrast to the tight binding determined with rEch (1–49) M28L, the mutant D26A exhibited minimal interactions, even at 120 nM (Fig. 1, black triangles, two experiments). This negative result is consistent with the conserved nature of this aspartate among small disintegrins (Calvete et al. 2005). However, we were surprised to find that the rEch mutant R24A (gray diamonds; pooled data from three experiments) exhibited a binding profile comparable to rEch (1–49) M28L, providing an early indication that the two charged components of echistatin's RGD integrin-recognition code contribute unequally. Moreover, rEch mutant D27W displayed even tighter binding than the wild-type protein, yielding Kd = 2 ± 1 nM (black hexagons; three experiments), suggesting that echistatin:integrin interactions can be enhanced by new, nonpolar contacts.
While the solid-phase binding assay has proven useful for screening, its reproducibility is limited by the small signal changes that result from biotin-labeled ligand binding to low-density immobilized receptors. Others have demonstrated that the amplification inherent in a biotin-stepavidin-based colorimetric readout can overestimate the affinity of integrin:ligand interactions (Tangemann and Engel 1995). Furthermore, αIIbβ3 undergoes conformational changes upon contact with artificial surfaces (Hussain and Siedlecki 2004), an effect that may explain our observations of the need for priming when it is immobilized on the wells of a polystyrene microtiter plate. Hence, we extended these initial observations by directly testing the ability of an expanded set of rEch mutants to perturb the resting conformation of micellar αIIbβ3 in solution. In addition, each rEch mutant was shown to exhibit a compact, folded conformation as evidenced by proton NMR spectra (demonstrating nativelike solution structures for all mutants), and thiol titrations (showing that >94% of the cysteines in each mutant were disulfide bonded). Furthermore, each rEch mutant eluted from a reversed-phase HPLC column as a single peak in a pattern that correlated with the hydrophobicity of their point mutations. These analytical separations were performed under conditions shown to separate the enzymatically active disintegrin, rBitstatin, from its reduced isomers (Knight and Romano 2005). Full details are presented in Materials and Methods as well as Supplemental Figures 1 and 2.
Differential effects of rEch mutants on αIIbβ3’s resting conformation
Sedimentation-velocity measurements coupled with time-derivative analyses (Stafford 1992) provided a sensitive, specific index of the ability of integrin ligands, ranging from synthetic peptides to echistatin, to shift the solution structure of micellar αIIbβ3 from a quiescent state to an open, slower-sedimenting conformation (Hantgan et al. 1999, 2001, 2004). This technology enabled us to demonstrate here that echistatin variants with point mutations in their integrin-targeting RGD site differ in their ability to promote this conformational change. As illustrated in Figure 2A, rEch mutant D26A had no significant effect on αIIbβ3 conformation, as indicated by the nearly superimposable distributions of sedimenting species observed in the presence (black triangles) and absence (open circles) of ligand. This echistatin variant also did not bind to immobilized αIIbβ3 in a solid-phase binding assay (Fig. 1). In contrast, an equivalent concentration of rEch mutant R24A, which bound to the immobilized integrin with an affinity comparable to the wild-type disintegrin (Fig. 1), induced a clear shift in g(S) distribution toward slower-sedimenting αIIbβ3 species (Fig. 2B, gray diamonds). More pronounced effects were observed with rEch mutant D27W (Fig. 2C, black hexagons), which caused conformational perturbations comparable to full-length rEch (1–49) M28L (Hantgan et al. 2004), although it bound approximately sevenfold more tightly to the immobilized receptor.
Figure 2.
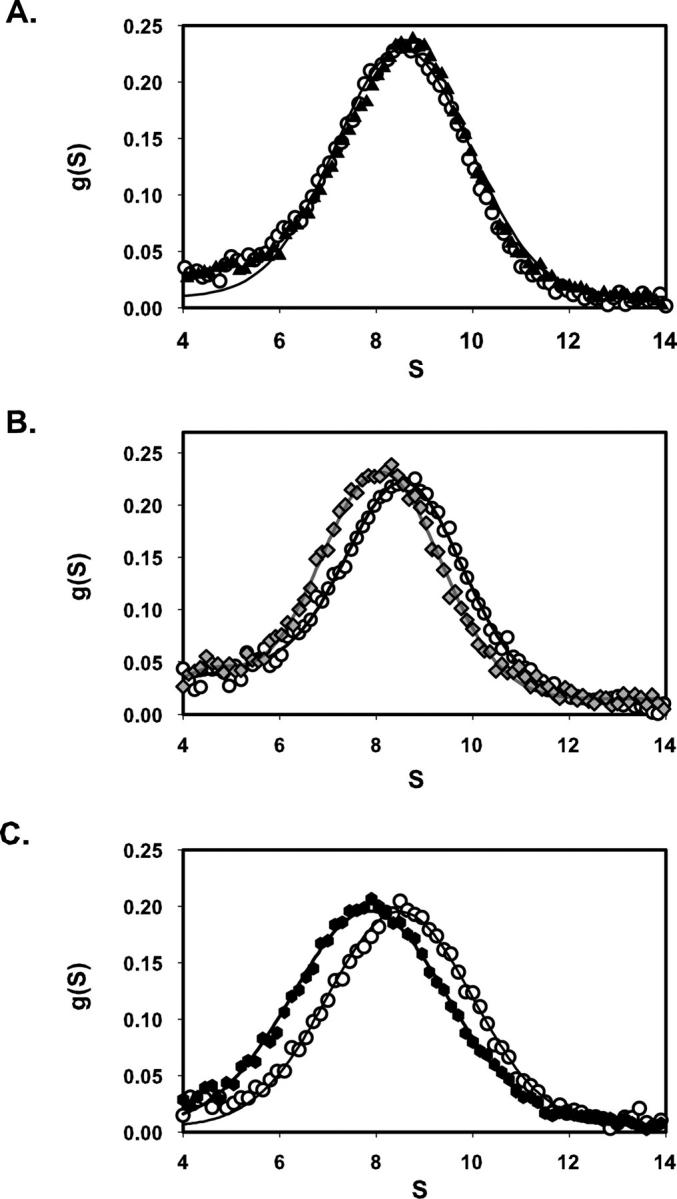
Effects of recombinant echistatin variants on the distribution of αIIbβ3 sedimenting species, g(S) versus S (time-derivative software; DCDT+; Philo 1997). Each panel compares the distribution obtained with ligand-free integrin (open circles) to a paired sample containing excess echistatin variant. Continuous lines were obtained by fitting the resultant distribution functions to a one- or two-species model, as required. (A) rEch (1–49) M28L D26A (black triangles). (B) rEch (1–49) M28L R24A (gray diamonds) (C) rEch (1–49) M28L D27W (black hexagons).
Extending this approach to an expanded set of rEch mutants yielded the data in Figure 3, where fractional changes in sedimentation coefficient are presented as a function of the mole ratio of ligand to receptor. These data demonstrate that conformational perturbation requires an aspartate at position 26; we note that mutating this residue to an alanine (black triangles) abrogated echistatin's effects. Similar results—i.e., a lack of conformational perturbation—were obtained with a glutamate mutant (open triangles).
Figure 3.

Concentration-dependent perturbation of αIIbβ3 solution conformation as measured by the fractional change in sedimentation coefficient, S/S0, versus the molar excess of echistatin variant, rEch:αIIbβ3. The dotted line denotes S/S0 = 1.0. Symbols as follows: rEch (1–49) M28L D26A (black triangles), rEch (1–49) M28L D26E (open triangles), rEch (1–49) M28L R24A (gray diamonds), rEch (1–49) M28L R24K (black diamonds), rEch (1–49) M28L (open circles), rEch (1–49) M28L D27W (black hexagons), rEch (1–49) M28L D27F (gray hexagons). The black and gray lines were obtained by fitting the data with full-length echistatin and the R24A mutant, respectively, to a hyperbolic inhibition model (Hantgan et al. 1999).
Conversely, these data indicate that the positive charge on R24 contributes to, but is not required for, ligand-induced conformation changes. Here, we found that rEch mutant R24A (gray diamonds) did affect αIIbβ3 conformation, although the midpoint of the concentration-dependence profile was shifted >10-fold compared to full-length rEch (1–49) M28L, and the maximum effect was ∼20% less. However, preserving the positive charge with rEch mutant R24K yielded a fully active variant (black diamonds). Sedimentation-velocity data also demonstrated the effects of introducing a nonpolar residue at position 27, immediately following echistatin's key aspartate 26. Here, we found that rEch mutants D27W (black hexagons) and D27F (gray hexagons) were potent perturbants with effects equivalent to full-length echistatin.
