

Abstract
Saposins A and C are sphingolipid activator proteins required for the lysosomal breakdown of galactosylceramide and glucosylceramide, respectively. The saposins interact with lipids, leading to an enhanced accessibility of the lipid headgroups to their cognate hydrolases. We have determined the crystal structures of human saposins A and C to 2.0 Å and 2.4 Å, respectively, and both reveal the compact, monomeric saposin fold. We confirmed that these two proteins were monomeric in solution at pH 7.0 by analytical centrifugation. However, at pH 4.8, in the presence of the detergent C8E5, saposin A assembled into dimers, while saposin C formed trimers. Saposin B was dimeric under all conditions tested. The self-association of the saposins is likely to be relevant to how these small proteins interact with lipids, membranes, and hydrolase enzymes.
Keywords: saposins, X-ray crystallography, analytical ultracentrifugation, protein–detergent interactions
Saposins A, B, C, and D are small, nonenzymatic proteins required for the breakdown of glycosphingolipids within the lysosome (Kolter and Sandhoff 2005). They are derived from the proteolytic processing of the precursor protein prosaposin, producing the four individual saposins. The four saposin domains contained within prosaposin most likely arose from two tandem duplications of an ancestral gene into one single copy gene (Hazkani-Covo et al. 2002). Each saposin “activates” the breakdown of particular lipid substrates by facilitating the access of the lipid headgroups to the active sites of cognate hydrolases. It is generally believed that in the absence of sphingolipid activator proteins, the oligosaccharide chains of the membrane-bound lipids do not extend far enough into the lysosomal lumen to be accessible to the active sites of the hydrolases. Mechanistically, the saposins appear to activate lipid hydrolysis by solubilizing the lipid substrates or possibly by destabilizing the membrane structure (Vaccaro et al. 1993, 1995, 1997; Wilkening et al. 1998; Salvioli et al. 2000). Human genetic defects in saposins B and C have confirmed the importance of their roles in the catabolism of cerebroside sulfate and glucosylceramide by arylsulfatase A and glucocerebrosidase, respectively (Kretz et al. 1990; Horowitz and Zimran 1994). A targeted disruption of saposin A in mice led to an accumulation of galactosylceramide, indicating its importance for galactocerebrosidase activity, while in vitro experiments have shown that saposin D may be involved in the degradation of ceramide by acid ceramidase in the breakdown pathway (Azuma et al. 1994; Matsuda et al. 2001). In addition to their glycosphingolipid degradation activities, the four saposins have lipid transfer activity in vitro (Conzelmann et al. 1982; Hiraiwa et al. 1992; Ciaffoni et al. 2006). This may be relevant for their roles in membrane maintenance and other lipid-associated activities. For example, the saposins are required for loading antigenic lipids onto CD1 proteins for presentation on T cells (Zhou et al. 2004; Kang and Cresswell 2004), and, in particular, saposin C is specifically required to load mycobacterial lipids onto CD1b (Winau et al. 2004).
The four saposins are members of the larger superfamily of saposin-like proteins (Bruhn 2005). The saposin-like motif is found in small α-helical proteins or in domains inserted within larger proteins and almost always has a characteristic set of six cysteines that form three intramolecular disulfide bonds. The members of this family are involved in a diverse range of functions, but most participate in membrane/lipid interactions. The structures of several saposin-like proteins are known, and most describe a compact, monomeric fold that buries a small hydrophobic core (Liepinsh et al. 1997; Kervinen et al. 1999; Gonzalez et al. 2000; Anderson et al. 2003; Hecht et al. 2004). In contrast, the crystal structure of saposin B revealed a major conformational variant that forms an open “V-shaped” monomer that self-associates into a dimer, resulting in a protein shell with a large hydrophobic cavity for lipid solubilization (Ahn et al. 2003a). Saposin B has lower sequence relatedness relative to the other three saposins (Hazkani-Covo et al. 2002), with pairwise sequence identities of 22%, 15%, and 21% with saposins A, C, and D, respectively. The latter three are more similar, with pairwise sequence identities ranging from 34% to 39% (Fig. 1).
Figure 1.

Sequence and structures of saposins A and C. (A) Multiple sequence alignment of the four human saposins. The six conserved cysteines are indicated along with their connectivity. Helices 1, 2, 3, and 4 from the known crystal structures are shaded blue, green, yellow, and red, respectively. A turn of 310 helix is present at the end of helix 2 in saposins A and B, and is shaded gray. The red box indicates the conserved N-linked glycosylation site located at the turn between helices 1 and 2. The green box indicates the conserved tyrosine 54 that is found at the position of the kink in helix 3 (see text). The designation of the “stem” and the “hairpin” regions is based on a comparison of related structures (see text). (B,C) Ribbon representation of saposins A and C, with helix coloring according to the scheme in A. (D) A superposition of saposin A (blue) and saposin C (red) shows slight differences in the three loop regions and in α2.
In order to further explore the range of conformations that can be adopted within the saposin family of proteins, we determined the high-resolution crystal structures of human saposins A and C to 2.0 Å and 2.4 Å resolution, respectively. Both structures revealed the monomeric form of the saposin fold, as previously seen in the NMR structure of saposin C (de Alba et al. 2003). We also observed pH and detergent-induced oligomerization of saposins A and C by analytical ultracentrifugation, suggesting that environmental triggers can induce structural changes within these proteins. In contrast, we did not detect a change in the quaternary structure of saposin B under the tested conditions. The differential effects of pH and detergent on the particular members of the saposin family are likely to be functionally important for their individual activities.
Results and Discussion
Expression and purification
Saposins are cysteine-rich human glycoproteins and, as such, present a challenging recombinant expression problem. We used our previously reported system for saposin B (Ahn et al. 2003b) to produce saposins A and C at levels of 2–3 mg of purified protein per liter of culture in Escherichia coli. Proper connectivity of the three disulfide bonds was verified by mass spectrometry (Ahn et al. 2003b) and crystallography (Ahn et al. 2003a; this work). We produced saposins A, B, and C without any additional purification tags, and the recombinant proteins differ from protein isolated from natural sources only by the addition of two N-terminal residues (Met–Gly for saposin A and Met–Asp for saposin C) that were introduced as the result of the cloning strategy. The saposins are heat-stable proteins, and the first purification step was to heat crude cell extracts at 85°C for 10 min. This resulted in the precipitation of most native E. coli proteins, leaving properly folded saposin in the soluble fraction of the heated extract. We did not detect any saposin in either inclusion bodies or in the pellet resulting from the heat treatment. Two additional chromatographic steps resulted in highly purified protein that produced crystals. We obtained our crystals using PEG precipitants at pHs ranging from 6 to 7. Glycosylation of the protein is not essential for the protein activity, and bacterially expressed saposin B had a specific activity that was similar to that of protein isolated from natural sources (Whitelegge et al. 2003; Norris et al. 2005).
Description of the structures
Both saposin A and saposin C adopt the monomeric saposin fold in these crystal structures and consist of four amphipathic α-helices folded into a oblate ellipsoid with approximate dimensions 26 × 28 × 16 Å (Figs. 1,2). As with the other saposin-fold proteins, the nonconserved charged residues are located on the surfaces, with the conserved hydrophobic residues pointing in toward a small core. The backbone atoms from the saposin A and saposin C crystal structures superimpose well, with the exception of a small but significant deviation of helix 2 relative to the rest of the structure (Fig. 1D). There is also very good agreement between the saposin C crystal structure presented here and the solution structure of saposin C previously determined by NMR (de Alba et al. 2003). The rmsd between the lowest energy saposin C NMR structure and the crystal structures is 0.86 Å over 78 Cα atoms (Asp2–Ser79). This increases to only 1.53 Å when the 609 main-chain and side-chain atoms are included.
Figure 2.
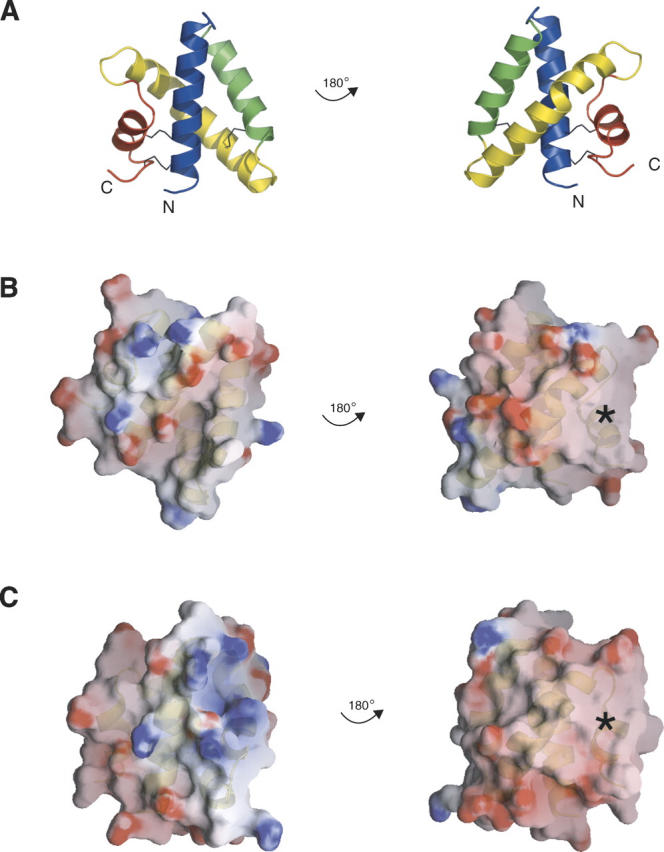
Electrostatic surface representations. The left and right columns show the two flat faces of the disc-shaped monomers. Ribbon representations of saposin A are shown in A as a guide to the surfaces shown for saposin A and saposin C in panels B and C. The surfaces are colored by electrostatic potential from –10 kT to 10 kT, where k is the Boltzmann constant and T is temperature in Kelvin (red = negative, blue = positive, white = neutral) as calculated with the program GRASP. The asterisks indicate an uncharged surface patch that is common to saposins A and C.