Prerequisites for αIIbβ3 integrin interactions with fibrinogen's γ-module
We next turned our attention to fibrinogen's γ-module (Medved et al. 1997), a 30-kDa recombinant protein with the unique KQAGDV sequence implicated in αIIbβ3 recognition (Hawiger 1995). While echistatin and the γ-module are structurally distinct, both display their integrin-recognition sites on flexible regions (Donahue et al. 1994; Yee et al. 1997; Monleon et al. 2005), suggesting that both ligands could interact with the resting receptor.
Hydrodynamic approaches
Sedimentation velocity measurements were used to test the hypothesis that the γ-module (Medved et al. 1997) would, like echistatin, form a complex with the initially resting micellar αIIbβ3 integrin. We first measured the distributions of sedimenting species with αIIbβ3 (Fig. 4A, open circles), γ148–411 (gray triangles), and an equimolar mixture (black squares). While a bimodal distribution was obtained with both receptor and ligand present, there was no evidence of complex formation because the g(S) profile appeared to be the sum of the individual species: αIIbβ3; 8.49 S, αIIbβ3 + γ148–411: 8.48, 2.48 S, γ148–411: 2.05 S. In contrast to the data with rEch mutants, no shift in αIIbβ3’s sedimentation velocity profile was observed, nor was there any significant decrease in the intensity of the γ-module peak, an effect anticipated for complex formation, as the 30-kDa fragment would then sediment at a higher S value. Similar results were obtained with micellar αIIbβ3 in the presence of 1 mM CaCl2 and 1 mM MgCl2 (data not shown), indicating that divalent cations alone are insufficient to promote complex formation.
Figure 4.

Effects of recombinant 30-kDa γ-module (γ 148–411) on the distribution of αIIbβ3 sedimenting species, g(S) versus S, as determined with time-derivative software (DCDT+; Philo1997). Each panel depicts the distributions obtained with ligand-free integrin (open circles; left axis), αIIbβ3 + γ 148–411 (black squares; right axis), and γ 148–411 alone (gray triangles; right axis). Continuous lines were obtained by fitting the resultant distribution functions to a one- or two-species model, as required. (A) Resting αIIbβ3 + γ 148–411; (B) primed αIIbβ3* + γ 148–411.
We next tested the hypothesis that transient exposure of αIIbβ3 with cHarGD, a ligand-mimetic peptide we have shown to promote formation of an open integrin (Hantgan et al. 2003), would yield a more reactive species through a process termed priming (Humphries et al. 2003). Here, a sample of αIIbβ3 was incubated with an equimolar concentration of cHarGD for 30 min; then excess ligand was removed by a rapid gel filtration step (Hantgan et al. 1999). Figure 4B presents sedimentation velocity data obtained with resting αIIbβ3 + γ148–411 (open circles), primed αIIbβ3* + γ148–411(black squares), and γ148–411 alone (gray triangles); both 1 mM CaCl2 and 1 mM MgCl2 were present in all these samples. Given the similarity of the profiles obtained with resting and primed receptors, we considered that either only weak interactions between αIIbβ3 and the γ-module were taking place or that the anticipated ∼9% increase in S for formation of an αIIbβ3: γ-module complex had been offset by the ∼7% decrease in S shown to accompany priming by cHarGD (Hantgan et al. 2003). Clearly a fresh approach, one not dependent on hydrodynamics, was called for to resolve this dilemma.
Spectroscopic approaches
Hydrodynamic computations using SOMO (Spotorno et al. 1997; Rai et al. 2005) indicated that fluorescence measurements are well suited to detect binding of fluorophore-labeled ligands to the αIIbβ3 integrin through the increase in rotational correlation time (τrot) and the corresponding increase in fluorescence anisotropy (A). For example, τrot for echistatin, labeled with Oregon Green (fluorescence lifetime ∼4 nsec), should increase from 3 nsec to >600 nsec upon binding to an elongated integrin, resulting in a nearly twofold increase in A; likewise, τrot for Oregon Green-γ 148–411 should increase from 16 nsec to >600 nsec, yielding an ∼25% increase in A. Indeed, independent of our work, another group (Wang et al. 2005) recently reported their development of a fluorescence polarization-based binding assay for monitoring RGD peptides binding to integrin αvβ3.
Monitoring echistatin binding by fluorescence anisotropy
The utility of this approach was demonstrated using Oregon Green-labeled rEch mutant D27W (OrGrn D27W), selected for its tight binding to αIIbβ3 in the solid-phase assay. As anticipated, the fractional change in anisotropy, expressed as A − A0/A0, increased in a hyperbolic manner, when increasing concentrations of (unlabeled) integrin were added to OrGrn D27W (0.5 μM) in HSC-OG buffer (Fig. 5A). Fitting these data to a single-site saturable binding model (Sevenich et al. 1998) yielded a predicted 1.8-fold increase in A; the curve's midpoint occurred at a ratio of 0.9 ± 0.3 mol integrin/mol ligand, indicative of a Kd ∼ 0.4 μM. We note that the affinity determined here for micellar αIIbβ3:D27W interactions is considerably weaker than the ∼2 nM we measured in a solid-phase binding assay; we anticipate that the amplification inherent in biotin-labeled assays is, in large measure, responsible for this difference (Tangemann and Engel 1995).
Figure 5.

Integrin:echistatin interactions measured in solution by changes in fluorescence anisotropy with Oregon Green-labeled echistatin. (A) Data obtained in a receptor:ligand titration are expressed as the fractional change in anisotropy, A − A0/A0 versus the mole ratio of αIIbβ3 to fluorophore-labeled rEch (1–49) M28L D27W (black hexagons). The black line was obtained by fitting the data to a single-site saturable binding model. (B) Size exclusion chromatography profile (G-75 Sephadex) of a mixture of αIIbβ3 and excess Oregon Green-rEch (1–49) M28L D27W with detection by fluorescence intensity (open triangles, continuous line) and fluorescence anisotropy (black hexagons, dashed line). Complex formation is detected in the early-eluting fraction by the increase in anisotropy compared to the later eluting free ligand.
Isolation and characterization of an αIIbβ3:echistatin complex
Based on these observations, we reasoned that it should be possible to isolate an integrin:echistatin complex by size-exclusion chromatography, starting with a sample containing a twofold molar excess of OrGrn D27W over αIIbβ3. The profile presented in Figure 5B demonstrates such a separation, monitored by changes in Oregon Green fluorescence emission (open triangles) and anisotropy (black hexagons). The integrated area of the early eluting, high-A peak corresponds to ∼19% of the total signal, indicating that ∼50% of the input rEch mutant formed a stable complex with αIIbβ3. This result is in reasonable agreement with the anisotropy titration data, which predicted 75% complex formation, yielding ∼38% of the input twofold excess fluorophore-labeled ligand in the early-eluting fraction.
Monitoring γ-module binding by fluorescence anisotropy
Extending these concepts to fibrinogen's γ-module, we added increasing concentrations of micellar αIIbβ3 to OrGrn-γ148–411 and monitored complex formation by changes in fluorescence anisotropy. We found that priming the integrin by a 30-min incubation with 1.3-fold molar excess of the ligand mimetic peptide cHarGD at 35°C, followed by a rapid gel filtration step to remove excess cHarGD (Hantgan et al. 1999), resulted in substantially enhanced binding. As illustrated in Figure 6A, minimal interactions resulted with resting αIIbβ3 isolated in the presence of either 1 mM Ca++/1 mM Mg++ (open circles) or 1 mM Ca++ (open squares) and incubated for 60 min at 23°C with OrGrn-γ148–411, where the titration curve showed a shallow, linear increase in anisotropy. In contrast, data obtained following a 60-min incubation of primed receptors with OrGrn-γ148–411 at 35°C (filled symbols) demonstrated a hyperbolic anisotropy increase of ∼25%, consistent with the predictions of hydrodynamic modeling. Comparable binding profiles resulted in the presence of 1 mM Ca++/1 mM Mg++ (black circles) or 1 mM Ca++ (gray squares). Fitting these data to a single-site saturable model yielded half-maximal binding at 1.9 ± 0.4 mol integrin/mol γ-module, consistent with a Kd ∼ 0.6 μM.
Figure 6.