Helix 3, shown in yellow in Figure 1, is sometimes considered as two different helices in related structures (Liepinsh et al. 1997; Anderson et al. 2003; de Alba et al. 2003), but we choose to describe this region as a single helix with a highly localized kink. This kink is remarkably similar in the saposin A and saposin C structures and in chain B of the saposin B dimer (Ahn et al. 2003a). In these three cases, the main-chain torsion angles deviate from canonical α-helical values only at the central conserved tyrosine 54 (Table 1), and the kinks are associated with a localized disruption of two main-chain H bonds at residues preceding the kink site. Thus, the following i, i + 4 H-bonds are not formed in our structures: S52 to P56 and S53 to V57 in saposin A, S52 to E56 and Q53 to I57 in saposin B (chain B), and D52 to S56 and T53 to S57 in saposin C. All of the remaining regions of the helix have the expected (ϕ,φ) values and regular (i,i + 4) main-chain hydrogen bonds. In the case of saposin A, the kink is a classic example of a disruption in an α-helix caused by a proline residue (in this case, Pro56) due to the steric clash of the ring and the loss of an H-bond donor (Barlow and Thornton 1988). However, the only other human saposin that has an equivalent proline at this position is SapD (Fig. 1; Bruhn 2005), so the structurally conserved kink cannot be solely attributed to the presence of the proline in these proteins. The bend in the helix axis is ∼52° in saposin A and saposin B (chain B), and slightly larger at 64° in saposin C. This feature is of considerable interest, since the crystal structure of saposin B showed that helix 3 could exist in either a kinked conformation (chain B) or a relatively straight conformation (chains A and C) (Ahn et al. 2003a). The reversible kinking of this helix is a critical feature of the structural change in saposin B from a lipid-bound dimer to an unliganded dimer (Table 1; Ahn et al. 2003a). A related effect appears to be important for saposin C, since the NMR structure of this activator in the presence of SDS reveals an open form of the protein in which this helix straightens relative to the detergent-free state, in which it is kinked (de Alba et al. 2003; Hawkins et al. 2005). The crystal structures presented here provide a high-resolution view of this region of the saposin proteins in the kinked state.
Table 1.
Main-chain torsion angles in helix 3

Electrostatic surfaces
The exposed residues are the least conserved among the family of saposin-like proteins and lead to significantly different surface charge distributions, and this likely results in their different functions and mechanisms of action (Bruhn 2005). For instance, the known structures of the saposin-like proteins with lytic ability, including NK-lysin, granulysin, bacteriocin AS-48, and amoebapore A, carry a net positive charge at neutral pH. However, each reveals different charged and hydrophobic surface features, resulting in different modes of action for their membrane-permeabilizing activities (Liepinsh et al. 1997; Gonzalez et al. 2000; Anderson et al. 2003; Hecht et al. 2004).
The isoelectric point of saposins A, B, and C are 4.5, 4.6, and 4.6, respectively, and the proteins are calculated to have net charges of −8.2, −8.0, and −9.1 at pH 7. At a pH of 4.8, which is representative of the pH in the late lysosome, all three of these proteins are expected to have a net charge of approximately −3. Inspection of the electrostatic surfaces shows that the negative charges are distributed over most of the surface of the disc-shaped proteins, but there is a significant uncharged zone on both saposin A and saposin C (Fig. 2). Negatively charged lipids are known to be important for saposin–membrane interactions, and there are a few positively charged surface clusters on the proteins, but these surfaces are not conserved across the saposins. The different surface charge distributions of saposins A and C may explain their different bilayer interactions, as this likely plays a key role in their membrane interactions and specificity (Vaccaro et al. 1997; Qi and Grabowski 2001; de Alba et al. 2003). The locations of initial membrane approach are not immediately obvious, but the lysine-rich N-terminal helices 1 and 2 in saposin C have been proposed as initial membrane contact sites (Qi and Chu 2004; Liu et al. 2005).
Comparison with saposin B
The crystal structures of saposins A and C are monomeric, in contrast to the saposin B dimer (Ahn et al. 2003a). As with NK-lysin and the other known saposin-fold structures, the saposin A and saposin C structures are essentially “closed” versions of the “open” saposin B conformation. In the saposin B dimer, two V-shaped monomers clasp together to form a shell with a large internal hydrophobic cavity (Ahn et al. 2003a). In contrast, the monomeric form of the saposin fold is collapsed onto itself and forms a small hydrophobic core. Saposin C has been shown to undergo a large change in tertiary structure from a closed monomer to an open V-shaped form in the presence of SDS (Hawkins et al. 2005). In addition to the kinked and straight forms of helix 3 described above, additional differences between the open conformation and the closed form of the fold can be localized to two hinge points between the “stem” and the “hairpin” regions of the proteins. The stem region consists of helices 1 and 4, which are linked by two disulfide bonds. The hairpin region consists of helices 2 and 3, which are also disulfide-linked. A difference distance matrix analysis of saposin B (chain B) and saposin A structures clearly reveals that the proteins differ by the relative orientations of these two constituent blocks (Fig. 3A). Remarkably, the base and the hairpin regions of saposin A and saposin B (chain B) can be independently fit to each other with excellent agreement (Fig. 3B,C). The fit of saposin A to chains A or C of saposin B does not agree as well, since helix 3 in the latter subunits are not kinked. Overall, superposition analysis points to three “hinge” areas in the saposins: the loops between helices α1/α2 and α3/α4, as well as the kink site in helix α3.
Figure 3.
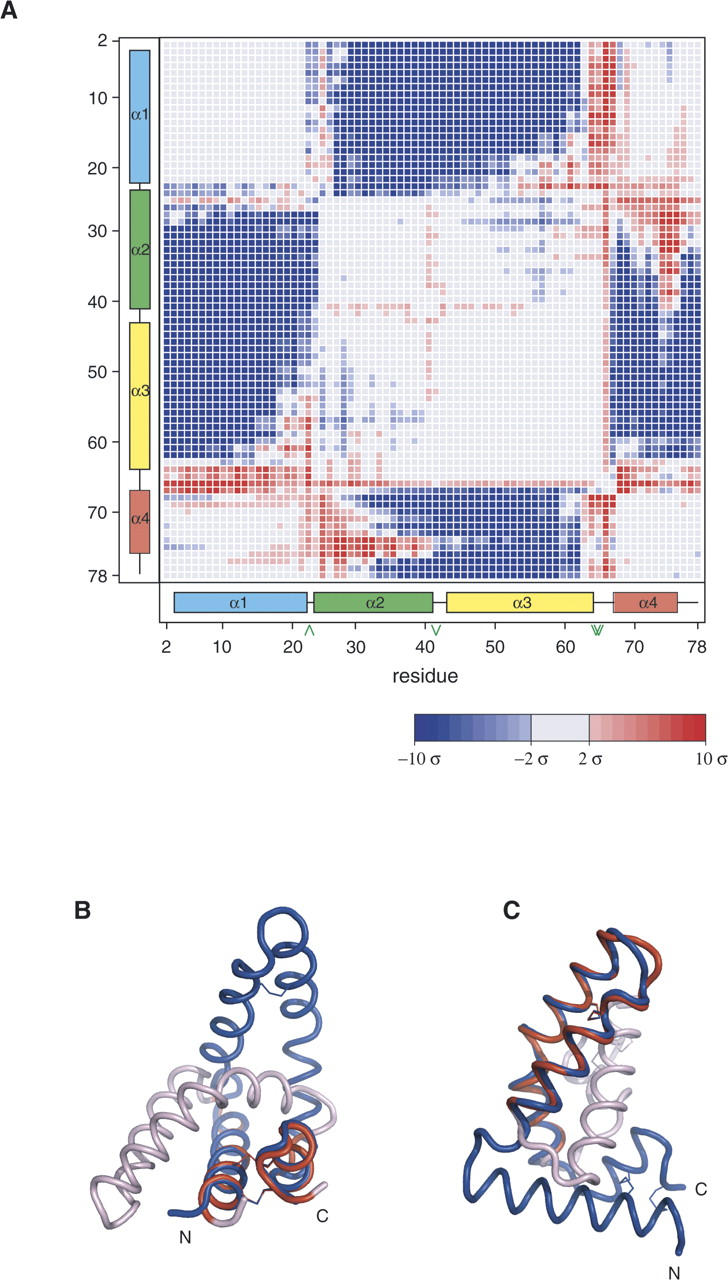
Structural comparison of saposins A and B. (A) Error-scaled difference distance matrix (Schneider 2000) between saposin A (this work) and chain B of saposin B (Ahn et al. 2003a). Blue coloring indicates pairs of Cα atoms that are closer together in the saposin A structure relative to the saposin B structure. Large empty blocks in the matrix indicate regions that are not significantly different between the two structures. (B) Superposition of the stem region of saposin B chain B (blue) and saposin A. Red coloring indicates the saposin A residues 2–21 and 67–80 that were fit to residues 2–21 and 65–78 of saposin B. The remaining residues of saposin A are colored light pink. (C) Saposin A hairpin residues 25–62 were fit to saposin B resides 26–62 and colored red. The remaining saposin A residues that were not used in this superposition are colored light pink. The view of saposin B is rotated 90° about a vertical axis relative to panel B.
Saposin B appears to solubilize its substrate sulfatide most actively at acidic pH (Fluharty et al. 2001). A similar pH-dependent change in membrane binding activity was also reported for saposins A and C (Vaccaro et al. 1995). Increased exposure of hydrophobic residues due to flexibility between the base and the hairpin regions of the monomers would facilitate membrane interactions. A similar mechanism is predicted for the membrane lysis activity of granulysin (Anderson et al. 2003). This type of motion may be generally important in both membrane binding and perturbation for increasing substrate accessibility by saposins and saposin-like proteins.
Detergent-induced self-association of saposins A and C
In order to further investigate the effect of pH and detergent on the self-association properties of the saposins, we carried out sedimentation equilibrium analyses of saposins A, B, and C at neutral and acidic pH, and in the presence and the absence of the polyoxyethyleleglycol detergent C8E5 (Table 2; Fig. 4). We used C8E5 as the detergent since it has a partial specific volume that is closely matched to that of aqueous buffers. This simplifies the use of the analytical centrifuge to measure the oligomeric state of proteins in the presence of amphiphiles (Ludwig et al. 1982). Saposin B was dimeric under the four tested conditions, in agreement with previous results (Whitelegge et al. 2003). In contrast, saposins A and C were monomeric in the absence of detergent at pH 7.0 and 4.8, but formed dimers and trimers, respectively, at low pH with added C8E5. The sedimentation analysis presented here does not depend on the shape of the protein particle and is an accurate measurement of the true molecular weight. Note that for saposin C, a monomer–trimer association model gave the lowest residual errors for the pH 7.0 C8E5 data, but under acidic conditions in the presence of the detergent, only trimers could be detected.
Table 2.
Analytical ultracentrifugation

Figure 4.

Sedimentation equilibrium analysis of saposin C. The lower panels represent the protein concentration gradients, and the upper panels represent the residuals of the fit to the sedimentation equilibrium equation. (A) Sedimentation data for 20 μM saposin C in 50 mM Tris (pH 7.0), 150 mM NaCl at 35,000 rpm. The solid line represents the expected curve for a single ideal species of molecular of 10,873 Da resulting from a global analysis from 10 profiles collected at two concentrations and five rotor speeds. (B) Sedimentation data for 20 μM saposin C in 50 mM NaAc (pH 4.8), 150 mM NaCl, 10 mM C8E5 at 35,000 rpm. The solid line represents the expected curve for a single ideal species of molecular weight of 28,078 Da resulting from a global analysis from eight profiles collected at two concentrations and four rotor speeds.