Integrin:γ-module interactions measured in solution by changes in fluorescence anisotropy with Oregon Green-labeled ligands. (A) Data obtained in a receptor:ligand titration with primed αIIbβ3* are expressed as fractional change in anisotropy, A − A0/A0 versus the mole ratio of αIIbβ3 to fluorophore-labeled γ148–411 in buffer containing 1 mM Ca++/1 mM Mg++ (black circles) or 1 mM Ca++ (gray squares) and γ148–392 in the presence of 1 mM Ca++/1 mM Mg++ (dark gray triangles). The black line was obtained by fitting the data to a single-site saturable binding model. Data obtained with these same γ-module ligands and resting αIIbβ3 are denoted by open symbols and the dashed line. (B) Size exclusion chromatography profile (G-75 Sephadex) of a mixture of primed αIIbβ3* and excess Oregon Green-γ148–411 with detection by fluorescence intensity (open diamonds, continuous line) and fluorescence anisotropy (black circles, dashed line). Complex formation is detected in the early-eluting fraction by the increase in anisotropy compared to the later eluting free ligand.
Isolation and characterization of an αIIbβ3:γ-module complex
We were also able to isolate a stable αIIbβ3: γ148–411 complex by size-exclusion chromatography, as shown in Figure 6B (black circles, anisotropy data; open diamonds, fluorescence intensity). Starting with a threefold excess of ligand over primed αIIbβ3, we recovered an early eluting fraction with increased anisotropy (A = 138 ± 8) and a later peak corresponding to free ligand (A = 100 ± 2). In this case, the early eluting peak contained ∼13% of the total fluorescence signal. This value is somewhat lower than expected from the anisotropy titration data, which predicted 65% complex formation, yielding ∼21% of the input threefold excess fluorophore-labeled ligand in the early-eluting fraction. The decreased yield suggests that partial dissociation of the αIIbβ3: γ148–411 complex may have occurred during chromatography.
Monitoring αIIbβ3:γ148–392 fragment Interactions
Fluorescence anisotropy titrations were also performed with a truncation mutant of fibrinogen's γ-module, γ148–392 fragment, which lacks the carboxy-terminal HHLGGAKQAGDV sequence implicated in αIIbβ3 recognition. As illustrated in Figure 6A, this variant exhibited a pattern of priming-dependent binding (dark gray triangles, primed αIIbβ3*; open triangles, resting) that was quite similar to the full-length γ-module. Taken together, these observations demonstrate that while priming is a prerequisite for αIIbβ3’s interactions with fibrinogen's γ-module, complex formation is not directly affected by truncation of the γ-module's flexible carboxy terminus. Our biophysical approach toward measuring αIIbβ3’s interactions with the γ148–392 fragment complements and extends the observations of other investigators, who have identified novel integrin recognition sites on fibrinogen's γ-module (Lounes et al. 2002; Remijn et al. 2002; Podolnikova et al. 2003, 2005).
Discussion
Defining the electrostatic contributions to echistatin-induced αIIbβ3 priming
The combination of site-directed mutagenesis and biophysical characterizations enables us to explore the relative contributions of both charged residues on echistatin's RGD integrin-targeting sequence to this disintegrin's self-priming activity. We observed that rEch mutants R24K and R24A were each capable of shifting the resting αIIbβ3 complex to a slower sedimenting conformer. In contrast, we observed no significant integrin conformational changes with D26A or D26E (Fig. 3).
Additional insights into the electrostatic contributions to echistatin's self-priming activity were obtained by a thermodynamic analysis. Here, we simulated the changes in αIIbβ3’s solution conformation as a function of the echistatin:integrin mole ratio, using a single-site binding equation that describes the changes in the concentrations of free and bound receptor as a function of the molar excess of added ligand (echistatin). The black line shown in Figure 3 was obtained using Kd = 400 nM (determined by fluorescence anisotropy) and a limiting value of ΔS/S0 = 0.080 (obtained by nonlinear regression). We note that this simulation provides a reasonable approximation of the data obtained with rEch (1–49) M28L as well as the mutants D27F and D27W. Calculating the free energy change for priming (i.e., the energy change for echistatin binding to the open integrin conformer minus the energy required to induce its conformation), we obtain ΔGpriming = −8.5 kcal/mol.
Extending this simulation to the partially active variant R24A, we obtained the gray line shown in Figure 3, using Kd = 10 μM (empirical) and a limiting value of ΔS/S0 = 0.064 (nonlinear regression). We obtained ΔGpriming = −6.7 kcal/mol for rEch mutant R24A. Comparing these values results in an estimate of ΔΔGpriming = −1.8 kcal/mol for Arg-24, suggesting that this residue contributes ∼20% of the maximum free energy change. Our observations with the R24A rEch mutant suggest that a positive charge at position 24 makes a substantial, though not essential, contribution to echistatin's disintegrin activity. We also observed that the rEch mutant R24K retained full conformational perturbing activity. Here we note that the disintegrin barbourin, which is highly selective for αIIbβ3 over αvβ3, displays a KGD motif (Scarborough et al. 1991).
Since the D26A and D26E variants are essentially inactive, we estimate that the negatively charged aspartate residue at position 26, conserved among small and dimeric disintegrins (Calvete et al. 2005), contributes the remaining ∼80% of echistatin's self-priming energy. However, the consequences of extending the side chain by only one carbon atom are somewhat surprising. While an aspartate is commonly found in the integrin-recognition motif of small and dimeric disintegrins, an equivalent glutamate is found in six of the nine known members of the hemorrhagin class, proteins comprised of a metalloproteinase domain as well as disintegrin and cysteine-rich domains (Calvete et al. 2005).
Molecular basis of echistatin:αIIbβ3 integrin interactions
We next employed a molecular template approach to further understand the relative contributions of electrostatic and hydrophobic interactions in stabilizing echistatin:integrin complexes. We started with the 2.7 Å resolution structure of an αIIbβ3 ectodomain construct, in particular that with the ligand-mimetic peptide eptifibatide bound (Xiao et al. 2004). Eptifibatide, a cyclic peptide patterned after the KGDW integrin-targeting sequence found in the disintegrin barbourin (Scarborough et al. 1991), binds tightly and selectively to the αIIbβ3 integrin (Scarborough et al. 1993a,b; Phillips and Scarborough 1997), shifting its conformation to an open form (Hantgan et al. 2001). As illustrated in Figure 7A, the crystallographic data indicate that eptifibatide binds in a crevice in αIIbβ3’s ectodomain, stabilized, in part, by electrostatic interactions; i.e., its homoarginine residue nearly contacts αIIb's Asp 224 (3.2 Å distant), while its aspartate moiety interacts with the β3 subunit's MIDAS Mg++ cation (2.0 Å away).
Figure 7.

Modeling eptifibatide/echistatin:αIIbβ3 interactions. (A) This figure is based on the crystallographically determined structure of the eptifibatide:αIIbβ3 ectodomain complex, 1TY6.pdb (Xiao et al. 2004). The αIIb subunit is shown in light blue, the β3 subunit in red, and eptifibatide in yellow with its functional groups colored as follows: nitrogen, blue; oxygen, red; sulfur, orange. Selected αIIb residues that interact with eptifibatide are highlighted including Asp 224, located 3.2 Å distant from the ligand's positively charged guanidinium moiety, and Phe 160, Tyr 190, and Phe 231, which pack around the homoarginine's aliphatic segment. Likewise, eptifibatide aspartate, which interacts with the β3 subunit's MIDAS Mg++ cation (2.0 Å away), is emphasized. (B) This figure compares the structures of eptifibatide (as in panel A) and echistatin, based on the NMR determined structure, 1R03.pdb (Monleon et al. 2005). The distances between key electrostatic residues on each ligand, eptifbatide's homoarginine (Cζ) and its aspartate (Cδ), echistatin's Arg 24 (Cζ) and Asp 26 (Cδ), are indicated by dashed lines. Echistatin's polypeptide backbone is shown as a green tube with selected residues highlighted.
Given the overall similarity of the spatial distribution of charges in both eptifibatide and echistatin, as illustrated in Figure 7B, we employed the eptifibatide:αIIbβ3 ectodomain structure as a template for understanding our site-directed mutagenesis results. First we note that echistatin's compact structure can readily fit into a groove at the subunit interface, positioning its D26 residue to interact with the divalent cation at the MIDAS site, mimicking eptifibatide's electrostatic interactions. Indeed we found that replacing this key residue with a neutral alanine abrogates echistatin's ability to perturb αIIbβ3 conformation.