Induced oligomerization has been demonstrated for other small membrane-active proteins. For example, the three-fingered β-sheet protein cardiotoxin A3 exists as a monomer in solution but crystallizes as a dimer in the presence of SDS. The dimers are thought to be intermediates in the eventual formation of higher order oligomers that are necessary for pore formation, resulting in membrane depolarization, leakage, and fusion (Forouhar et al. 2003). Similarly, the cyclic protein bacteriocin AS-48 adopts different oligomeric structures when in a membrane-bound or a soluble form. AS-48 resembles the saposin-fold but falls outside the classification of saposin-like proteins. It exists as a dimer at physiological pH values between 4.5 and 8.5, but as a stable monomer at pH 3 as determined by chemical cross-linking studies, NMR, crystallography, and analytical ultracentrifugation (Gonzalez et al. 2000; Abriouel et al. 2001; Sanchez-Barrena et al. 2003). Similarly, the saposin-like protein amoebapore A appears to undergo pH-dependent dimerization and further assembly into membrane-spanning pores, as determined by size exclusion chromatography (Hecht et al. 2004). Likewise, saposin A and C oligomerization in the presence of amphiphiles may be a prerequisite for their membrane activities.
Conclusions
Saposins A and C both possess the typical closed monomeric saposin-fold conformation in the structures reported here, in contrast to the previously determined open dimer structure of saposin B (Ahn et al. 2003a). However, we have observed detergent and pH-induced self-association of saposins A and C, confirming that these proteins can exist in multiple structural states depending on the environmental conditions. It is difficult to propose reasonable models for lipid-binding based on closed-state structures, regardless of whether these associate into dimers, trimers, or higher order assemblies. Instead, we favor a model in which changes in quaternary structure are driven by significant structural changes within individual chains. We expect that the acidic and the lipid-rich environment within the lysosome can trigger conformational changes within the saposins A and C. The open-state subunits, such as those seen with saposin B, could bind directly to lipid bilayers and/or self-associate to form multimeric complexes with large hydrophobic cavities for the accommodation of lipidic ligands.
It is interesting to note that saposin B and the GM2 activator protein appear to activate glycosphingolipid degradation by a similar substrate solubilization mechanism despite having widely different structures. Both have large hydrophobic binding pockets and flexible entryways, but these are formed by either the α-helical saposin B homodimer or the single-chain β-cup fold in the GM2 activator protein (Wright et al. 2000, 2003; Ahn et al. 2003a). Further studies will be required to clarify the similarities and the differences in the mechanisms used by the four saposins to carry out their respective biological functions.
Materials and methods
Cloning, expression, and purification
Cloning and expression protocols for saposins A and C followed the protocol developed for saposin B (Ahn et al. 2003b). Briefly, the saposin A and C coding regions were amplified from a partial prosaposin cDNA (I.M.A.G.E. 3,615,413) by the polymerase chain reaction with primers that created 5′ NcoI and 3′ BamHI sites. The amplified fragments, which encoded additional N-terminal Met–Gly residues for saposin A and Met–Asp for saposin C, were digested and inserted into the pET-16 expression vector (Novagen). The resulting saposin A and saposin C vectors were used to transform E. coli BL21(DE3) Codon Plus and AD494(DE3) cells (Novagen), respectively. Cultures were grown at 37°C in Terrific Broth to an OD600 between 0.6 and 0.7, induced with 0.8 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG), and grown for an additional 4 h before harvesting by centrifugation. Cells were resuspended in anion-exchange column binding buffer (25 mM NaCl, 25 mM Tris-HCl at pH 7.5) and lysed by passage through an Avestin Emulsiflex-C5 at 20,000 lb/in2. The lysates were clarified by centrifugation at 26,000g for 20 min and the supernatants were heated at 85°C for ∼10 min. Precipitated protein was removed by centrifuged at 26,000g for 20 min. The resulting supernatant was applied directly to a Q-Sepharose column (BioRad) that had been pre-equilibrated in binding buffer, and bound protein was eluted with a linear gradient of elution buffer (1 M NaCl, 25 mM Tris-HCl at pH 7.5). The peak fractions containing saposins A or C were pooled, concentrated, and applied to a Superdex-75 size-exclusion column in 50 mM phosphate buffer (pH 6.9). The pooled saposin fractions were concentrated to 15–25 mg/mL and stored at 4°C.
The methionine-inhibition pathway was utilized for the expression of selenomethionine-substituted saposin A (Doublié 1997). Transformants were grown at 37°C in M9 medium supplemented with 0.4% glucose, 10 μg/mL vitamins (biotin and thiamine), 2 mM 1 M MgSO4, 0.1 mM CaCl2 to an OD600 of 0.3, at which time 100 mg/L of lysine, isoleucine, leucine, phenylalanine, valine, threonine, and 60 mg/L of selenomethionine were added; 0.8 mM IPTG was added when the cells reached an OD600 of 0.6, and cells were harvested 6 h later. The purification protocol was identical to the native protein, except that 3 mM β-Mercaptoethanol was included in all buffers to minimize selenomethionine oxidation. Saposin B was expressed and purified as described previously (Ahn et al. 2003b).