This template can also help to explain the complementary role played by the arginine residue on echistatin's RGD integrin-targeting sequence. As illustrated in Figure 7B, R24’s charged side chain is 14 Å distant from D26, some 1.5 Å shorter than the equivalent distance in eptifibatide; thus R24 will likely reside further from its potential salt-bridging partner, αIIb's Asp 224. Since electrostatic interactions are inversely proportional to distance, decreasing approximately sixfold at 10 Å (in 0.1 M salt) (Lee et al. 2002), this may explain the differential contributions of R24 (distant) and D26 (proximate) to the structure and stability of integrin:echistatin complexes. However, the charge-preserving Arg-to-Lys echistatin mutation, which would move the positive group ∼1 Å further from αIIb's D224, exhibited the same conformation-perturbing activity as rEch (1–49) M28L. Perhaps the increased charge density on lysine's amino group, compared to that distributed over arginine's guanidinium moiety (Creighton 1993), compensates for the increased distance.
Nonpolar interactions also contribute to eptifibatide binding, as three aromatic αIIb residues (F160, Y190, F231) pack around the homoarginine's aliphatic segment, and β3 F160 is nearly in van der Waals contact with the heterocyclic ring of the ligand's tryptophan moiety (Fig. 7A; Xiao et al. 2004). Hence, we anticipated that adding a nonpolar residue to echistatin's integrin-targeting triad would introduce new contacts, perhaps mimicking the KGDW sequence in barbourin (Minoux et al. 2000). However, this was not the case. Figure 7B suggests that the tryptophan on our engineered D27W mutant was not properly oriented for such interactions. This may explain why no additional conformational perturbations, beyond those seen with the wild-type disintegrin, were observed.
Molecular prerequisites for integrin:fibrinogen interactions
By focusing on fibrinogen's γ-module, a 30-kDa recombinant protein with the KQAGDV sequence implicated in αIIbβ3 recognition (Hawiger 1995), this study also provides fresh insights into the relationship between priming and ligand binding. Since structural studies suggest that both echistatin (Monleon et al. 2005) and the γ-module display integrin-targeting sites on flexible regions (Donahue et al. 1994; Yee et al. 1997), we reasoned that the γ-module could share with rEch the ability to bind to the resting αIIbβ3 complex and perturb its conformation. This concept was reinforced by our observation that mutating echistatin's RGD site to an AGD like that found in the γ-module (rEch mutant R24A, Fig. 3) did not abrogate its self-priming activity.
In contrast, our experimental observations demonstrate that incubating αIIbβ3 with a ligand-mimetic peptide at 35°C was required for formation of a high-affinity complex between the micellar integrin and γ148–411 (Fig. 6A). Thus, we distinguish echistatin, a self-priming ligand, from fibrinogen's γ-module, a regulated ligand whose interactions with αIIbβ3 are critically dependent on conformational rearrangements in the integrin's ectodomain that also lead to enhanced oligomerization (Hantgan et al. 2003). Studies are currently in progress to determine the on and off rates for binding echistatin, cHarGD, and the γ-module to αIIbβ3; obtaining those kinetic parameters will enable us to complete a numerical simulation of the multistep processes of integrin priming and macromolecular ligand binding.
We also observed similar priming-dependent binding profiles for γ148–411 and truncation mutant γ148–392, which lacks the carboxy-terminal HHLGGAKQAGDV sequence implicated in αIIbβ3 recognition (Hawiger et al. 1982; Hawiger 1995). While a role of this site in platelet aggregation and adhesion to immobilized fibrinogen has been clearly demonstrated (Farrell et al. 1992; Farrell and Thiagarajan 1994), other studies have shown that platelet-mediated clot retraction still occurs when fibrinogen's AGDV site has been deleted (Rooney et al. 1996, 1998). Binding of purified, radiolabeled αIIbβ3 to immobilized fibrinogen was also not blocked by synthetic RGD or HHLGGAKQAGDV peptides, suggesting the involvement of novel binding sites (Parise et al. 1993). Furthermore, variant fibrinogens with aspartate-to-alanine substitutions at positions γ318 and 320 exhibited defective platelet aggregation and adhesion to fibrinogen, indicating that regions spatially distinct from fibrinogen's carboxy-terminal γ-module are required for αIIbβ3 interactions (Lounes et al. 2002; Remijn et al. 2002). An additional integrin-binding site on fibrinogen's γ-module was recently localized to a cluster of four basic residues present within the sequence 370–381 (Podolnikova et al. 2003, 2005).
However, all those experimental approaches monitored complex biological phenomena in heterogeneous systems, where events beyond direct formation of receptor:ligand complexes are probably involved. Hence, our biophysical studies in a purified system of resting-yet-responsive integrins and recombinant γ-modules, demonstrating that γ148–411 and truncation mutant γ148–392 exhibited comparable priming-dependent micromolar affinity for αIIbβ3, add a new dimension to the description of integrin:fibrinogen interactions.
Our studies differentiate priming ligands, such as echistatin, which bind to the resting receptor and perturb its conformation, from regulated ligands, such as fibrinogen's γ-module, where binding-site remodeling must first occur. Echistatin's binding energy is sufficient to perturb the subunit interface, but regulated ligands like fibrinogen must rely on priming steps to overcome conformational barriers. We further propose that pharmacologic GpIIb/IIIa antagonists are also priming ligands, fundamentally limited in safety and efficacy (Leclerc 2002; Newby et al. 2002; Boersma and Westerhout 2004), because they mimic the effects of naturally occurring disintegrins.
Materials and methods
αIIbβ3 purification
Milligram quantities of highly purified αIIbβ3 were isolated from outdated human blood platelets (Blood Bank, North Carolina Baptist Hospital, Winston-Salem, NC) as previously described (Hantgan et al. 1993, 1999). Monodisperse samples of resting αIIbβ3 were obtained by size-exclusion chromatography at 4°C on a 0.9 × 85 cm column of Sephacryl S-300 equilibrated in a pH 7.4 buffer containing 0.13 mol/L NaCl, 0.01 mol/L HEPES, 3 × 10−8 mol/L basic trypsin inhibitor, 10−6 mol/L leupeptin, 0.02% sodium azide, 0.03 mol/L n-octyl-β-D-glucopyranoside, and 0.002 mol/L CaCl2 (HSC-OG), or 0.001 mol/L CaCl2/ 0.001 mol/L MgCl2 (HSCM-OG). Peak fractions were then concentrated in an Amicon pressure concentrator with a PLHK cellulose membrane, 100,000 Da retention limit.
Cloning, site-directed mutagenesis, and expression of echistatin variant:
This study used a plasmid constructed from the vector pGEX-KG, which contains the Glutathione-S-transferase (GST) gene and a thrombin-sensitive linker (Smith and Johnson 1988). An echistatin gene expressing the full-length protein with an M28L mutation, rEch (1–49) M28L, was inserted at a BamHI restriction site that added four N-terminal amino acids (Gly, Ser, Thr, Met) (Wierzbicka-Patynowski et al. 1999; Hantgan et al. 2004). Plasmids encoding rEch (1–49) M28L variants with point mutations in their amino acid sequence were prepared using the “quikchange” site-directed mutagenesis method (Stratagene) with the following primers:
R24A: CTGTAAGAGAGCTGCAGGTGACGACTTAG (forward)
CTAAGTCGTCACCTGCAGCTCTCTTACAG (reverse complement)
R24K: CTGTAAGAGAGCTAAGGGTGACGACTTAG
TAAGTCGTCACCCTTAGCTCTCTTACAG
D26A: CTGTAAGAGAGCTAGAGGTGCGGACTTAG
CTAAGTCCGCACCTCTAGCTCTCTTACAG
D26E: CTGTAAGAGAGCTAGAGGTGAGGACTTAG
CTAAGTCCTCACCTCTAGCTCTCTTACAG
D27W: CTGTAAGAGAGCTAGAGGTGACTGGTTAGACG
CGTCTAACCAGTCACCTCTAGCTCTCTTACAG
D27F: CTGTAAGAGAGCTAGAGGTGACTTCTTAGACG
CGTCTAAGAAGTCACCTCTAGCTCTCTTACAG
Plasmid DNA was amplified by transforming Escherichia coli strain HB101–1424471, and sequences of the resultant clones were verified (DNA Sequencing Core Laboratory of the Comprehensive Cancer Center of Wake Forest University School of Medicine). Each of these six rEch mutants was expressed in E. coli BL21, then isolated from cell lysates by adsorption to Glutathione-agarose followed by thrombin-cleavage of the bead-bound constructs (Smith and Johnson 1988).
rEch mutant purification and characterizations
Following procedures optimized with rEch (1–49) M28L to achieve a fully active disintegrin (Hantgan et al. 2004), each mutant was purified by size-exclusion chromatography on Sephadex G-75 in 0.05 M ammonium acetate buffer (pH 7). Elution was monitored by absorbance at 276 nm; peak fractions were combined, lyophilized, and stored at −20°C. Recoveries were typically 0.5–1 mg of rEch mutant from 500 mL of cell lysate. Recombinant protein purity was assessed by SDS-PAGE and by reversed-phase HPLC on a C18 column eluted with a gradient of 0.1% TFA in water/0.1% TFA in acetonitrile (Knight and Romano 2005). Full-length rEch (1–49) M28L and the rEch mutants each eluted as a single peak. The order of elution correlated with the hydrophobicity of the single-site mutations: R24K < D26E ~ rEch (1–49) M28L < D26A < D27F < D27W (Supplemental Material). rEch concentrations were initially determined by quantitative amino acid composition analysis (Hantgan et al. 1999).
Spectral characterizations
These samples were also subjected to UV-visible spectral measurements in a Beckman diode array spectrophotometer, enabling determination of the molar extinction coefficients for each mutant, as previously described (Hantgan et al. 2004). Then concentrations of the reconstituted rEch mutants were determined from spectral data, using the molar extinction coefficient data reported below; while D27W exhibited λmax at 280 nm, the other rEch mutants exhibited λmax at 276 nm, as expected for proteins containing a single tyrosine.
Mass determinations
Mass spectrometry (MALDI-TOF performed by the Biomolecular Resource Facility of Wake Forest University, as previously described; Hantgan et al. 2004) yielded the average mass data shown in Table 1; we note the close agreement between the experimentally determined and calculated masses.
Table 1.
Average mass data from mass spectrometry analysis

Thiol titers
Thiol titers were also determined for each rEch mutant using 5,5′-Dithiobis(2-nitrobenzoic acid) (DTNB) as a colorimetric sulfhydryl reagent. Free cysteine concentrations were determined with rEch mutant samples denatured in 4 M guanidinium chloride (to maximize the accessibility of any buried thiols) by the increased absorbance at 412 nm in the presence of excess DTNB as described by Riddles et al. (1979). Combining these data with rEch mutant concentrations determined from their UV spectra, we obtained the thiol titer data cited in the table above, expressed as moles free cysteine/mole protein. We note that the wild-type echistatin rEch (1–49) M28L and each of the mutants exhibited substoichiometric thiol titers with >94% of their eight cysteine residues oxidized to cystines, as expected for folded proteins.
Reduced/alkylated echistatin
Denatured rEch (1–49) M28L was obtained by complete disulfide bond reduction (incubated for 18 h at 37°C in 4 M guanidinium chloride in the presence of excess dithiothreitol) followed by alkylation (excess iodoacetamide). This reduced/alkylated conformer exhibited a red-shifted UV spectrum (λmax 280 nm), consistent with that expected for a denatured protein containing a single tyrosine residue (Hantgan et al. 1974).
NMR measurements
Proton NMR spectral data were also obtained for each construct as an additional, sensitive probe for proper folding. Hence, we examined rEch (1–49) M28L in both its folded, biologically active conformation (Hantgan et al. 2004), as well as the maximally unfolded form. Proton NMR spectra were acquired at 25°C on a Bruker Avance 600 MHz NMR spectrometer equipped with a Cryoprobe. Spectra for each echistatin mutant were highly similar to that for native wild type, indicating that each mutant was properly folded. In contrast, reduced and alkylated wild type exhibited a markedly different spectrum (Supplemental Material).
Expression and purification of recombinant fibrinogen γ-domain fragments
The recombinant fibrinogen γ-chain fragment including residues γ148–411 (γ-module) and its truncated variant lacking γ-chain residues 393–411 (γ148–392 fragment) were produced in E. coli using the pET-20b expression vector; following cell lysis and solubilization in guanidine hydrochloride, each γ-chain fragment was purified by size exclusion chromatography in urea, then refolded as previously described (Medved et al. 1997; Yakovlev et al. 2000). Mass spectrometry (MALDI-TOF) yielded an average mass of 29,750 ± 25 Da for the γ-module, in close agreement with the value of 29.7 kDa derived from amino acid composition data (Medved et al. 1997) and the calculated average mass of 29,743 Da. Concentrations of the γ-module and the γ148–392 fragment were determined spectrophotometrically using extinction coefficients (E280, 1%) equal to 24.8 and 26.5, respectively (Medved et al. 1997; Yakovlev et al. 2000).
Synthetic peptide integrin antagonist
Cyclo(S,S)-L-Lysyl-L-Tyrosyl-Glycyl-L-Cystinyl-L-Homoarginyl-Glycyl-L-Aspartyl-L-Trytophanyl-L-Prolyl-L-Cystine (cHArGD) (Hantgan et al. 2001) was synthesized and purified by the Protein Analysis Core Laboratory of the Comprehensive Cancer Center of Wake Forest University (Winston-Salem, NC), using previously described protocols (Hantgan et al. 1992). cHArGD was shown to have the correct amino acid sequence and the correct molecular mass, using an Applied Biosystems 475 automated peptide synthesizer and a Quattro II triple quadropole mass spectrometer (Micromass, Inc.), respectively (Hantgan et al. 2003).
Biotin labeling
Biotin was covalently coupled to lysine residues on selected rEch mutants using EZ-Link Sulfo-NHS-biotin (Pierce) in a 2-h reaction carried out in phosphate-buffered saline (pH 7) at 0°C. The labeled protein was separated from excess biotin reagent by extensive dialysis against HEPES-buffered saline (pH 7.4). Aliquots were subjected to quantitative amino acid analysis to determine protein concentrations. The degree of labeling, typically 1–3 mol biotin/mol protein, was determined by difference spectroscopy, using an avidin displacement assay (Pierce).
Solid-phase binding assay
Purified αIIbβ3, isolated in a resting conformation in octyl glucoside micelles, was coated in microtiter plate wells by overnight incubation at 4°C at low concentrations (5–10 μg/mL) to achieve a monolayer (Tangemann and Engel 1995). Priming was achieved by coating with αIIbβ3 in the presence of the synthetic peptide integrin antagonist cHArGD at 10 μM (Hantgan et al. 2003). Selected wells were incubated with HSC-OG only for subsequent determination of the extent of nonspecific binding. After extensive washing, each well was blocked with 30 mg/mL BSA in HSC-OG buffer; all subsequent binding steps were carried out with 1 mg/mL BSA. Selected concentrations of biotin-labeled rEch mutant were added in quadruplicate to the integrin-coated and blank wells, then incubated for 1 h at 37°C followed by extensive washing with HSC-OG buffer. Next, a streptavidin-HRP conjugate was added (1:15,000 dilution) and incubated for 1 h at 37°C.
Following extensive washing, Sure Blue colorimetric reagent was added, and color developed for 30 min at room temperature with moderate agitation; next, stop solution was added to quench the reaction and the absorbance measured at 450 nm in a Vmax microtiter plate reader. The specific signal for each input ligand concentration was determined by subtracting the average absorbance of each set of blank wells from the corresponding average absorbance of each set of integrin-coated wells. Data obtained from duplicate or triplicate experiments were pooled and analyzed by nonlinear regression (Sigma Plot, Jandel Scientific) to determine the dissociation constants (Kd, mol/L) for binding biotin-labeled rEch mutants to immobilized αIIbβ3 integrins.
Fluorophore labeling
Oregon Green succinimidyl ethyl ester (Molecular Probes) was covalently coupled to lysine residues on selected rEch mutants in a 45-min reaction at pH 8.3 and room temperature. The labeled protein was separated from excess dye by overnight dialysis versus 0.05 M ammonium acetate buffer (pH 7.0), followed by size-exclusion chromatography on G-75 Sephadex in this volatile buffer. Samples were lyophilized and then reconstituted in HSC-OG buffer. A similar labeling protocol was followed with γ 148–411 and γ148–392; excess dye was removed by extensive dialysis versus HEPES-buffered saline (pH 7.4). Samples of Oregon Green-labeled γ148–411or γ148–392 were then equilibrated with HSCM-OG buffer by sedimenting through G-25 Sephadex at 1000 g for 2 min (Hantgan et al. 1999). The reconstituted protein concentrations and the degree of labeling, typically 1–3 mol Oregon Green/mol protein, were determined by UV-vis spectroscopy.