Saposin A crystallization and structure determination
Selenomethionine-substituted saposin A was crystallized by vapor diffusion by mixing 1 μL of the protein stock solution with an equal volume of a precipitant solution consisting of 19% PEG 8K, 0.2 M calcium acetate and 0.1 M MES (pH 6.0) or sodium cacodylate (pH 6.5), and equilibrating the droplets with 600 μL of the precipitant solution. Mother liquor containing 15% PEG 400 was used as a cryoprotectant. Multiwavelength anomalous diffraction (MAD) data were collected at 100 K at beamline F2 at Cornell High Energy Synchrotron Source (CHESS), Cornell University. Data processing and reduction were done with DENZO/SCALEPACK (Otwinowski and Minor 1997). Crystals belonged to the orthorhombic space group P21212, with unit cell dimensions of a = 45.68 Å, b = 50.04 Å, c = 33.82 Å. Three out of the four expected selenium sites were located with SOLVE (Terwilliger and Berendzen 1999), leading to interpretable electron density maps. DM and O were used for density modification and model building (Jones et al. 1991; Collaborative Computational Project 1994). Refinement and addition of water molecules were done with CNS (Brünger et al. 1998).
Saposin C crystallization and structure determination
Saposin C was crystallized by vapor diffusion by equilibration against a reservoir solution of 24% PEG 400, 0.4 M calcium chloride and 0.1 M sodium HEPES (pH 7.0) or sodium cacodylate (pH 6.0). Mother liquor with 10% glycerol was used as a cryoprotectant. Data were collected at CHESS F2 as described above for saposin A. Saposin C crystals belonged to space group P63 with unit cell dimensions of a = b = 53.20 Å, c = 52.46 Å. The structure was solved by molecular replacement with a saposin C model from NMR data (de Alba et al. 2003). To circumvent the lack of B-factors, the program MULTI PROBE (written by Gerard Kleywegt, Uppsala University, Sweden) was used to prepare ensemble models. An “all atom” and a poly-Ser-Ala-Gly (poly-SAG) model with B-factors set to 20 Å2 were used in AmoRe calculations (Collaborative Computational Project 1994). Each of the ensemble models contained residues 3–74 from the NMR structure, and both resulted in equivalent, unambiguous solutions. After initial rigid-body refinement in AmoRe, the best representative conformer from the NMR ensemble was chosen as starting model for atomic refinement. REFMAC5 refinement proceeded with cycles of manual rebuilding with O (Jones et al. 1991; Collaborative Computational Project 1994). There was significant diffraction anisotropy in the data, and TLS refinement (Winn et al. 2001) led to a small but significant improvement in the R and Rfree refinement statistics. The crystallographic statistics for the saposins A and C structures are shown in Table 3.
Table 3.
Crystallographic data

Analytical ultracentrifugation
Sedimentation equilibrium experiments were performed at 20°C on a Beckman XLI Analytical Ultracentrifuge (Beckman Instruments) using an AN50-Ti rotor, quartz windows, and standard six-sector charcoal-filled Epon centerpieces. The samples were prepared at concentrations ranging from 5 to100 μM saposin in pH 7 (150 mM NaCl, 50 mM Tris-HCl at pH 7.0) and pH 4.8 (150 mM NaCl, 50 mM NaAc, at pH 4.8) buffers with and without 10 mM penta-ethylene glycol mono-octyl ether (C8E5). The centrifugation runs were carried out at 30,000 rpm, 35,000 rpm, 40,000 rpm, 45,000 rpm, and 50,000 rpm for 18 h at each speed to ensure equilibrium was reached before absorbance measurements were taken. Molecular weights were determined by a global fit of the sedimentation data for two to three different initial sample concentrations at four to five different rotor speeds using Beckman XLI data analysis software. In all cases but one, the data were best fit as single ideal species, resulting in a determination of the molecular weight of the species in the solution. In the case of saposin C at pH 7.0 in C8E5 slightly lower residuals were obtained for a monomer–trimer self-association model with a subunit molecular weight calculated from the sequence (9138 Da) (see Table 2 and text). The partial specific volume and density of the sample were calculated using the program SEDNTERP from the amino acid sequence and buffer composition, respectively. A partial specific volume of 0.993 was used for the detergent C8E5 (Ludwig et al. 1982).
Data deposition
Atomic coordinates and diffraction data have been deposited with the PDB, with ID code 2DOB for saposin A and 2GTG for saposin C.
Acknowledgments
This work was supported by a CIHR grant to G.G.P. We thank Avi Chakrabartty and Sylvia Ho for the analytical ultracentrifugation measurements, and the Cornell High Energy Synchrotron Source and Advanced Photon Source for access to synchrotron facilities.
Footnotes
Reprint requests to: Gilbert G. Privé, Division of Cancer Genomics and Proteomics, Ontario Cancer Institute, 101 College Street, Toronto, Ontario M5G 1L7, Canada; e-mail: prive@uhnres.utoronto.ca; fax: (416) 581-7562.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062256606.
References
- Abriouel H., Valdivia E., Galvez A., Maqueda M. 2001. Influence of physico-chemical factors on the oligomerization and biological activity of bacteriocin AS-48. Curr. Microbiol. 42: 89–95. [DOI] [PubMed] [Google Scholar]
- Ahn V.E., Faull K.F., Whitelegge J.P., Fluharty A.L., Privé G.G. 2003a. Crystal structure of saposin B reveals a dimeric shell for lipid binding. Proc. Natl. Acad. Sci. 100: 38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn V.E., Faull K.F., Whitelegge J.P., Higginson J., Fluharty A.L., Privé G.G. 2003b. Expression, purification, crystallization, and preliminary X-ray analysis of recombinant human saposin B. Protein Expr. Purif. 27: 186–193. [DOI] [PubMed] [Google Scholar]
- Anderson D.H., Sawaya M.R., Cascio D., Ernst W., Modlin R., Krensky A., Eisenberg D. 2003. Granulysin crystal structure and a structure-derived lytic mechanism. J. Mol. Biol. 325: 355–365. [DOI] [PubMed] [Google Scholar]
- Azuma N., O'Brien J.S., Moser H.W., Kishimoto Y. 1994. Stimulation of acid ceramidase activity by saposin D. Arch. Biochem. Biophys. 311: 354–357. [DOI] [PubMed] [Google Scholar]
- Barlow D.J. and Thornton J.M. 1988. Helix geometry in proteins. J. Mol. Biol. 201: 601–609. [DOI] [PubMed] [Google Scholar]
- Bruhn H. 2005. A short guided tour through functional and structural features of saposin-like proteins. Biochem. J. 389: 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Ciaffoni F., Tatti M., Boe A., Salvioli R., Fluharty A., Sonnino S., Vaccaro A.M. 2006. Saposin B binds and transfers phospholipids. J. Lipid Res. 47: 1045–1053. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (CCP4). 1994. The CCP4 Suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Conzelmann E., Burg J., Stephan G., Sandhoff K. 1982. Complexing of glycolipids and their transfer between membranes by the activator protein for degradation of lysosomal ganglioside GM2. Eur. J. Biochem. 123: 455–464. [DOI] [PubMed] [Google Scholar]
- de Alba E., Weiler S., Tjandra N. 2003. Solution structure of human saposin C: pH-Dependent interaction with phospholipid vesicles. Biochemistry 42: 14729–14740. [DOI] [PubMed] [Google Scholar]
- Doublié S. 1997. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 236: 523–530. [PubMed] [Google Scholar]
- Fluharty C.B., Johnson J., Whitelegge J., Faull K.F., Fluharty A.L. 2001. Comparative lipid binding study on the cerebroside sulfate activator (saposin B). J. Neurosci. Res. 63: 82–89. [DOI] [PubMed] [Google Scholar]
- Forouhar F., Huang W.N., Liu J.H., Chien K.Y., Wu W.G., Hsiao C.D. 2003. Structural basis of membrane-induced cardiotoxin A3 oligomerization. J. Biol. Chem. 278: 21980–21988. [DOI] [PubMed] [Google Scholar]
- Gonzalez C., Langdon G.M., Bruix M., Galvez A., Valdivia E., Mazueda M., Rico M. 2000. Bacteriocin AS-48, a microbial cyclic polypeptide structurally and functionally related to mammalian NK-lysin. Proc. Natl. Acad. Sci. 97: 11221–11226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins C.A., de Alba E., Tjandra N. 2005. Solution structure of saposin C in a detergent environment. J. Mol. Biol. 346: 1381–1392. [DOI] [PubMed] [Google Scholar]
- Hazkani-Covo E., Altman N., Horowitz M., Graur D. 2002. The evolutionary history of prosaposin: Two successive tandem-duplication events gave rise to the four saposin domains in vertebrates. J. Mol. Evol. 54: 30–34. [DOI] [PubMed] [Google Scholar]
- Hecht O., Van Nuland N.A., Schleinkofer K., Dingley A.J., Bruhn H., Leippe M., Grotzinger J. 2004. Solution structure of the pore-forming protein of Entamoeba histolytica. J. Biol. Chem. 279: 17834–17841. [DOI] [PubMed] [Google Scholar]
- Hiraiwa M., Soeda S., Kishimoto Y., O'Brien J.S. 1992. Binding and transport of gangliosides by prosaposin. Proc. Natl. Acad. Sci. 89: 11254–11258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz M. and Zimran A. 1994. Mutations causing Gaucher disease. Hum. Mutat. 3: 1–11. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou J.Y., Cowan S.W., Kjeldgaard M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47: 110–119. [DOI] [PubMed] [Google Scholar]
- Kang S.J. and Cresswell P. 2004. Saposins facilitate CD1d-restricted presentation of an exogenous lipid antigen to T cells. Nat. Immunol. 5: 175–181. [DOI] [PubMed] [Google Scholar]
- Kervinen J., Tobin G.J., Costa J., Waugh D.S., Wlodawer A., Zdanov A. 1999. Crystal structure of plant aspartic proteinase prophytepsin: Inactivation and vacuolar targeting. EMBO J. 18: 3947–3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolter T. and Sandhoff K. 2005. Principles of lysosomal membrane digestion: Stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu. Rev. Cell Dev. Biol. 21: 81–103. [DOI] [PubMed] [Google Scholar]