Fluorescence spectroscopy
Fluorescence intensity and anisotropy measurements were carried out on a Safire II (Tecan Instruments) equipped with dual monochromators and the ability to rapidly collect data from quadruplicate 20-μL samples in the wells of a microtiter plate. Samples of Oregon Green-labeled rEch mutants or γ-module construct, alone or in complex with αIIbβ3, were excited at 475 nm and emission measured at 525 nm, typically using 5-nm slits. The instrument computes anisotropy data from samples illuminated with vertically polarized light from the vertically and horizontally polarized components of the emitted light:
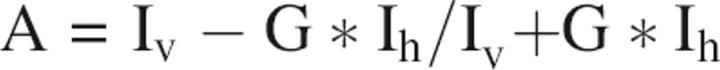 |
The G-factor, which corrects for differential detector responses to the vertically and horizontally polarized emitted light, was determined from samples with minimal anisotropy, e.g., free Oregon Green fluorophore or a rEch mutant conjugate.
Fluorescence detection of integrin:ligand interactions
Fluorescence emission and anisotropy measurements were used to monitor the elution of mixtures of fluorophore-labeled ligand and integrin from an analytical-scale size-exclusion column (0.9 × 15 mm, Sephacryl S-300) with the goal of detecting complex formation. Anisotropy measurements also formed the basis of a fluid-phase binding assay in which increasing concentrations of αIIbβ3 were added to Oregon Green-labeled rEch or γ-module construct. In both cases, complex formation was detected by the increased anisotropy that resulted when the fluorophore-labeled ligand formed a high-molecular-weight complex with the (unlabeled) integrin.
Analytical ultracentrifugation
Sedimentation velocity and equilibrium measurements were performed in a Beckman Optima XL-A analytical ultracentrifuge (Beckman Instruments) equipped with absorbance optics and an An60 Ti rotor (Hantgan et al. 1999, 2001). Sedimentation-velocity data were collected with the αIIbβ3 complex alone and in the presence of rEch mutants or γ 148–411 at 20°C at a rotor speed of 35,000 rpm and analyzed using both SVEDBERG (version 1.04) and DCDT+ (version 6.31) software (J. Philo, Thousand Oaks, CA) to obtain the weight-average sedimentation coefficient (Sw) and distribution of sedimenting species, g (s*), respectively (Stafford 1992). All sedimentation coefficients have been corrected for solvent density and viscosity to obtain S20, w values.
Modeling integrin:ligand complexes
PyMol molecular graphics software was used to visualize and measure interatomic distances within the 2.7-Å resolution structure of the αIIbβ3 headpiece, crystallized in the presence of the ligand-mimetic peptide eptifibatide, 1TY6.pdb (Xiao et al. 2004). Similarly, PyMol was used to visualize and measure the distance between key integrin-recognition residues on echistatin, using the NMR determined structure, 1R03.pdb (Monleon et al. 2005).
Electronic supplemental material
Supplemental Figure 1 shows the analytical characterization of echistatin mutants by reversed-phase HPLC, and Figure 2 shows the 1H NMR spectra of wild-type and mutant echistatins.
Acknowledgments
The contributions of the Wake Forest University Biomolecular Resource Facility, Dr. Mark O. Lively, Director, and J. Mark Morris, Research Technician, to this work, including mass spectrometry, amino acid analyses, and peptide synthesis, are gratefully acknowledged. Thanks are due to Gregory J. Pomper, M.D., and Rita Joseph, R.N., for their assistance in obtaining outdated platelets from the Blood Bank of the North Carolina Baptist Hospital. Special thanks are expressed to Dr. Douglas S. Lyles for stimulating scientific discussions and Dr. Julie Edelson for her skilled editing. This research was supported by Grants-in-Aid 0355869U and 0555527U from the American Heart Association, Mid-Atlantic Affiliate (to R.R.H.) and by National Institutes of Health Grant HL-56051 (L.M.).
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Roy R. Hantgan, Ph.D., Department of Biochemistry, Wake Forest University School of Medicine, Medical Center Boulevard, Winston-Salem, NC 27157-1019, USA; e-mail: rhantgan@wfubmc.edu; fax: (336) 716-7671.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.052049506.
References
- Adair B.D. and Yeager M. 2002. Three-dimensional model of the human platelet integrin αIIbβ3 based on electron cryomicroscopy and x-ray crystallography. Proc. Natl. Acad. Sci. 99: 14059–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adair B.D., Xiong J.P., Maddock C., Goodman S.L., Arnaout M.A., Yeager M. 2005. Three-dimensional EM structure of the ectodomain of integrin αVβ3 in a complex with fibronectin. J. Cell Biol. 168: 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaout M.A., Goodman S.L., Xiong J.-P. 2002. Coming to grips with integrin binding to ligands. Curr. Opin. Cell Biol. 14: 641–651. [DOI] [PubMed] [Google Scholar]
- Beglova N., Blacklow S.C., Takagi J., Springer T.A. 2002. Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat. Struct. Biol. 9: 282–287. [DOI] [PubMed] [Google Scholar]
- Boersma E. and Westerhout C.M. 2004. Intravenous glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: Lessons from recently conducted randomized clinical trials. Curr. Opin. Investig. Drugs 5: 313–319. [PubMed] [Google Scholar]
- Calderwood D.A., Shattil S.J., Ginsberg M.H. 2000. Integrins and actin filaments: Reciprocal regulation of cell adhesion and signaling. J. Biol. Chem. 275: 22607–22610. [DOI] [PubMed] [Google Scholar]
- Calvete J.J., Marcinkiewicz C., Monleon D., Esteve V., Celda B., Juarez P., Sanz L. 2005. Snake venom disintegrins: Evolution of structure and function. Toxicon 45: 1063–1074. [DOI] [PubMed] [Google Scholar]
- Carrell N.A., Fitzgerald L.A., Steiner B., Erickson H.P., Phillips D.R. 1985. Structure of human platelet glycoproteins IIb and IIIa as determined by electron microscopy. J. Biol. Chem. 260: 1743–1749. [PubMed] [Google Scholar]
- Cook J.J., Bednar B., Lynch J., Gould R.J., Egbertson M.S., Halczenko W., Duggan M.E., Hartman G.D., Lo M.-W., Murphy G.M. et al. 1999. Tirofiban (Aggrastat). Cardiovasc. Drug Rev. 17: 199–224. [Google Scholar]
- Creighton T.E. 1993. Chemical properties of polypeptides. In Proteins. Structures and molecular properties (ed. Creighton T.E.) . pp. 1–48. 2nd ed W.H. Freeman, New York.
- Critchley D.R., Holt M.R., Barry S.T., Priddle H., Hemmings L., Norman J. 1999. Integrin-mediated cell adhesion: The cytoskeletal connection. Biochem. Soc. Symp. 65: 79–99. [PubMed] [Google Scholar]
- Donahue J.P., Patel H., Anderson W.F., Hawiger J. 1994. Three-dimensional structure of the platelet integrin recognition segment of the fibrinogen γ chain obtained by carrier protein-driven crystallization. Proc. Natl. Acad. Sci. 91: 12178–12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell D.H. and Thiagarajan P. 1994. Binding of recombinant fibrinogen mutants to platelets. J. Biol. Chem. 269: 226–231. [PubMed] [Google Scholar]
- Farrell D.H., Thiagarajan P., Chung D.W., Davie E.W. 1992. Role of fibrinogen α and γ chain sites in platelet aggregation. Proc. Natl. Acad. Sci. 89: 10729–10732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faull R.J. and Ginsberg M.H. 1996. Inside-out signaling through integrins. J. Am. Soc. Nephrol. 7: 1091–1097. [DOI] [PubMed] [Google Scholar]
- Giancotti F.G. and Ruoslahti E. 1999. Integrin signaling. Science 285: 1028–1032. [DOI] [PubMed] [Google Scholar]
- Gottschalk K.E., Adams P.D., Brunger A.T., Kessler H. 2002. Transmembrane signal transduction of the αIIbβ3 integrin. Protein Sci. 11: 1800–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantgan R.R., Hammes G.G., Scheraga H.A. 1974. Pathways of folding of reduced bovine pancreatic ribonuclease. Biochemistry 13: 3421–3431. [DOI] [PubMed] [Google Scholar]
- Hantgan R.R., Endenburg S.C., Cavero I., Marguerie G., Uzan A., Sixma J.J., De Groot P.G. 1992. Inhibition of platelet adhesion to fibrin(ogen) in flowing whole blood by Arg-Gly-Asp and fibrinogen γ-chain carboxy terminal peptides. Thromb. Haemost. 68: 694–700. [PubMed] [Google Scholar]
- Hantgan R.R., Braaten J.V., Rocco M. 1993. Dynamic light scattering studies of αIIbβ3solution conformation. Biochemistry 32: 3935–3941. [DOI] [PubMed] [Google Scholar]
- Hantgan R.R., Paumi C., Rocco M., Weisel J.W. 1999. Effects of ligand-mimetic peptides Arg-Gly-Asp-X (X = Phe, Trp, Ser) on αIIbβ3 integrin conformation and oligomerization. Biochemistry 38: 14461–14464. [DOI] [PubMed] [Google Scholar]
- Hantgan R.R., Rocco M., Nagaswami C., Weisel J.W. 2001. Binding of a fibrinogen mimetic stabilizes integrin αIIbβ3’s open conformation. Protein Sci. 10: 1614–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantgan R.R., Stahle M.C., Jerome W.G., Nagaswami C., Weisel J.W. 2002. Tirofiban blocks platelet adhesion to fibrin with minimal perturbatin of GpIIb/IIIa structure. Thromb. Haemost. 87: 910–917. [PubMed] [Google Scholar]
- Hantgan R.R., Lyles D.S., Mallett T.C., Rocco M., Nagaswami C., Weisel J.W. 2003. Ligand binding promotes the entropy-driven oligomerization of integrin αIIbβ3. J. Biol. Chem. 278: 3417–3426. [DOI] [PubMed] [Google Scholar]
- Hantgan R.R., Stahle M.C., Connor J.H., Lyles D.S., Horita D.A., Rocco M., Nagaswami C., Weisel J.W., McLane M.A. 2004. The disintegrin echistatin stabilizes integrin αIIbβ3’s open conformation and promotes its oligomerization. J. Mol. Biol. 342: 1625–1636. [DOI] [PubMed] [Google Scholar]
- Hartwig J.H., Barkalow K., Azim A., Italiano J. 1999. The elegant platelet: Signals controlling actin assembly. Thromb. Haemost. 82: 392–398. [PubMed] [Google Scholar]
- Hawiger J. 1995. Adhesive ends of fibrinogen and its antiadhesive peptides: The end of a saga. Semin. Hematol. 32: 99–109. [PubMed] [Google Scholar]
- Hawiger J., Timmons S., Kloczewiak M., Strong D.D., Doolittle R.F. 1982. γ and α chains of human fibrinogen possess sites reactive with human platelet receptors. Proc. Natl. Acad. Sci. 79: 2068–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes P.E. and Pfaff M. 1998. Integrin affinity modulation. Trends Cell Biol. 8: 359–364. [DOI] [PubMed] [Google Scholar]
- Humphries M.J. 2004. Monoclonal antibodies as probes of integrin priming and activation. Biochem. Soc. Trans. 32: 407–411. [DOI] [PubMed] [Google Scholar]
- Humphries M.J., McEwan P.A., Barton S.J., Buckley P.A., Bella J., Paul M.A. 2003. Integrin structure: Heady advances in ligand binding, but activation still makes the knees wobble. Trends Biochem. Sci. 28: 313–320. [DOI] [PubMed] [Google Scholar]
- Hussain M.A. and Siedlecki C.A. 2004. The platelet integrin α(IIb) β(3) imaged by atomic force microscopy on model surfaces. Micron 35: 565–573. [DOI] [PubMed] [Google Scholar]
- Hynes R.O. 2002. Integrins: Bidirectional, allosteric signaling machines. Cell 110: 673–687. [DOI] [PubMed] [Google Scholar]
- Iwasaki K., Mitsuoka K., Fujiyoshi Y., Fujisawa Y., Kikuchi M., Sekiguchi K., Yamada T. 2005. Electron tomography reveals diverse conformations of integrin αIIbβ3 in the active state. J. Struct. Biol. 150: 259–267. [DOI] [PubMed] [Google Scholar]
- Kim M., Carman C.V., Springer T.A. 2003. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 301: 1720–1725. [DOI] [PubMed] [Google Scholar]
- Knight L.C. and Romano J.E. 2005. Functional expression of bitistatin, a disintegrin with potential use in molecular imaging of thromboembolic disease. Protein Expr. Purif. 39: 307–319. [DOI] [PubMed] [Google Scholar]
- Leclerc J.R. 2002. Platelet glycoprotein IIb/IIIa antagonists: Lessons learned from clinical trials and future directions. Crit. Care Med. 30: S332–S340. [DOI] [PubMed] [Google Scholar]
- Lee K.K., Fitch C.A., Garcia-Moreno E.B. 2002. Distance dependence and salt sensitivity of pairwise, coulombic interactions in a protein. Protein Sci. 11: 1004–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddington R.C. 2002. Will the real integrin please stand up? Structure 10: 605–607. [DOI] [PubMed] [Google Scholar]
- Liddington R.C. and Bankston L.A. 2000. The structural basis of dynamic cell adhesion: Heads, tails, and allostery. Exp. Cell Res. 261: 37–43. [DOI] [PubMed] [Google Scholar]
- Litvinov R.I., Nagaswami C., Vilaire G., Shuman H., Bennett J.S., Weisel J.W. 2004. Functional and structural correlations of individual αIIbβ3 molecules. Blood 104: 3979–3985. [DOI] [PubMed] [Google Scholar]
- Liu Q.D., Rooney M.M., Kasirerfriede A., Brown E., Lord S.T., Frojmovic M.M. 1998. Role of the γ chain Ala-Gly-Asp-Val and Aα chain Arg-Gly-Asp-Ser sites of fibrinogen in coaggregation of platelets and fibrinogen-coated beads. Biochim. Biophys. Acta 1385: 33–42. [DOI] [PubMed] [Google Scholar]
- Lounes K.C., Ping L., Gorkun O.V., Lord S.T. 2002. Analysis of engineered fibrinogen variants suggests that an additional site mediates platelet aggregation and that “B-b” interactions have a role in protofibril formation. Biochemistry 41: 5291–5299. [DOI] [PubMed] [Google Scholar]
- Luo B.H., Springer T.A., Takagi J. 2004. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biol. 2:–E153. [DOI] [PMC free article] [PubMed]
- Medved L., Litvinovich S., Ugarova T., Matsuka Y., Ingham K. 1997. Domain structure and functional activity of the recombinant human fibrinogen γ-module (γ148–411). Biochemistry 36: 4685–4693. [DOI] [PubMed] [Google Scholar]
- Minoux H., Chipot C., Brown D., Maigret B. 2000. Structural analysis of the KGD sequence loop of barbourin, an αIIbβ3-specific disintegrin. J. Comput. Aided Mol. Des. 14: 317–327. [DOI] [PubMed] [Google Scholar]
- Monleon D., Esteve V., Kovacs H., Calvete J.J., Celda B. 2005. Conformation and concerted dynamics of the integrin-binding site and the C-terminal region of echistatin revealed by homonuclear NMR. Biochem. J. 387: 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mould A.P. and Humphries M.J. 2004. Cell biology: Adhesion articulated. Nature 432: 27–28. [DOI] [PubMed] [Google Scholar]
- Mould A.P., Symonds E.J., Buckley P.A., Grossmann J.G., McEwan P.A., Barton S.J., Askari J.A., Craig S.E., Bella J., Humphries M.J. 2003. Structure of an integrin–ligand complex deduced from solution x-ray scattering and site-directed mutagenesis. J. Biol. Chem. 278: 39993–39999. [DOI] [PubMed] [Google Scholar]