- Kretz K.A., Carson G.S., Morimoto S., Kishimoto Y., Fluharty A.L., O'Brien J.S. 1990. Characterization of a mutation in a family with saposin B deficiency: A glycosylation site defect. Proc. Natl. Acad. Sci. 87: 2541–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liepinsh E., Andersson M., Ruysschaert J.M., Otting G. 1997. Saposin fold revealed by the NMR structure of NK-lysin. Nat. Struct. Biol. 4: 793–795. [DOI] [PubMed] [Google Scholar]
- Liu A., Wenzel N., Qi X. 2005. Role of lysine residues in membrane anchoring of saposin C. Arch. Biochem. Biophys. 443: 101–112. [DOI] [PubMed] [Google Scholar]
- Ludwig B., Grabo M., Gregor I., Lustig A., Regenass M., Rosenbusch J.P. 1982. Solubilized cytochrome c oxidase from Paracoccus denitrificans is a monomer. J. Biol. Chem. 257: 5576–5578. [PubMed] [Google Scholar]
- Matsuda J., Vanier M.T., Saito Y., Tohyama J., Suzuki K. 2001. A mutation in the saposin A domain of the sphingolipid activator protein (prosaposin) gene results in a late-onset, chronic form of globoid cell leukodystrophy in the mouse. Hum. Mol. Genet. 10: 1191–1199. [DOI] [PubMed] [Google Scholar]
- Norris A.J., Whitelegge J.P., Yaghoubian A., Alattia J.R., Privé G.G., Toyokuni T., Sun H., Brooks M.N., Panza L., Matto P. et al. 2005. A novel mass spectrometric assay for the cerebroside sulfate activator protein (saposin B) and arylsulfatase A. J. Lipid Res. 46: 2254–2264. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Qi X. and Chu Z. 2004. Fusogenic domain and lysines in saposin C. Arch. Biochem. Biophys. 424: 210–218. [DOI] [PubMed] [Google Scholar]
- Qi X. and Grabowski G.A. 2001. Differential membrane interactions of saposins A and C: Implications for the functional specificity. J. Biol. Chem. 276: 27010–27017. [DOI] [PubMed] [Google Scholar]
- Salvioli R., Tatti M., Ciaffoni F., Vaccaro A.M. 2000. Further studies on the reconstitution of glucosylceramidase activity by Sap C and anionic phospholipids. FEBS Lett. 472: 17–21. [DOI] [PubMed] [Google Scholar]
- Sanchez-Barrena M.J., Martinez-Ripoll M., Galvez A., Valdivia E., Maqueda M., Cruz V., Albert A. 2003. Structure of bacteriocin AS-48: From soluble state to membrane bound state. J. Mol. Biol. 334: 541–549. [DOI] [PubMed] [Google Scholar]
- Schneider T.L. 2000. Objective comparison of protein structures: Error-scaled difference distance matrices. Acta Crystallogr. D Biol. Crystallogr. 56: 714–721. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55: 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccaro A.M., Tatti M., Ciaffoni F., Salvioli R., Maras B., Barca A. 1993. Function of saposin C in the reconstitution of glucosylceramidase by phosphatidylserine liposomes. FEBS Lett. 336: 159–162. [DOI] [PubMed] [Google Scholar]
- Vaccaro A.M., Ciaffoni F., Tatti M., Salvioli R., Barca A., Tognozzi D., Scerch C. 1995. pH-dependent conformational properties of saposins and their interactions with phospholipid membranes. J. Biol. Chem. 270: 30576–30580. [DOI] [PubMed] [Google Scholar]
- Vaccaro A.M., Tatti M., Ciaffoni F., Salvioli R., Barca A., Scerch C. 1997. Effect of saposins A and C on the enzymatic hydrolysis of liposomal glucosylceramide. J. Biol. Chem. 272: 16862–16867. [DOI] [PubMed] [Google Scholar]
- Whitelegge J.P., Ahn V., Norris A.J., Sung H., Waring A., Stevens R.L., Fluharty C.B., Privé G., Faull K.F., Fluharty A.L. 2003. Characterization of a recombinant molecule covalently indistinguishable from human cerebroside-sulfate activator protein (CSAct or Saposin B). Cell. Mol. Biol. (Noisy-le-grand) 49: 799–807. [PubMed] [Google Scholar]
- Wilkening G., Linke T., Sandhoff K. 1998. Lysosomal degradation on vesicular membrane surfaces. Enhanced glucosylceramide degradation by lysosomal anionic lipids and activators. J. Biol. Chem. 273: 30271–30278. [DOI] [PubMed] [Google Scholar]
- Winau F., Schwierzeck V., Hurwitz R., Remmel N., Sieling P.A., Modlin R.L., Porcelli S.A., Brinkmann V., Sugita M., Sandhoff K. et al. 2004. Saposin C is required for lipid presentation by human CD1b. Nat. Immunol. 5: 169–174. [DOI] [PubMed] [Google Scholar]
- Winn M.D., Isupov M.N., Murshudov G.N. 2001. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57: 122–133. [DOI] [PubMed] [Google Scholar]
- Wright C.S., Li S.C., Rastinejad F. 2000. Crystal structure of human GM2-activator protein with a novel β-cup topology. J. Mol. Biol. 304: 411–422. [DOI] [PubMed] [Google Scholar]
- Wright C.S., Zhao Q., Rastinejad F. 2003. Structural analysis of lipid complexes of GM2-activator protein. J. Mol. Biol. 331: 951–964. [DOI] [PubMed] [Google Scholar]
- Zhou D., Cantu C. III, Sagiv Y., Schrantz N., Kulkarni A.B., Qi X., Mahuran D.J., Morales C.R., Grabowski G.A., Benlagha K. et al. 2004. Editing of CD1d-bound lipid antigens by endosomal lipid transfer proteins. Science 303: 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]