- Nermut M.V., Green N.M., Eason P., Yamada S.S., Yamada K.M. 1988. Electron microscopy and structural model of human fibronectin receptor. EMBO J. 7: 4093–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newby L.K., Califf R.M., White H.D., Harrington R.A., van De W.F., Granger C.B., Simes R.J., Hasselblad V., Armstrong P.W. 2002. The failure of orally administered glycoprotein IIb/IIIa inhibitors to prevent recurrent cardiac events. Am. J. Med. 112: 647–658. [DOI] [PubMed] [Google Scholar]
- Parise L.V., Steiner B., Nannizzi L., Criss A.B., Phillips D.R. 1993. Evidence for novel binding sites on the platelet glycoprotein IIb and IIIa subunits and immobilized fibrinogen. Biochem. J. 289: 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips D.R. and Scarborough R.M. 1997. Clinical pharmacology of eptifibatide. Am. J. Cardiol. 80: 11B–20B. [DOI] [PubMed] [Google Scholar]
- Philo J.S. 1997. An improved function for fitting sedimentation velocity data for low-molecular-weight solutes. Biophys. J. 72: 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plow E.F., Haas T.A., Zhang L., Loftus J., Smith J.W. 2000. Ligand binding to integrins. J. Biol. Chem. 275: 21785–21788. [DOI] [PubMed] [Google Scholar]
- Podolnikova N.P., Yakubenko V.P., Volkov G.L., Plow E.F., Ugarova T.P. 2003. Identification of a novel binding site for platelet integrins αIIbβ3 (GPIIbIIIa) and α5 β1 in the γ C-domain of fibrinogen. J. Biol. Chem. 278: 32251–32258. [DOI] [PubMed] [Google Scholar]
- Podolnikova N.P., Gorkun O.V., Loreth R.M., Yee V.C., Lord S.T., Ugarova T.P. 2005. A cluster of basic amino acid residues in the γ 370-381 sequence of fibrinogen comprise a binding site for platelet integrin αIIbβ3 (glycoprotein IIb/IIIa). Biochemistry 44: 16920–16930. [DOI] [PubMed] [Google Scholar]
- Rai N., Nollmann M., Spotorno B., Tassara G., Byron O., Rocco M. 2005. SOMO (SOlution MOdeler) differences between X-ray- and NMR-derived bead models suggest a role for side chain flexibility in protein hydrodynamics. Structure 13: 723–734. [DOI] [PubMed] [Google Scholar]
- Remijn J.A., Ijsseldijk M.J., van Hemel B.M., Galanakis D.K., Hogan K.A., Lounes K.C., Lord S.T., Sixma J.J., De Groot P.G. 2002. Reduced platelet adhesion in flowing blood to fibrinogen by alterations in segment γ316–322, part of the fibrin-specific region. Br. J. Haematol. 117: 650–657. [DOI] [PubMed] [Google Scholar]
- Riddles P.W., Blakeley R.L., Zerner B. 1979. Ellman's reagent: 5, 5′-dithiobis(2-nitrobenzoic acid)–A reexamination. Anal. Biochem. 94: 75–81. [DOI] [PubMed] [Google Scholar]
- Rooney M.M., Parise L.V., Lord S.T. 1996. Dissecting clot retraction and platelet aggregation—Clot retraction does not require an intact fibrinogen γ chain C terminus. J. Biol. Chem. 271: 8553–8555. [DOI] [PubMed] [Google Scholar]
- Rooney M.M., Farrell D.H., van Hemel B.M., De Groot P.G., Lord S.T. 1998. The contribution of the three hypothesized integrin-binding sites in fibrinogen to platelet-mediated clot retraction. Blood 92: 2374–2381. [PubMed] [Google Scholar]
- Scarborough R.M., Rose J.W., Hsu M.A., Phillips D.R., Fried V.A., Campbell A.M., Nannizzi L., Charo I.F. 1991. Barbourin. A GPIIb-IIIa-specific integrin antagonist from the venom of Sistrurus m. barbouri. J. Biol. Chem. 266: 9359–9362. [PubMed] [Google Scholar]
- Scarborough R.M., Naughton M.A., Teng W., Rose J.W., Phillips D.R., Nannizzi L., Arfsten A., Campbell A.M., Charo I.F. 1993a. Design of potent and specific integrin antagonists. Peptide antagonists with a high specificity for glycoprotein IIb-IIIa. J. Biol. Chem. 268: 1066–1073. [PubMed] [Google Scholar]
- Scarborough R.M., Rose J.W., Naughton M.A., Phillips D.R., Nannizzi L., Arfsten A., Campbell A.M., Charo I.F. 1993b. Characterization of the integrin specificities of disintegrins isolated from American pit viper venoms. J. Biol. Chem. 268: 1058–1065. [PubMed] [Google Scholar]
- Sevenich F.W., Langowski J., Weiss V., Rippe K. 1998. DNA binding and oligomerization of NtrC studied by fluorescence anisotropy and fluorescence correlation spectroscopy. Nucleic Acids Res. 26: 1373–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattil S.J. 1999. Signaling through platelet integrin αIIbβ3: Inside-out, outside-in, and sideways. Thromb. Haemost. 82: 318–325. [PubMed] [Google Scholar]
- Shattil S.J. and Newman P.J. 2004. Integrins: Dynamic scaffolds for adhesion and signaling in platelets. Blood 104: 1606–1615. [DOI] [PubMed] [Google Scholar]
- Shattil S.J., Kashiwagi H., Pampori N. 1998. Integrin signaling: The platelet paradigm. Blood 91: 2645–2657. [PubMed] [Google Scholar]
- Shimaoka M., Takagi J., Springer T.A. 2002. Conformational regulation of integrin structure and function. Annu. Rev. Biophys. Biomol. Struct. 31: 485–516. [DOI] [PubMed] [Google Scholar]
- Smith D.B. and Johnson K.S. 1988. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67: 31–40. [DOI] [PubMed] [Google Scholar]
- Spotorno B., Piccinini L., Ruggiero C., Nardini M., Molina F., Rocco M. 1997. BEAMS (beads modeling system): A set of computer programs for the visualization and the computation of hydrodynamic and conformational properties of beads models of proteins. Eur. Biophys. J. 25: 373–384. [Google Scholar]
- Stafford III W.F. 1992. Boundary analysis in sedimentation transport experiments: A procedure for obtaining sedimentation coefficient distributions using the time derivative of the concentration profile. Anal. Biochem. 203: 295–301. [DOI] [PubMed] [Google Scholar]
- Takagi J., Petre B.M., Walz T., Springer T.A. 2002. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 110: 599–611. [DOI] [PubMed] [Google Scholar]
- Takagi J., Strokovich K., Springer T.A., Walz T. 2003. Structure of integrin αI5β1 in complex with fibronectin. EMBO J. 22: 4607–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangemann K. and Engel J. 1995. Demonstration of non-linear detection in ELISA resulting in up to 1000-fold too high affinities of fibrinogen binding to integrin αIIbβ3. FEBS Lett. 358: 179–181. [DOI] [PubMed] [Google Scholar]
- Wang W., Wu Q., Pasuelo M., McMurray J.S., Li C. 2005. Probing for integrin αvβ3 binding of RGD peptides using fluorescence polarization. Bioconjug. Chem. 16: 729–734. [DOI] [PubMed] [Google Scholar]
- Weisel J.W., Nagaswami C., Vilaire G., Bennett J.S. 1992. Examination of the platelet membrane glycoprotein IIb-IIIa complex and its interaction with fibrinogen and other ligands by electron microscopy. J. Biol. Chem. 267: 16637–16643. [PubMed] [Google Scholar]
- Wierzbicka-Patynowski I., Niewiarowski S., Marcinkiewicz C., Calvete J.J., Marcinkiewicz M.M., McLane M.A. 1999. Structural requirements of echistatin for the recognition of αvβ3 and α5β1 integrins. J. Biol. Chem. 274: 37809–37814. [DOI] [PubMed] [Google Scholar]
- Xiao T., Takagi J., Coller B.S., Wang J.H., Springer T.A. 2004. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature 432: 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J.P., Stehle T., Diefenbach B., Zhang R., Dunker R., Scott D.L., Joachimiak A., Goodman S.L., Arnaout M.A. 2001. Crystal structure of the extracellular segment of integrin αvβ3. Science 294: 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J.P., Stehle T., Zhang R., Joachimiak A., Frech M., Goodman S.L., Arnaout M.A. 2002. Crystal structure of the extracellular segment of integrin αvβ3 in complex with an Arg-Gly-Asp ligand. Science 296: 151–155. [DOI] [PubMed] [Google Scholar]
- Xiong J.P., Stehle T., Goodman S.L., Arnaout M.A. 2003. New insights into the structural basis of integrin activation. Blood 102: 1155–1199. [DOI] [PubMed] [Google Scholar]
- Yakovlev S., Litvinovich S., Loukinov D., Medved L. 2000. Role of the β-strand insert in the central domain of the fibrinogen γ-module. Biochemistry 39: 15721–15729. [DOI] [PubMed] [Google Scholar]
- Yee V.C., Pratt K.P., Côté H.C.F., Le Trong I., Chung D.W., Davie E.W., Stenkamp R.E., Teller D.C. 1997. Crystal structure of a 30 kDa C-terminal fragment from the γ chain of human fibrinogen. Structure 5: 125–138. [DOI] [PubMed] [Google Scholar]